

Abstract
Nonalcoholic steatohepatitis (NASH) is a progressive liver disease projected to become the leading cause of cirrhosis and liver transplantation in the next decade. Cenicriviroc (CVC), a dual chemokine receptor 2 and 5 antagonist, prevents macrophage trafficking and is under clinical investigation for the treatment of human NASH fibrosis. We assessed the efficacy and durability of short and prolonged CVC therapy in a diet‐induced mouse model of NASH, the choline deficient, L‐amino acid‐defined, high‐fat diet (CDAHFD) model. C57BL/6 mice received 4 or 14 weeks of standard chow or the CDAHFD. CVC (10 mg/kg/day and 30 mg/kg/day for 4 weeks and 20 mg/kg/day and 30 mg/kg/day for 14 weeks) was initiated simultaneously with the CDAHFD. At 4 and 14 weeks, livers were harvested for histology and flow cytometric analyses of intrahepatic immune cells. High‐dose CVC (30 mg/kg/day) therapy in CDAHFD mice for 4 or 14 weeks inhibited intrahepatic accumulation of Ly6Chigh bone marrow‐derived macrophages. Prolonged CVC therapy (14 weeks) yielded no significant differences in the total intrahepatic macrophage populations among treatment groups but increased the frequency of intrahepatic anti‐inflammatory macrophages in the high‐dose CVC group. Despite ongoing steatohepatitis, there was significantly less fibrosis in CDAHFD mice receiving high‐dose CVC for 14 weeks based on histologic and molecular markers, mirroring observations in human NASH CVC trials. CVC also directly inhibited the profibrotic gene signature of transforming growth factor‐β‐stimulated primary mouse hepatic stellate cells in vitro. Conclusion: CVC is a novel therapeutic agent that is associated with reduced fibrosis despite ongoing steatohepatitis. Its ability to alter intrahepatic macrophage populations and inhibit profibrogenic genes in hepatic stellate cells in NASH livers may contribute to its observed antifibrotic effect. (Hepatology Communications 2018;2:529‐545)
Abbreviations
- α‐SMA
alpha‐smooth muscle actin
- ACTA2
α‐2 actin
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ATP
adenosine triphosphate
- CCL
chemokine ligand
- CCR
chemokine receptor
- CD
clusters of differentiation
- CDAHFD
choline‐deficient, L‐amino acid‐defined, high‐fat diet model
- COL1A1
collagen 1 alpha 1
- CVC
cenicriviroc
- FACS
fluorescence‐activated cell sorting buffer
- FBS
fetal bovine serum
- FFPE
fixed in formalin and paraffin embedded
- HSC
hepatic stellate cell
- IP
intraperitoneal
- MCP
monocyte chemotactic protein
- MDC
macrophage‐derived chemokine
- MGH
Massachusetts General Hospital
- MIG
monokine induced by gamma interferon
- MIP
macrophage inflammatory protein
- NASH
non‐alcoholic steatohepatitis
- NK
natural killer
- NKT cell
natural killer T cell
- NPC
nonparenchymal cell
- pHSC
primary hepatic stellate cell
- TGF‐β
transforming growth factor‐β
Nonalcoholic steatohepatitis (NASH) is a severe form of nonalcoholic fatty liver disease characterized by steatosis, inflammation, and fibrosis that can progress to liver failure and hepatocellular carcinoma. NASH affects 4 million‐6 million Americans1, 2 and is projected to become the leading cause of cirrhosis and liver transplantation in the next decade.3, 4 The unmet need for NASH therapeutics is vast as currently no approved therapies for this condition exist.
Histopathologic analyses of human livers with progressive NASH have identified mixed inflammatory infiltrates5 primarily composed of macrophages and T cells at all stages of disease.5, 6 Monocytes and neutrophils are the earliest cells migrating to sites of injured, ballooning hepatocytes in early NASH where they phagocytose debris and recruit the adaptive immune system.7 Damage induced by the innate and adaptive immune response in addition to ongoing lipotoxic insults is believed to activate hepatic stellate cells (HSCs) and induce liver fibrosis, asserting a causal role for inflammation in hepatic fibrogenesis. This argument is supported by observations that short‐term chemical blockade or genetic knockdown of individual chemokine ligands (CCLs) or receptors (CCRs), e.g., CCR2, CCL2, and chemokine C‐X‐C motif ligand 16, prevents steatohepatitis and fibrosis in mice fed a steatotic diet by inhibiting inflammatory monocyte trafficking.8, 9, 10
Although chemokine antagonism is an attractive therapeutic target, it is also a highly redundant system, raising concerns that a singular CCR/CCL blockade may be easily surmounted, mitigating any antirecruitment effects. In this study, we evaluate the intrahepatic efficacy and durability of blocking two macrophage chemotactic receptor proteins simultaneously in both a short‐term and longer term trial in a diet‐induced mouse model of NASH.
Cenicriviroc (CVC) is a dual CCR2/CCR5 chemokine antagonist currently in phase 3 evaluation in adults with NASH and NASH fibrosis (https://clinicaltrials.gov/ct2/show/NCT03028740). CVC has previously been studied in other dietary murine models of NASH, the methionine‐ and choline‐deficient diet and the streptozotocin plus high‐fat diet.11, 12 Although it reproduces the histology of NASH, the methionine‐ and choline‐deficient diet model is not associated with weight gain or metabolic syndrome. The streptozotocin plus high‐fat diet model is limited by a lack of progressive fibrosis, and CVC therapy was only continued for a short‐term period (9 weeks) in this dietary model.12 Because future NASH therapies in humans, including CVC, may be required for the long term, due to the challenge of adherence to long‐term lifestyle measures, more protracted studies of CVC in mice are warranted to evaluate the durability of drug efficacy.
In the present study, we characterize the effect of daily intraperitoneal (IP) CVC to compare differences in steatosis, inflammation, and fibrosis after a short (4 weeks) and prolonged (14 weeks) treatment course in an improved dietary mouse model of NASH, known as the choline‐deficient, L‐amino acid‐defined, high‐fat diet (CDAHFD). This dietary model is superior to other NASH models because CDAHFD mice gain weight,13, 14 develop aspartate and alanine aminotransferase (AST and ALT) abnormalities similar to those observed in human NASH, and have progressive fibrosis resulting in cirrhosis.13 We also tested the effects of CVC on primary human and mouse HSCs in vitro.
Materials and Methods
ANIMALS
Five‐week‐old male C57BL/6N mice were purchased from Charles River Laboratories (Wilmington, MA) and housed in a specific pathogen‐free environment (maximum four per cage). After 1 week of acclimation, mice were initiated on either standard chow (Prolab Isopro 3000; Scotts Distributing #8670) or CDAHFD (L‐amino acid diet with 60 kcal% fat with 0.1% methionine without added choline; Research Diets A06071302) ad libitum. CDAHFD mice were simultaneously initiated on diet and treatment (diet alone, vehicle control, CVC 10 mg/kg/day intraperitoneally, or CVC 30 mg/kg/day intraperitoneally for 4 weeks; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full), dosages previously evaluated in animal models. Because CVC 10 mg/kg/day did not have a discernable effect on hepatic macrophages in the 4‐week trial, doses of 20 mg/kg/day and 30 mg/kg/day were administered in the 14‐week trial (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full). The 4‐week time point was chosen to explore the effects of CVC on the distribution and phenotypes of intrahepatic immune cells before fibrosis onset; 14 weeks was chosen once fibrosis was firmly established without the presence of cirrhosis, when hepatic immune cell infiltration is diminished. The number of mice per group for each trial is detailed in http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full. After the 4 or 14 weeks of therapy, mice were anesthetized using 100 mg/kg of ketamine and 10 mg/kg of xylazine intraperitoneally followed by terminal cardiac puncture. Animals received humane care per criteria outlined in the Guide for the Care and Use of Laboratory Animals by the National Academy of Sciences (National Institutes of Health publication 86‐23, revised 1985) and in accordance with the Massachusetts General Hospital (MGH) Institutional Animal Care and Use Committee guidelines (Protocol 2009N000207).
CVC PREPARATION AND DELIVERY
CVC was kindly provided by Tobira Therapeutics, a subsidiary of Allergan Plc. (South San Francisco, CA), as a lyophilized powder and was reconstituted for IP injection as per the manufacturer's recommendations. Briefly, lyophilized CVC mesylate was dissolved in vehicle (10% hydroxypropyl‐beta‐cyclodextrin and 5% solutol in sterile water) by sonication. Vehicle without CVC was used as the vehicle control. Mice were weighed weekly, with the appropriate weight‐based dose of CVC administered daily. For in vitro studies, CVC was dissolved in dimethyl sulfoxide to a stock concentration of 50 μM and stored at –20oC until its use in cell culture when it was diluted to the appropriate concentrations with culture media.
BLOOD AND TISSUE PROCESSING
Serum/Plasma
Following cardiac puncture, harvested blood was processed for collection of serum and plasma and stored at –80oC until future testing. Serum was used to quantify chemokine levels, AST, ALT, total bilirubin, blood urea nitrogen, creatinine, glucose, total cholesterol, and triglycerides at the MGH Animal Facility Core Laboratories. Plasma was used to determine CVC levels (performed by Tobira).
Liver
Livers of mice from the 4‐ and 14‐week trials were harvested at the time of sacrifice and weighed. A small section of the left liver lobe was fixed in formalin and paraffin embedded (FFPE) for histology. The remaining liver was manually dispersed in a sterile petri dish over a mesh gauze (Seward stomacher strainer; FermionX, Worthing, United Kingdom), digested with collagenase B (0.5 mg/mL with approximately 0.180 units/mg; Roche Diagnostics, Indianapolis, IN) and deoxyribonuclease 1 (20 units/mL, Roche Diagnostics) at 37oC for 20 minutes with gentle agitation, followed by collagenase neutralization with R10 media (Roswell Park Memorial Institute with 10% 4‐(2‐hydroxyethyl)‐1‐piperazine ethanesulfonic acid, 10% heat inactivated fetal bovine serum [FBS], 1% penicillin 5,000 units/mL, and streptomycin 5,000 μg/mL), and then centrifuged at 50g for 3 minutes to pellet hepatocytes. The supernatant containing the nonparenchymal cells (NPCs) was harvested and filtered through a 70‐μm cell strainer to remove cell debris. NPCs were isolated using histopaque density gradient centrifugation and cryopreserved in 80% FBS with 20% dimethyl sulfoxide until flow cytometric analysis.
FLOW CYTOMETRIC ANALYSIS
Cryopreserved NPCs were rapidly thawed, washed with fluorescence‐activated cell sorting buffer (FACS) (phosphate‐buffered saline with 2% heat‐inactivated FBS), and quantified with viability staining using Moxicyte and Moxiflow cytometry (Orflo, Ketchum, IN). A total of 1.5 million NPCs were placed into three tubes and stained with the viability dye live dead blue (Molecular Probes, Eugene, OR). NPCs were then washed, stained with three surface antibody panels detailed in http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full, fixed with 1% paraformaldehyde, and run on the LSRII flow cytometer (BD Biosciences, San Jose, CA). Data were acquired using BD FACSDIVA software (BD Biosciences). The gating strategies are described in http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full. FACS data were analyzed using FlowJo, version 10.1 (Ashland, OR).
HISTOLOGY
FFPE liver tissue was sectioned at a thickness of 4 μm. Slides were stained with hematoxylin and eosin (for steatosis and inflammation scoring) or with trichrome (for fibrosis staging) at the MGH Cytopathology Core. Slides were evaluated by a blinded expert MGH liver pathologist for steatosis, inflammation, and fibrosis, using criteria specifically developed for murine NASH15 and modeled on human NASH histologic scoring systems.
HYDROXYPROLINE QUANTIFICATION
Hydroxyproline was quantified from FFPE liver tissue as described.16
RNA EXTRACTION AND QUANTITATIVE POLYMERASE CHAIN REACTION
RNA was extracted from FFPE tissue using the Qiagen RNeasy FFPE mini kit and from cultured cells using the Qiagen RNeasy mini kit (Qiagen, Hilden, Germany). Complementary DNA was generated using the High Capacity Kit (Applied Biosystems, Foster City, CA). Quantitative polymerase chain reaction was performed in duplicate for the human profibrotic genes collagen 1 alpha 1 (COL1A1) and alpha‐smooth muscle actin (α‐SMA) and for the mouse profibrotic genes COL1A1 and α‐2 actin (ACTA2), using published primer sequences (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full).
IN VITRO HSC STUDIES
Human NPCs (obtained from a 4‐month‐old Caucasian male infant) were purchased from TRL (a subsidiary of Lonza) and cultured in Dulbecco's modified Eagle's medium supplemented with 1% penicillin/streptomycin and 10% heat‐inactivated FBS. Primary HSCs (pHSCs) were isolated from these NPCs following prolonged culture for 5 days. Residual contaminating macrophages were removed by positive selection with anti‐clusters of differentiation (CD)14 magnetic beads (Miltenyi Biotech, Bergisch Gladbach, Germany) thereby allowing enrichment of pHSCs in the negatively selected flow‐through solution. Greater than 95% purity of isolated pHSCs was confirmed using autofluorescent ultraviolet analysis and bodipy staining for vitamin A droplets. Mouse pHSCs were derived from livers of 5‐week‐old male C57BL/6N mice fed CDAHFD for 4 weeks. NPCs were isolated from the livers as described above and sequentially passaged in Dulbecco's modified Eagle's medium with 10% heat‐inactivated FBS and 0.4% L‐glutamine for up to 6 passages until contaminating macrophages and endothelial cells were eliminated by attrition. Human and mouse pHSCs at passage 6‐9 were seeded in a 12‐well tissue culture plate, allowed to adhere overnight, and treated for 48 hours as follows: (a) media alone; (b) CVC 0.5 μg/mL; (c) CVC 5 μg/mL; (d) CVC 10 μg/mL; (e) CVC 15 μg/mL; (f) CVC 20 μg/mL; (g) transforming growth factor‐β (TGF‐β) 10 ng/mL; (h) TGF‐β 10 ng/mL + CVC 0.5 μg/mL; (i) TGF‐β 10 ng/mL + CVC 5 μg/mL; (j) TGF‐β 10 ng/mL + CVC 10 μg/mL; (k) TGF‐β 10 ng/mL + CVC 15 μg/mL; (l) TGF‐β 10 ng/mL + CVC 20 μg/mL. Cells were harvested for quantitative polymerase chain reaction studies, and data were acquired from four independent in vitro experiments for each cell type. Cell viability was measured in triplicate using the CellTiter Glo luminescent cell viability assay (Promega, Madison, WI), which measures total adenosine triphosphate (ATP) levels in each treatment well using a chemiluminescent assay. Cell viability is represented as percentage of ATP in the treatment group divided by ATP in mock cells.
MOUSE SERUM CHEMOKINE ANALYSIS
Circulating mouse chemokines were quantified in serum/plasma using the MAP Mouse Cytokine/Chemokine Magnetic Bead Panel per the manufacturer's instructions (EMD Millipore, Billerica, MA). Chemokines tested included eotaxin, fractalkine, interferon‐gamma‐inducible protein 10, keratinocyte chemoattractant, monocyte chemotactic protein (MCP)‐1 (CCL2), MCP‐5, monokine induced by gamma interferon (MIG), macrophage‐derived chemokine (MDC), macrophage inflammatory protein (MIP)‐1α (CCL3), MIP1β (CCL4), MIP‐2, MIP‐3α, MIP‐3β, and thymus and activation regulated chemokine. Data were acquired on the Luminex200 platform (Luminex, Austin, TX) and analyzed using XPONENT, version 3.1 software (Luminex).
STATISTICAL ANALYSES
All statistical analyses were performed using GraphPad Prism, version 7.0 (GraphPad Software, La Jolla, CA). Comparisons between groups of nonparametric, unpaired, continuous variables were performed using the Mann–Whitney U test or a Fisher's exact test. Statistically significant (P < 0.05) and near significant (P < 0.1) findings are reported where pertinent.
Results
CDAHFD‐FED MICE GAIN WEIGHT BUT DO NOT DEVELOP CLASSIC HUMAN METABOLIC FEATURES OF NASH
CDAHFD mice gained weight during the 14‐week trial irrespective of treatment group (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full). Liver weight, as a percentage of total body weight, in all CDAHFD mice was significantly higher than standard chow mice (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full), highlighting that the CDAHFD results in significant hepatic steatosis (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full). Additionally, livers of CVC‐treated mice tended to weigh less than CDAHFD untreated controls. Serum glucose, triglycerides, and cholesterol were normal in all groups in the 4‐ and 14‐week trials, demonstrating that some of the key metabolic features of human NASH, namely hyperglycemia and hyperlipidemia, are not observed in this murine NASH model (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full).
SERUM ALT AND AST LEVELS ARE LOWER AFTER 4 WEEKS OF HIGH‐DOSE CVC
Serum ALT and AST values were normal in standard chow mice but were significantly elevated in CDAHFD untreated controls in 4‐ and 14‐week trials (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full). In contrast, there was a dose‐dependent trend toward lower ALT levels in CDAHFD mice treated with CVC 10 mg/kg/day and 30 mg/kg/day for 4 weeks compared with CDAHFD untreated and vehicle controls. Interestingly, at 14 weeks, there was no significant difference in ALT levels in CDAHFD mice regardless of treatment group; in fact, the median ALT was highest in the CVC 30 mg/kg/day group. No significant differences were observed in other biochemical parameters during either the 4‐ or 14‐week trials. At 4 weeks of CVC therapy, plasma CVC levels at 4 and 24 hours after IP injection were within the expected therapeutic range (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full).
HISTOLOGIC SCORES FOR STEATOSIS, INFLAMMATION, AND HEPATOCYTE BALLOONING ARE UNCHANGED AFTER 4 OR 14 WEEKS OF CVC
Representative hematoxylin and eosin‐ and trichrome‐stained livers from each treatment group are shown in http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full for the 4‐ and 14‐week trials. Histologic evaluation demonstrated no significant differences in hepatic steatosis (Fig. 1A) or inflammation (Fig. 1B) grades between any of the CDAHFD treatment groups at 4 or 14 weeks. All CDAHFD mice exhibited hepatocyte ballooning at 4 and 14 weeks, but there was no difference in the degree of ballooning between control and CVC‐treated groups as all groups received a mean score of 1 for ballooning.
Figure 1.
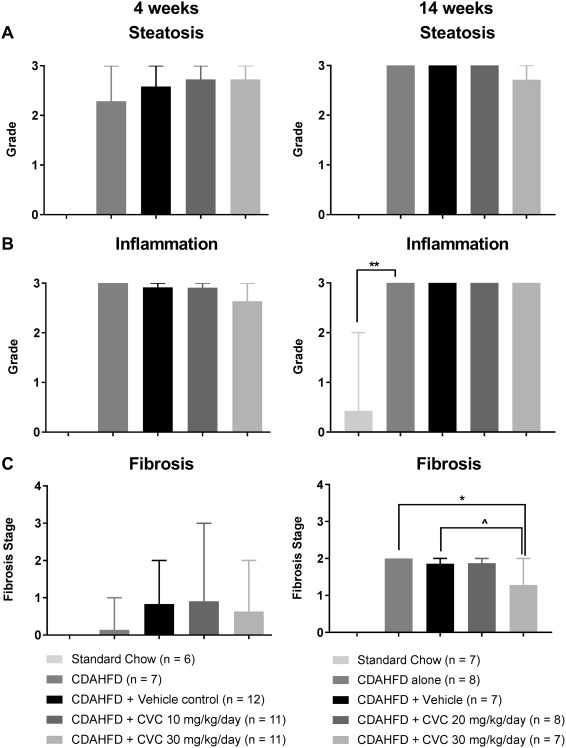
Liver histology. Data represent mean scores with range for (A) steatosis, (B) inflammation, and (C) fibrosis by treatment group at 4 and 14 weeks. **P = 0.0002, *P = 0.001, ∧ P = 0.01.
HIGH‐DOSE CVC DECREASES TOTAL HEPATIC LEUKOCYTES AND TOTAL HEPATIC MACROPHAGES AFTER 4 BUT NOT 14 WEEKS OF THERAPY
Current validated histologic inflammation grading scores are limited to a 3‐point scoring system, and therefore modest but significant changes in the intrahepatic inflammatory infiltrates may not be readily discernable by a narrow histologic scoring system. Therefore, we performed in‐depth characterization of intrahepatic leukocyte populations from liver single‐cell homogenates using flow cytometry. The major immune cell populations isolated from the NPCs of each of the five groups in our study for the 4‐ and 14‐week treatment trials are depicted in Fig. 2; this specifically demonstrates that, similar to mice fed standard chow, CDAHFD‐fed mice are not devoid of any major immune cell subpopulation. Flow cytometric analyses of the liver NPC fraction in each of the five groups at 4 weeks revealed that the intrahepatic immune cell population, as represented by the frequency of live CD45+ cells (a common leukocyte antigen), was significantly lower in the high‐dose CVC group (30 mg/kg/day) compared with CDAHFD‐untreated mice or vehicle‐treated controls (Fig. 2A, left column). This decrease paralleled the decrease in the frequency of all macrophages (CD45+ F4/80+ CD11b+ cells; see http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full for gating strategy) in the CVC 30‐mg/kg/day group (Fig. 2B, left column). The significant decrease in total CD45+ cells was observed despite a small but statistically significant increase in the frequencies of all CD3+ T cells and CD8+ T cells in the CVC 30‐mg/kg/day treated mice (Fig. 2C, left column). Other intrahepatic immune cell populations, including natural killer (NK) cells and NK T (NKT) cells, did not change significantly at 4 weeks (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full).
Figure 2.
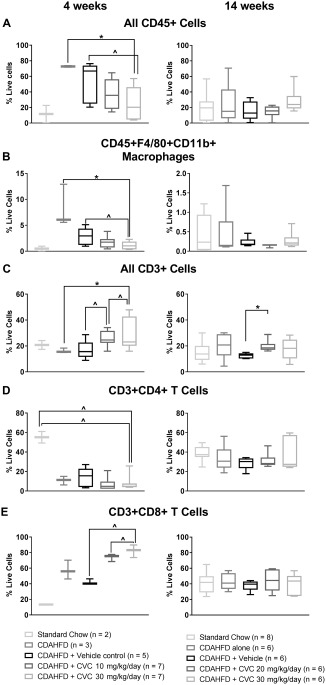
Leukocyte subtypes isolated from mouse livers in the 4‐ and 14‐week trials, analyzed by flow cytometry. Data represent median with IQR, min and max. (A) All CD45+ leukocytes, (B) CD45+F4‐80+CD11b+ macrophages, (C) all CD3+ cells, (D) CD3+CD4+ T cells, (E) CD3+CD8+ T cells. *P ≤ 0.05, ∧ P ≤ 0.10.
In contrast, following 14 weeks of CDAHFD plus CVC therapy, there were no significant differences in the frequencies of total leukocytes (CD45+ cells) or macrophage populations among any of the treatment groups (Fig. 2A,B, right column). In addition, there were no significant differences in the NK and NKT cell populations (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full), and the increase in CD3+ and the CD8+ T‐cell frequencies observed with high‐dose CVC at 4 weeks was no longer observed at 14 weeks (Fig. 2C,E, right column).
HIGH‐DOSE CVC LOWERS THE FREQUENCY OF INTRAHEPATIC BONE MARROW‐DERIVED (TRAFFICKING) MACROPHAGES AFTER 4 AND 14 WEEKS
CVC is known to specifically inhibit the CCL2‐CCR2‐mediated activation and homing of bone marrow‐derived monocytes.12, 17 Therefore, we hypothesized that CVC blockade should prevent trafficking of bone marrow‐derived (defined as Ly6Chigh) monocytes to the liver. We confirmed that after 4 weeks of high‐dose CVC, there was a significant decrease in the frequency of intrahepatic Ly6Chigh macrophages relative to the vehicle control (Fig. 3A, left column). Only data for vehicle controls and CVC‐treated mice are represented in Fig. 3 because the vehicle control is the most relevant group for comparison to assess the effect of CVC. This decrease in bone marrow‐derived macrophages was dose dependent, with mice receiving CVC 10 mg/kg/day having an intermediate frequency of Ly6Chigh macrophages relative to the vehicle and high‐dose CVC mice. This decrease in Ly6Chigh macrophage frequency was accompanied by an increase in the Ly6Clow macrophages in the CVC 30‐mg/kg/day group relative to vehicle controls, again in a dose‐dependent manner (Fig. 3B, left column). Ly6Clow macrophages represent either native tissue resident macrophages or infiltrating monocytes that have become tissue resident macrophages by down‐regulating Ly6C antigens.
Figure 3.
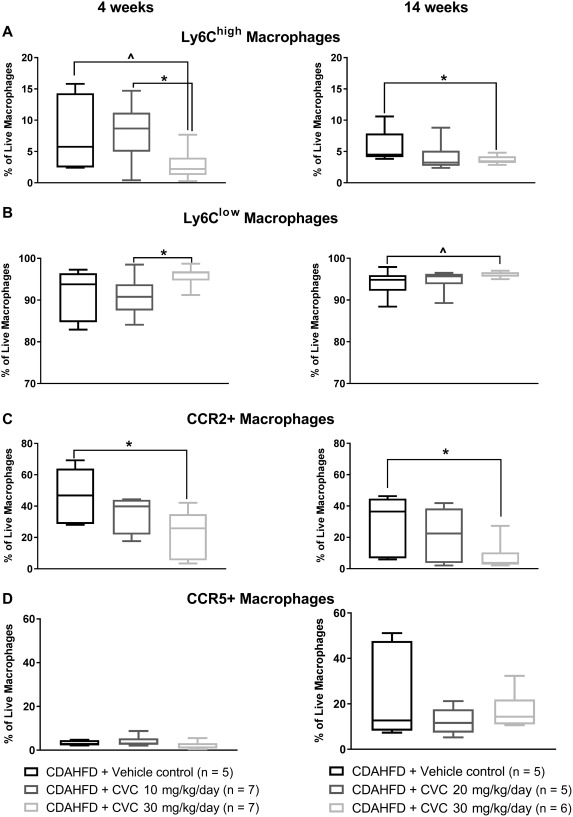
Macrophage subpopulations isolated from mouse livers in the 4‐ and 14‐week trials, analyzed by flow cytometry. Data represent median with IQR, min and max. (A) Ly6Chigh macrophages, (B) Ly6Clow macrophages, (C) CCR2+ macrophages, (D) CCR5+ macrophages. *P ≤ 0.05, ∧ P ≤ 0.10.
Following prolonged high‐dose CVC for 14 weeks, the frequency of hepatic Ly6Chigh macrophages remained significantly lower relative to vehicle controls; however, the effect size was attenuated compared to that observed after 4 weeks of high‐dose CVC (Fig. 3A, right column). These data suggest that prolonged high‐dose CVC is still able to inhibit trafficking bone marrow‐derived macrophages to the liver in the face of continued CDAHFD hepatic insult, although the effect size was attenuated.
HIGH‐DOSE CVC DECREASES THE FREQUENCY OF INTRAHEPATIC CCR2+ MACROPHAGES AT 4 AND 14 WEEKS
In line with the known effects of CVC on CCR2 and CCR5, we observed a significant and dose‐dependent decrease in the frequency of intrahepatic CCR2+ macrophages (Fig. 3C, left column) but not CCR5+ macrophages with 4 weeks of CVC (Fig. 3D, left column). CVC has greater affinity for CCR2 than CCR5 in mice,18 and the selective reduction in CCR2+ hepatic macrophages suggests that antagonism of CCR5 may be limited. After 14 weeks of CDAHFD plus CVC, there remained a significant difference in the frequency of CCR2+ hepatic macrophages compared with vehicle controls (Fig. 3C, right column) without any significant decrease in CCR5+ intrahepatic macrophages after high‐dose CVC compared with vehicle controls even after 14 weeks of therapy (Fig. 3D, right column).
CVC REDUCES INTRAHEPATIC M1 PROINFLAMMATORY MACROPHAGES AT 4 WEEKS AND INCREASES INTRAHEPATIC M2 ANTI‐INFLAMMATORY MACROPHAGES AT 14 WEEKS
Given the observed reduction in total intrahepatic and bone marrow‐derived macrophages observed after 4 weeks of high‐dose CVC, we sought to determine the phenotype of the remaining intrahepatic macrophages following CVC therapy. To achieve this, we stained intrahepatic macrophages for the M1 proinflammatory markers CD80+ and CD86+ and the M2 anti‐inflammatory markers CD169+ and CD206+ (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full for gating strategy). At 4 weeks, there was a significantly lower frequency of macrophages expressing M1 markers (Fig. 4A,B,E, left column) in mice receiving high‐dose CVC compared with vehicle controls but there was no significant difference in M2 marker expression (Fig. 4C‐E, left column). In contrast, no significant differences in M1 marker (CD80+, CD86+) expression existed between groups after 14 weeks of therapy (Fig. 4A,B, right column). There was, however, a modest but significant increase in the frequency of intrahepatic macrophages expressing the M2 marker CD206+ (Fig. 4D, right column) in the high‐dose CVC mice relative to vehicle controls at 14 weeks, which, in addition to having anti‐inflammatory effects, are also known to possess scavenger functions and can resorb extracellular matrix from hepatic scar tissue.19, 20 Mean cell counts for major cell subsets depicted in Figs. 2, 3, 4 by treatment group are detailed in http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full.
Figure 4.
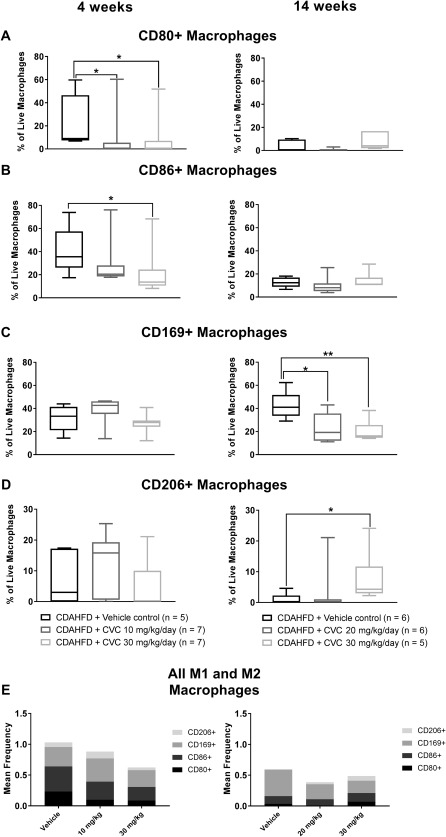
M1 and M2 macrophage subpopulations isolated from mouse livers in the 4‐ and 14‐week trials, analyzed by flow cytometry. Data represent median with IQR, min and max. (A) CD80+ macrophages, (B) CD86+ macrophages, (C) CD169+ macrophages, (D) CD206+ macrophages, (E) all M1 and M2 macrophages. *P ≤ 0.05, **P = 0.009.
CVC THERAPY RESULTS IN A SIGNIFICANT INCREASE IN SEVERAL MACROPHAGE‐RELATED CHEMOKINES AT 14 BUT NOT 4 WEEKS
After 14 weeks of high‐dose CVC, we obtained incongruous findings of reduced intrahepatic CCR2+ trafficking macrophages, without changes in the total intrahepatic macrophage population. We hypothesized that the robust hepatic inflammation observed with high‐dose CVC may result from up‐regulation of alternative chemokine/chemokine ligand pathways that circumvent the CCR2/CCR5 blockade to recruit inflammatory cells to the liver. If this hypothesis were true, these circulating chemokine levels would be similar among all treatment groups at 4 weeks but significantly increased in CVC‐treated mice at 14 weeks compared with vehicle controls. Therefore, we quantified a panel of chemokines in sera at 4 and 14 weeks. We found that eotaxin, MDC, MIP‐1β, and MIG levels were unchanged between treatment groups at 4 weeks but were significantly elevated in CVC‐treated mice at 14 weeks in a dose‐dependent manner (Fig. 5). Interestingly, we also observed that MCP‐1 (CCL2, the ligand for CCR2) and MCP‐5 were significantly elevated in the sera of CVC‐treated mice compared with the vehicle at both 4 and 14 weeks, suggesting that chronic inhibition of CCR2/CCR5 could promote sustained up‐regulation of cognate ligands (Fig. 6). Overall, these data suggest that several chemokines are up‐regulated in response to CVC to compensate for CCR2/CCR5 blockade and may contribute to hepatocyte leukocyte infiltration by alternative pathways.
Figure 5.
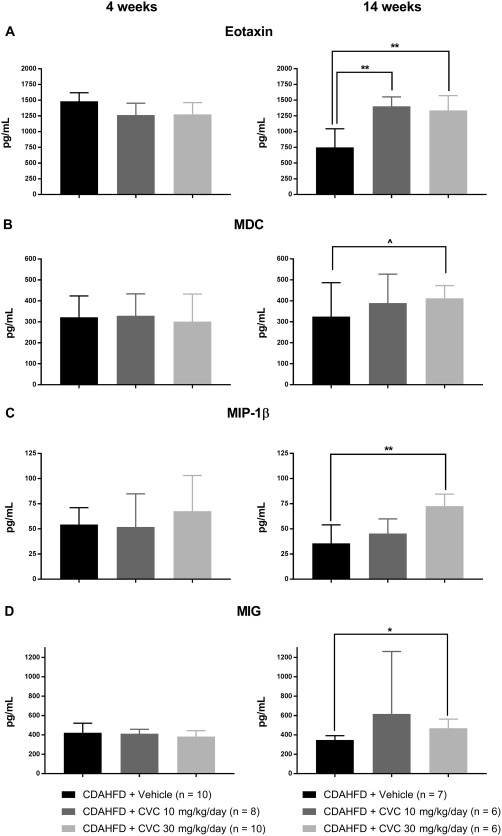
Serum chemokines unchanged at 4 weeks but increased after 14 weeks of CVC therapy. Data represent mean ± SD. (A) Eotaxin; (B) macrophage‐derived chemokine; (C) macrophage inflammatory protein‐1β; (D) monocyte induced by gamma interferon. ∧ P = 0.07, *P = 0.02, **P < 0.005.
Figure 6.
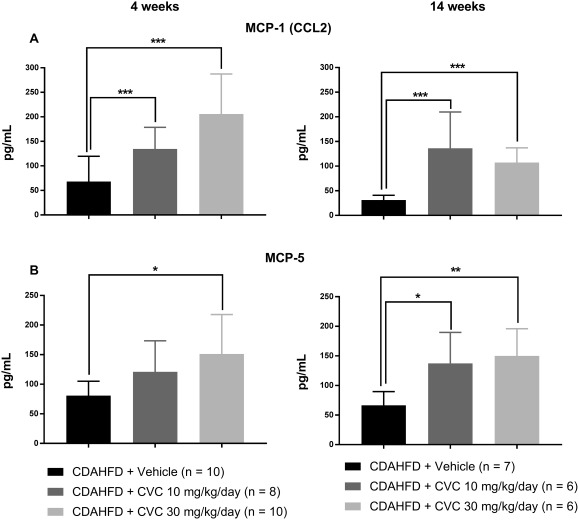
Serum chemokines increased at both 4 and 14 weeks of CVC therapy. Data represent mean ± SD. (A) Monocyte chemotactic protein 1/chemokine ligand 2. (B) Monocyte chemotactic protein 5. *P = 0.02, **P = 0.01, ***P ≤ 0.006.
HIGH‐DOSE CVC FOR 14 WEEKS SIGNIFICANTLY REDUCES HISTOLOGIC AND MOLECULAR MARKERS OF HEPATIC FIBROSIS
We did not observe any significant differences in histologic fibrosis scores at 4 weeks by treatment group (Fig. 1C, left column); however, surprisingly, there was a significant decrease in the histologic fibrosis scores of mice treated with high‐dose CVC compared with CDAHFD alone and a strong trend toward reduced fibrosis compared with vehicle controls (P = 0.10) after 14 weeks of therapy (Fig. 1C, right column). To confirm this decrease in fibrosis, we quantified hepatic hydroxyproline concentrations and COL1A1 gene expression at 14 weeks. Hepatic hydroxyproline levels were significantly lower in CVC‐treated mice compared with controls in a dose‐dependent manner (Fig. 7A). COL1A1 messenger RNA expression paralleled these findings, with the lowest expression observed in high‐dose CVC mice relative to standard chow mice (Fig. 7B). These data demonstrate a clear discordance between the extent of hepatic inflammation and fibrosis following 14 weeks of high‐dose CVC, with lower fibrosis despite a robust hepatic inflammatory infiltrate. Similar findings were recently observed in phase 2b human clinical trials of CVC.21
Figure 7.
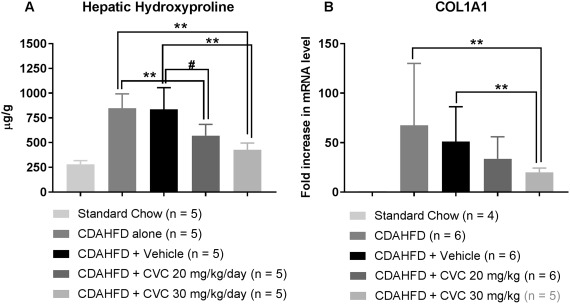
Hepatic hydroxyproline protein concentrations and hepatic COL1A1 mRNA levels are maximally decreased with high‐dose CVC. Data represent mean and SD for (A) and (B): (A) Total hepatic hydroxyproline levels quantified from formalin‐fixed paraffin‐embedded liver tissue at 14 weeks. (B) Fold increase in total hepatic COL1A1 mRNA expression in CDAHFD mice relative to standard chow at 14 weeks, normalized to GAPDH. *P = 0.001, **P < 0.005, ∧ P = 0.01, # P = 0.06. Abbreviations: GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; mRNA, messenger RNA.
CVC INHIBITS PROFIBROGENIC SIGNALS IN PRIMARY HSCs IN VITRO
Given the improvement in fibrosis that we observed after 14 weeks of CVC without significant improvements in hepatic inflammation, we questioned whether CVC could directly alter HSC function. We hypothesized that HSCs, which are known to express CCR5 and can be activated in the presence of steatosis and inflammation, may experience inhibition of these functions in the presence of CVC due to CCR5 blockade. We tested this hypothesis by determining if CVC could inhibit up‐regulation of a profibrogenic gene program induced by TGF‐β in mouse and human pHSCs. We found that plate‐activated human and mouse pHSCs treated for 48 hours with increasing doses of CVC ranging from 0.5 μg/mL to 20 μg/mL (concentrations spanning the trough and peak levels observed in the sera of CVC‐treated mice; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full) decreased their gene expression of COL1A1 and α‐SMA in human pHSCs and COL1A1 and ACTA2 in mouse pHSCs (Fig. 8A,B, respectively, for human; Fig. 8D,E, respectively, for mouse). TGF‐β stimulation of plate‐activated pHSCs increased the expression of α‐SMA and COL1A1 from human pHSCs and ACTA2 and COL1A1 from mouse pHSCs, but this increase could be inhibited with concomitant CVC treatment in a dose‐dependent manner (Fig. 8G,H [human]; Fig. 8J,K [mouse]). The antifibrogenic effect of CVC was more significantly apparent on mouse pHSCs (Fig. 8D,E) than human pHSCs (Fig. 8A,B), although similar trends were apparent in both cell types. Cell viability was not significantly affected by the doses of CVC used in these experiments (Fig. 8C,F).
Figure 8.
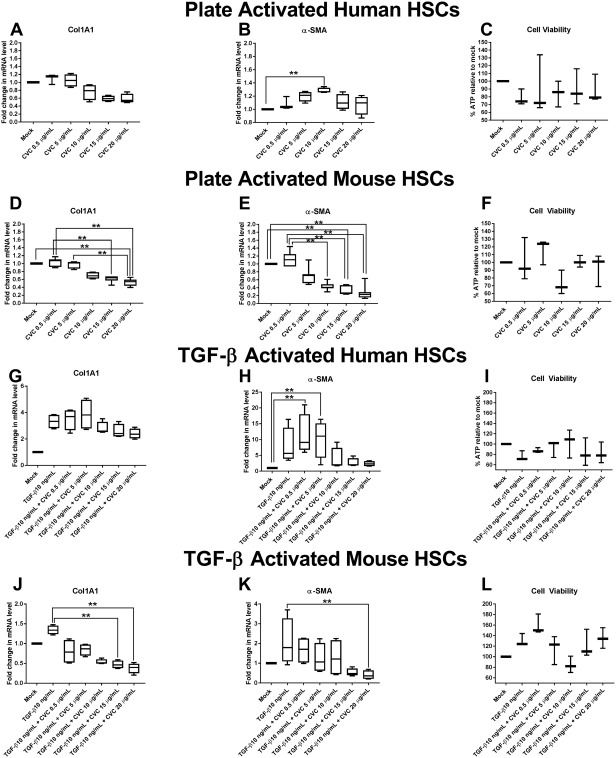
Quantitative polymerase chain reaction of profibrogenic gene transcripts from in vitro plate‐activated (A,B) human pHSCs or (D,E) mouse pHSCs or in vitro TGF‐β‐stimulated (G,H) human pHSCs or (J,K) mouse pHSCs in the presence or absence of CVC at increasing doses ranging from 0.5 μg/mL to 20 μg/mL. mRNA expression is reported as fold increase compared with mock‐treated cells and normalized to GAPDH for each treatment condition. Data represent four independent experiments in human pHSCs and six independent experiments in mouse pHSCs. Data represent median with IQR, min and max. Cell viability for each experiment type was performed in triplicate and (C,F,I,L) is represented as percent ATP in cells in each treatment condition relative to mock‐treated cells. Abbreviations: GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; mRNA, messenger RNA **P ≤ 0.05.
Discussion
Chemokine antagonism, specifically CCR2/CCR5 dual antagonism, is a novel therapeutic strategy to prevent inflammation and fibrosis in NASH. A recent phase 2b study of CVC offers promise for its future treatment of NASH.21 The most important finding in our study of CVC in the murine CDAHFD model of NASH is that 14 weeks of concomitant diet and high‐dose CVC (30 mg/kg/day) leads to a significant improvement in hepatic fibrosis (based on histologic score, hepatic hydroxyproline levels, and hepatic COL1A1 messenger RNA expression), despite the lack of significant improvement in hepatic inflammation, mirroring human NASH CVC clinical trial results. This effect may be accomplished by a combination of mechanisms, including preventing accumulation of proinflammatory macrophages while enriching anti‐inflammatory macrophages and directly inhibiting profibrogenic gene responses from HSCs. Our study is also the longest trial of CVC in a mouse model to date.
Our study findings and human trials raise several important questions: 1) Are the anti‐inflammatory properties of CVC durable with prolonged therapy? 2) What is the mechanism underlying fibrosis reduction despite ongoing steatohepatitis? These questions must be addressed, particularly if prolonged CVC therapy is expected in NASH patients. The CDAHFD model allows us to further explore the biological effect of CVC to gain insight into the importance of the hepatic immune cell composition in preventing fibrogenesis. If such a mechanism can be identified, it could be harnessed for novel therapeutic strategies in NASH and other chronic inflammatory liver diseases associated with progressive fibrosis. This mouse model is ideally suited for such exploratory studies, first, because it recapitulates features of human NASH (robust steatosis within days of CDAHFD exposure, significant inflammation within weeks, and significant fibrosis by 6 weeks), and second, it allows adequate hepatic tissue sampling, which is not feasible in human studies.
Several interesting findings about hepatic inflammation emerged from our study. First, we found that mice receiving high‐dose CVC had decreased frequencies of intrahepatic bone marrow‐derived Ly6Chigh macrophages at 4 weeks, a reduction that was still observed, albeit attenuated, after 14 weeks of high‐dose CVC therapy despite no change in total intrahepatic macrophage or leukocyte populations. This finding suggests that alternative macrophage populations may be enriched in the liver following 14 weeks of CVC, possibly differentiating from infiltrating non‐bone marrow‐derived monocytes (e.g., peritoneal, given the route of administration of CVC during this study, lymph node or splenic macrophages), proliferating resident hepatic macrophages, or other trafficking monocytes that become liver resident macrophages by down‐regulating Ly6C surface expression. We are currently characterizing this interesting macrophage subtype and exploring whether the enriched hepatic Ly6Clow population observed with high‐dose CVC at 14 weeks contributes to its antifibrotic effect.
Second, we observed a reduced frequency of intrahepatic CCR2+ macrophages at both 4 and 14 weeks of high‐dose CVC, suggesting ongoing effective receptor blockade of CCR2 in high‐dose CVC‐treated mice. In contrast, intrahepatic CCR5+ macrophage frequency was not different between groups at either 4 or 14 weeks, reflecting the less potent receptor antagonistic effect of CVC on CCR5 or host compensatory mechanisms over time. We also observed differences in levels of several other chemokines at 14 weeks not observed at 4 weeks, namely eotaxin, MDC, MIP‐1β, and MIG. Together, these data suggest that CCR2 antagonism is sustained with prolonged high‐dose CVC for 14 weeks but that CCR5 blockade can be overcome due to decreased potency of the drug against this receptor, potentially accounting for the attenuated reduction in the intrahepatic Ly6Chigh macrophages at 14 weeks.
Third, the phenotype of intrahepatic macrophages was altered following treatment with high‐dose CVC. The frequency of macrophages expressing proinflammatory M1 markers was significantly lower after 4 weeks of high‐dose CVC, and by 14 weeks we observed an increase in intrahepatic macrophages displaying the anti‐inflammatory M2 marker CD206, which is known to have anti‐inflammatory properties (e.g., secreting amphiregulin to promote immunosuppressive activity by intrahepatic regulatory T cells and inhibiting cytolytic CD8+ T cells22) and antifibrotic properties (resorption of extracellular matrix from hepatic scar tissue19, 20). Krenkel and colleagues23 recently reported the enrichment of CD206+ macrophages in livers of mice fed an alternate NASH‐inducing diet, the Western diet, and treated with CVC for 5 weeks following a 9‐week dietary lead‐in period, adding credibility to our findings. A decrease in the frequency of CD169+ cells in the high‐dose CVC group at 14 weeks may have contributed to decreasing intrahepatic CD8+ T‐cell activity (e.g., invariant NKT cell activity).24 Together, these data suggest that the composition of the inflammatory infiltrate is more important than total hepatic inflammation in determining the fate of liver injury and fibrogenesis. Finally, our in vitro studies are the first to demonstrate that CVC exerts a direct effect on mouse and human pHSCs by decreasing their TGF‐β‐induced profibrogenic program, an effect that was more pronounced in mouse pHSCs. Ongoing studies in our laboratory are addressing whether the decrease in the profibrogenic gene expression from mouse pHSCs results in lower protein levels of collagen and actin produced by these cells and whether these effects are mediated through inhibition of the TGF‐ β receptor or downstream proteins in the canonical or noncanonical TGF‐ β pathways.
There are limitations to the CDAHFD model. First, CDAHFD mice do not exhibit some of the key components of metabolic syndrome often observed in human NASH, including hypercholesterolemia, hypertriglyceridemia, and hyperglycemia. Nevertheless, they develop steatosis, inflammation, and fibrosis at a relatively rapid rate, suggesting that the metabolic syndrome is not required to develop NASH in this model. Second, CVC was only administered intraperitoneally in our study while it is delivered orally in humans. IP administration may recruit cells with anti‐inflammatory or antifibrotic properties to the liver that were not targeted in our flow panels (e.g., peritoneal macrophages or neutrophils). Third, our trial design whereby CVC was administered concomitantly with CDAHFD without a dietary lead‐in was deliberately chosen to maximize any observed effects of CVC on prevention or attenuation of NASH. We also continued the diet for the duration of CVC therapy because adherence to dietary measures in human NASH is poor. Because findings of our current study could represent delayed fibrosis progression, future mouse studies should also employ a dietary lead‐in to allow fibrosis development prior to CVC administration to determine if CVC promotes fibrosis regression. Finally, although this mouse study represents the longest duration of CVC administration in animal models of NASH, the ideal duration of therapy remains unknown. Studies with more protracted administration of CVC are needed to determine the durability of the changes in inflammation and fibrosis that we observed.
In conclusion, we evaluated CVC in the CDAHFD mouse model where CVC demonstrated a potent antifibrotic effect after 14 weeks, consistent with human phase 2b NASH studies. This model is an ideal system for evaluating the long‐term efficacy and durability of CVC as well as for dissecting the observed discordance between inflammation and fibrosis. Of greatest concern regarding any long‐term therapy for NASH is whether ongoing dietary habits that continue to promote hepatocyte injury and apoptosis could thwart even the best therapy from achieving its goal. Furthermore, long‐term blockade of innate immune responses that can prevent fibrogenesis but are required to fight infection may prove challenging. Our study highlights that not all inflammation is equal and that cellular composition and phenotypes of inflammatory infiltrates may be more critical in determining liver disease progression than total hepatic inflammation. Therefore, drugs that influence the immune composition of hepatic inflammation may be successful in the future treatment of this disease.
Authors names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full.
Supporting Information 1
Acknowledgment
Cenicriviroc was kindly donated by Tobira Therapeutics (subsidiary of Allergan Plc.). We thank Maris Handley and Amy Galvin (MGH Flow Cytometry Core) for their generous assistance with flow antibody panel design, LSRII cytometer and DIVA software training, and troubleshooting.
Potential conflict of interest: Dr. Chung received grants from Gilead, Bristol‐Myers Squibb, AbbVie, Merck, Janssen, Boehringer Ingelheim, Kaleido, and Synlogic. Dr. Caravan consults and owns stock in Collagen Medical, consults for Bayer and Guerbet, received grants from Pfizer and Pliant, and owns stock in Reveal and Factor 1A. Dr. Vig and Dr. Lefebvre are employed by Allergan.
Supported by National Institutes of Health (NIH) awards F32DK10958‐01 (to A.J.K.), DK078772, DK108370, DK098079 (to R.T.C.), and DK104956 (to B.C.F.) and an American Association for the Study of Liver Diseases Foundation Pinnacle Research Award (to R.M.).
REFERENCES
- 1. Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA 2015;313:2263‐2273. [DOI] [PubMed] [Google Scholar]
- 2. Kabbany MN, Conjeevaram Selvakumar PK, Watt K, Lopez R, Akras Z, Zein N, et al. Prevalence of nonalcoholic steatohepatitis‐associated cirrhosis in the United States: an analysis of National Health and Nutrition Examination Survey data. Am J Gastroenterol 2017;112:581‐587. [DOI] [PubMed] [Google Scholar]
- 3. Pais R, Barritt AS, Calmus Y, Scatton O, Runge T, Lebray P, et al. NAFLD and liver transplantation: current burden and expected challenges. J Hepatol 2016;65:1245‐1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zezos P, Renner EL. Liver transplantation and non‐alcoholic fatty liver disease. World J Gastroenterol 2014;20:15532‐15538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kleiner DE, Makhlouf HR. Histology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in adults and children. Clin Liver Dis 2016;20:293‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Magee N, Zou A, Zhang Y. Pathogenesis of nonalcoholic steatohepatitis: interactions between liver parenchymal and nonparenchymal cells. Biomed Res Int 2016;2016:5170402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in NAFLD/NASH. Dig Dis Sci 2016;61:1294‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol 2012;302:G1310‐G1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wehr A, Baeck C, Ulmer F, Gassler N, Hittatiya K, Luedde T, et al. Pharmacological inhibition of the chemokine CXCL16 diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. PLoS One 2014;9:e112327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP‐1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012;61:416‐426. [DOI] [PubMed] [Google Scholar]
- 11. Puengel T, Krenkel O, Mossanen JC, Longerich T, Lefebvre E, Trautwein C, et al. The dual CCR2/CCR5 antagonist cenicriviroc ameliorates steatohepatitis and fibrosis in vivo by inhibiting the infiltration of inflammatory monocytes into injured liver. In: EASL International Liver Congress; 2016; Barcelona, Spain.
- 12. Lefebvre E, Moyle G, Reshef R, Richman LP, Thompson M, Hong F, et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS One 2016;11:e0158156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matsumoto M, Hada N, Sakamaki Y, Uno A, Shiga T, Tanaka C, et al. An improved mouse model that rapidly develops fibrosis in non‐alcoholic steatohepatitis. Int J Exp Pathol 2013;94:93‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Machado MV, Michelotti GA, Xie G, Almeida Pereira T, Boursier J, Bohnic B, et al. Mouse models of diet‐induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One 2015;10:e0127991. Erratum in: PLoS One 2015;10:e0132315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander‐Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 1999;94:2467‐2474. [DOI] [PubMed] [Google Scholar]
- 16. Farrar CT, DePeralta DK, Day H, Rietz TA, Wei L, Lauwers GY, et al. 3D molecular MR imaging of liver fibrosis and response to rapamycin therapy in a bile duct ligation rat model. J Hepatol 2015;63:689‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jung H, Mithal DS, Park JE, Miller RJ. Localized CCR2 activation in the bone marrow niche mobilizes monocytes by desensitizing CXCR4. PLoS One 2015;10:e0128387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Byron MM, D'Antoni M, Premeaux T, Lefebvre E, Ndhlovu LC. Dual CCR2/CCR5 antagonism by cenicriviroc efficiently inhibits both MCP‐1 and RANTES induced chemokine receptor internalization in murine pro‐inflammatory monocytes. In: Immunology 2015‐The American Association of Immunologists (AAI) Annual Meeting; May 11, 2015; New Orleans, LA.
- 19. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol 2014;14:181‐194. [DOI] [PubMed] [Google Scholar]
- 20. Adhyatmika A, Putri KS, Beljaars L, Melgert BN. The elusive antifibrotic macrophage. Front Med (Lausanne) 2015;2:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanyal A, Ratziu V, Harrison S, Abdelmalek MF, Aithal GP, Caballeria J, et al. Cenicriviroc versus placebo for the treatment of nonalcoholic steatohepatitis with liver fibrosis: results from the year 1 primary analysis of the phase 2b CENTAUR study. In: AASLD The Liver Meeting; November 2016; Boston, MA.
- 22. Dai K, Huang L, Sun X, Yang L, Gong Z. Hepatic CD206‐positive macrophages express amphiregulin to promote the immunosuppressive activity of regulatory T cells in HBV infection. J Leukoc Biol 2015;98:1071‐1080. [DOI] [PubMed] [Google Scholar]
- 23. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2017; doi: 10.1002/hep.29544. [DOI] [PubMed] [Google Scholar]
- 24. Barral P, Polzella P, Bruckbauer A, van Rooijen N, Besra GS, Cerundolo V, et al. CD169(+) macrophages present lipid antigens to mediate early activation of iNKT cells in lymph nodes. Nat Immunol 2010;11:303‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1160/full.
Supporting Information 1