

Summary
Aggregates of human islet amyloid polypeptide (IAPP) in the pancreas of patients with type-2 diabetes (T2D) are thought to contribute to β-cell dysfunction and death. To understand how IAPP harms cells and how this might be overcome, we created a yeast model of IAPP toxicity. Ste24, an evolutionarily conserved protease that was recently reported to degrade peptides stuck within the translocon between the cytoplasm and the endoplasmic reticulum, was the strongest suppressor of IAPP toxicity. By testing variants of the human homolog, ZMPSTE24, with varying activity levels, the rescue of IAPP toxicity proved to be directly proportional to the declogging efficiency. Clinically relevant ZMPSTE24 variants identified in the largest database of exomes sequences derived from T2D patients were characterized using the yeast model, revealing 14 partial loss-of-function variants, which were enriched among diabetes patients over 2-fold. Thus, clogging of the translocon by IAPP oligomers may contribute to β-cell failure.
Graphical abstract
Introduction
Type-2 diabetes (T2D) is a widespread and growing metabolic disease that currently afflicts nearly 415 million people worldwide. The pathogenesis of T2D is multi-faceted with at least two fundamental components: 1) development of insulin resistance and 2) pancreatic β-cell failure. In patients with T2D, a 37-amino acid hormone that is co-secreted with insulin, islet amyloid polypeptide (IAPP), typically misfolds and forms amyloid deposits in pancreatic islets (Clark et al., 1988; Cooper et al., 1987; Westermark, 1972).
In health, IAPP plays a role in glycemic regulation. It inhibits insulin secretion, slows gastric emptying, and suppresses appetite. In disease, the buildup of toxic IAPP aggregates likely contribute to β-cell failure (Jurgens et al., 2011; Lorenzo et al., 1994; Mukherjee et al., 2015). Supporting the hypothesis that toxic IAPP aggregates play a role in diabetes, a mutation in IAPP which converts serine at position 20 to glycine has been shown to increase the amyloidogenicity and toxicity of the peptide (Ma et al., 2001; Sakagashira et al., 2000). This mutation has been associated with premature onset diabetes (Sakagashira et al., 1996) and more rapid deterioration of insulin secretion (Morita et al., 2011). Although mouse IAPP is not amyloidogenic, transgenic mice overexpressing human IAPP developed spontaneous diabetes, with mice expressing higher levels of IAPP progressed to diabetes more rapidly than those expressing lower levels of IAPP (Zhang et al., 2014). IAPP aggregates have recently been shown to behave as prions in mice, signifying that IAPP aggregation could accelerate and spread once it has started (Mukherjee et al., 2017). Thus, it will be necessary intervene at the earliest onset of aggregation to avoid irreparable damage to β-cells.
Similar to findings for neurodegeneration-associated protein aggregates, the consensus on what constitutes the toxic form of IAPP has shifted from amyloid deposits, to smaller, diffusible oligomers. Indeed, the extent of IAPP amyloid deposition often does not correlate with β-cell death (Butler et al., 2004; Janson et al., 1996). IAPP oligomers and many toxic oligomers share a common epitope (Gurlo et al., 2010; Ritzel et al., 2006), which is detectable by an antibody raised against Aβ oligomers (Kayed, 2003). Staining human IAPP-expressing mouse islets with this oligomer-specific antibody has revealed that IAPP oligomers are predominantly found intracellularly and independent of IAPP amyloid deposits (Lin et al., 2007). IAPP oligomers likely disrupt multiple aspects of β-cell biology. They induce endoplasmic reticulum (ER) stress (Huang et al., 2007), proteasome deficiency (Costes et al., 2010), defects in autophagy (Rivera et al., 2014), and cell membrane permeabilization (Janson et al., 1999). Yet, we do not have a complete understanding of how IAPP aggregates cause toxicity, preempting our ability to identify ways to avert it. Thus, it is critical that we develop new tools in order to discover potential avenues for therapeutic intervention.
The remarkable evolutionary conservation of proteostasis mechanisms among eukaryotes makes Saccharomyces cerevisiae a powerful model system for interrogating proteotoxicities. Comprehensive, genome-wide screens in yeast expressing α-synuclein (Outeiro, 2003), TDP-43 (Kim et al., 2013), Aβ (Treusch et al., 2011), FUS (Ju et al., 2011), polyglutamine (Kayatekin et al., 2014; Ripaud et al., 2014; Wolfe et al., 2014), and other toxic proteins (Couthouis et al., 2011) have identified distinct sets of toxicity modifiers for each protein.
Building on the successes of these yeast models, we have created the first yeast model of IAPP toxicity. Strains expressing IAPP oligomers experienced a strong growth defect enabling genome-wide overexpression and deletion screens for modifiers of this defect. The known functions of the genetic modifiers recovered from these screens provided insights into pathways perturbed by IAPP. The strongest suppressor recovered was Ste24, a protease that is highly conserved from yeast to humans (ZMPSTE24). Mutations in ZMPSTE24 have been previously shown to cause laminopathies such as mandibuloacral dysplasia (Agarwal et al., 2003) and restrictive dermopathy (Navarro et al., 2004), as well as maturity-onset diabetes of the young (Flannick et al., 2016). However, ZMPSTE24 had never been associated with IAPP toxicity. As Schuldiner and colleagues recently demonstrated, Ste24 and ZMPSTE24 clear translocons of polypeptides that have stalled during translocation (Ast et al., 2016). Our results, therefore, revealed a previously unidentified proteotoxic mechanism for IAPP oligomers: clogging of the ER translocon.
Results
The yeast model of IAPP toxicity
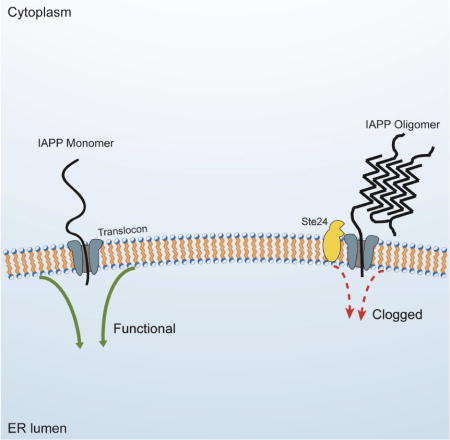
IAPP is post-translationally processed from 89 to 37 amino acids while transiting the secretory pathway of pancreatic β-cells, at the end of which it is stored in secretory granules. To mimic endogenous IAPP trafficking in yeast, we adapted a strategy successfully employed for our yeast model of Aβ toxicity, which exposes the cytoplasm, as well as much of the secretory and endocytic pathways, to the peptide (Treusch et al., 2011). We fused the ER signal peptide of the Kar2 protein (Kar2SS) to the N-terminus of IAPP. Once this fusion protein enters the ER, the signal peptide is removed. IAPP is then trafficked through the secretory pathway to the plasma membrane, where it is retained by the cell wall. There, IAPP can undergo endocytosis and traffic through endocytic compartments.
Overexpressing IAPP monomers did not produce sufficient toxicity under conditions required for screening (Figure S1A, B). This is in keeping with previous work indicating that IAPP oligomers, not monomers, are responsible for toxicity. To increase the production of proteotoxic IAPP molecules, we created a genetically encoded IAPP oligomer, which we expected would model some aspects of IAPP proteotoxicity. This approach enabled us to bypass a rate-limiting step of protein aggregation: primary nucleation. We linked six IAPP monomers into a single polypeptide and fused it to the Kar2 signal peptide forming a construct we termed 6xIAPP (Figure S1). Expression of 6xIAPP produced protein of the expected size and elicited a strong growth defect from a single-copy insertion into the genome (Figure 1A, B). The two bands visible by western blot resulted from the presence or absence of the Kar2 signal peptide. 6xIAPP tagged with monomeric super folder GFP (msfGFP, (Landgraf et al., 2016)) retained its toxicity (Figure S1C) and its pattern of distribution in the cell was consistent with localization to secretory compartments (Figure 1C). Since 6xIAPP was properly localized and produced a growth defect strong enough for screening, we used the 6xIAPP expressing yeast as our model of IAPP toxicity.
Figure 1. Growth defects and ER stress in yeast expressing 6xIAPP.
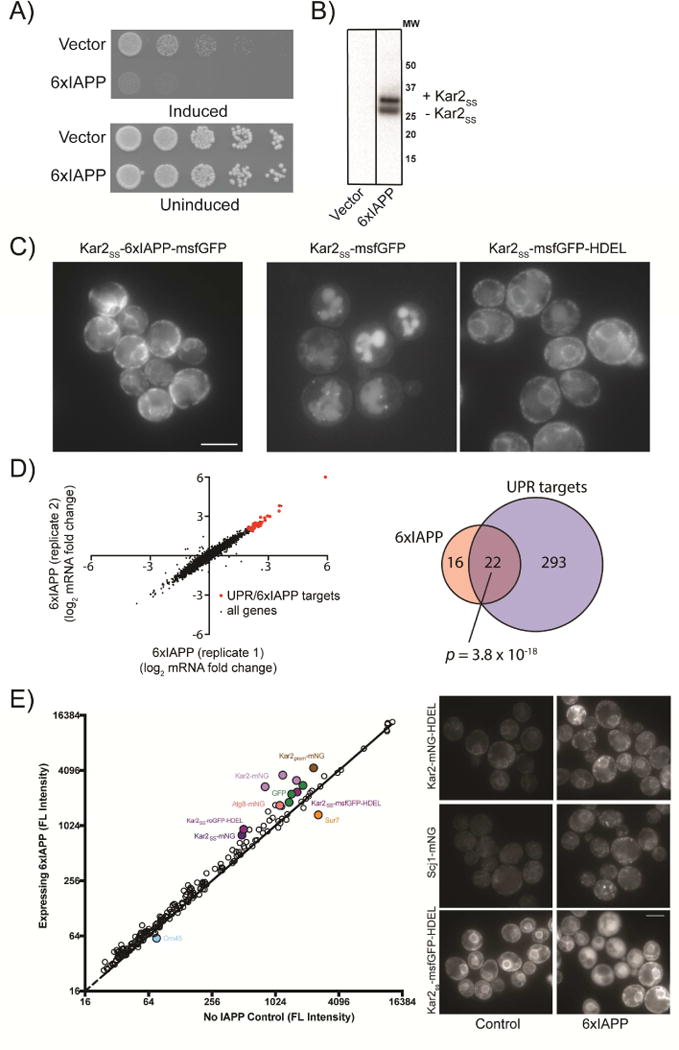
A) Yeast harboring empty vector or 6xIAPP were induced with 100 nM estradiol and grown on agar plates for 24 hrs. B) Western blot of cells expressing 6xIAPP. C) Cells expressing 6xIAPP-msfGFP fused to a Kar2 signal peptide (Kar2SS) (panel 1), msfGFP fused to Kar2SS (panel 2), and msfGFP fused to Kar2SS and an ER retention peptide (HDEL) (Panel 3). Scale bar = 5 μm. D) RNAseq data from 2 biological replicates of strains harboring a single copy of 6xIAPP. In red are genes upregulated in both the IAPP strains and in the unfolded protein response. The Venn diagram shows the genes typically upregulated in the UPR (blue) and those upregulated >4 fold when 6xIAPP was expressed (pink). The overlap between the two sets is shown in dark pink, along with the p-value calculated using Fisher’s exact test. E) Mean GFP intensity comparison across all fluorescent reporter strains for cells expressing 6xIAPP and controls. The solid line represents equal GFP expression. Filled circles are reporter strains with large intensity changes upon 6xIAPP expression. The most highly perturbed strains (Kar2ss-GFP-HDEL, Scj1-mNG, Kar2-mNG-HDEL) were measured independently and are shown on the right. Scale bar = 5 μm. See also Figure S1.
IAPP oligomers induced the unfolded protein response, ER stress, and a translocation defect
Before making genetic manipulations, we observed the effects of IAPP oligomer overexpression in yeast. First, we measured changes in the transcriptome of 6xIAPP-expressing yeast using RNAseq (Figure 1D). Relative to control cells, 6xIAPP-expressing cells had 38 genes with >4-fold increased expression. Of these, 22 were targets of the unfolded protein response (UPR) (Pincus et al., 2014), representing a highly significant enrichment for this pathway (p = 3.8 × 10−18, Fisher’s exact test). We confirmed the activation of the UPR by expressing 6xIAPP in a reporter strain harboring GFP under the control of a UPR-inducible promoter (Figure S1D). These results gave us confidence that our 6xIAPP-expressing yeast were modeling a biologically relevant toxicity, as ER stress is also a phenotype of IAPP toxicity in other model systems (Huang et al., 2007; Matveyenko et al., 2009).
We employed a fluorescent reporter library recently constructed in our lab (Landgraf et al., manuscript in preparation) to further probe the mechanisms of IAPP toxicity (Figure 1E). This library allowed us to examine changes in the levels and localization of key proteins. Furthermore, the sensitivity of this library to proteotoxic stresses allowed us to interrogate cellular responses at low levels of toxicity (Figure S1E). For most reporter strains in the library, cells expressing 6xIAPP had higher mean fluorescence intensity relative to controls, potentially due to the slower growth rate of the 6xIAPP-expressing cells or lower protein turnover in these cells. ER-stress reporter strains (such as those expressing monomeric Neon Green (mNG) fusions of Kar2 and Scj1, tagged at their endogenous loci) had greatly elevated fluorescence, consistent with the ER stress signal in the RNAseq data. The most striking changes were in strains that overexpressed ER-targeted GFP (Kar2SS-mNG, Kar2SS-msfGFP-HDEL, etc.). In IAPP-expressing cells, the ER-targeted GFP molecules were localized to the cytoplasm instead of the ER. Therefore, we concluded that 6xIAPP induces ER stress and impairs transport into the ER.
Genome-wide screens identified protective pathways and highlighted the translocon as a focus of IAPP oligomer toxicity
To identify modifiers of IAPP toxicity, we performed a genome-wide overexpression screen. We created a library of diploid yeast carrying two copies of estradiol-inducible 6xIAPP and 5532 yeast open reading frames (ORFs) on low-copy plasmids under the control of the galactose-inducible promoter (Hu et al., 2007). This diploid, two-copy 6xIAPP strain experienced significant IAPP toxicity but not lethality, allowing for simultaneous screening for enhancers and suppressors of IAPP toxicity. An ORF was defined as modifier if it changed the growth of IAPP-expressing cells while producing no change in growth on its own (Figure 2A). After extensive secondary validation, we confirmed 29 suppressors (Figure S2A, Table S1) and 3 enhancers (Figure S2B) as bona fide modifiers of 6xIAPP toxicity.
Figure 2. Genome-wide screens for modifiers of IAPP toxicity.
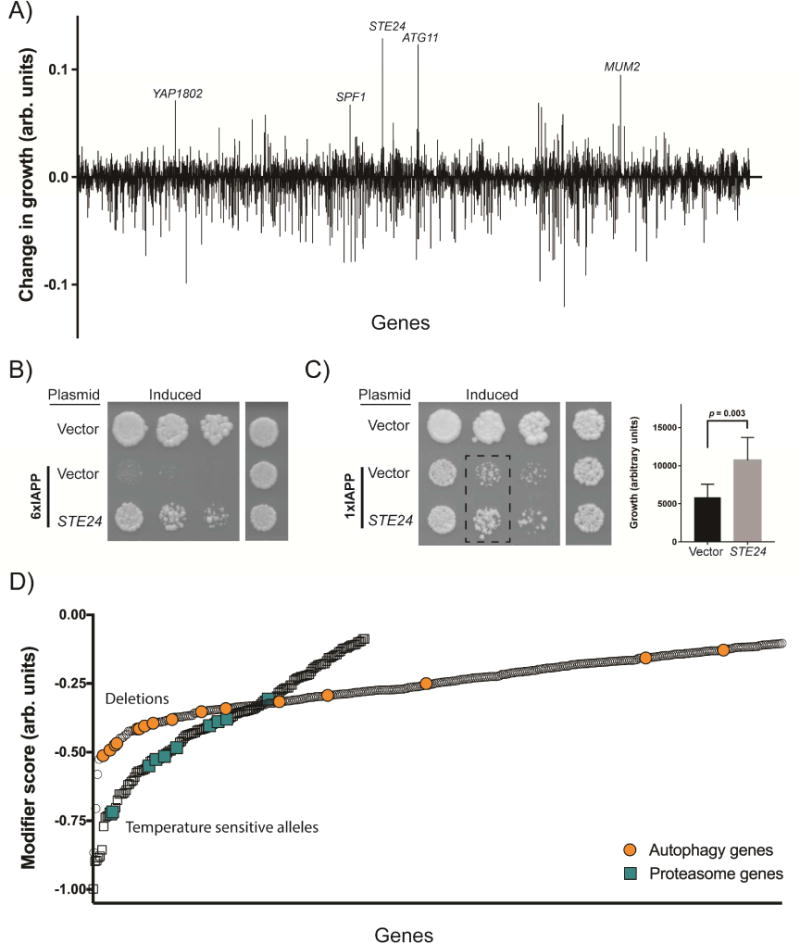
A) The suppressors and enhancers recovered from the overexpression screen, highlighting the strongest suppressors. A positive score indicates a suppressor of toxicity, while a negative score indicates an enhancer of toxicity. B) Spotting assays demonstrating independent verification of the Ste24 suppressor on both the 6xIAPP and C) 1xIAPP yeast models. 6xIAPP strains and uninduced 1xIAPP strains were grown for 48 hours. The induced 1xIAPP strains were grown for 72 hours. The bar chart is a quantification of the 1xIAPP dilution assay (dashed rectangle) with cells co-overexpressing control vector represented by the black bar (n=6) and cells co-overexpressing STE24 represented by the gray bar (n=9). Statistical analysis was performed using Student’s t-test. D) The enhancers of toxicity recovered from the genome wide deletion of non-essential genes (circles) and temperature-sensitive allele screen of essential genes (squares). A more negative score indicates a stronger enhancer of toxicity. The enrichment for autophagy genes (orange circles) and proteasome genes (turquoise squares) among the enhancers is highlighted. See also Figures S2, S3, and Tables S1-S3.
Two strong suppressors from the overexpression screen were related to protein degradation. These were Rpn4, a transcription factor that promotes the expression of proteasomal subunits, and Atg17, a phagophore assembly scaffold protein involved in autophagy. These findings were consistent with the proposed roles of autophagy and the proteasome in other models of IAPP toxicity (Costes et al., 2010; Rivera et al., 2014).
The strongest suppressor of 6xIAPP toxicity was Ste24, a highly conserved transmembrane protease of the ER with the active site in the cytoplasm. Ste24 overexpression suppressed both 6xIAPP (Figure 2B) and 1xIAPP toxicity (Figure 2C), with the 6xIAPP being rescued more strongly than the 1xIAPP. These results indicated that while direct expression of IAPP oligomers produced greater amounts of the toxic species that is affected by Ste24, overexpression of IAPP monomers could also produce this toxic form.
In yeast, the annotated function of Ste24 is the processing of a-factor, a mating pheromone. The annotated function of the human homolog ZMPSTE24 is the processing of lamin A, a nuclear membrane protein not present in yeast. Notably, Schuldiner and colleagues recently discovered that Ste24 and ZMPSTE24 clear the ER translocon of trapped polypeptides, identifying a shared function between the two proteins (Ast et al., 2016). If translocon clearing was indeed responsible for relieving IAPP oligomer toxicity, then IAPP oligomers might clog the translocon, providing a mechanistic explanation for our observed ER defects.
To expand our knowledge of the genetic modifiers of IAPP toxicity, we followed up on our overexpression screen with a genome-wide deletion screen (Baryshnikova et al., 2010; Costanzo et al., 2010; Kuzmin et al., 2016). We measured colony sizes for 1,388 temperature-sensitive alleles of essential genes and 4,635 viable deletion mutants with and without 6xIAPP overexpression (Tables S2 and S3). There were no overrepresented biological pathways among suppressors of IAPP toxicity (www.pantherdb.org), but there were several overrepresented pathways among enhancers of IAPP toxicity (Figure S3A). In concordance with the overexpression screen, two of the overrepresented pathways were macroautophagy and the ubiquitin proteasome pathway (Figure 2D). Furthermore, although interactions identified in the deletion screen were not systematically confirmed, four of the top 11 suppressors (STE24, MUM2, MSN5, RPN4) recovered from our overexpression screen were recovered as enhancers from the deletion screen.
Many genes related to translocation were recovered as modifiers of IAPP toxicity. Two components of the translocon complex, Sec72 and Sec62, were suppressors of IAPP toxicity. We confirmed this finding by recreating the SEC72 deletion in an independent strain (Figure S3B). Furthermore, there was a strong overrepresentation of physical and genetic interactors of Sec61 (www.yeastgenome.org), the protein-conducting subunit of the ER translocon, among modifiers of IAPP toxicity (p = 4.6×10−9, hypergeometric test). Among the strongest enhancers were two proteins that have been proposed to function in concert with Ste24 to clear clogged translocons: Cdc48, an AAA ATPase that generates a pulling force to extract proteins from the ER, and Dfm1, the recruitment factor for Cdc48 (Ast et al., 2016). These connections between translocon declogging machinery and IAPP toxicity further supported the hypothesis that IAPP oligomers clog the translocon.
ZMPSTE24 protects mammalian cells against IAPP oligomer toxicity
Taking advantage of the high degree of conservation between yeast Ste24 and mammalian ZMPSTE24, we probed the interaction between ZMPSTE24 and 6xIAPP in a biologically-relevant mammalian cell line to test the evolutionary conservation of this mechanism. We used a doxycycline-inducible promoter to drive the expression of 6xIAPP in INS-1 823/13, a rat insulinoma line widely used as a model for pancreatic β cells (Hohmeier et al., 2000). As a control, we expressed an identically constructed rat IAPP oligomer. The mature rat and human IAPP differ at just six residues, all in the amyloid-forming region. These amino acid differences prevent amyloid formation and toxicity of the rat protein. Both the rat and the human 6xIAPP were fused to the human IAPP signal peptide (Figure S1A).
To test the interaction between IAPP oligomers and ZMPSTE24, we sought a synergistic effect between 6xIAPP expression and ZMPSTE24 inhibition. We used the human immunodeficiency virus protease inhibitor lopinavir, which also inhibits ZMPSTE24 (Clark et al., 2017; Coffinier et al., 2007). The expression of 6xIAPP (human or rat) did not cause overt toxicity in INS-1 823/13 cells (Figure 3). However, human 6xIAPP expression was toxic when combined with lopinavir, suggesting that ZMPSTE24 inhibition sensitized the cells to 6xIAPP toxicity. Importantly, the synergistic effect between ZMPSTE24 inhibition and IAPP expression was specific to human IAPP and not observed with rat IAPP. These results confirmed the mammalian relevance of the genetic interaction between IAPP and Ste24 first uncovered in yeast.
Figure 3. ZMPSTE24 inhibition unmasked IAPP oligomer toxicity in a pancreatic cell line.
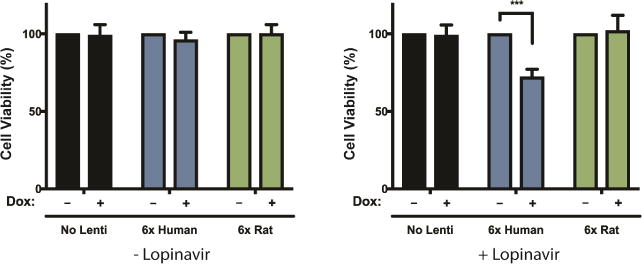
6x human IAPP and 6x rat IAPP were introduced into INS-1 823/13 cells by lentiviral infection and induced with doxycycline. Inhibition of ZMPSTE24 with 20 uM lopinavir reduced the survival of cells expressing 6x human IAPP (***p < 0.001, Student’s t-test) but not cells expressing 6x rat IAPP.
Ste24 overexpression uniquely suppressed the toxicity of IAPP directed to the secretory pathway
Having validated the interaction between IAPP oligomers and Ste24 in a mammalian system, we returned to yeast to investigate whether the translocon clearing activity of Ste24 might suppress IAPP toxicity. Ste24 is a protease, so overexpression of Ste24 might generally increase degradation of highly expressed proteins. While Ste24 overexpression reduced IAPP levels somewhat (Figure 4A), the magnitude of the reduction was insufficient to fully account for the massive reduction in toxicity. None of the other suppressors recovered from the overexpression screen were as potent as Ste24, yet many lowered 6xIAPP levels much more than Ste24 (Figure S4A). Furthermore, the suppression of toxicity by Ste24 overexpression was specific to IAPP toxicity. Ste24 did not rescue the toxicity of either TDP-43 or α- synuclein (Figure 4B). This indicated that Ste24 does not act as a generic protease or interfere with overexpression of toxic proteins. Additionally, Ste24 overexpression did not rescue the toxicity of 6xIAPP lacking a secretory signal peptide (Figure S4B), indicating that it did not degrade IAPP unless it was directed to the secretory pathway. Since Ste24 does not degrade IAPP in the cytoplasm and cannot once it passes into the ER membrane, the most likely place where Ste24 can act on IAPP is at the interface between the ER and the cytoplasm, the translocon.
Figure 4. Suppression of toxicity by Ste24 overexpression was not due to reduced IAPP levels and is specific for IAPP.
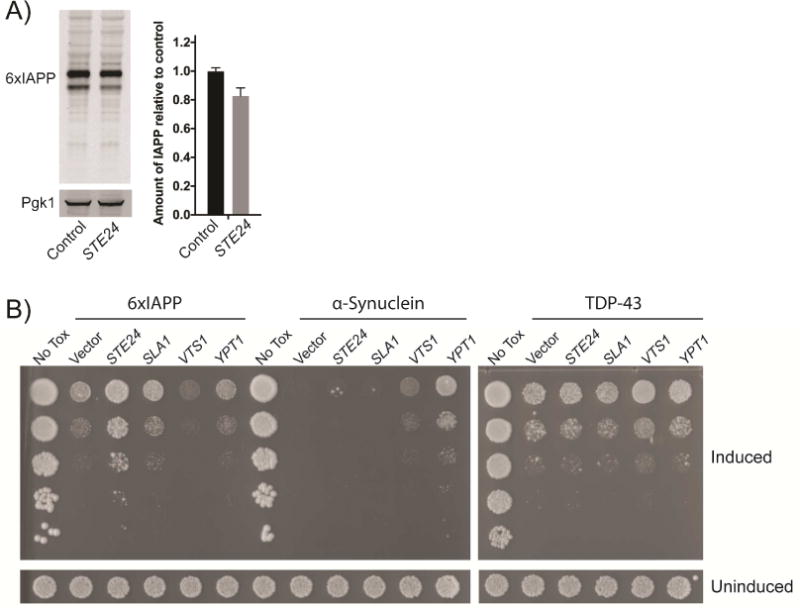
A) Western blot of IAPP expression measured in whole cell lysates prepared after 6 hours of 6xIAPP expression, with and without co-overexpression of STE24. The sum of the upper and lower bands was quantified and normalized to control cells. Overexpression of Ste24 modestly reduced 6xIAPP levels. Pgk1 was the loading control. B) STE24 overexpression rescued 6xIAPP toxicity but not α-synuclein or TDP-43 toxicities. Effects of STE24 overexpression are shown in spotting assays alongside SLA1 (Aβ suppressor), VTS1 (TDP-43 suppressor), and YPT1 (α-synuclein suppressor). 6xIAPP and α-synuclein were induced with 100 nM estradiol. TDP43 was induced with 5 nM estradiol. All strains were grown for 48 hours.
Ste24 overexpression relieved 6xIAPP induced ER-stress and translocation defect
We hypothesized that the ER-stress in 6xIAPP-expressing cells was due to IAPP clogging the translocon. Therefore, if IAPP oligomers clogging the ER translocon are the cause of the ER stress, then overexpression of Ste24 should relieve the ER stress phenotypes. To test this, we explored whether Ste24 overexpression could rescue the 6xIAPP-induced phenotypes in our fluorescent reporter strains. Specifically, we examined the increased expression of Kar2-mNG and Scj1-mNG, as well as the re-localization of an ER-directed GFP with an ER retention sequence, Kar2SS-mNG-HDEL (Figure 5). Ste24 overexpression attenuated ER stress phenotypes at both high and low levels of toxicity, returning the GFP expression to levels resembling those in the absence of 6xIAPP expression. Ste24 overexpression was an especially potent suppressor of the translocation defect in cells expressing Kar2SS-msfGFP-HDEL, almost completely reversing the defect. Since Ste24 overexpression relieved both the translocation defect and the ER stress phenotypes, it is likely that both phenotypes were the consequence of a single toxic event.
Figure 5. Ste24 overexpression attenuated 6xIAPP-induced changes in Kar2 and Scj1 expression and Kar2SS-msfGFP-HDEL localization.
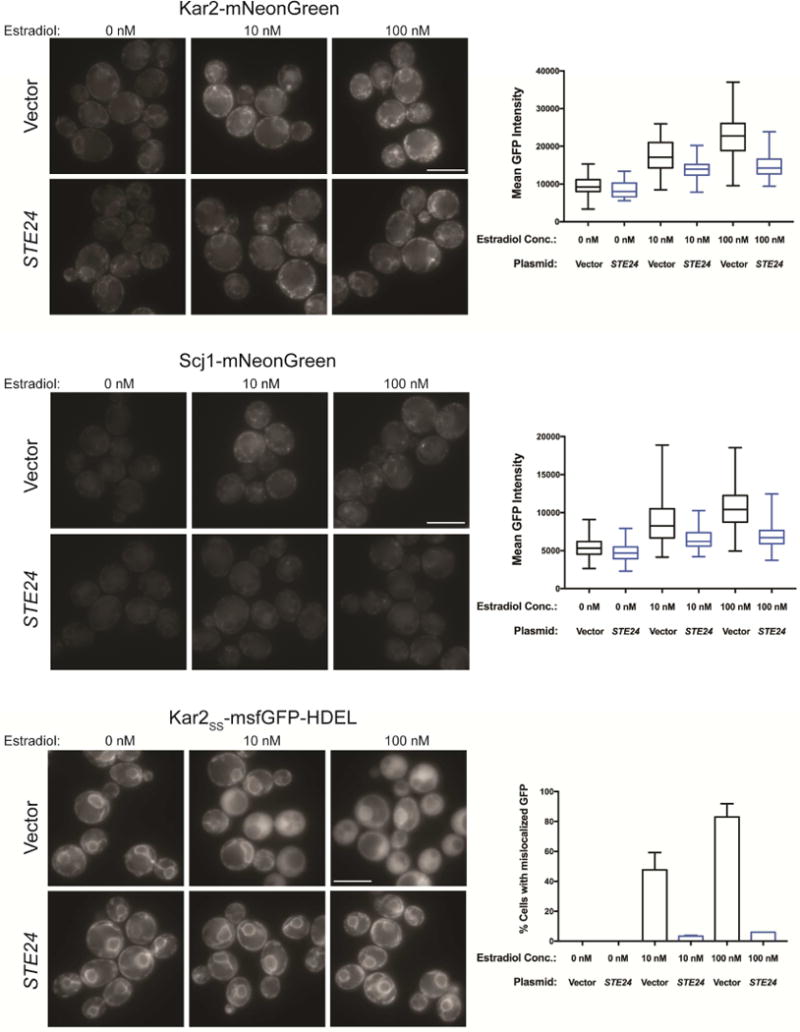
Representative fluorescence images for cells co-expressing 6xIAPP in combination with either STE24 or a vector control and either Kar2 protein C-terminally fused to mNG at its endogenous locus (Kar2-mNG, top panels); Scj1 protein C-terminally fused to mNG at its endogenous locus (Scj1-mNG, middle panels); or overexpressed msfGFP N-terminally fused to the Kar2ss and C-terminally fused to the ER-retention sequence HDEL (Kar2ss-msfGFP-HDEL, bottom panels). The three estradiol doses are for uninduced (0 nM), low toxicity (10 nM), and high toxicity (100 nM) conditions. For Kar2 and Scj1 the mean GFP intensity of the cells was quantified, while the fraction of cells with cytoplasmic, rather than ER-localized GFP was quantified for Kar2ss-msfGFP-HDEL. At least 100 cells were quantified per replicate and experiments were performed in biological triplicate. Error bars = SD of biological replicates. Scale bar = 5 μm.
ZMPSTE24 mutants rescued IAPP toxicity proportionately to their declogging activity
The evolutionary conservation of Ste24 provided an opportunity to test whether the human protein, ZMPSTE24, could also rescue IAPP toxicity in yeast. We first confirmed that the deletion of endogenous STE24, while conferring no growth defect on its own, caused an increase in 6xIAPP toxicity (Figure 6A). This was evident from comparing the Δste24 strain expressing 6xIAPP to the same strain transformed with a single-copy plasmid expressing STE24. While reintroduction of wild-type (WT) Ste24 lessened IAPP toxicity, a protease-impaired E298G variant of Ste24 had little effect (Figure 6A) (Ast et al., 2016). Next, we introduced ZMPSTE24 into the Δste24 strain. Like Ste24, ZMPSTE24 expression alone did not affect the growth of the yeast yet suppressed 6xIAPP toxicity (Figure 6B).
Figure 6. Loss of STE24 enhanced IAPP oligomer toxicity.
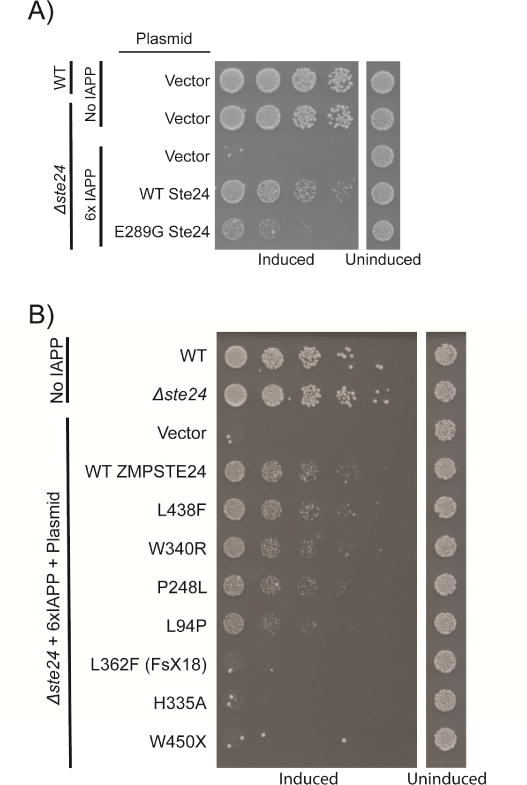
A) Deletion of STE24 produced no growth defect on its own but greatly enhanced 6xIAPP toxicity. This was rescued by reintroducing STE24 on a plasmid under the control of its endogenous promoter. A catalytically inactive E289G Ste24 was much less effective in rescuing the enhanced IAPP toxicity of the Δste24 strain. B) WT human ZMPSTE24 functionally substituted for Ste24 and relieved 6xIAPP toxicity in the Δste24 strain. The effects of substitution with ZMPSTE24 mutants possessing a range of declogging activities are shown in order of decreasing activity (for a measurement of their declogging activity please see (Ast et al., 2016)).
We used the ZMPSTE24-complemented Δste24 yeast strain to test the functional consequences of ZMPSTE24 mutations. We postulated that amino acid replacements that reduce ZMPSTE24 translocon-clearing activity should also reduce its capacity to suppress 6xIAPP toxicity. Several mutants identified from patients with ZMPSTE24-related laminopathies had been previously characterized for their ability to unclog the translocon (Ast et al., 2016; Barrowman et al., 2012). These variants possessed a spectrum of translocon declogging activities, ranging from WT-like to completely inactive. In our assay, the level of declogging activity possessed by each variant was in complete agreement with its rescue of IAPP toxicity (Figure 6B). These results supported the hypothesis that IAPP oligomers clog the translocon and that ZMPSTE24/Ste24 protects cells from IAPP toxicity by declogging the translocon.
The yeast IAPP model enabled functional testing of ZMPSTE24 mutants identified in the population
The direct correlation between the declogging activity of ZMPSTE24 and the growth rate of IAPP-expressing yeast provided an opportunity to perform a comprehensive functional analysis on ZMPSTE24 polymorphisms discovered in the human population. Importantly, since yeast lack lamin, ZMPSTE24 activity on IAPP could be measured independently of the laminopathy phenotypes that ZMPSTE24 variants might produce in mammalian cells. Using the resources compiled by the T2D-GENES/GoT2D consortium (manuscript in preparation), we identified and created plasmids to express 111 single nucleotide missense mutants of ZMPSTE24 found in either the T2D population or non-diabetic controls. We introduced these plasmids into the Δste24 strain harboring a single copy of 6xIAPP and measured the growth rate of this strain upon 6xIAPP expression (Figure 7A). We then retested variants that had an apparent growth defect (Figure 7B). The variants that grew more poorly than WT ZMPSTE24 expressing cells in this secondary analysis (One sided ANOVA followed by Dunnett’s test, threshold was p < 0.01) were considered loss-of-function variants.
Figure 7. Analysis of the IAPP oligomer toxicity-rescuing ability of 111 ZMPSTE24 missense mutants.
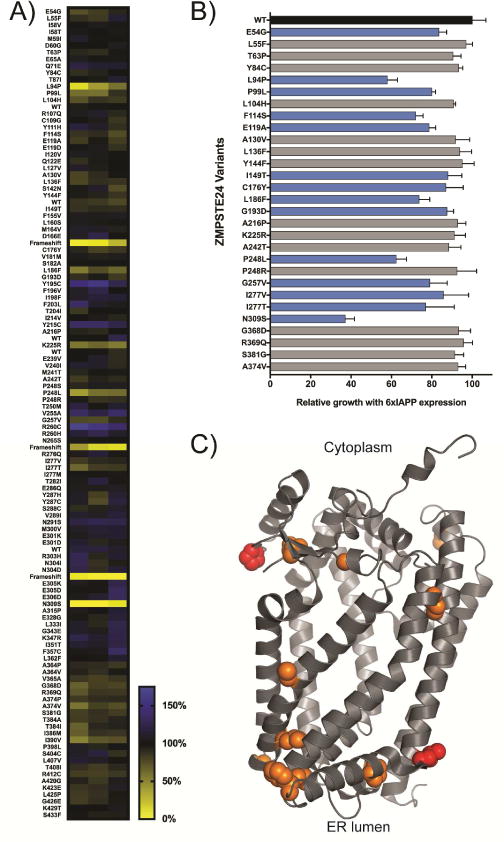
ZMPSTE24 mutants identified from the sequencing of diabetes patients and healthy controls were tested for their ability to rescue 6xIAPP toxicity in the Δste24 yeast strain. The growth of 6xIAPP strains expressing ZMPSTE24 variants was quantified as the area under the growth curve at 48 hours. A) Each cell of the heat map is the average growth of four biological replicates, while each column is a technical replicate. B) Retest of poorly growing variants. Variants meeting statistical significance were deemed loss-of-function variants (One-sided ANOVA followed by Dunnett’s test, cutoff: p < 0.01) and are shown in blue bars. Error bars = SD. C) Residues in ZMPSTE24 where amino acid changes produced lower 6xIAPP toxicity rescuing activity are shown on the crystal structure (PDB: 5SYT) in orange, with laminopathy-causing variants shown in red.
We identified 14 loss-of-function variants, including P94L and P248L, which are known laminopathy-causing variants of ZMPSTE24 (Figure 7C). To our knowledge, none of the other loss-of-function alleles are known to cause laminopathies and had milder defects than the laminopathy variants. Since clinical outcomes data were available, we examined whether mutants with reduced 6xIAPP-rescuing function were enriched among the diabetic population. Because all the mutations were found in singletons, statistical power was limited. Though it did not reach conventional statistical significance (p=0.075), we observed an intriguing trend where ZMPSTE24 mutants with loss-of-function were enriched among T2D patients ~2-fold.
Discussion
Deposits of aggregated human IAPP are frequently present in the pancreatic islets of patients with T2D and are thought to contribute to β-cell dysfunction and death. To understand how IAPP oligomers disrupt the biology of eukaryotic cells we created a yeast model of IAPP toxicity. Through genome-wide screens in this model, we found that Ste24 was the most potent suppressor of IAPP toxicity and validated this finding in mammalian cell lines. Ste24 is a protease responsible for clearing peptides that clog the cytoplasm-to-ER protein-conducting channel (Ast et al., 2016). The strong evolutionary conservation of this protein from yeast to human enabled the functional replacement of yeast Ste24 with human ZMPSTE24, creating a platform to measure the consequence of missense mutations identified in humans. We found many previously uncharacterized loss-of-function variants, many of which clustered on the ER-lumen facing side of ZMPSTE24. Furthermore, these variants were more likely to be found in the diabetic population.
The use of yeast as a model system has produced a wealth of data on the cell autonomous toxicities of many misfolding-prone proteins. Here, we presented the first yeast model of IAPP toxicity. A growing body of evidence has implicated IAPP oligomers as the cytotoxic agent. IAPP oligomers are found intracellularly, while the amyloid deposits are extracellular (Gurlo et al., 2010; Lin et al., 2007). In vitro preparations of IAPP aggregates are most toxic when freshly prepared, while the end-stage aggregates are inert (Abedini et al., 2016). In mice, the frequency of β-cell death is correlated with the rate of increase in amyloid deposition and not the presence of amyloids, suggesting that molecules preceding amyloid formation are responsible (Butler et al., 2003). Based on this body of work, for our yeast model of IAPP toxicity, instead of overexpressing monomers with the expectation that a small population would form toxic oligomers, we directly expressed an IAPP oligomer consisting of six repeats of IAPP. We hypothesize that this generated much greater toxicity than 1xIAPP because the direct expression of an oligomer bypassed the nucleation barrier for protein aggregation, thereby producing far more toxic particles than monomer overexpression. While our use of 6xIAPP made it difficult to study cellular mechanisms that might prevent the nucleation process, it highlighted pathways that were perturbed following IAPP oligomer formation.
Our genetic screens yielded numerous suppressors and enhancers, highlighting various pathways of IAPP toxicity. Genes involved in autophagy and the proteasome were recovered from both the deletion and overexpression screens. We were reassured to capture these pathways in our yeast model as the proteasome and autophagy are documented pathways involved in IAPP toxicity in other model systems (Costes et al., 2010; Rivera et al., 2014). We also uncovered a novel proteotoxic mechanism for IAPP oligomers, translocon clogging. To our knowledge, this is the first demonstration of an oligomer that induces cytotoxicity through this mechanism. This cytotoxic mechanism may be particularly relevant in the context of pancreatic β-cells, which are dedicated secretory cells and critically dependent on an efficient secretory pathway. Our yeast model could be used in high-throughput drug screens to identify modulators of any of these pathways or even IAPP itself.
Our genetic analyses in yeast strongly supported the hypothesis that the translocon-clearing activity of Ste24 rescues IAPP toxicity. Ste24 protease activity was required to relieve IAPP toxicity and Ste24 did not rescue 6xIAPP without a secretory targeting peptide, i.e. 6xIAPP in the cytoplasm. Since the protease domain of Ste24 is in the cytoplasm, the likely place where Ste24 could act on IAPP is at the translocon. Ste24 is thought to clear translocons in concert with Cdc48 and its recruitment factor Dfm1 (Ast et al., 2016). These two genes were among the strongest enhancers of toxicity in our deletion screen. Moreover, in strains deleted for Ste24 and complemented with laminopathy-causing variants of ZMPSTE24, rescue of IAPP toxicity was directly correlated to the declogging activity of the ZMPSTE24 variant. The L438F variant was especially interesting. This variant is strongly defective in CaaX-processing activity, but close to WT in translocon clearing activity. In our assays, this L438F ZMPSTE24 was closer to WT in its rescue of IAPP toxicity. These results support the hypothesis that the lamin-cleaving activity of ZMPSTE24 may be partially separable from its translocon-clearing activity (Ast et al., 2016).
Translocon clogging is thought to occur when a polypeptide prematurely folds before it fully passes through the translocon. Monomeric IAPP is a small and intrinsically disordered molecule, making it an unlikely clogger. We hypothesize that newly synthesized IAPP could clog the translocon by forming misfolded oligomers in the cytoplasm prior to translocation. In human islets cultured in high glucose, IAPP is upregulated even more than insulin (Novials et al., 1993). This increased production of IAPP would increase its likelihood of aggregating following synthesis. Intriguingly, intrinsically disordered proteins like IAPP may be inherently difficult to translocate into the ER (Gonsberg et al., 2017). Such failed translocation events, which return the polypeptide into the cytoplasm with an intact signal peptide (Gonsberg et al., 2017), could contribute to a buildup of IAPP in the cytoplasm. Furthermore, pre-existing IAPP aggregates could actively template the aggregation of newly synthesized IAPP, especially since IAPP aggregates have recently been shown to have prion-like, self-templating properties (Mukherjee et al., 2017). Thus, there are multiple complementary mechanisms by which IAPP oligomers could be formed before they are directed to the secretory pathway.
The ever-expanding availability of human genetic information combined with associated clinical data has enhanced our ability to link genetic polymorphisms to disease. One challenge in this endeavor is determining which mutations are benign and which mutations are deleterious. Here, we used the association between ZMPSTE24 function and IAPP toxicity to search for loss-of-function mutations in ZMPSTE24 and assess enrichment among people with T2D. Using the largest available database of matched genetic and clinical data on T2D to find ZMPSTE24 mutants, we examined 111 single nucleotide changes. Most of these mutants had no measurable defect. Of those that did, half of amino acid changes were not in the protease domain. Rather, they were located on the side of ZMPSTE24 facing the ER lumen. These amino acid changes might modify the interaction of ZMPSTE24 with other components of the translocon or translocon declogging machinery, or potentially diminish protease activity in an allosteric manner. The loss-of-function alleles were extremely rare, with only a single person harboring any given mutation. Nevertheless, we observed an intriguing trend where these loss-of-function alleles were more likely to be found among patients with diabetes. Though this did not reach conventional statistical significance, the odds ratio of 2.19 was far greater than those typically observed for T2D risk factors (Billings and Florez, 2010). These results suggest that the declogging activity of ZMPSTE24 is protective against T2D. Therefore, defects in this protein quality control pathway may contribute, in a subset of cases, to the onset of diabetes or the progression from a prediabetic state to a diabetic one. More study of the variation in ZMPSTE24 and associated proteins, including non-coding mutations and epigenetic modifications that may alter protein expression levels, and their roles in diabetes are warranted as data become available.
Of the many treatments to address the end result of hyperglycemia in diabetes, few can be considered as disease modifying in that they target the underlying causes. This is partly because we do not yet have a complete grasp of the basic biology involved. Here we have presented a new model system for dissecting the cell-autonomous toxicity associated with a diabetic proteinopathy, IAPP aggregation. Our yeast model provides a new a platform for identifying genetic and pharmaceutical targets to combat the toxic protein aggregates that build up in the pancreas of patients with T2D.
STAR Methods
Contact for Reagent and Resource Sharing
Requests for materials should be addressed to the Lead Contact, Can Kayatekin (can.kayatekin@gmail.com)
Experimental Model and Subject Details
Saccharomyces cerevisiae strains used in this study are described in Table S4. Unless otherwise stated, strains were grown with shaking at 30°C in appropriate synthetic media purchased from Sunrise Science Products, made up as per their instructions, and supplemented with 2% glucose, raffinose, or galactose.
The INS-1 823/13 cells used in this study were maintained at 37°C in T75 flasks in RPMI-1640 medium supplemented with 10% fetal calf serum, 10mM HEPES, 2nM L-glutamine, 1mM sodium-pyruvate and 0.05mM 2-mercaptoethanol.
Method Details
Estradiol-inducible yeast models
Many previous yeast studies relied on the galactose-inducible GAL1 promoter to overexpress genes. The GAL1 promoter is extremely strong, but its induction requires a carbon source shift. In addition to overexpressing the gene of interest, this dramatically alters the expression of many unintended genes. This could obscure salient aspects of IAPP-related toxicity. To overcome this dilemma, we adopted an estradiol-responsive zinc-finger based expression system to effectively and specifically regulate expression of IAPP (Aranda-Díaz et al., 2016; McIsaac et al., 2012). The system employs a transcriptional regulator (TR) consisting of a chimeric transcription factor containing the Zif268-DNA binding domain fused to the human estradiol receptor and the Msn2 activation domain (ZEM). Expression of ZEM was driven by the constitutive EIF1α promoter from S. paradoxus. Estradiol-bound ZEM translocates to the nucleus, binds and activates transcription from pZ, a modified pGAL1 promoter containing Zif268 binding sites and lacking Gal4 binding sites.
Isolation, sequencing, and analysis of yeast mRNA
Yeast cells were grown in 5 mL of minimal media with appropriate prototrophic selection to logarithmic growth phase and IAPP expression was induced for 6 hours with 100 nM estradiol. RNA was isolated using a standard hot acid phenol-chloroform method. The cells were pelleted and resuspended in 400 μL of TES buffer (10 mM Tris-HCl, 10 mM EDTA, 0.5% SDS, pH 7.5). An equal volume of acid phenol was added and the sample was briefly shaken for 10 s and then incubated at 65 °C with occasional vortexing for 1 hour. Samples were then cooled on ice for 5 min and centrifuged for 5 min at 10000g at 4 °C. The aqueous phase was combined with 400 μL of chloroform, vigorously vortexed and centrifuged for 5 min at 10000g at 4°C. The aqueous phase was isolated and combined with 40 μL of 3 M sodium acetate, pH 5.3, and 1 ml of cold 100% ethanol and precipitated for 60 min. After another 5 min centrifugation step (10000g at 4°C), the pellet was isolated, washed once with 70% ethanol, and resuspended in 50 μL of RNAse-free water. The concentration of RNA was calculated using absorbance measurements at 260 and 280 nm. The RNA was then analyzed to confirm sample integrity using a BioAnalyzer (Agilent). Sequencing libraries were prepped using the TruSeq RNA Library Prep Kit (Ilumina) according to manufacturer’s instructions. Sequencing was performed on an Illumina HiSeq 2500 sequencing platform and multiplexed with 8-9 samples per lane. This yielded 13.4 and 19.6 million reads per sample with a complexity of 15-20%.
Reads were mapped with TopHat v2.0.13 (Trapnell et al., 2009) against the sacCer3 version of the yeast genome using the following parameters: “-I 2500, –solexa1.3-quals, –segment-length 20, –no-novel-juncs, –no-coverage-search, –b2-very-sensitive, -g 1, and -G gtf file”. We used the annotation file “Saccharomyces_cerevisiae.R64-1-1.80.gtf.gz” downloaded from ENSEMBL. We assigned reads to genes with htseq-count (Anders et al., 2015) using the options “–mode=intersection-strict, –stranded=reverse”, and the same gtf file used in the mapping. Then, EdgeR (Robinson et al., 2010) was run to assay differential gene expression.
FACS analysis of UPR activation
Cells were inoculated from saturated cultures into synthetic complete media at an OD of 0.025 and incubated for 5 to 6 hours at 30°C to reach exponential growth (OD of 0.2). Cells were then split into 3 conditions: treated with 2 mM DTT (to induce UPR activation), or 100 nM estradiol, or untreated for 4 hours before measurement. 10,000 events were acquired using a MACSQuant VYB cytometer with a 96-well plate platform (Miltenyi Biotech) and data was processed using FlowJo. The B1 channel (525/50 filter) was used to measure green fluorescence. To exclude dead cells, the cultures were treated with 10 ug/mL propidium iodide and the Y3 channel (661/20 filter) was used to measure propidium iodide staining. Events were gated by forward and side scatter, and median fluorescence values were calculated. Experiments were performed in biological duplicate.
Screening the library of fluorescent reporters of yeast biology
All strains were constructed in the using the W303 genetic background yeast strain. The EIF1α-promoter driven ZEM was integrated at the YBR032W locus and pZ-driven 6xIAPP or empty control vector was integrated at the LEU2 locus. This strain was mated to the reporter library which consisted of strains harboring fluorescent protein fusions of genes at their endogenous loci or fluorescent reporters that were integrated as single copies into the yeast genome at the TRP1 locus. The resulting diploid cells harboring the single copy 6xIAPP plasmid were mixed at a 2:1 ratio with the cells harboring the control empty plasmid and grown for 4.5 hours from a starting density of OD600 = 0.08 in synthetic complete media with 2% glucose and 5nM estradiol. Cells were then diluted to OD600 of 0.15 in the same media and transferred to a concanavalin A coated plate that was then spun down at 2000g for 5 minutes to attach the cells to the surface. Using an automated microscope, the reporter strains were imaged sequentially with 25 independent fields acquired for each well. Total acquisition time for one 96-well plate was ~3 hours. Control cells were differentiated from 6xIAPP-expressing cells by the presence of a constitutively expressed blue fluorescent protein driven by the TDH3 promoter integrated at the LEU2 locus. Quantification of the GFP intensity was performed using MATLAB. This library, acquisition, and analysis platform will be described in a separate manuscript currently under preparation (Landgraf et al., in preparation). This screen of IAPP toxicity across all the reporter strains was performed a single time.
Genome wide overexpression screen
The yeast strains for the overexpression screen were generated by mating a BY strain harboring 2 copies of 6xIAPP (at HIS3 and LEU2) and the EIF1α-driven ZEM integrated at YBR032W to an arrayed overexpression library containing 5532 single-copy plasmids containing ORFs under the control of the GAL1 promoter (Hu et al., 2007). The mated cultures were pinned from the plates into synthetic media lacking histidine and uracil with 2% glucose media in 96-well plates and grown to saturation. They were then diluted into the same media with 2% raffinose and grown to saturation once again to relieve glucose repression of the GAL1 promoter. These cultures were then re-diluted to an OD of 0.2 in fresh media and grown for 6-7 hours until the OD reached ~0.5. The cultures were then pinned onto agar plates with synthetic media lacking histidine and uracil with 2% galactose, with and without 100nM estradiol. The strains were grown for 48 hours and imaged using a scanner.
We used the program Cell Profiler (Kamentsky et al., 2011) (cellprofiler.org) to quantify all estradiol-containing plates. The first 20 analyzed plates were averaged at each of the 96-well plate positions (omitting any clear outliers) to generate an average growth value, which was used to correct for positional growth differences. Enhancers were checked manually against the corresponding plates lacking estradiol to determine whether they were specific to IAPP expression or generically reduced growth. Any putative enhancers that caused a growth defect in the absence of IAPP expression were eliminated from consideration. All hits were confirmed by sequencing and validated by isolating each plasmid from an independent bacterial stock and retesting the rescue in a naïve IAPP yeast strain.
Genome wide deletion screen
The query strain was constructed in a haploid BY4741 background, which produced an intermediate a level of toxicity such that both suppressors and enhancers could be identified. A single copy of 6xIAPP was introduced into the LEU2 locus under the control of the pZ promoter. The EIF1α-promoter driven ZEM was integrated at the YBR032W locus with nourseothricin selection. This strain also carried Synthetic Genetic Array (SGA) compatible selectable markers and reporters (Kuzmin et al., 2016) allowing for the inducible 6xIAPP gene to be introduced into arrayed collections of nonessential gene deletion alleles and temperature-sensitive alleles of essential genes (Li et al., 2011) using the SGA method. The query strain was crossed to an ordered array of mutant strains. The resulting diploids were pinned to a sporulation-inducing medium, after which the cells were transferred to synthetic medium lacking histidine but containing canavanine and thialysine to allow for selective germination of haploid meiotic progeny. These haploids were then transferred to a medium containing genticin to select for array mutants, and then to a medium containing both genticin and lacking leucine to also select for 6xIAPP.
To identify modifiers of 6xIAPP toxicity, mutant arrays were grown on plates with 2% raffinose in 1536-density arrays, with 4 independent isolates per array mutant, in the presence of 100nM estradiol. The strains were grown for 2 days and resultant colony sizes were measured and compared to the colony sizes of the same mutants derived from SGA screens using an isogenic query control strain, which did not express 6xIAPP. All plates were imaged and quantified using an automated, high-resolution imaging system. Colony size and interaction measurements were performed using the SGA score algorithm (Baryshnikova et al., 2010; Costanzo et al., 2010).
Cell culture experiments
The rat insulinoma clonal cell line INS-1 823/13 was kindly provided by the Newgard laboratory at Duke University (Hohmeier et al., 2000). Cells were maintained in T75 flasks in RPMI-1640 medium supplemented with 10% fetal calf serum, 10mM HEPES, 2nM L-glutamine, 1mM sodium-pyruvate and 0.05mM 2-mercaptoethanol. The IAPP constructs were introduced in INS-1 823/13 cells by lentiviral infection (2nd generation, pMD2.G envelope and psPAX2 packaging vectors) with doxycycline-inducible expression vector selectable by puromycin at 0.5 ug/mL final concentration. The viability experiments were performed as follows: 105 INS-1 cells were plated in 96-well plates on day 1, doxycycline was added on day 2 at 50 ng/mL final concentration, lopinavir was added on day 4 and cell viability was assayed by Alamar Blue assay on day 5.
Functional testing of ZMPSTE24 mutants
All ZMPSTE24 mutants were cloned into uracil selectable low-copy plasmids bearing the constitutive TDH3 promoter with a three-piece Gibson assembly reaction. Using complementary PCR primers containing the mutated nucleotide sequence, the ZMPSTE24 ORF was cloned as two fragments and assembled with the plasmid backbone. All clones were verified by PCR to confirm the size of the insert and sequenced with a single primer to verify the presence of the mutation. The constructs were transformed into a ∆ste24 BY4741 strain, harboring a single insertion of pZ driven 6xIAPP at the LEU2 locus and EIF1α promoter driven ZEM at the YBR032W locus. Cells were grown in 70uL volumes in 384-well plates inoculated from a saturated culture with a 300:1 dilution. Each variant was tested in biological quadruplicate, with three technical replicates. Cells were grown in synthetic media lacking uracil with 2% glucose and 100 nM estradiol. Growth was quantified as the area under the growth curve at 2500 minutes. To correct for edge effects, this value was normalized to that of cells harboring a WT ZMPSTE24 plasmid grown in the same position on a different plate. The corrected areas under the curve were then normalized to a 0-100% scale where the poorest growing frameshift ZMPSTE24 variant = 0% and the mean WT ZMPSTE24 = 100%.
Variants that grew worse than WT in 2 out of 3 technical replicates were retested in biological and technical quadruplicate. These experiments were performed in the same manner as the primary screen, with the exception that the edges of the plate (2 wells deep on either side) were left empty to avoid edge effects. Variants that grew worse than WT with p ≤ 0.01 were considered bona fide loss-of-function variants (One sided ANOVA followed by Dunnett’s test). For the graph, the mean area under the curve of cells harboring the WT plasmids was defined as 100%, while the mean area under the curve of cells harboring frameshift plasmids was defined as 0%. All analysis was performed with the Prism 7 software package.
Statistical Analysis of ZMPSTE24 mutants
To test for association between variants characterized as loss-of-function and type-2 diabetes, sequence data from 45,231 subjects (20,791 T2D cases and 24,440 controls), drawn from five different ancestries, were aggregated at the Broad Institute. The vast majority of sequence data was generated at the Broad, using a standard Illumina sequencing protocol: genomic DNA was sheared, end repaired, ligated with barcoded Illumina sequencing adapters, amplified, size selected, and subjected to in-solution hybrid capture using either the Agilent SureSelect or Illumina Nexome bait set. Resulting Illumina exome sequencing libraries were qPCR quantified, pooled, and sequenced with 76-bp paired-end reads using HiSeq 2000 sequencers to ~82-fold mean coverage. Details of the full sequence dataset will be described elsewhere (manuscript in preparation). Sequencing reads were then processed and aligned to the human genome (build hg19) using the Picard (http://broadinstitute.github.io/picard/), BWA, and GATK software packages, following best-practice pipelines (https://software.broadinstitute.org/gatk/best-practices/). Single nucleotide and short indel variants were then called using a series of GATK commands (version nightly-2015-07-31-g3c929b0): ApplyRecalibration, CombineGVCFs, CombineVariants, GenotypeGVCFs, HaplotypeCaller, SelectVariants, and VariantFiltration. Default parameter values were used for each command. Hard calls (the GATK-called genotypes but set as missing at a genotype quality [GQ] <20 threshold) and dosages (the expected value of the genotype, defined as Pr(RX|data) + 2Pr(XX|data), where R is the reference and X the alternative allele) were computed for each individual at each variant site. Hard calls were used only for quality control, while dosages were used in downstream association analyses. Quality control was performed on samples by first calculating a range of metrics measuring sample sequencing quality, stratifying samples by ancestry and sequence capture technology, and excluding outlier samples according to any metric based on visual inspection by comparison to other samples within the same stratum. After exclusion of samples, we calculated an additional set of variant metrics and excluded any variant with overall call rate <0.3, average non-reference genotype quality <10 (e.g. an estimated error rate >90% for non-reference genotypes), heterozygosity of 1, or heterozygote allele balance of 0 or 1 (e.g. 100% or 0% of reads called non-reference for heterozygous genotypes).
Variants were annotated with the Variant Effect Predictor (VEP) (McLaren et al., 2016), and all variants classified as IMPACT=MODERATE (e.g. nonsynonymous variants) were subjected to experimental characterization. Variants considered functionally deficient, together with variants classified by the VEP as IMPACT=HIGH (19 variants in total), were used to conduct a burden test for association with T2D. Although E119A, I277T, and N309S were experimentally found to have loss of function, they were omitted from this analysis because either the sequencing quality was not sufficient or we did not have information on the diabetes status of the patient. Each sample was assigned the number of variants carried in the set, and Firth logistic regression was performed using EPACTS to estimate association with (p-values) and effect sizes on (odds ratios) T2D. Covariates measuring ancestry (as computed from 10 principal components, calculated using the EIGENSTRAT software package, of common coding variation exome-wide) as well as sequencing batch and cohort of collection were included in the model.
Western Blotting
For all western blots, yeast cultures were diluted from saturated overnight cultures to around OD600 = 0.1. After 6 hours of growth in synthetic media with or without 100 nM estradiol, 1.5 OD600 of cells were collected for each sample and total protein extracts were precipitated using trichloroacetic acid. Precipitates were resuspended in 50 uL of HU buffer (2% SDS, 7M urea, 1 mM TCEP, 200 mM TRIS pH 6.8) and incubated at 65 °C for 15 minutes. Gels were transferred to nitrocellulose membranes using the iBlot2 dry blotting system (Thermo Fisher Scientific). IAPP was probed overnight at 4 °C, using the rabbit anti-IAPP antibody from Sigma-Aldrich (Catalog number: HPA053194, 1:5000 dilution). Quantitative measurements were performed on the LI-COR Odyssey imaging platform, using fluorescent secondary antibodies. The sum of both the higher and lower bands intensities was used when quantifying 6xIAPP levels.
Standard fluorescence microscopy
For fluorescent imaging, all strains were grown to saturation in synthetic complete media with 2% glucose, diluted to OD600 = 0.2, and induced for 6 hours with estradiol. All images were acquired with a Nikon Plan Apo 100x oil objective (NA 1.4) using a Nikon Eclipse Ti-E inverted microscope and a CCD camera (Andor technology). The fluorescent reporter strains were identical to those used in for the high content microscopy. Specifically, the Kar2SS-msfGFP-HDEL gene was driven by the TDH3 promoter and integrated in single copy at the TRP1 locus. Both Kar2 and Scj1 were C-terminally fused to mNG at their endogenous locus. Total cellular GFP fluorescence was quantified manually using ImageJ. The fraction of cells with mislocalized GFP (i.e. GFP in the cytoplasm instead of the ER) was determined visually. For the experiments demonstrating STE24 rescue of the IAPP-induced phenotypes, STE24 was inserted with a single-copy integrating plasmid at the URA3 locus under the control of the pZ promoter. Due to the additional estradiol-inducible promoter binding site introduced by integrating STE24 in this manner, for the low toxicity experiments the concentration of estradiol used was double that of the screening experiments. For each data point, the fluorescence intensity of at least 100 cells was quantified using ImageJ. Experiment were performed in biological triplicate.
Quantification and Statistical Analysis
Microsoft Excel and GraphPad Prism software were used to process data. Images were manipulated using ImageJ. Software for specialized analyses can be found in the appropriate section of the Method Details and in the Key Resources Table. Statistical details such as the number of replicates, error calculations, and significance calculations are provided in figure legends and in the appropriate section of the Method Details.
Data and Software Availability
Requests for additional data should be directed to the Lead Contact, Can Kayatekin (can.kayatekin@gmail.com).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-PGK1 polyclonal | Antibodies-online.com | Cat# ABIN568371 |
| Rabbit anti-IAPP polyclonal | Sigma Aldrich | Cat# HPA053194 |
| Donkey anti-mouse secondary 800CW dye conjugate | LI-COR | Cat# 926-32212 |
| Donkey anti-rabbit secondary 680CW dye conjugate | LI-COR | Cat# 926-68073 |
| Rabbit anti-mouse secondary polyclonal HRP conjugate | Sigma Aldrich | Cat# A-9044 |
| Bacterial and Virus Strains | ||
| Escherichia coli DH5alpha | ATCC | Cat# 69021 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nourseothricin dihydrogen sulfate (ClonNAT) | Werner BioAgents | Cat# 5.005.000 |
| G418 (geneticin) | Life Technologies | Cat# 10131027 |
| B-Estradiol | Sigma Aldrich | Cat# E8875-5G |
| Propidium iodide | Sigma Aldrich | Cat# P4864 |
| Lopinavir | Sigma Aldrich | Cat# SML1222-10MG |
| Yeast nitrogen base | VWR | Cat# 90004-146 |
| Complete supplement mixture (CSM) media | Sunrise Science Products | Cat# 1001-100 |
| CSM lacking uracil | Sunrise Science Products | Cat# 1004-100 |
| CSM lacking leucine | Sunrise Science Products | Cat# 1005-100 |
| CSM lacking histidine | Sunrise Science Products | Cat# 1006-100 |
| CSM lacking uracil and histidine | Sunrise Science Products | Cat# 1009-100 |
| Critical Commercial Assays | ||
| TruSeq RNA Library Prep Kit | Ilumina | |
| Deposited Data | ||
| RNASeq | GEO | GSE104172 |
| Experimental Models: Cell Lines | ||
| INS-1 823/13 | Newgard Lab | CVCL_7226 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae: Strain background: W303 | ATCC | Cat# 201238 |
| S. cerevisiae: Strain background: BY4741 | ATCC | Cat# 201388 |
| Oligonucleotides | ||
| Please see Table S5 | ||
| Recombinant DNA | ||
| Please see Table S6 for all plasmids | ||
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| SGA Score algorithm | Baryshnikova et al, 2010 | |
| Cell Profiler | Kamentsky et al., 2011 | cellprofiler.org |
| TopHat | Trapnell et al., 2009 | https://ccb.jhu.edu/software/tophat/index.shtml |
| htseq-count | Anders et al., 2015 | |
| EdgeR | Robinson et al., 2010 | |
| Picard | Broad Institute | http://broadinstitute.github.io/picard/ |
| Burrows-Wheeler Aligner (BWA) | http://bio-bwa.sourceforge.net/ | |
| GATK | Broad Institute | https://software.broadinstitute.org/gatk/ |
| Variant Effect Predictor (VEP) | McLaren et al., 2016 | https://useast.ensembl.org/info/docs/tools/vep/index.html |
| EPACTS | http://csg.sph.umich.edu/kang/epacts/download/index.html | |
| EIGENSTRAT | https://github.com/DReichLab/EIG/tree/master/EIGENSTRAT | |
| MATLAB | The Mathworks Inc. | https://www.mathworks.com/products/matlab.html |
| FlowJo v8 | FlowJo, LLC. | https://www.flowjo.com/solutions/flowjo/downloads |
| ImageJ | https://imagej.net/Welcome | |
| Other | ||
Supplementary Material
Table S1. IAPP overexpression suppressors. Related to Figure 2. The verified suppressors recovered from the genome-wide overexpression screen.
Table S2. Temperature sensitive alleles that modify IAPP toxicity. Related to Figure 2. The statistically significant 6xIAPP suppressors and enhancers recovered from the screen of 1,388 temperature sensitive alleles.
Table S3. Deletions that modify IAPP toxicity. Related to Figure 2. The statistically significant 6xIAPP suppressors and enhancers recovered from the screen of 4,635 viable genetic deletions.
Table S4. Yeast strains used in this study. Related to STAR Methods: Experimental Model and Subject Details
Table S5. Oligonucleotides used in this study. Related to Key Resources Table: Oligonucleotides
Table S6. Plasmids used in this study. Related to Key Resources Table: Recombinant DNA
Figure S1. IAPP constructs used. Related to Figure 1. A) Amino acid sequences of IAPP constructs employed in this work. The highlighted elements are the signal peptide (orange), human IAPP pro peptide (red), mature IAPP hormone (blue), linker residues (black). B) Spotting assay showing growth defect induced by expression of 1xIAPP from a 2μ high-copy plasmid, driven by the estradiol-inducible promoter. This result is representative of the strongest growth defect we achieved with monomer IAPP overexpression. Yeast were grown on agar plates, with and without 100 nM estradiol. C) Spotting assay showing growth defect induced by a single copy insertion of 6xIAPP fused to msfGFP driven by the estradiol-inducible promoter. Yeast were grown on SD CSM plates, with and without 100 nM estradiol. D) A measurement of UPR activation by 6xIAPP expression in a strain bearing a UPR-inducible GFP reporter is shown. Expression of GFP was measured by flow cytometry following 4 hours of growth with 100 nM estradiol, 2 mM dithiothreitol (DTT), or without any treatment. E) Estradiol-dose response growth curves showing 6xIAPP toxicity in the diploid strain used for fluorescent microscopy screening. Yeast were grown in liquid media with the indicated dose of estradiol. The 5 nM dose was chosen for the high-throughput high-content microscopy experiments.
Figure S2. Validation of modifiers recovered from the overexpression screen. Related to Figure 2A. A) Spotting assays demonstrating representative verification data for 6xIAPP suppressors identified in the overexpression screen. Strains were diploid W303 and contain two copies of 6xIAPP. Strains were grown for 48 hours on the indicated media, with the exception of Rpn4 and Ydl129W, which were grown for 24 hours. B) Spotting assays demonstrating verification data for the 6xIAPP enhancers identified in the overexpression screen. Strains were diploid W303 containing one copy of 6xIAPP and grown for 48 hours on the indicated media.
Figure S3. Functional enrichment and validation of enhancers recovered from the deletion screen. Related to Figure 2C. A) Functional enrichment analysis of the 6xIAPP enhancers from yeast deletion and temperature-sensitive allele screens (performed with pantherdb.org). B) Spotting assay verification of Δsec72 as a suppressor of 6xIAPP toxicity. The Δsec72 strain was constructed in the W303 1-copy 6xIAPP background and cells were grown for 48 hours on agar plates with 0 or 100 nM estradiol.
Figure S4. Ste24 acts on 6xIAPP directed to the secretory pathway. Related to Figure 4. A) Western blot of IAPP in whole cell extracts from yeast co-overexpressing 6xIAPP and each of the 16 strongest hits from the overexpression screen. Pgk1 is used as a loading control. B) Yeast spotting assays demonstrating the toxicity of 6xIAPP without the Kar2 signal peptide compared to 6xIAPP with the Kar2 signal peptide as well as the effects of Ste24, Atg11, and Yap1802 overexpression on the toxicity of 6xIAPP without the Kar2 signal peptide. The strains were on the BY4741 background and grown on agar plates for 48 hours with (induced) and without (uninduced) 100nM estradiol.
Highlights.
Developed a yeast model of IAPP proteotoxicity.
IAPP oligomers clogged the ER to cytoplasm translocon
Genetic screens identified STE24/ZMPSTE24 as a strong suppressor of IAPP proteotoxicity
Loss-of-function mutations in ZMPSTE24 were more often found in diabetes patients.
A combination of yeast and human genetics studies explains how aggregates of human islet amyloid polypeptide interfere with protein translocon function to drive β-cell dysfunction in type 2 diabetes
Acknowledgments
This work is dedicated to the memory of our dear friend and mentor, Susan Lindquist, from whom we have learned so much and without whom this work would not have been possible. We thank A. Muthukumar, D. Melton, Y. Freyzon, L. Bayru, L. Clayton, B. Bevis, B.K. Wagner, A. Vetere, A. Haque, E. Hallaçlı, G. Newby, I. Oderberg, and G. Bell, along with members of the Lindquist, Florez, and Melton laboratories for materials, insightful discussions, and comments. We thank the Schuldiner lab for generously providing ZMPSTE24 plasmids and the El-Samad lab for generously providing plasmids for the estradiol-inducible expression system.
Funding
This work was supported by grants from the Whitehead Institute for Biomedical Research, the Picower Institute at MIT, the University of Texas, M.D. Anderson Center, the Howard Hughes Medical Institute, the Glenn Foundation for Medical Research, the Eleanor Schwartz Charitable Foundation, the Edward N. and Della L. Thome Foundation, the JPB Foundation, the Robert A. and Renee E. Belfer Foundation, National Institutes of Health (NIH) grant GM025874, NIH grant R01HG005084, NIH grant R01HG005853, Canadian Institute of Health Research grant FDN-143264, the Department of Defense grant W81XWH14-1-0157. G. Gaglia was supported by a fellowship from the American Italian Cancer Foundation and NIH grant R0 CA175744. D. Landgraf was supported by a Fellowship from the American Parkinson’s Disease Foundation and NIH grant R21 NS087557. P. Narayan was supported by a Fellowship from the Helen Hay Whitney Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, C.K., J.C.F., and S.L.; Methodology, C.K., G.G., J.F., M.I.B., D.L., W.R.H., M.C., C.L.M., C.B., J.C.F., and S.L.; Analysis, C.K., J.F., M.I.B., D.P., J.N., M.C., and C.L.M.; Investigation, C.K., A.A., G.G., J.F., J.M.B., S.F., P.N., J.N., and M.C.; Resources, J.F., J.M.B., D.L., W.R.H., M.C., T2D-GENES Consortium, C.B., J.C.F.; Writing-Original Draft, C.K.; Writing-Review & Editing, all authors; Supervision, J.C.F., S.L.; Funding acquisition, C.L.M., C.B., J.C.F., S.L.
Declaration of Interests
The authors declare no competing interests.
References
- Abedini A, Plesner A, Cao P, Ridgway Z, Zhang J, Tu LH, Middleton CT, Chao B, Sartori DJ, Meng F, et al. Time-resolved studies define the nature of toxic IAPP intermediates, providing insight for anti-amyloidosis therapeutics. eLife Sciences. 2016;5:e12977. doi: 10.7554/eLife.12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda-Díaz A, Mace K, Zuleta I, Harrigan P, El-Samad H. Robust Synthetic Circuits for Two-Dimensional Control of Gene Expression in Yeast. ACS Synth Biol. 2016;6:545–554. doi: 10.1021/acssynbio.6b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ast T, Michaelis S, Schuldiner M. The Protease Ste24 Clears Clogged Translocons. Cell. 2016;164:103–114. doi: 10.1016/j.cell.2015.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrowman J, Wiley PA, Hudon-Miller SE, Hrycyna CA, Michaelis S. Human ZMPSTE24 disease mutations: residual proteolytic activity correlates with disease severity. Hum Mol Genet. 2012;21:4084–4093. doi: 10.1093/hmg/dds233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baryshnikova A, Costanzo M, Kim Y, Ding H, Koh J, Toufighi K, Youn JY, Ou J, San Luis BJ, Bandyopadhyay S, et al. Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat Methods. 2010;7:1017–1024. doi: 10.1038/nmeth.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings LK, Florez JC. The genetics of type 2 diabetes: what have we learned from GWAS? Annals of the New York Academy of Sciences. 2010;1212:59–77. doi: 10.1111/j.1749-6632.2010.05838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AE, Jang J, Gurlo T, Carty MD, Soeller WC, Butler PC. Diabetes Due to a Progressive Defect in -Cell Mass in Rats Transgenic for Human Islet Amyloid Polypeptide (HIP Rat): A New Model for Type 2 Diabetes. Diabetes. 2004;53:1509–1516. doi: 10.2337/diabetes.53.6.1509. [DOI] [PubMed] [Google Scholar]
- Butler AE, Janson J, Soeller WC, Butler PC. Increased β-Cell Apoptosis Prevents Adaptive Increase in β-Cell Mass in Mouse Model of Type 2 Diabetes: Evidence for Role of Islet Amyloid Formation Rather Than Direct Action of Amyloid. Diabetes. 2003;52:2304–2314. doi: 10.2337/diabetes.52.9.2304. [DOI] [PubMed] [Google Scholar]
- Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, Cooper GJ, Holman RR, Turner RC. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988;9:151–159. [PubMed] [Google Scholar]
- Clark KM, Jenkins JL, Fedoriw N, Dumont ME. Human CaaX protease ZMPSTE24 expressed in yeast: Structure and inhibition by HIV protease inhibitors. Protein Science. 2017;26:242–257. doi: 10.1002/pro.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffinier C, Hudon SE, Farber EA, Chang SY, Hrycyna CA, Young SG, Fong LG. HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells. Pnas. 2007;104:13432–13437. doi: 10.1073/pnas.0704212104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Pnas. 1987;84:8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JLY, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costes S, Huang CJ, Gurlo T, Daval M, Matveyenko AV, Rizza RA, Butler AE, Butler PC. -Cell Dysfunctional ERAD/Ubiquitin/Proteasome System in Type 2 Diabetes Mediated by Islet Amyloid Polypeptide-Induced UCH-L1 Deficiency. Diabetes. 2010;60:227–238. doi: 10.2337/db10-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, Erion R, Oristano R, Liu AX, Ramos D, Jethava N, Hosangadi D, et al. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci USA. 2011;108:20881–20890. doi: 10.1073/pnas.1109434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannick J, Johansson S, Njølstad PR. Common and rare forms of diabetes mellitus: towards a continuum of diabetes subtypes. Nat Rev Endocrinol. 2016;12:394–406. doi: 10.1038/nrendo.2016.50. [DOI] [PubMed] [Google Scholar]
- Gonsberg A, Jung S, Ulbrich S, Origi A, Ziska A, Baier M, Koch HG, Zimmermann R, Winklhofer KF, Tatzelt J. The Sec61/SecY complex is inherently deficient in translocating intrinsically disordered proteins. J Biol Chem. 2017 doi: 10.1074/jbc.M117.788067. jbc.M117.788067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurlo T, Ryazantsev S, Huang CJ, Yeh MW, Reber HA, Hines OJ, O’Brien TD, Glabe CG, Butler PC. Evidence for Proteotoxicity in β Cells in Type 2 Diabetes. The American Journal of Pathology. 2010;176:861–869. doi: 10.2353/ajpath.2010.090532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 2000;49:424–430. doi: 10.2337/diabetes.49.3.424. [DOI] [PubMed] [Google Scholar]
- Hu Y, Rolfs A, Bhullar B, Murthy TVS, Zhu C, Berger MF, Camargo AA, Kelley F, McCarron S, Jepson D, et al. Approaching a complete repository of sequence-verified protein-encoding clones for Saccharomyces cerevisiae. Genome Res. 2007;17:536–543. doi: 10.1101/gr.6037607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CJ, Haataja L, Gurlo T, Butler AE, Wu X, Soeller WC, Butler PC. Induction of endoplasmic reticulum stress-induced β-cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide. American Journal of Physiology - Endocrinology and Metabolism. 2007;293:E1656–E1662. doi: 10.1152/ajpendo.00318.2007. [DOI] [PubMed] [Google Scholar]
- Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes. 1999;48:491–498. doi: 10.2337/diabetes.48.3.491. [DOI] [PubMed] [Google Scholar]
- Janson J, Soeller WC, Roche PC, Nelson RT, Torchia AJ, Kreutter DK, Butler PC. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Pnas. 1996;93:7283–7288. doi: 10.1073/pnas.93.14.7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju S, Tardiff DF, Han H, Divya K, Zhong Q, Maquat LE, Bosco DA, Hayward LJ, Brown RH, Jr, Lindquist S, et al. A Yeast Model of FUS/TLS-Dependent Cytotoxicity. PLOS Biology. 2011;9:e1001052. doi: 10.1371/journal.pbio.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens CA, Toukatly MN, Fligner CL, Udayasankar J, Subramanian SL, Zraika S, Aston-Mourney K, Carr DB, Westermark P, Westermark GT, et al. β-Cell Loss and β-Cell Apoptosis in Human Type 2 Diabetes Are Related to Islet Amyloid Deposition. The American Journal of Pathology. 2011;178:2632–2640. doi: 10.1016/j.ajpath.2011.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics. 2011;27:1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayatekin C, Matlack KES, Hesse WR, Guan Y, Chakrabortee S, Russ J, Wanker EE, Shah JV, Lindquist S. Prion-like proteins sequester and suppress the toxicity of huntingtin exon 1. Pnas. 2014;111:12085–12090. doi: 10.1073/pnas.1412504111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VMY, Finkbeiner S, Gitler AD, Bonini NM. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2013;46:152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin E, Costanzo M, Andrews B, Boone C. Synthetic Genetic Array Analysis. Cold Spring Harb Protoc. 2016;2016 doi: 10.1101/pdb.prot088807. pdb.prot088807. [DOI] [PubMed] [Google Scholar]
- Landgraf D, Huh D, Hallacli E, Lindquist S. Scarless Gene Tagging with One-Step Transformation and Two-Step Selection in Saccharomyces cerevisiae and Schizosaccharomyces pombe. PLoS ONE. 2016;11:e0163950. doi: 10.1371/journal.pone.0163950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Vizeacoumar FJ, Bahr S, Li J, Warringer J, Vizeacoumar FS, Min R, VanderSluis B, Bellay J, Devit M, et al. Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat Biotechnol. 2011;29:361–367. doi: 10.1038/nbt.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Gurlo T, Kayed R, Butler AE, Haataja L, Glabe CG, Butler PC. Toxic Human Islet Amyloid Polypeptide (h-IAPP) Oligomers Are Intracellular, and Vaccination to Induce Anti-Toxic Oligomer Antibodies Does Not Prevent h-IAPP–Induced β-Cell Apoptosis in h-IAPP Transgenic Mice. Diabetes. 2007;56:1324–1332. doi: 10.2337/db06-1579. [DOI] [PubMed] [Google Scholar]
- Lorenzo A, Razzaboni B, Weir GC, Yankner BA. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature. 1994;368:756–760. doi: 10.1038/368756a0. [DOI] [PubMed] [Google Scholar]
- Ma Z, Westermark GT, Sakagashira S, Sanke T, Gustavsson A, Sakamoto H, Engström U, Nanjo K, Westermark P. Enhanced in vitro production of amyloid-like fibrils from mutant (S20G) islet amyloid polypeptide. Amyloid. 2001;8:242–249. doi: 10.3109/13506120108993820. [DOI] [PubMed] [Google Scholar]
- Matveyenko AV, Gurlo T, Daval M, Butler AE, Butler PC. Successful Versus Failed Adaptation to High-Fat Diet–Induced Insulin Resistance: The Role of IAPP-Induced β-Cell Endoplasmic Reticulum Stress. Diabetes. 2009;58:906–916. doi: 10.2337/db08-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIsaac RS, Oakes BL, Wang X, Dummit KA, Botstein D, Noyes MB. Synthetic gene expression perturbation systems with rapid, tunable, single-gene specificity in yeast. Nucleic Acids Research. 2012;41:e57–e57. doi: 10.1093/nar/gks1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:S8. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita S, Sakagashira S, Ueyama M, Shimajiri Y, Furuta M, Sanke T. Progressive deterioration of insulin secretion in Japanese type 2 diabetic patients in comparison with those who carry the S20G mutation of the islet amyloid polypeptide gene: A long-term follow-up study. Journal of Diabetes Investigation. 2011;2:287–292. doi: 10.1111/j.2040-1124.2011.00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Morales-Scheihing D, Butler PC, Soto C. Type 2 diabetes as a protein misfolding disease. Trends in Molecular Medicine. 2015;21:439–449. doi: 10.1016/j.molmed.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Morales-Scheihing D, Salvadores N, Moreno-Gonzalez I, Gonzalez C, Taylor-Presse K, Mendez N, Shahnawaz M, Gaber AO, Sabek OM, et al. Induction of IAPP amyloid deposition and associated diabetic abnormalities by a prion-like mechanism. Journal of Experimental Medicine. 2017;214 doi: 10.1084/jem.20161134. jem.20161134–jem.20162610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro CL, De Sandre-Giovannoli A, Bernard R, Boccaccio I, Boyer A, Geneviève D, Hadj-Rabia S, Gaudy-Marqueste C, Smitt HS, Vabres P, et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004;13:2493–2503. doi: 10.1093/hmg/ddh265. [DOI] [PubMed] [Google Scholar]
- Novials A, Sarri Y, Casamitjana R, Rivera F, Gomis R. Regulation of Islet Amyloid Polypeptide in Human Pancreatic Islets. Diabetes. 1993;42:1514–1519. doi: 10.2337/diab.42.10.1514. [DOI] [PubMed] [Google Scholar]
- Outeiro TF. Yeast Cells Provide Insight into Alpha-Synuclein Biology and Pathobiology. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus D, Aranda-Díaz A, Zuleta IA, Walter P, El-Samad H. Delayed Ras/PKA signaling augments the unfolded protein response. Proc Natl Acad Sci USA. 2014;111:14800–14805. doi: 10.1073/pnas.1409588111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripaud L, Chumakova V, Antonin M, Hastie AR, Pinkert S, Körner R, Ruff KM, Pappu RV, Hornburg D, Mann M, et al. Overexpression of Q-rich prion-like proteins suppresses polyQ cytotoxicity and alters the polyQ interactome. Pnas. 2014;111:18219–18224. doi: 10.1073/pnas.1421313111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzel RA, Meier JJ, Lin CY, Veldhuis JD, Butler PC. Human Islet Amyloid Polypeptide Oligomers Disrupt Cell Coupling, Induce Apoptosis, and Impair Insulin Secretion in Isolated Human Islets. Diabetes. 2006;56:65–71. doi: 10.2337/db06-0734. [DOI] [PubMed] [Google Scholar]
- Rivera JF, Costes S, Gurlo T, Glabe CG, Butler PC. Autophagy defends pancreatic β cells from human islet amyloid polypeptide-induced toxicity. J Clin Invest. 2014;124:3489–3500. doi: 10.1172/JCI71981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakagashira S, Hiddinga HJ, Tateishi K, Sanke T, Hanabusa T, Nanjo K, Eberhardt NL. S20G Mutant Amylin Exhibits Increased in Vitro Amyloidogenicity and Increased Intracellular Cytotoxicity Compared to Wild-Type Amylin. The American Journal of Pathology. 2000;157:2101–2109. doi: 10.1016/S0002-9440(10)64848-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakagashira S, Sanke T, Hanabusa T, Shimomura H, Ohagi S, Kumagaye KY, Nakajima K, Nanjo K. Missense Mutation of Amylin Gene (S20G) in Japanese NIDDM Patients. Diabetes. 1996;45:1279–1281. doi: 10.2337/diab.45.9.1279. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treusch S, Hamamichi S, Goodman JL, Matlack KES, Chung CY, Baru V, Shulman JM, Parrado A, Bevis BJ, Valastyan JS, et al. Functional Links Between Aβ Toxicity, Endocytic Trafficking, and Alzheimer’s Disease Risk Factors in Yeast. Science. 2011;334:1241–1245. doi: 10.1126/science.1213210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark P. Quantitative studies on amyloid in the islets of Langerhans. Ups J Med Sci. 1972;77:91–94. doi: 10.1517/03009734000000014. [DOI] [PubMed] [Google Scholar]
- Wolfe KJ, Ren HY, Trepte P, Cyr DM. Polyglutamine-Rich Suppressors of Huntingtin Toxicity Act Upstream of Hsp70 and Sti1 in Spatial Quality Control of Amyloid-Like Proteins. PLoS ONE. 2014;9:e95914. doi: 10.1371/journal.pone.0095914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Liu H, Chuang CL, Li X, Au M, Zhang L, Phillips ARJ, Scott DW, Cooper GJS. The pathogenic mechanism of diabetes varies with the degree of overexpression and oligomerization of human amylin in the pancreatic islet β cells. Faseb J. 2014;28:5083–5096. doi: 10.1096/fj.14-251744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. IAPP overexpression suppressors. Related to Figure 2. The verified suppressors recovered from the genome-wide overexpression screen.
Table S2. Temperature sensitive alleles that modify IAPP toxicity. Related to Figure 2. The statistically significant 6xIAPP suppressors and enhancers recovered from the screen of 1,388 temperature sensitive alleles.
Table S3. Deletions that modify IAPP toxicity. Related to Figure 2. The statistically significant 6xIAPP suppressors and enhancers recovered from the screen of 4,635 viable genetic deletions.
Table S4. Yeast strains used in this study. Related to STAR Methods: Experimental Model and Subject Details
Table S5. Oligonucleotides used in this study. Related to Key Resources Table: Oligonucleotides
Table S6. Plasmids used in this study. Related to Key Resources Table: Recombinant DNA
Figure S1. IAPP constructs used. Related to Figure 1. A) Amino acid sequences of IAPP constructs employed in this work. The highlighted elements are the signal peptide (orange), human IAPP pro peptide (red), mature IAPP hormone (blue), linker residues (black). B) Spotting assay showing growth defect induced by expression of 1xIAPP from a 2μ high-copy plasmid, driven by the estradiol-inducible promoter. This result is representative of the strongest growth defect we achieved with monomer IAPP overexpression. Yeast were grown on agar plates, with and without 100 nM estradiol. C) Spotting assay showing growth defect induced by a single copy insertion of 6xIAPP fused to msfGFP driven by the estradiol-inducible promoter. Yeast were grown on SD CSM plates, with and without 100 nM estradiol. D) A measurement of UPR activation by 6xIAPP expression in a strain bearing a UPR-inducible GFP reporter is shown. Expression of GFP was measured by flow cytometry following 4 hours of growth with 100 nM estradiol, 2 mM dithiothreitol (DTT), or without any treatment. E) Estradiol-dose response growth curves showing 6xIAPP toxicity in the diploid strain used for fluorescent microscopy screening. Yeast were grown in liquid media with the indicated dose of estradiol. The 5 nM dose was chosen for the high-throughput high-content microscopy experiments.
Figure S2. Validation of modifiers recovered from the overexpression screen. Related to Figure 2A. A) Spotting assays demonstrating representative verification data for 6xIAPP suppressors identified in the overexpression screen. Strains were diploid W303 and contain two copies of 6xIAPP. Strains were grown for 48 hours on the indicated media, with the exception of Rpn4 and Ydl129W, which were grown for 24 hours. B) Spotting assays demonstrating verification data for the 6xIAPP enhancers identified in the overexpression screen. Strains were diploid W303 containing one copy of 6xIAPP and grown for 48 hours on the indicated media.
Figure S3. Functional enrichment and validation of enhancers recovered from the deletion screen. Related to Figure 2C. A) Functional enrichment analysis of the 6xIAPP enhancers from yeast deletion and temperature-sensitive allele screens (performed with pantherdb.org). B) Spotting assay verification of Δsec72 as a suppressor of 6xIAPP toxicity. The Δsec72 strain was constructed in the W303 1-copy 6xIAPP background and cells were grown for 48 hours on agar plates with 0 or 100 nM estradiol.
Figure S4. Ste24 acts on 6xIAPP directed to the secretory pathway. Related to Figure 4. A) Western blot of IAPP in whole cell extracts from yeast co-overexpressing 6xIAPP and each of the 16 strongest hits from the overexpression screen. Pgk1 is used as a loading control. B) Yeast spotting assays demonstrating the toxicity of 6xIAPP without the Kar2 signal peptide compared to 6xIAPP with the Kar2 signal peptide as well as the effects of Ste24, Atg11, and Yap1802 overexpression on the toxicity of 6xIAPP without the Kar2 signal peptide. The strains were on the BY4741 background and grown on agar plates for 48 hours with (induced) and without (uninduced) 100nM estradiol.