

Abstract
Progressive familial intrahepatic cholestasis (PFIC) with normal circulating gamma‐glutamyl transpeptidase levels can result from mutations in the ATP8B1 gene (encoding familial intrahepatic cholestasis 1 [FIC1] deficiency) or the ABCB11 gene (bile salt export protein [BSEP] deficiency). We investigated the outcomes of partial external biliary diversion, ileal exclusion, and liver transplantation in these two conditions. We conducted a retrospective multicenter study of 42 patients with FIC1 deficiency (FIC1 patients) and 60 patients with BSEP deficiency (BSEP patients) who had undergone one or more surgical procedures (57 diversions, 6 exclusions, and 57 transplants). For surgeries performed prior to transplantation, BSEP patients were divided into two groups, BSEP‐common (bearing common missense mutations D482G or E297G, with likely residual function) and BSEP‐other. We evaluated clinical and biochemical outcomes in these patients. Overall, diversion improved biochemical parameters, pruritus, and growth, with substantial variation in individual response. BSEP‐common or FIC1 patients survived longer after diversion without developing cirrhosis, being listed for or undergoing liver transplantation, or dying, compared to BSEP‐other patients. Transplantation resolved cholestasis in all groups. However, FIC1 patients commonly developed hepatic steatosis, diarrhea, and/or pancreatic disease after transplant accompanied by biochemical abnormalities and often had continued poor growth. In BSEP patients with impaired growth, this generally improved after transplantation. Conclusion: Diversion can improve clinical and biochemical status in FIC1 and BSEP deficiencies, but outcomes differ depending on genetic etiology. For many patients, particularly BSEP‐other, diversion is not a permanent solution and transplantation is required. Although transplantation resolves cholestasis in patients with FIC1 and BSEP deficiencies, the overall outcome remains unsatisfactory in many FIC1 patients; this is mainly due to extrahepatic manifestations. (Hepatology Communications 2018;2:515‐528)
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BSEP
bile salt export protein
- CI
confidence interval
- FIC1
familial intrahepatic cholestasis 1
- FLLN
fold of the lower limit of the normal range
- FULN
fold of the upper limit of the normal range
- GGT
gamma‐glutamyl transpeptidase
- HCC
hepatocellular carcinoma
- HR
hazard ratio
- IE
ileal exclusion
- IQR
interquartile range
- LTX
liver transplantation
- OR
odds ratio
- PEBD
partial external biliary diversion
- PFIC
progressive familial intrahepatic cholestasis
- sAT
serum aminotransferase
- sBA
serum bile acid
Familial intrahepatic cholestasis 1 (FIC1) and bile salt export protein (BSEP) deficiencies are the two most prevalent identified forms of progressive familial intrahepatic cholestasis (PFIC) with normal circulating gamma‐glutamyl transpeptidase (GGT) activity.1, 2, 3 FIC1 deficiency results from mutations in the ATPase phospholipid transporting 8B1 (ATP8B1) gene, and BSEP deficiency results from mutations in the ATP binding cassette subfamily B member 11 (ABCB11) gene.1, 2, 4, 5, 6 The severe forms of both conditions, termed PFIC, present in early childhood with progressive liver disease. We have reported differences in presentation and progression between the two conditions.3 PFIC often resists medical therapy and requires surgical intervention. Orthotopic liver transplantation (LTX) is widely employed as are techniques to reduce bile salt recirculation, including partial external biliary diversion (PEBD) and, less frequently, ileal exclusion (IE).7, 8 PEBD interposes a small bowel segment between the gallbladder and anterior abdominal wall, and bile and bile salts are lost through this segment.7 IE anastomoses proximal ileum to caecum, shunting chyme with bile salts past distal ileum into feces where bile salts are lost.9 Case reports and case series suggest that outcomes of these procedures vary,9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 but a comprehensive comparison stratified by genetic diagnosis has been lacking. Here, we present clinical and laboratory data from 102 patients with PFIC, genetically defined as FIC1 or BSEP deficiency, who underwent one or more of these procedures.
Patients and Methods
PATIENTS
Clinical data were collected from a large cohort of patients with PFIC through centers with approved human study protocols and conforming to the ethical guidelines of the 1975 Declaration of Helsinki, as described.3 Participants had surgeries between 1985 and 2004 inclusive.3 Criteria for inclusion were a clinical diagnosis of low‐GGT PFIC and mutation(s) in ATP8B1 (FIC1 patients) or ABCB11 (BSEP patients). From this database of 145 patients, we identified patients who had undergone PEBD, IE, and/or LTX; 57 patients underwent PEBD, 6 patients underwent IE, and 64 patients underwent LTX. Data from 7 patients (5 BSEP, 2 FIC1) who underwent LTX were excluded from this analysis as substantially described elsewhere.22 A total of 42 FIC1 and 60 BSEP patients underwent one or more of these surgeries and were included. The reported race of these 102 participants was as follows: 85 white, 10 mixed ancestry (7 Hispanic, 2 Asian/Hispanic, and 1 African American), 3 Native American, 2 Asian, and 2 unknown. Subsequent to our prior publication,3 additional mutation screening was performed (when DNA was available) in patients in whom only one mutated allele had previously been found. Consequently, only 5 BSEP patients and no FIC1 patients included in this study have a mutation detected on only one allele.
Data collection was retrospective, and not all requested information was available for each patient. The number of patients (n) for whom data were available is indicated for each variable. We analyzed findings to assess the outcomes after surgery.
PRURITUS
Pruritus data were collected as written descriptions by clinicians completing the study data collection form. We resolved these comments into the following categories: no improvement, partial improvement, or complete resolution, and subclassified as either transient or sustained until the most recent data collection. For statistical analysis, outcomes were dichotomized to sustained improvement (partial or complete) versus no sustained improvement (no or transient improvement).
GROWTH
Presurgical height and weight were the last available values prior to the indicated surgery. Postsurgical height and weight were the last available values at least 1 year after surgery.
PUBERTY
Patients for whom data were available were subdivided into those who completed puberty before age 18 years (boys) and 16 years (girls) (normal), those who completed puberty after those ages (delayed; but now complete), and those who had not completed puberty at last data collection despite being past the normal age window (delayed; still not complete). Only girls aged at least 16 years and boys aged at least 18 years were included.
CLINICAL CHEMISTRY
For PEBD, blood chemistry values used were the most recent available value prior to surgery and the first measurement from at least 2 months after surgery. For LTX, only post‐LTX values were used, again from at least 2 months after surgery; where a patient had more than one measurement available at least 2 months after LTX, the median value was used. To adjust for differences in reference ranges between sites, most analyte values were transformed into multiples (fold) of the upper or lower limit of the normal range (FULN or FLLN, respectively). Total bilirubin values were not normalized but were converted to mg/dL. Alkaline phosphatase values were not normalized as age‐specific normal ranges were only available from some centers; absolute values in IU/L were used. Alkaline phosphatase values reported as μkat/L from one center were converted to IU/L by multiplying by 59. Platelet levels were grouped into below normal, normal, or above normal categories. For analysis of transaminase activities post‐PEBD compared to pre‐PEBD, we defined the variable serum aminotransferase (sAT) as the FULN for alanine aminotransferase (ALT) or for aspartate aminotransferase (AST) if ALT values were unavailable.3
ANALYSIS
Median and interquartile range (IQR) were calculated for continuous variables. Where necessary to normalize distributions, continuous variables were log‐transformed before analysis, and t tests, paired t tests, analysis of variance, or mixed‐effects linear regression were used, as appropriate. Analyses were performed using Graphpad Prism and Quickcalc (Graphpad Software, La Jolla, CA) and Statistical Analysis System versions 9.3 and 9.4 (SAS Institute, Cary, NC). Where transformed data remained non‐normally distributed, appropriate nonparametric methods were used, including Mann‐Whitney and Kruskal‐Wallis tests. Where estimated effects and confidence intervals (CIs) were needed despite persistent non‐normality, bias‐corrected accelerated bootstrapping was used to obtain valid CIs, with P values defined as 1 minus the highest confidence level for which the interval excluded an effect of zero. Categorical variables were assessed using Fisher's exact test or logistic regression, as appropriate. We report nominal P values without adjustment for multiple testing; such adjustment is unhelpful here because biological relationships among the parameters mean that coherent sets of results often reinforce rather than detract from one another.3
Our analyses focused on assessment of univariate associations with gene and for PEBD also with mutation group. For PEBD and LTX, we also assessed these potential confounding factors: sex, year of birth, year of surgery, and age at surgery. For LTX, we additionally assessed living related versus nonrelated donor and reported medication use after LTX as potential confounding factors. For post‐LTX blood biochemistry, linear‐mixed models analysis was performed, factoring in gene and time post‐LTX (the latter as both fixed and random effects). Dichotomized outcomes, such as whether an analyte was normal, were analyzed by logistic regression models, with random intercept terms included when using multiple measurements from the same patients. Additional analyses assessed the influence of potential confounders. For analyses yielding small P values for both genetic category and potential confounding factors, we tested the robustness of gene and mutation effects with respect to the potential confounders, using bivariate and multivariate analyses (where computationally feasible).
For survival analysis, poor outcome after PEBD was defined as progression to histologically proven cirrhosis, listing for or receiving LTX, or death. (One patient had PEBD replaced with IE due to recurrent cholangitis [Fig. 1; patient #3] and was not included in this analysis.) Time was measured from PEBD to the first poor outcome or last follow‐up. We used the Kaplan‐Meier method to plot post‐PEBD survival without a poor outcome by mutation group. Risk estimates, including hazard ratio (HR) and 95% CI, were determined using Cox proportional hazards regression.
Figure 1.
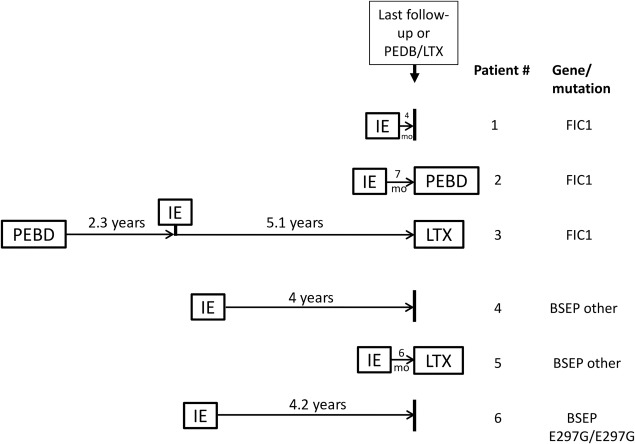
Timeline of surgeries in patients receiving IE. Of 6 patients who underwent IE, 3 (patient numbers 3, 4, and 6, spread across gene and mutation categories) remained with IE for ≥4 years. Of the remaining 3, 2 (patient numbers 3 and 5) proceeded to PEBD or LTX within 6‐7 months and the third (patient number 1) had a short follow‐up time. A brief summary of outcomes for each patient follows, incorporating available information: For patient 1, partial improvement in pruritus was reported but follow‐up time was short. For patient 2, incomplete response to IE was reported and improvement was noted after PEBD was performed. sBAs were normal for a time after IE in patient 3, but this patient experienced complications (recurrent cholangitis, pancreatitis, pain, weight loss, requirement for total parenteral nutrition) as well as worsening cholestasis and jaundice and was ultimately referred for LTX. For patient 4, sBA normalized after IE and remained normal at the last available testing date, >3.5 years after IE; some pruritus was reported, and the patient was on ursodeoxycholate. Patient 5 was referred for LTX soon after IE. For patient 6 post‐IE, minimal pruritus was reported and sBA demonstrated a sustained improvement of >50% (although remaining elevated).
Results
Two common European mutations, D482G and E297G, have been identified in BSEP. Laboratory data suggest that these mutations may result in partial function of BSEP and thus a less severe phenotype than those that wholly eliminate BSEP expression or function.23, 24, 25, 26, 27, 28, 29 Before surgical intervention, disease in PFIC patients with the D482G mutation tends to progress less rapidly than in patients with other BSEP mutations.3 Therefore, we analyzed patients with either of these two mutations separately before LTX. The designation BSEP‐D482G refers to patients heterozygous or homozygous for this mutation; likewise for BSEP‐E297G. One patient was a compound heterozygote for both mutations and was included in the BSEP‐D482G group. These two groups are combined in some analyses as BSEP‐common. BSEP patients without these mutations are referred to as BSEP‐other. No similar subgroups were identified within the FIC1 deficiency group.
PEBD
Of the 145 PFIC patients with FIC1 or BSEP disease included in the original study cohort,3 57 underwent PEBD during the study period (36% of all FIC1 patients and 42% of all BSEP patients) (Table 1). Of the 51 BSEP‐common patients, 49% underwent PEBD while 30% of the 33 BSEP‐other patients did (Fisher's exact test; P = 0.11).
Table 1.
Demographic and Genetic Features of Patients Who Underwent PEBD
| BSEP | ||||||
|---|---|---|---|---|---|---|
| Common | P a | |||||
| Feature | FIC1 | All BSEP | D482G | E297G | Other | (FIC1 vs. All BSEP) |
| Patients (n) | 22 | 35 | 11 | 14 | 10 | |
| Median year of birth (range) | 1995 (1977‐2002) | 1994 (1969‐2001) | 1994 (1980‐1997) | 1993 (1987‐2001) | 1997 (1969‐2000) | 0.41 |
| Median age in years at PEBD (IQR) | 1.6 (0.8‐6.5) | 2.3 (1.4‐4.8) | 2.9 (2.1‐10.7) | 2.3 (1.1‐3.1) | 2.0 (1.4‐4.8) | 0.38 |
| % With older affected sibling | 32% | 29% | 18% | 36% | 30% | 1.00 |
| % Male | 32% | 60% | 64% | 64% | 50% | 0.057 |
| Median years with PEBD (IQR) | 2.4 (1.3‐4.6) | 4.6 (1.2‐6.2) | 5.2 (4.8‐6.2) | 5.0 (1.6‐9.2) | 1.0 (0.7‐1.2) | 0.34 |
These data were available for all participants.
Fisher's exact test (2‐tailed), Wilcoxon rank sum, or Kruskal‐Wallis tests, as appropriate; a number of subgroup comparisons were significant; see text.
The year of birth, age at PEBD, and proportion of patients with older affected siblings were comparable between FIC1 and BSEP deficiencies (Table 1). Of those having PEBD, a greater proportion of FIC1 patients were female, while a greater proportion of BSEP patients were male (P = 0.042 in the subgroup comparison FIC1 versus BSEP‐common; P = 0.057 overall). Median time with PEBD was defined as time between PEBD and LTX, IE, or last follow‐up, whichever came first. In one case, IE was undertaken after PEBD due to recurrent cholangitis. BSEP‐common had the longest median time with PEBD, while BSEP‐other had the shortest (P = 0.0009); FIC1 patients were intermediate (P = 0.020 versus BSEP‐common; P = 0.039 versus BSEP‐other) (Table 1). Medication data were available for 21 patients of whom 9 (43%) received choleretic medication after PEBD (4/7 FIC1, 1/3 BSEP‐D482G, 2/7 BSEP‐E297G, 2/4 BSEP‐other).
EFFECT OF PEBD ON PRURITUS
Overall, no difference in the proportion of FIC1 versus BSEP patients with sustained improvement in pruritus was detected (P = 0.58). Most FIC1 patients (81%) experienced subjective improvement of pruritus after PEBD, with sustained improvement in 57% (Table 2). All but 1 BSEP‐D482G patient had sustained pruritus improvement as did 64% of BSEP‐E297G patients. When combined, BSEP‐common showed a 76% sustained response; in contrast, this was reported in only 33% of BSEP‐other patients (Table 2). Collectively, sustained response was more likely among BSEP‐common than BSEP‐other patients (odds ratio [OR], 6.3; 95% CI, 1.2‐33.4; P = 0.030). Testing for potential confounding factors revealed that older age at PEBD and earlier year of birth were associated with greater chances of sustained improvement in pruritus (P = 0.0062, using ln[age] and 0.018, respectively). In multivariate analysis incorporating age at surgery and year of birth as well as genetic category, BSEP‐common remained more likely than BSEP‐other to have sustained response (OR, 8.1; 95% CI, 1.2‐55.9; P = 0.035).
Table 2.
Pruritus Response to PEBD
| Impact on Pruritus |
FIC1 (n = 21) |
BSEP | ||||
|---|---|---|---|---|---|---|
|
All BSEP (n = 34) |
Common |
Other (n = 9) |
||||
|
D482G (n = 11) |
E297G (n = 14) |
|||||
| No improvement | 4 (19%) | 8 (24%) | 1 (9%) | 3 (21%) | 4 (44%) | |
| Partial improvement | Transient | 4 (19%) | 4 (12%) | 0 (0%) | 2 (14%) | 2 (22%) |
| Sustained | 8 (38%) | 11 (32%) | 4 (36%) | 6 (43%) | 1 (11%) | |
| Complete resolution | Transient | 1 (5%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Sustained | 4 (19%) | 11 (32%) | 6 (55%) | 3 (21%) | 2 (22%) | |
Two patients are excluded from the table: 1 FIC1 patient had complete resolution of pruritus, but the surgery was performed shortly before completion of data collection so it is unknown whether the response was transient or sustained. One other BSEP patient had no information on pruritus response provided.
PATIENT AND LIVER SURVIVAL AFTER PEBD
No difference in the frequency of clinically important poor outcomes of PEBD was detected between FIC1 and BSEP patients (Table 3). A greater proportion of BSEP‐other compared to FIC1 patients progressed to cirrhosis. BSEP‐other patients were also more likely to be listed for or to undergo LTX than FIC1 or BSEP‐common patients. Survival with PEBD without any of the three poor outcomes was evaluated in univariate analysis (Fig. 2), and BSEP‐other patients fared worst. Compared with BSEP‐other patients, risk of poor outcome was reduced for FIC1 (HR, 0.16; 95% CI, 0.05‐0.51; P = 0.0021), BSEP‐D482G (HR, 0.09; 95% CI, 0.02‐0.44; P = 0.0032), and BSEP‐E297G patients (HR, 0.20; 95% CI, 0.06‐0.64; P = 0.0067). Older age at PEBD was associated with decreased risk of poor outcome (using ln[age], P = 0.03). Both mutation group (P = 0.003 or lower for all comparisons) and age (P = 0.005) remained significant in bivariate analyses.
Table 3.
PEBD Outcomes
| Feature |
FIC1 (n = 22) |
BSEP |
P*,
Common BSEP vs. Other BSEP |
P*, FIC1 vs. Other BSEP |
|||
|---|---|---|---|---|---|---|---|
|
All BSEP (n = 35) |
Common |
Other (n = 10) |
|||||
|
D482G (n = 11) |
E297G (n = 14) |
||||||
| Progressed to cirrhosis‡, | 0/20 (0%) | 5/30 (17%) | 0/10 (0%) | 2/11 (18%) | 3/9 (33%) | 0.143 | 0.023 |
| LTX or listing | 6/22 (27%) | 11/35 (31%) | 1/11 (9%) | 3/14 (21%) | 7/10 (70%) | 0.0039 | 0.049 |
| Deceased | 0/22 (0%) | 3/35 (9%) | 1/11 (9%) | 2/14 (14%) | 0/10 (0%) | 0.542 | † |
| Poor outcome§ | 6/22 (27%) | 14/35 (40%) | 2/11 (18%) | 5/14 (36%) | 7/10 (70%) | 0.053 | 0.049 |
Each cell shows total n (denominators). P values <0.05 are noted in bold. For all features in this table, P values for all BSEP versus FIC1 and for common BSEP versus FIC1 did not reach significance. *Two‐tailed Fisher's exact tests; † P value not calculable. ‡,Progression to cirrhosis excludes patients who had cirrhosis at the time of diversion. Six patients (1 D482G carrier, 3 E297G carriers, and 2 FIC1 patients) had been diagnosed with cirrhosis prior to diversion. §Poor outcome was defined as one or more of the following: progression to cirrhosis, listing for or performance of LTX, or death. Three patients, all with one or two copies of E297G, developed HCC after PEBD; of these patients, 2 died and 1 had LTX. A third patient who was homozygous for D482G died due to end‐stage liver disease approximately 5 years after PEBD.
Figure 2.
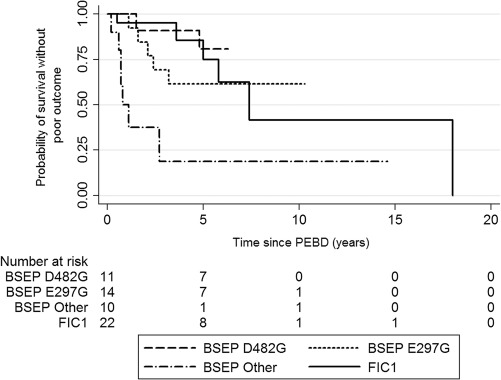
Probability of survival without a poor outcome after PEBD, by mutation group. Patients were followed from PEBD to the first poor outcome (progression to cirrhosis, listing for or receiving a liver transplant, or dying) or last follow‐up. Number at risk reflects the number of patients remaining at risk for a poor outcome at each time point.
GROWTH
Height and weight were assessed where data were available before and at least 1 year after surgery (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1168/full). A majority of FIC1 and BSEP patients were lower than the tenth percentile for height and weight before surgery; both height and weight improved in most of these patients in both diseases. Nearly all available BSEP data were from BSEP‐common patients. (No statistical analysis was attempted.)
BLOOD BIOCHEMISTRY
We assessed the impact of PEBD on circulating biochemistry, using the last available presurgical value and the first value available at least 2 months after surgery. Three variables showed statistically and clinically relevant changes (Table 4). Bilirubin and serum bile acid (sBA) fell after PEBD in FIC1 and BSEP patients. Bilirubin and sBA fell in BSEP‐D482G and BSEP‐E297G patients but not in the few BSEP‐other patients for whom data were available. Before surgery, values for sAT were less elevated in FIC1 than in BSEP patients. There was little change in sAT for FIC1 patients, but sAT fell in BSEP patients overall as well as in BSEP‐D482G and BSEP‐E297G patients. For these analytes, we also determined the proportion of patients with clinically meaningful changes (see http://onlinelibrary.wiley.com/doi/10.1002/hep4.1168/full). Because patients with good outcomes were more likely to have pre‐PEBD and post‐PEBD sBA data available (P = 0.005), improvement in sBA may be overestimated.
Table 4.
Blood Biochemistry Pre‐PEBD and Post‐PEBD
| FIC1 | BSEP | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| All BSEP | Common | Other | |||||||||
| D482G | E297G | ||||||||||
| Serum Assay | Time | Median (IQR) |
P
n |
Median (IQR) |
P
n |
Median (IQR) |
P
n |
Median (IQR) |
P
n |
Median (IQR) |
P
n |
| Total bilirubin (mg/dL) | Presurgical | 5.4 (2.5‐8.9) |
P = 0.002
n = 19 |
6.3 (2.0‐10.2) |
P = 0.0001
n = 26 |
3.7 (1.7‐12.2) |
P = 0.027
n = 11 |
6.2 (2.2‐10.2) |
P = 0.0097
n = 11 |
8.0 (3.6‐10.6) |
P = 0.18 n = 4 |
| Postsurgical | 1.0 (0.3‐3.9) | 1.0 (0.6‐4.1) | 1.0 (1.0‐2.0) | 1.0 (0.4‐4.7) | 2.4 (0.6‐5.5) | ||||||
| sBA (FULN) | Presurgical | 13.4 (6.0‐20.8) |
P = 0.037
n = 9 |
35.0 (27.3‐49.1) |
P<0.0001
n = 18 |
31.1 (23.0‐35.0) |
P = 0.010
n = 8 |
43.7 (32.8‐55.6) |
P = 0.0003
n = 8 |
40.6 (22.4‐58.8) |
P = 0.61 n = 2 |
| Postsurgical | 3.3 (0.3‐7.1) | 0.7 (0.5‐17.9) | 0.7 (0.5‐20.2) | 0.6 (0.2‐2.6) | 54.9 (49.4‐60.3) | ||||||
| sAT (FULN) | Presurgical | 1.6 (0.9‐2.1) |
P = 0.57 n = 19 |
2.4 (1.5‐5.3) |
P<0.0001
n = 27 |
2.4 (1.1‐4.0) |
P = 0.034
n = 11 |
2.7 (1.6‐8.9) |
P = 0.003
n = 12 |
4.0 (1.2‐9.8) |
P = 0.10 n = 4 |
| Postsurgical | 1.7 (1.0‐2.9) | 1.0 (0.6‐1.9) | 0.7 (0.6‐1.4) | 1.1 (0.6‐2.0) | 1.9 (0.6‐8.8) | ||||||
P values <0.05 are noted in bold. P‐values compare pre‐ and post‐surgical values for each serum assay.
Medians, IQR, and sample sizes (n) are shown. Presurgical values are the last available before surgery. Postsurgical values are the first available values at least 2 months after surgery.
ELECTROLYTE IMBALANCE AND OTHER COMPLICATIONS
An open‐ended question about complications of PEBD yielded reports of electrolyte imbalance and/or dehydration related to high bile output in 3 BSEP patients (1 accompanied by bleeding from stoma) and 3 FIC1 patients. In 1 FIC1 patient, this led to cardiac arrest. Also reported were cholangitis (1 BSEP and 2 FIC patients), no or low bile flow or bile stagnation (2 FIC1 patients), stone/obstruction/occlusion (1 BSEP and 2 FIC1 patients), and high bile output with bleeding from the stoma but without reported electrolyte imbalance or dehydration (1 BSEP patient).
IE
Six patients underwent IE. Reported responses varied with too few patients to compare between categories. For a summary of outcomes, see Fig. 1.
LTX
Of the 145 patients with severe FIC1 or BSEP deficiency in the study cohort,3 64 underwent LTX during the study period; our analysis here includes 57 of these patients. The median follow‐up time after LTX was similar between diseases, but BSEP patients had LTX at a younger age than FIC1 patients; consistent with this, the BSEP patients were born more recently (Table 5). Of the 33 BSEP patients with transplants, 18 were BSEP‐common and 15 BSEP‐other. BSEP patients were analyzed as a single group as the protein is thought to have significant function only in the liver and therefore genotype is not expected to influence LTX outcome. The exception would be the possibility of alloantibody formation (see Discussion).
Table 5.
LTX: Demographics and Outcomes
| Feature | FIC1 | BSEP | P |
|---|---|---|---|
| # Undergoing LTX | 24 | 33 | |
| Median years with LTX (IQR) | 3.6 (1.1‐7.1) n = 24 | 3.6 (1.1‐6.1) n = 33 | 0.99 |
| Median age (years) at LTX (IQR) | 5.8 (3.0‐12.1) n = 24 | 3.1 (1.9‐5.8) n = 33 | 0.010 |
| Median year of birth (full range) | 1991 (1976‐1999) n = 24 | 1996 (1978‐2003) n = 33 | 0.030 |
| Proportion male | 8/24 (33%) | 16/33 (48%) | 0.29 |
| # With older affected sibling | 4/24 (17%) | 5/33 (15%) | 1.00 |
| # Died post‐LTX | 3/24 (13%) | 0/33 (0%) | 0.07 |
| # Requiring retransplant | 1/24 (4%) | 4/33 (12%) | 0.39 |
| # With rejection | 15/21 (71%) | 22/31 (71%) | 1.0 |
| # With hepatic steatosis | 19/21 (90%) | 2/31 (6%) | <0.0001 |
| # With diarrhea | 17/21 (81%) | 2/29 (7%) | <0.0001 |
| # Requiring parenteral nutrition | 3/20 (15%) | 0/29 (0%) | 0.062 |
| # With pancreatitis or pancreatic insufficiency |
8/20 (40%) 7 pancreatitis 1 insufficiency |
0/29 (0%) | 0.0003 |
P values <0.05 are noted in bold.
Overall, outcomes after transplantation were good with few deaths (5%, all FIC1 patients) or patients requiring retransplantation (9%) (Table 5). Two of the three deaths were due to nonspecific complications of LTX (1 had respiratory failure, another had multiple surgical complications). The third child died, aged 12, of complications following subsequent small bowel transplantation.
The overall reported incidence of rejection (chronic, acute, or unspecified) was not significantly different between the two groups (Table 5). Retransplantation was undertaken in 4 BSEP patients due to surgical complications in 1, chronic rejection in a second, and multiple problems in the remaining 2, including autoimmune hepatitis in 1 and giant cell hepatitis in the other. One FIC1 patient underwent retransplantation twice for technical complications within 1 week of the initial LTX.
Five BSEP and 4 FIC1 patients underwent living‐related‐donor transplantation (7 from obligate heterozygote parents, 1 from an uncle, and 1 from an unspecified relative). Such transplantation was not associated with detectably different outcomes from unrelated‐donor LTX: 1 such FIC1 patient and no BSEP patients required retransplantation. All living‐related‐donor transplant recipients were alive at last follow‐up.
Management after LTX varied according to the local protocol (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1168/full).
LIVER FUNCTION AFTER LTX
Allograft steatosis was reported in only 2 of 31 BSEP patients for whom biopsy reports after LTX were available but was found in 19 of 21 such FIC1 patients (Table 5). An additional FIC1 patient was diagnosed with steatosis based upon ultrasonographic findings unconfirmed by biopsy.
Consistent with the finding of steatosis in FIC1 patients, serum levels of ALT and AST (≥2 months after LTX) were mildly elevated for most FIC1 patients but were in the normal range for most BSEP patients (Table 6). A small number of FIC1 patients had elevated platelet counts (4/18) suggestive of inflammation, whereas no BSEP patients did (0/27) (likelihood ratio P = 0.015; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1168/full). Bilirubin levels normalized in nearly all patients and were similar between diseases.
Table 6.
Post‐LTX Blood Biochemistry: Median Values per Patient for Each Disease
| Serum Assay |
FIC1 Median (IQR) n |
BSEP Median (IQR) n |
P for FIC1 vs. BSEP* |
Effect/Trend (95% CI) |
|---|---|---|---|---|
| ALT (FULN) |
1.6 (1.1‐2.3) n = 16 |
0.8 (0.5‐0.9) n = 23 |
<0.0001 |
97% higher in FIC1 (30%‐190% higher) |
| AST (FULN) |
1.6 (1.1‐2.2) n = 18 |
0.9 (0.7‐1.0) n = 27 |
0.0083 |
61% higher in FIC1 (17%‐107% higher) |
| Total bilirubin (mg/dL) |
0.4 (0.3‐0.6) n = 18 |
0.9 (0.5‐1.0) n = 26 |
0.46 |
0.2 mg lower in FIC1 (0.5 mg lower to 0.3 mg higher) |
| GGT† (FULN) |
0.7 (0.3‐1.6) n = 16 |
0.3 (0.2‐0.8) n = 27 |
0.037 |
77% higher in FIC1 (4%‐274% higher) |
| Albumin‡ (FLLN) |
1.0 (0.8‐1.0) n = 18 |
1.1 (1.0‐1.2) n = 18 |
0.0052 |
17% lower in FIC1 (7%‐24% lower) |
| Total protein (FLLN) |
1.0 (1.0‐1.2) n = 17 |
1.2 (1.1‐1.2) n = 17 |
0.06 |
9% lower in FIC1 (16% lower to 0.5% higher) |
| Cholesterol (FULN) |
0.4 (0.3‐0.6) n = 8 |
0.6 (0.5‐0.7) n = 19 |
0.027 |
37% lower in FIC1 (8%‐57% lower) |
| Hemoglobin (FLLN) |
1.0 (0.9‐1.1) n = 16 |
1.1 (1.1‐1.2) n = 24 |
0.0007 |
16% lower in FIC1 (5%‐27% lower) |
| ALP (IU/L) |
292 (199‐445) n = 19 |
255 (212‐319) n = 20 |
0.43 |
14% higher in FIC1 (20% lower to 70% higher) |
| Hematocrit (FLLN) |
1.0 (1.0‐1.1) n = 11 |
1.1 (1.0‐1.3) n = 21 |
0.24 |
8% lower in FIC1 (20% lower to 6% higher) |
| Triglycerides (FULN) |
0.3 (0.3‐0.5) n = 8 |
0.3 (0.2‐1.0) n = 12 |
0.95 |
7% lower in FIC1 (58% lower to 118% higher) |
P values <0.05 are noted in bold.
n, number of patients for whom data were available. Median value per patient was used for patients for whom results of multiple tests were available. *Comparison of geometric means, factoring in time. †The apparent gene effect on GGT may be due to effects of year of LTX and age at LTX. ‡Additional analysis indicates some of the apparent gene effect may be accounted for by an effect of steroid use. Abbreviation: ALP, alkaline phosphatase.
Circulating GGT levels were also higher in FIC1 than in BSEP patients after LTX (Table 6; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1168/full); however, in testing for confounding variables, higher GGT levels were also associated with older age at transplant and earlier year of transplant. In multivariate analyses, the gene effect on GGT no longer attained P < 0.05, while age and year did.
INTESTINAL DISEASE AND EVIDENCE OF MALABSORPTION
Diarrhea is a known complication of LTX; it is also a frequent feature of FIC1 deficiency before LTX.3 Diarrhea after LTX was reported in only 7% of the BSEP‐deficient group but was reported in 81% of the FIC1‐deficient group (Table 5). Of the 17 FIC1 patients with diarrhea after LTX, only 8 had the problem before LTX. In 3 FIC1 patients, diarrhea after LTX had sufficient nutritional impact that parenteral nutrition was necessary (Table 5); 1 of these patients underwent a subsequent small bowel transplant.
Consistent with the much greater incidence of diarrhea after LTX in FIC1 patients, circulating albumin and total protein levels after LTX in these patients were more likely to be below normal range than in BSEP patients (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1168/full). Also consistent with poorer nutritional status, circulating cholesterol levels and hemoglobin levels were lower in FIC1 patients than in BSEP patients (Table 6).
PANCREATIC DISEASE
Pancreatic problems can occur in FIC1 deficiency.3 Seven FIC1 patients had pancreatitis after LTX and 1 had exocrine insufficiency (Table 5). Three of these 8 patients had known pancreatic disease before LTX. No BSEP patients had evidence of pancreatic disease after LTX.
GROWTH
Growth failure is a major problem in all forms of cholestatic liver disease in children. We previously demonstrated that it is notably worse in FIC1 deficiency than in BSEP deficiency before surgical intervention.3 Growth status was assessed using data collected >1 year after LTX to allow for catch‐up growth. Differences in both height and weight between the two conditions were marked. Only 35% and 31% of FIC1 patients were above the third percentile for height and weight, respectively, in contrast to 88% and 90% of BSEP patients (height, P = 0.0004 and weight P < 0.0001, FIC1 versus BSEP, each binned into 3 percentile bands; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1168/full). Among patients for whom growth data were available both before and after LTX, FIC1 patients were more likely to have weight below the tenth percentile before transplantation compared to BSEP patients (P = 0.0456, Fisher's exact test; Fig. 3). After LTX, there was a trend toward FIC1 patients being less likely than BSEP patients to experience catch‐up weight gain (P = 0.10, Fisher's exact test; Fig. 3).
Figure 3.
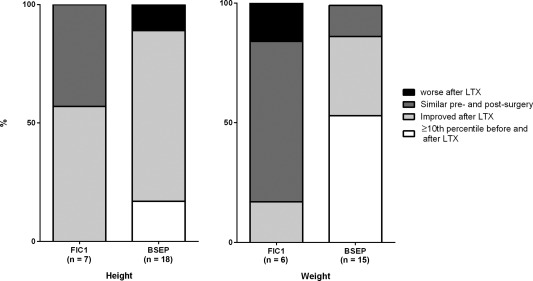
Comparison of growth before and after LTX. As with http://onlinelibrary.wiley.com/doi/10.1002/hep4.1168/full, patients' growth was binned into three categories: ≥10th percentile, 3rd‐10th percentile, and <3rd percentile. The categories in the graphs are as follows: patient already ≥10th percentile prior to surgery and remained in that category post‐LTX; patient growth improved by one to two categories post‐LTX; patient growth remained in the same category after LTX; patient growth category worsened post‐LTX.
PUBERTY IN PATIENTS WHO HAD UNDERGONE PEBD AND/OR LTX
At the time of the last data collection, 13 patients (10 girls and 3 boys) had undergone PEBD and/or LTX and were at or past the normal age for puberty completion (16 years, girls; 18 years, boys). Among FIC1 patients, 1/6 completed puberty within the normal age range, 2/6 completed puberty after the normal age window, and 3/6 had not completed puberty. Among BSEP patients, 5/7 completed puberty within the normal age range, 1/7 completed puberty after the normal age window, and 1/7 had not completed puberty (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1168/full). There was a trend (P = 0.10, Fisher's exact test) toward FIC1 patients being more likely to have delayed or incomplete puberty. When only patients who had undergone LTX were evaluated, 2/5 FIC1 patients had delayed but completed puberty and 3/5 had not completed puberty, while all 4 BSEP patients completed puberty within the normal window (P = 0.008, Fisher's exact test).
Discussion
PEBD
The aim of PEBD is to interrupt bile salt recirculation, to relieve pruritus, and potentially to improve liver function and overall clinical outcome. Similar proportions of FIC1 and BSEP patients from our original cohort underwent PEBD. Our retrospective data indicate that some patients in all genotype groups responded with improvements in pruritus and biochemistries.
A majority of patients with FIC1 or BSEP deficiencies had sustained subjective improvement in pruritus. However, patients with one of the BSEP‐common mutations (D482G or E297G) were more likely to have a sustained response than were BSEP patients with other mutations.
Blood levels of bilirubin, bile acids, and transaminases improved or remained good after PEBD in most FIC1 and BSEP patients; however, in FIC1 disease, transaminase activities were only modestly elevated before PEBD and did not detectably change after surgery. Insufficient biochemical data from before and after PEBD were available from BSEP‐other to allow comparison of results between BSEP‐common and BSEP‐other.
Clinical outcome of PEBD was assessed by avoidance of poor outcomes, including death, development of cirrhosis, LTX, or listing for LTX. On this basis, BSEP‐other patients fared worse than patients in other groups. The data presented here were collected prior to increased understanding of differing outcomes in BSEP versus FIC1 deficiency and before realization that mutation type within BSEP deficiency makes a difference to the risk of hepatocellular carcinoma (HCC).6 Patient management decisions were therefore likely based on clinical rather than genetic findings. Three patients developed HCC after PEBD and before the last data collection; 1 received a successful transplant, and the other 2 died. Interestingly, all 3 had at least one copy of E297G, which would have been predicted to confer a lower risk of HCC than some BSEP genotypes.
In this cohort, patients who underwent PEBD at an older age were more likely to have sustained improvement in pruritus and decreased risk of poor clinical outcome compared to patients who underwent PEBD at a younger age. Analyses taking both age and gene/mutation into account suggest that this effect may not be completely explained by patients with inherently milder disease having PEBD at a later age than those with more severe disease. Assessment of pruritus is, however, affected by patient age and communication skills.
PEBD results in clinical improvement in many patients with FIC1 or BSEP deficiency. However, our data support the clinical impression that PFIC due to BSEP deficiency can be divided into different clinical subgroups. It was previously suggested that patients bearing the E297G BSEP mutation may have a better response to PEBD than other BSEP patients.22 Results in our larger cohort indicate that PEBD may be especially helpful to those bearing mutation(s) likely to retain residual protein function, including either BSEP‐common mutation.
LTX
Liver transplantation is a major therapeutic tool in the management of PFIC. Unlike BSEP, FIC1 is widely expressed. It is not surprising, therefore, that responses to LTX differed between BSEP and FIC1 patients. Graft and patient survival were good in both groups. Patient survival trended worse in FIC1 deficiency; survival of the initial graft was, however, similar. The figures compare favorably with those in other pediatric LTX series from the late 1980s onward.30, 31
In this study, BSEP patients came to LTX younger than FIC1 patients. As the data for this study were assembled before the realization that BSEP patients may be at significant risk of native‐liver HCC, awareness of HCC risk is unlikely to explain this trend.6, 32 In this cohort, 2 patients received transplants after a diagnosis of HCC, 1 of whom had previously undergone PEBD (discussed above); both were well at last follow‐up. The patient without PEBD was from the BSEP‐other group.
LTX resolved cholestasis in both cohorts; however, as hypothesized based on tissue sites of protein expression, extrahepatic complications were worse with FIC1 deficiency than with BSEP deficiency. BSEP patients were free of pancreatic disease, while a higher proportion of FIC1 patients manifested pancreatic disease after LTX than before (40% versus 12%).3 This increase in pancreatic disease may be directly related to LTX or may simply reflect progression of extrahepatic disease in FIC1 patients over time.
FIC1 is highly expressed in the small and large intestine of healthy individuals. Diarrhea after the transplant was 12‐fold more common with FIC1 than BSEP deficiency. After LTX, FIC1 patients also often have biochemical markers of malnutrition and may have poorer catch‐up growth and more persistent pubertal delay.
Steatosis in the allograft liver has been reported in FIC1 patients but is now confirmed in this large genetically characterized cohort and contrasted with the status of livers transplanted into BSEP‐deficient patients. In this cohort, most FIC1‐deficient patients had graft steatosis and accompanying elevated transaminase activities with otherwise acceptable graft function.
The concept of alloantibody formation after LTX had not been proposed when collection of these data was completed. It is not clear if this complication contributed to the outcomes among the BSEP patients. Four BSEP patients underwent retransplantation, of whom only 1 was described as having giant cell hepatitis. None of the patients who received the retransplant had two protein truncating mutations, which would have put them at the highest risk of alloantibody formation.33, 34
Not only is the gastrointestinal tract abnormal in FIC1 deficiency but an abnormality in gut–liver signaling exists.35, 36 The situation appears to change when a genetically normal transplanted liver is coupled with a FIC1‐deficient intestine. The steatosis seen after LTX in FIC1 deficiency could be a manifestation of altered signaling in this unusual circumstance. Gastrointestinal dysfunction and pancreatic injury may contribute to diarrhea, poor growth, and low albumin and hemoglobin after LTX in FIC1 deficiency. There are several reports in the literature of different biliary diversion procedures at or after LTX in FIC1‐deficient patients37, 38, 39; PEBD seems to have been effective in mitigating the steatosis and diarrhea in most of these cases. Our data reinforce the opinion that FIC1 deficiency is a multisystem disease and that isolated LTX should not be considered a cure.
Several previous case series have presented data on the outcomes of surgical management of PFIC. Unfortunately, those studies have had small numbers or lacked systematic genetic diagnoses.7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 Our data comprise the largest set of outcomes of PEBD or LTX in genetically diagnosed PFIC. Results from this cohort can be used to help guide clinical management. In particular, the likely outcomes after different interventions can to some extent be anticipated. Patients with BSEP deficiency harboring either the E297G or D482G mutation can be expected to do well after PEBD, although 3 patients developed HCC in the E297G group. This contrasts with our previous finding that patients with missense mutations have a lower risk of HCC than those with two protein‐truncating mutations.6 PEBD can also be expected to result in a good outcome in most FIC1 patients. As FIC1 deficiency is a multisystem disease, extrahepatic manifestations and graft dysfunction complicate and limit the utility of LTX. BSEP deficiency, on the other hand, is a primarily hepatic disorder and generally responds well to LTX.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1168/full.
Supplementary Table 1. Growth post‐PEBD
Supplementary Table 2. Clinically meaningful improvement in blood biochemistry post‐PEBD.
Supplementary Table 3. Reported use of immunosuppressants post‐transplant.
Supplementary Table 4. Relative odds of blood chemistry being abnormal, after recovery from LTX
Supplementary Table 5. Height and weight after LTX
Supplementary Table 6. Puberty in Patients who have undergone PEBD and/or LTX and were past the normal age for puberty completion at last data collection.
Acknowledgment
We thank Peter Bacchetti and Chengshi Jin for assistance with statistical analysis, J. Vargas for assistance with data compilation, and A.S. Knisely for comments on the manuscript. For contribution of patient data, we thank D. Barton, B. Bourke, A. Devenyi, K. Foster, M. Gadomski, M. Gerrer, I. Gonçalves, P. Harren, D. Harris, S. Horslen, S. Kelly, S. Lidofsky, J. Lokar, D. Piccoli, C. Potter, S. Radwal, E. Rand, R. Redline, E. Roberts, H. Sharp, B. Shneider, Y. Sims, B. Stahulak, T. Stephen, L. Szonyi, and S. Tifft. We also thank the families who participated in this study.
Potential conflict of interest: Dr. Houwen has consulted for Albireo and Alexion. Dr. Lobritto consults for IPRO and received grants from AbbVie. Dr. Sokal consults, advises, received grants, holds intellectual property rights, and owns stock in Promethera. Dr. Rosenthal consults and is on the speakers' bureau for Retrophin; he consults and received grants from Gilead and AbbVie and consults for Intercept, Alexion, Albireo, and Audentes; he received grants from Bristol‐Myers Squibb and Roche‐Genentech. Dr. Thompson consults, is on the speakers' bureau, and received grants from Shire; he consults for Albireo, Alexion, Arcturus, GlaxoSmithKline, and Retrophin. The other authors have nothing to disclose with respect to this manuscript.
Supported by the National Institutes of Health (NIH) award numbers R01 DK094828 (to L.N.B. and R.J.T.), R01 DK058214 (to L.N.B.), U01 DK062500 (to P.R.), U01 DK062453 (to Ronald J. Sokol), U01 DK062436 (to P.F.W.), and NIH CTSI grant UL1 TR000004 (to UCSF); the University of California San Francisco (UCSF)‐King's College Health Partners Faculty Fellowship Travel Grant (UCSF Academic Senate to L.N.B.); and University Medical Center Utrecht and Utrecht University (AMvE). We also acknowledge the biostatistical services of the Clinical and Translational Core facility of the UCSF Liver Center (NIH P30 DK026743).
REFERENCES
- 1. Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, et al. A gene encoding a P‐type ATPase mutated in two forms of hereditary cholestasis. Nat Genet 1998;18:219‐224. [DOI] [PubMed] [Google Scholar]
- 2. Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, et al. A gene encoding a liver‐specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet 1998;20:233‐238. [DOI] [PubMed] [Google Scholar]
- 3. Pawlikowska L, Strautnieks S, Jankowska I, Czubkowski P, Emerick K, Antoniou A, et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J Hepatol 2010;53:170‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klomp LW, Bull LN, Knisely AS, van Der Doelen MA, Juijn JA, Berger R, et al. A missense mutation in FIC1 is associated with greenland familial cholestasis. Hepatology 2000;32:1337‐1341. [DOI] [PubMed] [Google Scholar]
- 5. Klomp LW, Vargas JC, van Mil SW, Pawlikowska L, Strautnieks SS, van Eijk MJ, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology 2004;40:27‐38. [DOI] [PubMed] [Google Scholar]
- 6. Strautnieks SS, Byrne JA, Pawlikowska L, Cebecauerova D, Rayner A, Dutton L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008;134:1203‐1214. [DOI] [PubMed] [Google Scholar]
- 7. Whitington PF, Whitington GL. Partial external diversion of bile for the treatment of intractable pruritus associated with intrahepatic cholestasis. Gastroenterology 1988;95:130‐136. [DOI] [PubMed] [Google Scholar]
- 8. Jericho HS, Kaurs E, Boverhof R, Knisely A, Shneider BL, Verkade HJ, et al. Bile acid pool dynamics in progressive familial intrahepatic cholestasis with partial external bile diversion. J Pediatr Gastroenterol Nutr 2015;60:368‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hollands CM, Rivera‐Pedrogo FJ, Gonzalez‐Vallina R, Loret‐de‐Mola O, Nahmad M, Burnweit CA. Ileal exclusion for Byler's disease: an alternative surgical approach with promising early results for pruritus. J Pediatr Surg 1998;33:220‐224. [DOI] [PubMed] [Google Scholar]
- 10. Emond JC, Whitington PF. Selective surgical management of progressive familial intrahepatic cholestasis (Byler's disease). J Pediatr Surg 1995;30:1635‐1641. [DOI] [PubMed] [Google Scholar]
- 11. Ismail H, Kalicinski P, Markiewicz M, Jankowska I, Pawlowska J, Kluge P, et al. Treatment of progressive familial intrahepatic cholestasis: liver transplantation or partial external biliary diversion. Pediatr Transplant 1999;3:219‐224. [DOI] [PubMed] [Google Scholar]
- 12. Melter M, Rodeck B, Kardorff R, Hoyer PF, Petersen C, Ballauff A, et al. Progressive familial intrahepatic cholestasis: partial biliary diversion normalizes serum lipids and improves growth in noncirrhotic patients. Am J Gastroenterol 2000;95:3522‐3528. [DOI] [PubMed] [Google Scholar]
- 13. Ng VL, Ryckman FC, Porta G, Miura IK, de Carvalho E, Servidoni MF, et al. Long‐term outcome after partial external biliary diversion for intractable pruritus in patients with intrahepatic cholestasis. J Pediatr Gastroenterol Nutr 2000;30:152‐156. [DOI] [PubMed] [Google Scholar]
- 14. Kalicinski PJ, Ismail H, Jankowska I, Kaminski A, Pawlowska J, Drewniak T, et al. Surgical treatment of progressive familial intrahepatic cholestasis: comparison of partial external biliary diversion and ileal bypass. Eur J Pediatr Surg 2003;13:307‐311. [DOI] [PubMed] [Google Scholar]
- 15. Kurbegov AC, Setchell KD, Haas JE, Mierau GW, Narkewicz M, Bancroft JD, et al. Biliary diversion for progressive familial intrahepatic cholestasis: improved liver morphology and bile acid profile. Gastroenterology 2003;125:1227‐1234. [DOI] [PubMed] [Google Scholar]
- 16. Arnell H, Bergdahl S, Papadogiannakis N, Nemeth A, Fischler B. Preoperative observations and short‐term outcome after partial external biliary diversion in 13 patients with progressive familial intrahepatic cholestasis. J Pediatr Surg 2008;43:1312‐1320. [DOI] [PubMed] [Google Scholar]
- 17. Halaweish I, Chwals WJ. Long‐term outcome after partial external biliary diversion for progressive familial intrahepatic cholestasis. J Pediatr Surg 2010;45:934‐937. [DOI] [PubMed] [Google Scholar]
- 18. Schukfeh N, Metzelder ML, Petersen C, Reismann M, Pfister ED, Ure BM, et al. Normalization of serum bile acids after partial external biliary diversion indicates an excellent long‐term outcome in children with progressive familial intrahepatic cholestasis. J Pediatr Surg 2012;47:501‐505. [DOI] [PubMed] [Google Scholar]
- 19. Jankowska I, Czubkowski P, Kalicinski P, Ismail H, Kowalski A, Ryzko J, et al. Ileal exclusion in children with progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr 2014;58:92‐95. [DOI] [PubMed] [Google Scholar]
- 20. Schukfeh N, Gerner P, Paul A, Kathemann S, Metzelder M. Laparoscopic button cholecystostomy for progressive familial intrahepatic cholestasis in two children. Eur J Pediatr Surg 2014;24:433‐436. [DOI] [PubMed] [Google Scholar]
- 21. Wang KS, Tiao G, Bass LM, Hertel PM, Mogul D, Kerkar N, et al.; Childhood Liver Disease Research Network (ChiLDReN) . Analysis of surgical interruption of the enterohepatic circulation as a treatment for pediatric cholestasis. Hepatology 2017;65:1645‐1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davit‐Spraul A, Fabre M, Branchereau S, Baussan C, Gonzales E, Stieger B, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma‐glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 2010;51:1645‐1655. [DOI] [PubMed] [Google Scholar]
- 23. Wang L, Soroka CJ, Boyer JL. The role of bile salt export pump mutations in progressive familial intrahepatic cholestasis type II. J Clin Invest 2002;110:965‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Plass JR, Mol O, Heegsma J, Geuken M, de Bruin J, Elling G, et al. A progressive familial intrahepatic cholestasis type 2 mutation causes an unstable, temperature‐sensitive bile salt export pump. J Hepatol 2004;40:24‐30. [DOI] [PubMed] [Google Scholar]
- 25. Hayashi H, Takada T, Suzuki H, Akita H, Sugiyama Y. Two common PFIC2 mutations are associated with the impaired membrane trafficking of BSEP/ABCB11. Hepatology 2005;41:916‐924. [DOI] [PubMed] [Google Scholar]
- 26. Noe J, Kullak‐Ublick GA, Jochum W, Stieger B, Kerb R, Haberl M, et al. Impaired expression and function of the bile salt export pump due to three novel ABCB11 mutations in intrahepatic cholestasis. J Hepatol 2005;43:536‐543. [DOI] [PubMed] [Google Scholar]
- 27. Mochizuki K, Kagawa T, Numari A, Harris MJ, Itoh J, Watanabe N, et al. Two N‐linked glycans are required to maintain the transport activity of the bile salt export pump (ABCB11) in MDCK II cells. Am J Physiol Gastrointest Liver Physiol 2007;292:G818‐G828. [DOI] [PubMed] [Google Scholar]
- 28. Kagawa T, Watanabe N, Mochizuki K, Numari A, Ikeno Y, Itoh J, et al. Phenotypic differences in PFIC2 and BRIC2 correlate with protein stability of mutant Bsep and impaired taurocholate secretion in MDCK II cells. Am J Physiol Gastrointest Liver Physiol 2008;294:G58‐G67. [DOI] [PubMed] [Google Scholar]
- 29. Byrne JA, Strautnieks SS, Ihrke G, Pagani F, Knisely AS, Linton KJ, et al. Missense mutations and single nucleotide polymorphisms in ABCB11 impair bile salt export pump processing and function or disrupt pre‐messenger RNA splicing. Hepatology 2009;49:553‐567. [DOI] [PubMed] [Google Scholar]
- 30. Farmer DG, Venick RS, McDiarmid SV, Ghobrial RM, Gordon SA, Yersiz H, et al. Predictors of outcomes after pediatric liver transplantation: an analysis of more than 800 cases performed at a single institution. J Am Coll Surg 2007;204:904‐914. [DOI] [PubMed] [Google Scholar]
- 31. Muiesan P, Vergani D, Mieli‐Vergani G. Liver transplantation in children. J Hepatol 2007;46:340‐348. [DOI] [PubMed] [Google Scholar]
- 32. Knisely AS, Strautnieks SS, Meier Y, Stieger B, Byrne JA, Portmann BC, et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 2006;44:478‐486. [DOI] [PubMed] [Google Scholar]
- 33. Keitel V, Burdelski M, Vojnisek Z, Schmitt L, Haussinger D, Kubitz R. De novo bile salt transporter antibodies as a possible cause of recurrent graft failure after liver transplantation: a novel mechanism of cholestasis. Hepatology 2009;50:510‐517. [DOI] [PubMed] [Google Scholar]
- 34. Jara P, Hierro L, Martinez‐Fernandez P, Alvarez‐Doforno R, Yanez F, Diaz MC, et al. Recurrence of bile salt export pump deficiency after liver transplantation. N Engl J Med 2009;361:1359‐1367. [DOI] [PubMed] [Google Scholar]
- 35. Lykavieris P, van Mil S, Cresteil D, Fabre M, Hadchouel M, Klomp L, et al. Progressive familial intrahepatic cholestasis type 1 and extrahepatic features: no catch‐up of stature growth, exacerbation of diarrhea, and appearance of liver steatosis after liver transplantation. J Hepatol 2003;39:447‐452. [DOI] [PubMed] [Google Scholar]
- 36. Chen F, Ananthanarayanan M, Emre S, Neimark E, Bull LN, Knisely AS, et al. Progressive familial intrahepatic cholestasis, type 1, is associated with decreased farnesoid X receptor activity. Gastroenterology 2004;126:756‐764. [DOI] [PubMed] [Google Scholar]
- 37. Usui M, Isaji S, Das BC, Kobayashi M, Osawa I, Iida T, et al. Liver retransplantation with external biliary diversion for progressive familial intrahepatic cholestasis type 1: a case report. Pediatr Transplant 2009;13:611‐614. [DOI] [PubMed] [Google Scholar]
- 38. Nicastro E, Stephenne X, Smets F, Fusaro F, de Magnee C, Reding R, et al. Recovery of graft steatosis and protein‐losing enteropathy after biliary diversion in a PFIC 1 liver transplanted child. Pediatr Transplant 2012;16:E177‐E182. [DOI] [PubMed] [Google Scholar]
- 39. Mali VP, Fukuda A, Shigeta T, Uchida H, Hirata Y, Rahayatri TH, et al. Total internal biliary diversion during liver transplantation for type 1 progressive familial intrahepatic cholestasis: a novel approach. Pediatr Transplant 2016;20:981‐986. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1168/full.
Supplementary Table 1. Growth post‐PEBD
Supplementary Table 2. Clinically meaningful improvement in blood biochemistry post‐PEBD.
Supplementary Table 3. Reported use of immunosuppressants post‐transplant.
Supplementary Table 4. Relative odds of blood chemistry being abnormal, after recovery from LTX
Supplementary Table 5. Height and weight after LTX
Supplementary Table 6. Puberty in Patients who have undergone PEBD and/or LTX and were past the normal age for puberty completion at last data collection.