

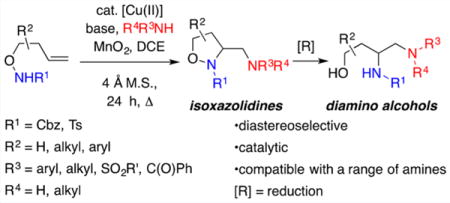
Abstract
A convenient copper-catalyzed intramolecular/intermolecular alkene diamination reaction to synthesize 3-aminomethyl-functionalized isoxazolidines under mild reaction conditions and with generally high levels of diastereoselectivity was achieved. This reaction demonstrates that previously underutilized unsaturated carbamates are good [Cu]-catalyzed diamination substrates. Sulfonamides, anilines, benzamide, morpholine, and piperidine can serve as the external amine source. This relatively broad amine range is attributed to the mild reaction conditions. Reduction of the N–O bond could also be achieved, revealing the corresponding 3,4-diamino-1-alcohols efficiently.
Keywords: isoxazolidine, diamination, copper, alkene, electron-rich amines
Graphical abstract
Because of the significant utility of vicinal 1,2-diamines, the diamination of alkenes is a reaction that has inspired the development of many synthetic methodologies.1 In particular, several powerful transition-metal-catalyzed alkene diamination reactions have been developed over the past decade.1 Palladium-catalyzed alkene diaminations have enabled the synthesis of cyclic ureas, amides, and carbamates with good regioselectivity and enantioselectivity.1b,c,2 Copper-catalyzed alkene diaminations reported by our group,3 and others,1b,4 have enabled both diastereoselective3a and enantioselective3a,4a synthesis of vicinal diamines containing sulfonamides and ureas. Enantioselective diamination of styrenes enabled by chiral hypervalent iodine catalysts have also been introduced.5 In the majority of examples, the exogenous amine nucleophile is uniformly electron-deficient. More recently, attention has been given to expansion of the alkene and amine scope, with an emphasis on the ease of amine functionalization. A [Rh]-catalyzed tandem aziridination/aziridine opening of a range of alkenes with sulfamides has contributed in this respect.6 More-recent copper-catalyzed alkene diaminations employing unsaturated oximes or hydroxylamines as the alkene component have enabled the scope of the amine nucleophile to be expanded to more electron-rich amines.7,8 Electron-rich amine nucleophiles pose a particular challenge to [Cu]-catalyzed diamination catalysis as they may succumb to oxidation themselves.9 Given the continued interest in vicinal diamines, we pursued expansion of our copper-catalyzed alkene diamination reaction to the synthesis of variously substituted 3-aminomethyl isoxazolidines with a range of external amine nucleophiles.
The synthesis of isoxazolidines is worth pursuing, because of their value as synthetic intermediates and their attractive biological activities.10 Until now, no alkene diamination strategy has been developed for the synthesis of isoxazolidine rings. Previous strategies employed for the synthesis of isoxazolidines include intermolecular [3 + 2] dipolar cycloaddition reactions11,12 and intramolecular addition of the hydroxylamines onto alkenes and allenes.13,14 However, these previous methods do not directly produce 3-aminomethyl isoxazolidines.
We have recently disclosed the stereoselecive synthesis of N-sulfonyl 3-methyleneoxy isoxazolidines via copper-catalyzed alkene aminooxygenation of N-sulfonyl-O-butenyl hydroxyl amines with yields up to 92% and diastereomeric ratio (dr) values up to >20:1.15 We observed these substrates to be reactive at much lower temperature (e.g., 60 °C) than other previously explored substrates including N-sulfonyl-2-allylanilines and N-sulfonyl-4-pentenylamines (which reacted at 100–110 °C).3 Herein, we report the first alkene diamination strategy to access 3-aminomethyl isoxazolidines. This reaction extends the scope of our diasteroselective copper-catalyzed intra/intermolecular alkene diamination reaction.3 With respect to both the intramolecular amine (alkenylamine) component, the use of unsaturated N-carbamoyl amines is enabled for the first time.16a,b Carbamates have demonstrated considerable utility in organic synthesis and medicinal chemistry.16c With respect to the exogenous amine component, the use of anilines, morpholine, piperidine, and benzamide under our [Cu]-catalyzed alkene diamination conditions3 was enabled for the first time. A key factor in the increased range observed in these reactions appears to be the innate superior reactivity of the unsaturated hydroxylamine substrates, which allows the comparatively less-reactive unsaturated carbamates to undergo copper-catalyzed cyclization and also enables an overall lower reaction temperature (60–85 °C, compared to 100 or 110 °C previously required with N-sulfonyl-2-allylanilines and 4-pentenylsulfonamides), which likely contributes to the better catalyst turnover and stability with otherwise challenging electron-rich amine nucleophiles (vide infra).
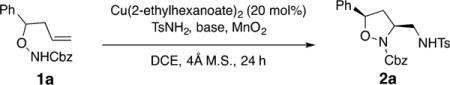
We launched our study by subjecting hydroxylamine 1a and TsNH2 to various reaction conditions (Table 1). To our delight, this Cbz-protected substrate 1a showed reactivity, for the first time, in copper-catalyzed alkene difunctionalization chemistry, and the desired 3-aminomethyl isoxazolidine 2a was obtained in 22% isolated yield (>90% conversion) using copper(II) 2-ethylhexanoate (20 mol %), MnO2 (85%, <5 μ) and 2,6-di-tert-butyl-4-methylpyridine at 95 °C (Table 1, entry 1). Decreasing the temperature to 85 °C enabled an increase in the isolated yield to 75% (Table 1, entry 2), which suggests that the substrate decomposes at higher temperatures.
Table 1.
Optimization of the Reaction Conditionsa
| ||||
|---|---|---|---|---|
|
| ||||
| entry | temperature (°C) | base | yieldb (%) | diastereomeric ratio, drc |
|
| ||||
| 1 | 95 | 2,6-di-t-Bu-4-Me-pyridine | 22 | 10:1 |
| 2 | 85 | 2,6-di-t-Bu-4-Me-pyridine | 75 | 10:1 |
| 3 | 85 | Cs2CO3 | 14 | 10:1 |
| 4 | 85 | K2CO3 | 48 | 10:1 |
| 5d | 85 | 2,6-di-t-Bu-4-Me-pyridine | 59 | 10:1 |
| 6e | 85 | 2,6-di-t-Bu-4-Me-pyridine | 53 | 10:1 |
| 7 | 75 | 2,6-di-t-Bu-4-Me-pyridine | 68 | 10:1 |
| 8f | 85 | 2,6-di-t-Bu-4-Me-pyridine | complex mixture | |
| 9g | 85 | 2,6-di-t-Bu-4-Me-pyridine | 15 | 10:1 |
| 10h | 85 | 2,6-di-t-Bu-4-Me-pyridine | 35 | 5:1 |
Reaction conditions: 1a (0.134 mmol), Cu(2-ethylhexanoate)2 (20 mol %), TsNH2 (300 mol %), MnO2 (270 mol %), base (100 mol %), DCE (0.2 M, 0.7 mL), flame activated 4 Å mol. sieves (13 mg), 85 °C, 24 h.
Isolated yield, chromatography by preparatory TLC on silica gel.
Determined by analysis of the crude 1H NMR.
Sulfonamide (200 mol %).
No molecular sieves.
DTBP (di-tert-butylperoxide) (300 mol %) was used instead of MnO2.
Oxygen (1 atm, balloon) was used instead of MnO2.
Cu(OTf)2 (20 mol %) and 1,10-phenanthroline (25 mol %) were used instead of Cu(2-ethylhexanoate)2.
The use of less-soluble bases such as Cs2CO3 or K2CO3 resulted in lower isolated yield (14% and 48%, respectively, Table 1, entries 3 and 4). In these examples, a hydroamination side product, likely the result of a Brønsted-acid-catalyzed alternative pathway, was observed (see Supporting Information (SI) for spectral data).3b Using 2 equiv instead of 3 equiv of the tosyl amine lowered the yield to 59% (Table 1, entry 5). Removal of 4 Å molecular sieves also diminished the isolated yield of 2a (Table 1, entry 6). Further lowering of the temperature to 75 °C reduced the yield to 68% for this substrate (Table 1, entry 7). No product was obtained by the use of di-tert-butylperoxide (DTBP) as an oxidant (Table 1, entry 8). Use of O2 (1 atm) as terminal oxidant enabled only 15% isolated yield of diamine 2a, along with multiple other products (Table 1, entry 9). When Cu(OTf)2 and 1,10-phenanthroline were used as catalyst and ligand, respectively, a 35% yield of 2a was obtained, along with at least two other side products (Table 1, entry 10).
With the optimal conditions in hand (Table 1, entry 2), we moved forward to establish the sulfonamide nucleophile scope (Chart 1). The N-Cbz α-phenyl substrate 1a underwent coupling with electron-rich (p-Me, p-OMe) as well as electron-deficient (p-CF3, p-NO2) aryl sulfonamides to afford the corresponding isoxazolidines 2a–2d in moderate to good yields. Alkyl sulfonamides 2-trimethylsilylethylsulfonamide and cyclopropylsulfonamide provided the corresponding isoxazolidines 2e and 2f in moderate yields. Isoxazolidines 2g and 2h were obtained from the N-tosyl substrate 1b. In all cases, good to excellent 3,5-cis diastereoselectivity was observed (relative stereochemistry assigned by NOE of 2b and by analogy).
Chart 1. Scope of the Sulfonamide Nucleophilesa,b,c,d.
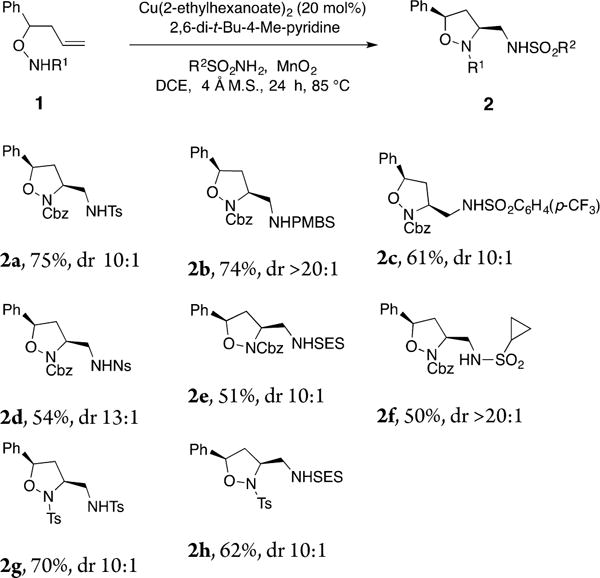
aFor reaction conditions, see Table 1, entry 2. bIsolated yield. cDiastereomeric ratios were determined by crude 1H NMR. dDiastereoselectivity assigned by NOE of 2b and by analogy.
We further expanded the scope of the diamination reaction with more electron-rich amines (Chart 2). Halogenated anilines (p-CF3, p-chloro, o-chloro, p-bromo, and p-fluoro) provided compounds 2i–2m in moderate to high yield and uniformly high diastereoselectivity. The secondary amine, N-methyl-p-chloroaniline, could also be used as a coupling partner, forming isoxazolidine 2n efficiently and selectively. The use of aniline as a nucleophile provided isoxazolidine 2o in moderate yield and good diastereoselectivity (54% yield, dr = 10:1). Use of the slightly more electron-rich p-t-butylaniline provides 2p in 40% yield, while the use of p-methoxyaniline gave <20% conversion (not shown).
Chart 2. External Amine Nucleophile Scopea,b,c,d.

aFor reaction conditions, see Table 1, entry 2. bIsolated yield. cDiastereomeric ratios were determined by crude 1H NMR. dRelative configuration assigned by NOE of 2n and 2t and by analogy. e200 mol % of amine nucleophile was used. fReaction run at 60 °C. gReaction was run with 40 mol % diethylsalicylamide in PhCF3 for 48 h.
When morpholine was used as the external amine, it provided isoxazolidines 2q and 2r in moderate yields (45% and 34%, respectively; see Chart 2). In these examples, the remainder of the mass was unreacted starting material, possibly due to catalyst poisoning by the exogenous amine. An initial attempt to couple piperidine with N-tosyl hydroxylamine 1b led to the formation of an undesired chlorine atom transfer side product (aminochlorination).14a Changing the solvent to PhCF3 enabled formation of the desired diamine 2s in low conversion. To minimize the ability of piperidine to complex with the catalyst, the copper(2-ethylhexanoate)2 (20 mol %) was treated with diethylsalicylamide (40 mol %) prior to addition of the substrate.16b,17 Gratifyingly, 50% of the desired diamine product 2s was then obtained (Chart 2). Finally, the use of benzamide as nucleophile afforded 2t in a modest 34% yield.
Many different heterocyclic and alkyl-substituted hydroxylamines 1 were also tested for their reactivity in our diamination reaction (Chart 3). Good yields were obtained for the formation of furanyl and the thienyl isoxazolidines 2u and 2v. Alkyl-substituted substrates were also amenable for the diamination reaction, although lower yields of their respective adducts 2u–2w were obtained. The TBDPS-protected substrate formed isoxazolidine 2z in good yield but moderate diastereoselectivity. While 3,5-cis-diastereoselectivity was favored with 2-substituted hydroxylamines, 3,4-trans-isoxazolidine 2aa was observed when a 3-phenyl hydroxylamine underwent cyclization and coupling (stereochemistry assigned by NOE). The unsubstituted hydroxylamine produced isolazolidine 2bb in 51% yield, indicating that backbone substituents are not required.
Chart 3. Alkene Scopea,b,c,d.
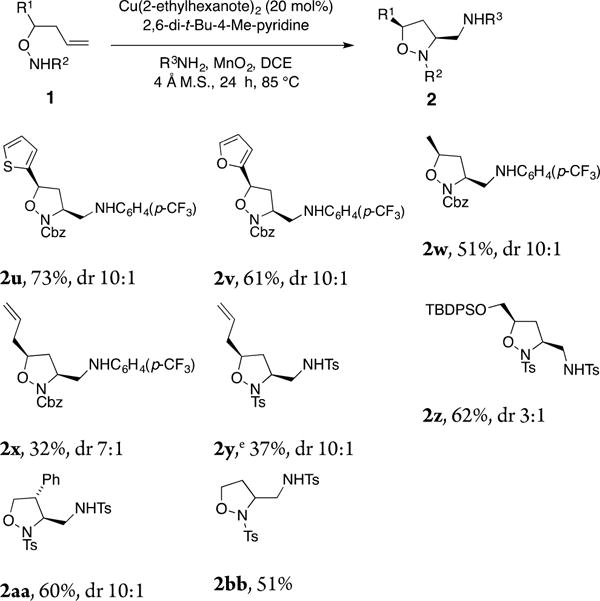
aFor reaction conditions, see Table 1, entry 2. bIsolated yield. cDiastereomeric ratios were determined by crude 1H NMR. dRelative configuration of 2z and 2aa assigned by NOE, or by analogy. eThe reaction was run for 6 h.
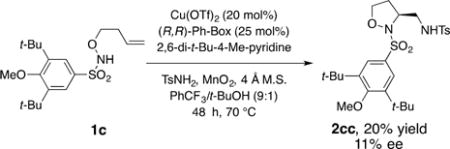
A catalytic enantioselective diamination protocol was also explored (eq 1). A brief substrate and solvent screen led us to
 |
(1) |
use 3,5-di-tert-butyl-4-methoxysulfonyl hydroxylamine 1c in PhCF3/t-BuOH (9:1) in an enantioselective diamination using [Cu(R,R)-Ph-box](OTf)2 as the chiral catalyst.3b While the yield and enantioselectivity of 2cc are low (20% yield, 11% ee), this result indicates feasibility and supports that the initial C–N bond formation could occur via aminocupration (vide infra, Scheme 1), in analogy to our previously reported diamination reactions,3,18 since some ligand-induced selectivity is observed. The comparatively low enantioselectivity may indicate some reversibility in the C–N bond-forming step (vide infra) or a small energy difference between competing pro-R and pro-S transition states.
Scheme 1.
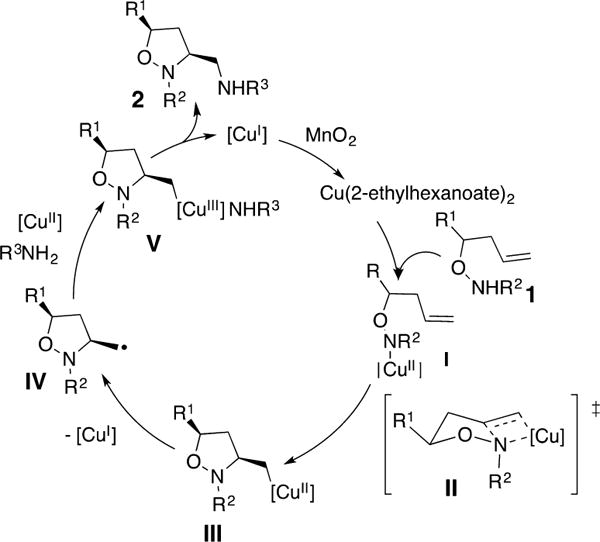
Proposed Reaction Mechanism
The proposed reaction mechanism is outlined in Scheme 1. Upon [N–Cu] bond formation to give I, a cis-aminocupration occurs via seven-membered chairlike transition state II that gives the unstable organocopper(II) intermediate III. The diastereoselectivity observed with substituted hydroxylamines originates from placement of the substituent in the pseudo-equatorial position in transition state II. Homolysis of the carbon–copper(II) bond gives [Cu(I)] and the carbon radical intermediate IV, which then adds to [Cu(II)]. The resulting amine-coordinated [Cu(III)] intermediate V then leads to 2 and [Cu(I)] via reductive elimination. Oxidation of [Cu(I)] to [Cu(II)] by MnO2 completes the catalytic cycle. The involvement of radical IV is implicated based on isotopic labeling studies performed in analogous diamination reactions.3a
One more clue regarding the reaction mechanism emerged from an attempted diamination of 1,1-disubstituted alkene 3 (Scheme 2). This alkene previously underwent aminooxygenation under copper catalysis in the presence of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) and O2 to provide isoxazolidine 4 (Scheme 2).15 To our surprise, when subjected to the copper-catalyzed diamination conditions, only oxidative amination product 5 was obtained (Scheme 2). Taken together, these results indicate that carbon radical intermediate IV′ could be in equilibrium with aminyl radical VI and that the relative rates of TEMPO radical trapping (of IV′) versus C–N bond formation (with regard to IV′, [Cu(II)] and TsNH2) are different and impact the product distribution. Thus, endo cyclization of aminyl radical VI onto its 1,1-disubstituted alkene to produce a tertiary carbon radical (not shown) that suffers oxidation to give alkene 5 is more favorable than the C–N bond formation involving carbon radical IV′, which would lead to a diamine product.19 The presence of a ring-opening pathway (such as IV′ to VI) could also account, at least in part, for the low observed enantioselectivity (eq 1), since the aminyl radical would produce a racemic product upon exocyclization to give a carbon radical like IV (Scheme 1). We have previously observed low enantioselectivity in related reactions where substrates can access a more-stabilized aminyl radical and where the carbon radical must engage in a relatively higher energy subsequent bond formation.17b
Scheme 2.
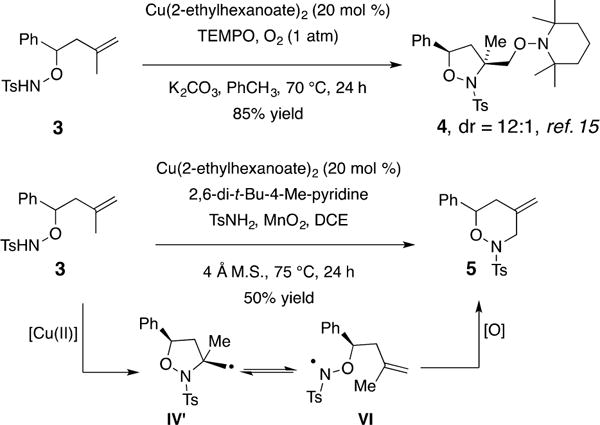
Attempted Diamination Gives Oxidative Amination
To demonstrate further synthetic utility, isoxazolidines 2b and 2z were converted to 3,4-diamino-1-alcohols 3a and 3b by reduction of the N–O bond with NaBH4 in the presence of Mo(CO)6 (Scheme 3).13e,20 These conditions proved far more effective than [Pd]-catalyzed hydrogenation.15 This new diamination methodology, in conjunction with N–O reduction, may well prove useful for the synthesis of 3,4-diamino-1-alcohols such as those found in bioactive natural products.21,22
Scheme 3.

N–O Bond Reduction
In summary, a broad range of 3-aminomethyl isoxazolidines 2 can be formed in moderate to good yields and with moderate to high diastereoselectivity via copper-catalyzed intramolecular/ intermolecular alkene diamination. The unsaturated hydroxylamine substrates 1 can bear Cbz and sulfonyl groups on the amine, and the intermolecular amine component can range from electron-deficient to more electron-rich. The higher reactivity of the hydroxylamine substrates 1, compared to 2-allylaniline and 4-pentenylamines previously explored,3 is possibly due to the greater nucleophilicity of the hydroxylamine, its greater ability to accommodate spin (or unpaired electron)18a or to a lower transannular strain at the transition state (e.g., II; see Scheme 1). This higher reactivity enables a lower reaction temperature that, in turn, appears to enable a broader amine scope. Thus, electron-rich amines are not inherently incompatible with copper-catalyzed alkene diaminations that occur under oxidative conditions; rather, milder reaction conditions, especially with respect to reaction temperature, can facilitate their successful application.23 The added ligand, diethylsalicylamide, also served to enable diamination with piperidine, perhaps by preventing catalyst poisoning. Further scope expansion and exploration of enantioselective conditions are in progress and will be reported in due course.
Supplementary Material
Acknowledgments
We thank the Egyptian government and Helwan University for the graduate school fellowship to Z.M.K.
Funding
We thank the National Institutes of Health for financial support (No. GM 078383).
ABBREVIATIONS
- DCE
1,2-dichloroethane
- Ts
toluene sulfonyl
- Ns
p-nitrobenzenesulfonyl
- PMBS
p-methoxybenzenesulfonyl
- TBDPS
t-Bu-diphenylsilyl
- Cbz
carboxybenzyl
- SES
2-trimethylsilylethylsulfonyl
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.7b01362.
Experimental procedures, tabulated characterization data and copies of NMR spectra for all new compounds (PDF)
ORCID
Sherry R. Chemler: 0000-0001-6702-5032
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.(a) De Jong S, Nosal DG, Wardrop DJ. Tetrahedron. 2012;68:4067–4105. doi: 10.1016/j.tet.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhu Y, Cornwall RG, Du H, Zhao B, Shi Y. Acc Chem Res. 2014;47:3665–3678. doi: 10.1021/ar500344t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Muniz K, Martinez C. J Org Chem. 2013;78:2168–2174. doi: 10.1021/jo302472w. [DOI] [PubMed] [Google Scholar]
- 2.Ingalls EL, Sibbald PA, Kaminsky W, Michael FE. J Am Chem Soc. 2013;135:8854–8856. doi: 10.1021/ja4043406. [DOI] [PubMed] [Google Scholar]
- 3.(a) Sequeira FC, Turnpenny BW, Chemler SR. Angew Chem, Int Ed. 2010;49:6365–6368. doi: 10.1002/anie.201003499. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Turnpenny BW, Chemler SR. Chem Sci. 2014;5:1786–1793. doi: 10.1039/C4SC00237G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Fu S, Yang H, Li G, Deng Y, Jiang H, Zeng W. Org Lett. 2015;17:1018–1021. doi: 10.1021/acs.orglett.5b00131. [DOI] [PubMed] [Google Scholar]; (b) Zhang B, Studer A. Org Lett. 2014;16:1790–1793. doi: 10.1021/ol500513b. [DOI] [PubMed] [Google Scholar]
- 5.(a) Roben C, Souto JA, Gonzalez Y, Lishchynskyi A, Muniz K. Angew Chem, Int Ed. 2011;50:9478–9482. doi: 10.1002/anie.201103077. [DOI] [PubMed] [Google Scholar]; (b) Muniz K, Barreiro L, Romero RM, Martinez C. J Am Chem Soc. 2017;139:4354–4357. doi: 10.1021/jacs.7b01443. [DOI] [PubMed] [Google Scholar]
- 6.Olson DE, Su JY, Roberts DA, Du Bois J. J Am Chem Soc. 2014;136:13506–13509. doi: 10.1021/ja506532h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Shen K, Wang Q. Chem Sci. 2015;6:4279–4283. doi: 10.1039/c5sc00897b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu RH, Wei D, Han B, Yu W. ACS Catal. 2016;6:6525–6530. [Google Scholar]
- 8.Metal-free alkene diaminations with electron-rich amines; see:; (a) Ortiz GX, Kang B, Wang Q. J Org Chem. 2014;79:571–581. doi: 10.1021/jo4022666. [DOI] [PubMed] [Google Scholar]; (b) Hong KB, Johnston JN. Org Lett. 2014;16:3804–3807. doi: 10.1021/ol501693j. [DOI] [PubMed] [Google Scholar]; (c) Danneman MW, Hong KB, Johnston JN. Org Lett. 2015;17:2558–2561. doi: 10.1021/acs.orglett.5b01177. [DOI] [PubMed] [Google Scholar]
- 9.Xu B, Hartigan EM, Feula G, Huang Z, Lumb J-P, Arndtsen BA. Angew Chem, Int Ed. 2016;55:15802–15806. doi: 10.1002/anie.201609255. and references therein. [DOI] [PubMed] [Google Scholar]
- 10.(a) Berthet M, Cheviet T, Dujardin G, Parrot I, Martinez J. Chem Rev. 2016;116:15235–15283. doi: 10.1021/acs.chemrev.6b00543. [DOI] [PubMed] [Google Scholar]; (b) Buhrlage SJ, Bates CA, Rowe SP, Minter AR, Brennan BB, Majmudar CY, Wemmer DE, Al-Hashimi H, Mapp AK. ACS Chem Biol. 2009;4:335–344. doi: 10.1021/cb900028j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huang SP, Koyama Y, Ikeda D, Kondo S, Takeuchi T. J Antibiot. 1992;45:1939–1948. doi: 10.7164/antibiotics.45.1939. [DOI] [PubMed] [Google Scholar]; (d) Khazir J, Singh PP, Reddy DM, Hyder I, Shafi S, Sawant SD, Chashoo G, Mahajan A, Alam MS, Saxena AK, Arvinda S, Gupta BD, Kumar HMS. Eur J Med Chem. 2013;63:279–289. doi: 10.1016/j.ejmech.2013.01.003. [DOI] [PubMed] [Google Scholar]; (e) Kokosza K, Andrei G, Schols D, Snoeck R, Piotrowska DG. Bioorg Med Chem. 2015;23:3135–3146. doi: 10.1016/j.bmc.2015.04.079. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Mancini F, Piazza MG, Trombini C. J Org Chem. 1991;56:4246–4252. [Google Scholar]; (g) Mullen GB, DeCory TR, Mitchell JT, Allen SD, Kinsolving CR, Georgiev V. J Med Chem. 1988;31:2008–2014. doi: 10.1021/jm00118a027. [DOI] [PubMed] [Google Scholar]; (h) Sethi N, Bhatti R, Ishar MPS. Anal Chem Lett. 2013;3:159–166. [Google Scholar]
- 11.(a) Brandi A, Cardona F, Cicchi S, Cordero FM, Goti A. Chem - Eur J. 2009;15:7808–7821. doi: 10.1002/chem.200900707. [DOI] [PubMed] [Google Scholar]; (b) Rueck-Braun K, Freysoldt THE, Wierschem F. Chem Soc Rev. 2005;34:507–516. doi: 10.1039/b311200b. [DOI] [PubMed] [Google Scholar]; (c) Gallos JK, Koumbis AE. Curr Org Chem. 2003;7:397–425. [Google Scholar]
- 12.(a) Jayaprakash Rao Y, Pravardhan Reddy E, Sridhar B, Lakshmi JK, Subba Reddy BV. Tetrahedron Lett. 2016;57:2853–2856. [Google Scholar]; (b) Barber JS, Styduhar ED, Pham HV, McMahon TC, Houk KN, Garg NK. J Am Chem Soc. 2016;138:2512–2515. doi: 10.1021/jacs.5b13304. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ma LL, Wang W, Wang GC. RSC Adv. 2016;6:53839–53851. [Google Scholar]
- 13.(a) Bates RW, Sa-Ei K. Org Lett. 2002;4:4225–4227. doi: 10.1021/ol026701d. [DOI] [PubMed] [Google Scholar]; (b) Dongol KG, Tay BY. Tetrahedron Lett. 2006;47:927–930. [Google Scholar]; (c) Lemen GS, Giampietro NC, Hay MB, Wolfe JP. J Org Chem. 2009;74:2533–2540. doi: 10.1021/jo8027399. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bates RW, Lu Y. Org Lett. 2010;12:3938–3941. doi: 10.1021/ol1016492. [DOI] [PubMed] [Google Scholar]; (e) Malkov AV, Lee DS, Barlog M, Elsegood MRJ, Kocovsky P. Chem - Eur J. 2014;20:4901–4905. doi: 10.1002/chem.201400123. [DOI] [PubMed] [Google Scholar]; (f) Lalonde RL, Wang ZJ, Mba M, Lackner AD, Toste FD. Angew Chem, Int Ed. 2010;49:598–601. doi: 10.1002/anie.200905000. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Wu L, Shi M. Chem -Eur J. 2011;17:13160–13165. doi: 10.1002/chem.201102159. [DOI] [PubMed] [Google Scholar]; (h) Cornil J, Gonnard L, Bensoussan C, Serra-Muns A, Gnamm C, Commandeur C, Commandeur M, Reymond S, Guérinot A, Cossy J. Acc Chem Res. 2015;48:761–773. doi: 10.1021/ar5004412. [DOI] [PubMed] [Google Scholar]
- 14.(a) Janza B, Studer A. Synthesis. 2002;2002:2117–2123. [Google Scholar]; (b) Moriyama K, Izumisawa Y, Togo H. J Org Chem. 2011;76:7249–7255. doi: 10.1021/jo201113r. [DOI] [PubMed] [Google Scholar]; (c) Egart B, Lentz D, Czekelius C. J Org Chem. 2013;78:2490–2499. doi: 10.1021/jo3026725. [DOI] [PubMed] [Google Scholar]
- 15.Karyakarte SD, Smith TP, Chemler SR. J Org Chem. 2012;77:7755–7760. doi: 10.1021/jo3013226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For reports where benzyl carbamate (RNH-Cbz) substrates do not work in copper-promoted and catalyzed reactions, see:; (a) Fuller PH, Kim JW, Chemler SR. J Am Chem Soc. 2008;130:17638–17639. doi: 10.1021/ja806585m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Casavant BJ, Khoder ZM, Berhane IA, Chemler SR. Org Lett. 2015;17:5958–5961. doi: 10.1021/acs.orglett.5b02833. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a review on utility of carbamates in medicinal chemistry, see:; (c) Ghosh AK, Brindisi M. J Med Chem. 2015;58:2895–2940. doi: 10.1021/jm501371s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Kwong FY, Buchwald SL. Org Lett. 2003;5:793–796. doi: 10.1021/ol0273396. [DOI] [PubMed] [Google Scholar]; (b) Zeng W, Chemler SR. J Am Chem Soc. 2007;129:12948–12949. doi: 10.1021/ja0762240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Paderes MC, Belding L, Fanovic B, Dudding T, Keister JB, Chemler SR. Chem - Eur J. 2012;18:1711–1726. doi: 10.1002/chem.201101703. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Paderes MC, Keister JB, Chemler SR. J Org Chem. 2013;78:506–515. doi: 10.1021/jo3023632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liwosz TW, Chemler SR. Chem - Eur J. 2013;19:12771–12777. doi: 10.1002/chem.201301800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang MXW, Warshakoon NC, Miller MJ. J Org Chem. 2005;70:2824–2827. doi: 10.1021/jo0484070. [DOI] [PubMed] [Google Scholar]
- 21.Iwatsuki M, Uchida R, Yoshijima H, Ui H, Shiomi K, Kim YP, Hirose T, Sunazuka T, Abe A, Tomoda H, Omura S. J Antibiot. 2008;61:230–236. doi: 10.1038/ja.2008.33. [DOI] [PubMed] [Google Scholar]
- 22.Harrak Y, Barra CM, Delgado A, Castano AR, Llebaria A. J Am Chem Soc. 2011;133:12079–12084. doi: 10.1021/ja202610x. [DOI] [PubMed] [Google Scholar]
- 23.For an example of a copper-catalyzed oxidative amination using electron-rich amines, see:; Evans RW, Zbieg JR, Zhu S, Li W, MacMillan DWC. J Am Chem Soc. 2013;135:16074–16077. doi: 10.1021/ja4096472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.