

Abstract
Microglia are emerging as a key cell type in neurodegenerative diseases, yet human microglia are challenging to study in vitro, especially in the large numbers of individuals needed for genetic studies. Here we demonstrate the effectiveness of an in vitro model system of human monocyte-derived microglia-like cells (MDMi) to recapitulate many aspects of microglia phenotype and function. We then used this model system to perform an expression quantitative trait locus (eQTL) study examining 94 genes from loci associated with Alzheimer’s disease, Parkinson’s disease and multiple sclerosis in 94 healthy individuals. We found six loci (CD33, PILRB, NUP160, LRRK2, RGS1, METTL21B) in which the risk haplotype drives the association with both disease susceptibility and altered expression of a nearby gene (cis-eQTL). In the PILRB and LRRK2 loci, the cis-eQTL is found in the MDMi cells but not in peripheral monocytes, suggesting that differentiation leads to the acquisition of a cellular state, which uncovers the functional consequence of certain genetic variants. We further validated the effect of risk haplotypes at the protein level for PILRB and CD33, and we confirmed that the CD33 risk haplotype alters a functional outcome, phagocytosis, in MDMi. Finally, we hypothesize that the MDMi-specific increased LRRK2 gene expression could be the key functional outcome of the GWAS Parkinson’s disease LRKK2 SNP, rs76904798.
Introduction
Genome-wide association studies (GWAS) and sequencing studies have implicated the innate immune system in neurodegenerative diseases, specifically Alzheimer’s disease (AD), Parkinson’s disease (PD) and multiple sclerosis (MS), in that an increasing number of identified disease-specific genetic loci contain innate immune specific genes. For example, CD33 is known to play a role in AD as shown by our group and others (1–3) and TREM2 has been implicated in frontotemporal dementia, PD and amyotrophic lateral sclerosis in addition to AD (4–9). In order to leverage GWAS findings for therapeutic targeting, the GWAS associations must first be translated to functional outcomes, as has been done for CD33, where we demonstrated that the risk allele leads to increased CD33 expression on the surface of monocytes and to diminished internalization of amyloid-β 42 peptide, supporting a robust genotype-to-phenotype association for this AD single-nucleotide polymorphism (SNP) (1). Mapping gene expression as a quantitative trait (eQTL analysis) can identify which genes in a locus modulate their expression in response to a SNP, and one can relate the direction of the association with expression to the direction of association with other traits, such as disease susceptibility. As the number of these eQTL studies has increased, it has become clear that many eQTLs are cell type, activation state or environment dependent. This observation reflects the fact that epigenomic features that are specific to a differentiation state, for example, influence access to the sequence variant and hence influence the transcriptional output of the locus (10–12).
We have previously reported a number of monocyte-specific eQTL associations for MS, AD and PD susceptibility variants (13). Since MS, AD and PD are all diseases of the central nervous system (CNS), we wanted to further examine these susceptibility loci in innate immune cells in the context of the CNS environment. Microglia are the resident innate immune cells of the CNS, and they are critical regulators of CNS homeostasis in health and of inflammatory responses in disease (14–16). There is also strong evidence, that in diseases such as stroke, brain trauma (17), AD (18, 19), PD (20, 21), and MS (22), peripheral leukocytes, including monocytes, infiltrate the brain and differentiate into effector cells such as macrophages that are found at the site of pathology. Therefore, for genetic studies, we would ideally isolate these resident microglia and infiltrating macrophages from a large number of subjects. However, due to the difficulty in acquiring these cells from the human brain, alternative methods of obtaining these cells are needed. In one approach, patient derived cells can be reprogrammed and differentiated into neural and certain glial cells (23–25); however, the differentiation of human induced pluripotent stem cells into microglia remains technically challenging, takes several months (26), and is currently not performed on the scale needed for genetic studies.
While the investigation of genetically-driven changes in microglial/infiltrating macrophage gene expression in neurodegenerative diseases is therefore limited by our lack of access to human microglia/infiltrating macrophages from the number of subjects required to perform well-powered genomic and functional analyses, a number of protocols do exist to differentiate human monocytes towards a microglia-like phenotype (27–32). These protocols have leveraged the plasticity of immune cells to direct primary bone marrow-derived monocytes towards a microglia-like phenotype, either by co-culturing with astrocytes, astrocyte-conditioned media, or via stimulation with recombinant human cytokines. These polarized cells have been shown to have a ramified morphology, phagocytic ability, engulf synapses, and have a CX3CR1high/CCR2low phenotype (27, 29, 32). They also express P2RY12 and TMEM119 at the protein level (32). Because these cells do not share the embryonic origin of the vast majority of resident microglia in the non-diseased brain (33), it is possible that they are more similar to infiltrating macrophages that have taken up residence in the CNS. As there is currently no robust marker capable of distinguishing these two cell types in humans, they currently cannot be differentiated. A study by Etemad et al., (2012) generated these cells by culturing ex vivo human monocytes in the presence of M-CSF, GM-CSF, NGF-β, and CCL2, all of which are important for microglia development and survival (33–37). In addition, Ohgidani et al., (2014) developed ramified microglia-like cells from human monocytes using a combination of GM-CSF and IL-34. IL-34 shares a receptor (CSF-1R) with M-CSF and is known to be important for directing the differentiation of microglia (38). These studies have shown that these inducible microglia/macrophage display features observed in resident microglia and that they are optimal for high throughput studies to enable genetic analyses since they work with recombinant cytokines and serum free media, reducing potential variation, which could arise from astrocytes or astrocyte conditioned media.
Here we used an in vitro model system of monocytes polarized with CNS cytokines that are critical for the microglia microenvironment in vivo, and we leveraged the capacity of this system to be deployed in moderate scale to perform a genetic study of genes associated with susceptibility to neurodegenerative disease. To validate this in vitro system, we compared our MDMi to induced microglia (26), ex vivo derived human microglia (39) and murine microglia (40). Protocols which polarize microglia and macrophages towards pro-inflammatory “M1” and anti-inflammatory “M2” phenotypes allow us to study molecular mechanisms that may help distinguish microglia from other myeloid cells. It has been shown that the M1 phenotype of human microglia is distinct from that of monocyte-derived macrophages (41), and we have leveraged this information to characterize our inducible microglia/macrophage cells. With the generation of a detailed transcriptomic reference and an understanding of the characteristics of our model system in hand, we then evaluated the effect of common genetic variation on the expression of genes found in susceptibility loci for MS, PD and AD. This study thus begins to investigate, at a larger scale, whether such disease susceptibility variants have effects that are present in the CNS milieu-polarized myeloid cells and to demonstrate that our in vitro generated MDMi system has a cell state that is distinct from that of ex vivo monocytes from which they are derived.
Results
Polarization of monocytes into monocyte-derived microglia-like cells (MDMi)
We first identified the genes (fold-change genes) that are more highly expressed in our MDMi model system when compared to a tissue-level profile from human dorsolateral prefrontal cortex (DLPFC). Specifically, human peripheral monocytes from five young healthy individuals were stimulated with GM-CSF, M-CSF, CCL2, NGF-β and IL-34 for 10 days and analyzed for gene expression using RNA-sequencing (RNA-seq). We then compared RNA-seq data from the MDMi to the RNA-seq data from the DLPFC of 248 individuals without a pathological diagnosis of AD, and we assembled the list of 368 genes who have a three-fold difference in expression in this analysis (fold-change genes). These fold-change genes were then compared to published lists of microglia-enriched genes from the ES/iPSC induced microglia (pMGL) (26), the enriched genes from ex vivo murine microglia (P60MG) (40) and ex vivo human microglia isolated from epilepsy/tumor surgery (39). Interestingly, there was much more overlap among the three human models than with the mouse microglia. We found that MDMi and pMGL share 203 genes, or 55% of the MDMi fold-change genes (Fig. 1A). Further, MDMi have 118 genes, or 32% of the fold-change genes, in common with the epileptic/tumor surgery-derived microglia enriched genes, and they share 100 genes, or 29%, of the pMGL enriched genes. In comparison, our MDMi only share 24 genes, or 6.5%, with the murine p60 microglia from the Bennett paper (40); similarly, the pMGL cells only share 19 genes, or 5.5%, and the surgery-derived microglia share 34 genes, or 5.4%, with these murine cells., highlighting the importance of working in the human system.
Fig. 1. Polarization of monocytes to MDMi induces a microglial gene expression and functional phenotype.

Peripheral human monocytes from young healthy subjects were incubated with polarizing cytokines and differentiated to MDMi. (A) The cell-type specific enriched genes for MDMi, ex vivo human microglia (Zhang), ex vivo murine microglia (P60MG) and iPSC/ES induced microglia were compared (pMGL). (B) Four genes defined as being microglia-specific in mice were significantly upregulated in MDMi (TGFβR1 ****P < 0.0001, PROS1 ***P = 0.0005, C1QB ***P = 0.0007, P2RX7 **P = 0.0058) at day 10 of differentiation compared to freshly isolated monocytes and MDM from the same 5 individuals. Gene expression was quantified via RNA-sequencing. One-way ANOVA with Tukey’s post hoc test. (C) P2RY12 and TMEM119 are highly expressed at the protein level in MDMi compared to monocytes. (D) MDMi functionally mimic human microglia in response to conditions that lead to M1 or M2 polarization. Under M1 conditions MDMi express significantly more IL-10 mRNA (**P < 0.01) compared to MDM from the same individuals. Student’s t-test, N = 14. For (B) and (D) each dot represents a biological replicate. Horizontal line denotes the mean.
The comparison of the datasets clearly demonstrates inter-mammalian differences in microglia gene expression; however, we also examined specific genes that have been reported to be key for microglial function in the murine system. For example, we found upregulation of genes such as TGFBR1 (P < 0.0001), PROS1 (P = 0.0005), P2RX7 (P = 0.0058) and C1QB (P = 0.0007) (Fig. 1B) in our MDMi relative to ex vivo human monocytes and monocyte-derived macrophages. Notably, P2RY12, and TMEM119, other murine microglia signature genes, are not upregulated compared to ex vivo monocytes, although P2RY12 mRNA is upregulated in MDMi compared to monocyte-derived macrophages (Fig. S1A). Therefore, we examined these molecules at the protein level, and determined that both TMEM119 and P2RY12 are highly expressed in MDMi compared to monocytes from the same individuals (Fig. 1C and Fig. S1B, C & D). Additionally, microglia are thought to be long-lived cells, while monocytes and monocyte-derived macrophages are thought to be more transient. To explore this, we kept the MDMi and the MDM in culture for 30 days and found that the apoptotic activator gene, BAX was highly expressed in ex vivo monocytes and MDM cultured for 30 days, but not MDMi (Fig. S2). Therefore, we conclude that MDMi may have a longer-lived phenotype that is similar to what is seen in vivo with microglia.
We also evaluated the function of MDMi and demonstrated that, like primary human microglia, MDMi differ from peripheral myeloid cells in respect to their response to environmental stimuli (41): under M1 but not M2 polarizing conditions, the level of IL10 expression was significantly higher (P = 0.0018) in MDMi compared to macrophages from the same individuals (Fig. 1D), which is concordant with a published report (41) in which purified human brain microglia from surgical tissue were polarized to an M1 or M2 state in vitro.
Functional consequences of genetic variants differs between monocytes and MDMi
Using a Fluidigm high-throughput qPCR chip, we measured 94 genes found in loci associated with one of three neurodegenerative diseases - AD, MS or PD - in MDMi from 95 young healthy subjects of European ancestry with genome-wide genotype data. From this mRNA data set, we performed an eQTL study to identify genotype-driven effects on the expression of nearby genes in MDMi (cis-expression quantitative trait locus analysis, cis-eQTL) (Fig. 2A). Specifically, for each gene measured, we evaluated whether SNPs found within 1 Mb of the transcription start site of the measured gene are associated with the gene’s expression level. At an FDR < 0.05, we identified cis-eQTL associations for 35 genes in MDMi (Table S1). After linkage disequilibrium (LD) pruning the list of SNPs with an MDMi eQTL (eliminating those SNPs with R2 > 0.2 with the top SNP in a locus), 141 SNPs with a cis-eQTL remained. We compared our MDMi eQTLs to our previously published monocyte eQTL data derived from 211 young, healthy subjects of European ancestry (13), and we found that 82 (58.1%) of these SNPs were also cis-eQTLs in monocytes (global FDR < 0.05) (Table S2). Of those eQTLs that are shared, 100% (82 out of 82) shared the same direction of effect in MDMi and monocytes, showing that differentiation does not affect the function of these variants. Correlations of the effect size (rho) of the SNPs in each gene (rho MDMi versus rho monocytes) were plotted for each lead SNP (Fig. S3).
Fig. 2. Genotype-induced differential gene expression varies between monocytes and MDMi.

Using a Fluidigm high-throughput qPCR chip, we measured the expression of 94 genes found in loci associated with susceptibility to AD, MS or PD in MDMi differentiated from the monocytes of 94 young healthy subjects of European ancestry with genome-wide genotype data. (A) Manhattan plot of eQTL results for the 94 genes measured in MDMi. N = 95 biological replicates per gene. Each dot is one SNP; selected SNPs include all of those found within 1 Mb of the transcription start site of the profiled gene. The x-axis denotes the physical position of the SNP, and the y axis reports the significance of the SNP’s association with the expression of the nearby gene (eQTL result). The red line highlights the threshold of significance in our analysis. (B) We compared our MDMi eQTL results to those of our previously published monocyte eQTL results derived from N = 211 young, healthy subjects of European ancestry (13). In the left panel, we plot the top eQTL SNP for each gene in the MDMi data; the x-axis reports the absolute value of the effect size (ρ) of the SNP in MDMI while the ρ in the monocyte data is presented in the y-axis. The threshold of significance (FDR<0.05) is shown by dotted lines in each dimension. The light red quadrant contains those loci with consistent effects in both cell types, while the light green quadrant contains those loci which have a significant association only in MDMi cells. The light blue quadrant contains those loci with an effect in monocytes that is not significant in our current MDMi analysis. The right panel displays the best SNP for each locus in the monocyte data. (C) Locus zoom plots highlight the regional distribution of associations in two loci, CD37 and CR1, which have very different eQTL associations in the two cell types. Each dot is one SNP in these figures, with the physical position captured on the x-axis and eQTL significance on the y-axis. The location of genes in this locus is shown below the SNPs. The top eQTL SNP for the MDMi data is shown as a purple diamond, and the other SNPs are colored by the extent of linkage disequilibrium (r2) with the lead SNP. In the monocyte plots, the SNP colors are defined by the lead MDMi SNP, highlighting the fact that the haplotype driving association in MDMi does not have a strong effect in monocytes. MDMi N = 95 biological replicates, monocytes N = 211 biological replicates.
In Fig. 2B, we show that of the best eQTL SNPs for each gene in the MDMi, some are shared with monocytes (NUP160 for example), while others are only found in the MDMi (ex: CD37) (Fig. 2B, left panel), and the same is true for the top eQTL SNPs in the monocytes (ex: CD40) (Fig. 2B, right panel). In Fig. 2C, regional plots of CD37 and CR1 illustrate the fact that certain haplotypes influence gene expression very differently in monocytes and MDMi, with clear peaks of associations in MDMi and no effect in our larger sample of subjects with profiled monocytes.
Association of disease-specific GWAS SNPs with gene expression in MDMi identifies different associations
To provide mechanistic insights into the effect of GWAS-derived SNPs on disease susceptibility, we merged our cis-eQTL results with a list of disease-associated SNPs influencing AD (42, 43), PD (44) and MS (45). Of the 70 disease-associated SNPs within 1 Mb of our genes of interest, we found seven eQTL associations at a secondary, targeted FDR of 0.05 (Table 1). The SNP/gene expression associations of rs10838725/NUP160, rs701006/METTL21B and rs1323292/RGS1 are also found in monocytes (13), while the associations for rs1476679/PILRB and rs76904798/LRRK2 appear to be unique to MDMi. Fig. 3A (upper panel) shows the cis-eQTL shared between monocytes and MDMi where the association for RGS1 and the MS SNP rs1323292 were similar in both cell types. Fig. 3A (lower panel) shows an example of a differentiation state-specific cis-eQTL in which PTK2B and its respective disease associated SNP rs28834970 was observed in the monocyte dataset, but not in the MDMi Fluidigm dataset. On the other hand, a significant association was found in MDMi for the PILRB gene (thought to be an activating immune receptor) and the AD SNP rs1476679 that was not seen in the monocyte dataset. Specifically, the rs1476679T risk allele was associated with increased PILRB expression in MDMi (P = 0.00084) but not in monocytes (P = 0.19) (Fig. 3B, upper panel), and these results were replicated via PCR in a smaller study examining an independent set of individuals from whom we assessed both monocytes and MDMi (Fig. 3B, middle panel). PILRB protein expression mirrored the mRNA data in both monocytes and MDMi (Fig. 3B, lower panel). In MDMi, a one-way ANOVA revealed a significant effect of genotype on PILRB mRNA [F(2, 34) = 6.78, P = 0.0034] and protein [F(2, 31) = 5.15, P = 0.0112] expression which was not present in monocytes [mRNA: F(2, 34) = 1.67, P = 0.2037; protein: F(2, 31) = 2.14, P = 0.1346].
Table 1.
MDMi exhibit significant disease associated cis-eQTLs
| SNP | Gene | CHR | MDMi rho | MDMi p-value | Monocyte rho | Monocyte p-value | Disease | MDMi Targeted FDR |
|---|---|---|---|---|---|---|---|---|
| rs3865444 | CD33_SHORT | 19 | −0.688 | 1.39E-14 | −0.254* | 1.96E-04* | AD | 1.38E-12 |
| CD33_LONG | 0.339 | 7.91E-04 | 1.30E-02 | |||||
| rs1476679 | PILRB | 7 | −0.337 | 8.42E-04 | −0.09 | 1.90E-01 | AD | 1.30E-02 |
| rs10838725 | NUP160 | 11 | −0.383 | 1.29E-04 | −0.241 | 4.08E-04 | AD | 3.22E-03 |
| rs76904798 | LRRK2 | 12 | −0.479 | 8.92E-07 | −0.163 | 1.77E-02 | PD | 2.96E-05 |
| rs1323292 | RGS1 | 1 | 0.516 | 8.62E-08 | 0.486 | 6.33E-14 | MS | 4.30E-06 |
| rs701006 | METTL21B | 12 | 0.335 | 9.14E-04 | 0.413 | 4.18E-10 | MS | 1.30E-02 |
Fig. 3. Association of disease-specific GWAS SNPs with gene expression in MDMi identifies different associations than in monocytes.
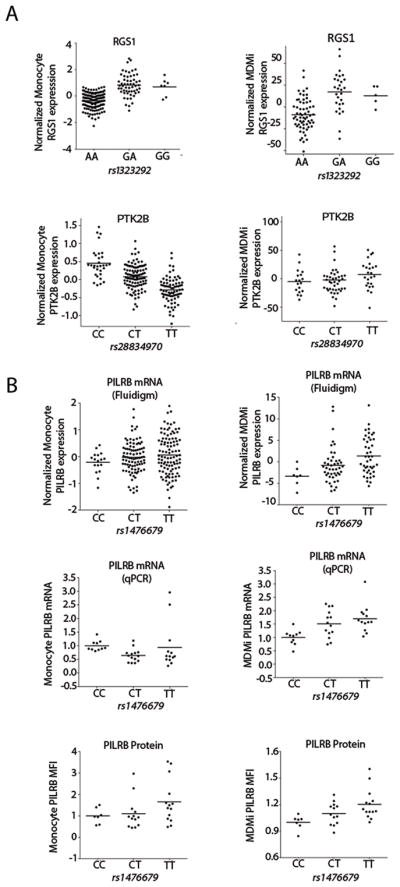
Each gene selected for the fluidigm experiment is in a locus associated with either AD, PD or MS. (A) Example of a cis-eQTL shared between monocytes and MDMi (upper pair of graphs): the association for RGS1 expression and the MS risk allele rs1323292G were similar in both cell types. With PTK2B (lower pair of graphs), the AD associated rs28834970C risk allele has an effect in monocytes but not in MDMi. N = 95 (MDMi), N = 211 (monocytes). (B) A significant eQTL was found in MDMi for the PILRB gene (P = 0.00084), illustrated with the AD SNP rs1476679 that was not seen in the monocyte dataset (P = 0.19) (upper row). N = 95 (MDMi), N = 211 (monocytes). This finding was replicated in an independent set of 37 individuals at the RNA level (P = 0.0034) using TaqMan PCR (middle row) and 34 individuals at the protein level (P = 0.0112) (bottom row). One-way ANOVA with Tukey’s post hoc test. Each dot represents a biological replicate. Horizontal line denotes the mean.
Of particular interest, we examined the GWAS SNP for PD at the LRRK2 locus (44). The role of LRRK2 in PD has been of great interest as dominant mutations in the gene have been associated with familial PD (46). The PD GWAS rs76904798T risk allele was associated with increased LRRK2 expression in MDMi (P = 8.92E-07), while there is a non-significant trend (P =0.0177) in monocytes (Fig. 4A). Thus, the polarization of the monocytes to MDMi appears to enhance the magnitude of the correlation between the PD SNP rs76904798 and LRRK2: the effect size of the SNP is significantly different between the two cell types (P < 0.05). Critically, using the colocalization method (47), we found that the PD association and the eQTL SNP colocalize in MDMi (posterior probability eQTL and PD associations driven by a shared causal variant=0.90) but not in monocytes (Fig. 4B), suggesting that the disease SNP regulates gene expression in the in vitro polarization model only.
Fig. 4. Significant association in MDMi between LRRK2 gene expression and the PD SNP rs7690479.

(A) The PD GWAS rs76904798T risk allele was associated with increased LRRK2 expression in MDMi (right) (P < 8.92E-07), while there is a non-significant trend in the monocyte data (left) (P < 1.77E-02). (B) Three regional association plots illustrate the colocalization of the PD susceptibility haplotype, tagged by rs76904798, and the eQTL haplotype in MDMi but not in monocytes. In the top panel, we present data from monocytes: each dot is one SNP and is colored in relation to the extent of linkage disequilibrium (r2) with the lead PD SNP (rs76904798). The color key is presented to the right of the panels. The x-axis presents the physical position of the SNP, and the y-axis presents the association between the SNP and the level of LRRK2 expression. In the middle panel, the same set of SNPs is presented; here, the association P-value is derived from the relation of each SNP to LRRK2 expression in MDMi cells. In the bottom panel, the results of the published PD GWAS (44) are presented for the same set of SNPs.
Differential expression of CD33 isoforms in MDMi
CD33 is a cell surface protein expressed by myeloid cells, and higher CD33 expression in the brain has been associated with more advanced cognitive decline and AD (2, 48). We recently demonstrated that individuals with the rs3865444CC risk genotype have increased CD33 on the surface of their monocytes compared to those with the rs3865444AA protective genotype (1). Furthermore, alternative splicing of CD33 generates two isoforms of the protein: full-length CD33M and truncated CD33m which lacks the Ig V-set domain encoded by exon 2 (49). The AD rs3865444 SNP or a SNP in high LD leads to alternative splicing of exon 2 as the primary mechanism of the genetically driven differential expression of CD33 (3, 13). Here we found a significant effect of genotype on CD33 expression: an increase in full-length CD33M mRNA expression [F(2, 75) = 7.74, P = 0.0009] and decreased CD33m mRNA expression [F(2, 75) = 42, P < 0.0001] in MDMi from subjects with the rs3865444CC risk genotype, in a dose dependent fashion (Table 1) (Fig. 5A). Similar findings were observed at the protein level (Fig. 5B). Densitometry analysis of the western blot revealed a significant effect of genotype on CD33M protein expression in monocytes (P = 0.034) and MDMi (P = 0.009) from the same individuals. Interestingly, the effect of genotype on CD33m protein expression was only observed in MDMi (P = 0.016) and not in monocytes (P = 0.124) (as we had previously reported (13)). The genotype-dependent difference in CD33 surface expression was further confirmed in MDMi using high content imaging (P = 0.001; Fig. 5C). Given that myeloid cells such as infiltrating macrophages or microglia are thought to be involved in AD by phagocytosis of amyloid-β, which accumulates in neuritic amyloid plaques (a neuropathologic feature of Alzheimer’s disease (50)), we tested the hypothesis that MDMi from subjects with the rs3865444C CD33 risk allele have a reduced state of activation and therefore a reduced phagocytic ability, compared with subjects with the protective allele. We found reduced uptake in MDMi bearing the rs3865444CC risk allele of fluorescently labeled dextran (P = 0.047; Fig. 5D), similar to what we had previously reported for monocytes (1).
Fig. 5. Differential expression of CD33 isoforms in MDM.

(A) In our model system, we found a significant effect of genotype on CD33 expression: an increase in CD33M mRNA expression (***P = 0.0009) and decreased CD33m mRNA expression (****P < 0.0001) in MDMi in a dose dependent fashion relative to the rs3865444C risk allele in the fluidigm dataset. One-way ANOVA with Tukey’s post hoc test, N = 95. (B) Western blot analysis shows a significant effect of genotype on CD33M protein expression in monocytes (*P = 0.034) and MDMi (**P = 0.009) from a second set of individuals (N = 16). However, a significant effect of genotype on CD33m protein expression was only observed in MDMi (*P = 0.016) and not monocytes (P = 0.124). A representative western blot displays the data from four subjects, with each subject being in a column. The genotype of the subject is included at the top of the column. (C) The genotype-dependent difference in CD33 surface expression (P = 0.001) and (D) FITC-dextran uptake (P = 0.047) was further confirmed in MDMi using high content imaging. Each dot represents a biological replicate. Student’s t-test performed for B–D.
Discussion
Here, we refine a translational research tool whereby microglia-like cells are generated from human peripheral blood monocytes in the presence of polarizing cytokines. The recent identification of microglia-specific gene signatures has enabled us to examine the phenotype of MDMi cells at the level of gene expression rather than relying on characteristics such as subtle shifts in mean fluorescence intensity for certain surface markers and morphology. We have shown for the first time that monocytes cultured for 10 days in the presence of polarizing cytokines upregulate microglia-specific genes when compared to monocytes, including TGFBR1, a key gene for microglial phenotype (51). Other highly upregulated genes in MDMi include C1QB, a critical mediator of synaptic refinement and plasticity (52) and PROS1 which directs microglia phagocytosis of apoptotic cells (53). A role for P2RX7 was described by Monif et al., (54) who found that P2RX7 drives microglia activation and proliferation. Thus, it is clear that MDMi express many genes that play important roles in microglia function.
There is no single functional assay that defines microglia. Several groups have used the ability to phagocytose material as a functional readout; however, other myeloid cells, including monocytes and macrophages, share this ability. Synaptic pruning is another characteristic function, but this is difficult to model in vitro. We leveraged a recent study (41) which characterized the functional properties of human microglia in vitro by assessing cytokine production under different inflammatory phenotypes, namely M1 and M2 conditions. Consistent with this study, we demonstrate that, under M1 conditions, MDMi express more IL-10 compared to MDM, with no difference seen under M2 conditions. Thus illustrating the distinctiveness of these cells relative to MDM. We also found that the MDMi survive up to 30 days in culture without upregulating the pro-apoptotic marker BAX, while the MDM do upregulate BAX and clearly have increased cell death, highlighting functional differences between these two in vitro-polarized cell types.
Currently, there is no method to differentiate microglia and CNS-infiltrating blood-derived macrophages in humans. The role of infiltrating blood-derived macrophages in the brain remains poorly understood and is most likely disease-dependent. In AD, inflammation and microglial activation have been extensively studied but the contribution of blood-derived cells was underestimated for many years. Nonetheless, these infiltrating cells have recently evoked a lot of attention. Recent data suggest that blood-derived monocytes play a role in amyloid-β clearance. For example, more perivascular amyloid-β was noted in CCR2 deficient Tg2576 AD mice, which have reduced early recruitment of monocytes (35). In support of this protective role, increasing monocyte recruitment may delay the progression of AD (55, 56). On the other hand, infiltrating monocytes may enhance tissue damage in MS. For example, monocyte-derived macrophages were found to initiate demyelination in the MS model experimental autoimmune encephalomyelitis (EAE) (57). Furthermore, blocking monocyte recruitment to the CNS blocked EAE progression, suggesting that these infiltrating cells are essential for disease progression (58). An argument could be made that our MDMi model system more likely represents CNS infiltrating blood-derived macrophages rather than embryonically derived microglia, but our current lack of knowledge in the comparison of human microglia and CNS infiltrating macrophages keep us from making that distinction at this time.
GWAS studies have identified novel loci associated with disease susceptibility. In determining the mechanistic outcomes of this genetic variation, it has been demonstrated that eQTLs are context dependent. In a number of studies it has become clear that cell type and activation state are important factors in identification of the modulation of gene expression by genetic variation (12, 13). To extend our understanding of the genetic modulation of gene expression in our in vitro model of microglia, we associated MDMi RNA expression with SNPs. We examined the expression of 94 genes in loci associated with increased risk of developing AD, MS or PD in MDMi from 94 young healthy individuals and compared their expression to microarray data from monocytes in 211 individuals generated in an earlier study (13). The expression of many of these genes is affected by genetic variation in the MDMi and it is clear that, for a number of genes, the genetic regulation of gene expression is determined by completely different SNPs in the MDMi and monocytes (Fig. 2). This demonstrates that the genotype driven differential gene expression between monocytes and MDMi can be context-specific and highlights the importance of examining genotype-induced gene expression differences in CNS relevant cell types.
A striking example of a cell-type-specific cis-eQTL was found in the PILRB gene. We found an association in MDMi for the PILRB gene and the AD SNP rs1476679 that was not seen in the monocyte dataset. However, based on a Bayesian colocalization analysis (47) that sought to determine whether our MDMi eQTL and the International Genomics of Alzheimer’s Project Stage 1 (42) AD susceptibility GWAS effects were driven by a shared causal variant, it appears more likely that the eQTL and AD GWAS effects are not driven by a shared variant (posterior probability eQTL and AD associations driven by a shared causal variant=0.04). This result suggests, that while the AD SNP displays some association with PILRB expression, the effect of rs1476679 is probably mediated by another mechanism, and the association may be the result of partial linkage disequilibrium between the AD and eQTL causal variants.
As activation of monocytes was shown to highlight different eQTL associations (12), we find that the MDMi differentiation also identifies additional associations, as is the case for PILRB, and may enhance some associations that are weakly seen in monocytes, as is the case for LRRK2. Pathogenic mutations in LRRK2 are the most common genetic cause of familial PD (59, 60). High LRRK2 expression has been discovered recently in myeloid cells but not in T-cells, suggesting a functional role for LRRK2 in the innate immune system (61). In addition, LRRK2 is expressed in microglia where it modulates the proinflammatory response in these cells (62). Interestingly, in our data, the monocyte eQTL SNP rs10784428 (56) is associated with LRRK2 expression in both monocytes and MDMi, while the PD-associated SNP rs76904798 is associated with LRRK2 expression only in MDMi (Fig. 4). This suggests that LRRK2 may have a context-specific role, which may also vary with the state of CNS. These results highlight the potential use of MDMi for studying the functional role of the LRRK2 variant in microglia.
Based on the expression of CD33 in human microglia (1) and our recent characterization of the AD risk gene CD33 in monocytes where we found increased CD33 surface protein in individuals with the CD33 rs3865444CC risk genotype, we explored whether this was also true in MDMi. We find that MDMi recapitulate the previously described expression and functional effects of the risk allele; however, interestingly, we also found differential protein expression of the truncated CD33m in MDMi, which was not seen in monocytes (63). We do note that while the CD33 locus has been associated with AD at a threshold of genome-wide significance (64), it fell below that threshold in a recent analysis (42). Further evaluation of these data and additional replication data supporting the role of CD33 in non-European populations (65–67) and AD-related endophenotypes (1) support its role; functional studies also link CD33 to TREM2, (68) complementing these genetic evaluations. Regardless of the association with AD, the CD33 variant has an important functional effect on myeloid cells and this report of differential effects in different myeloid contexts strengthens the argument that MDMi are distinct from monocytes and suggest that the disease-associated effect of rs3865444 may differ subtly between microglia and monocytes. To confirm this, future work should investigate whether the isoform-specific pattern is also seen in postmortem primary human microglia from genotyped individuals.
In summary, this report presents the detailed characterization of an in vitro model system that can be used to investigate the role of human innate immune cells in neurodegenerative disease. The characterization of eQTL effects with the MDMi illustrates the utility of the model system for moderate-throughput experiments in which 94 different subjects were examined in parallel and has returned very relevant findings, particularly for LRRK2 where the risk allele did not have an effect in peripheral monocytes. MDMi cells therefore offer an important tool for translational studies in neuroimmunology, one that has a place along with the characterization of biopsy- or autopsy-derived microglia and macrophage and, probably, human iPSC-derived cells that take much longer and are more costly to differentiate.
While MDMi are a promising translational research tool, some limitations must be considered. First, we must keep in mind that MDMi are derived from peripheral monocytes, which may be different from yolk sac derived microglia, especially in terms of epigenetic changes that occur over a lifetime. Despite this, our data suggest that MDMi may allow us to study cells that share many features with microglia; they may certainly be relevant to studying brain macrophages, as these are differentiated from infiltrating monocytes that are exposed to the CNS cytokine milieu. As we continue to use this cellular system to model human microglia and macrophages, we will identify additional strengths and limitations as a model system, as has been done for monocyte-derived dendritic cells which are routinely used to model in vivo dendritic cells by human immunologists (69).
Second, monolayers of MDMi do not resemble the 3 dimensional (3D) spatial arrangement of microglia in the CNS, and stiff plastic culture dishes do not resemble the physical environment of the brain. Therefore, a recently described 3D culture system using a collagen construct may allow us to study these cells in a more relevant tissue-like microenvironment (70). In addition, due to unknown reasons, we find that in approximately 10% of subjects the monocytes do not survive the differentiation process. This may have implications in studies where the sample size is low. To control for this, we excluded subjects from analysis who had poorly differentiated MDMi based on visual observations.
Of note, the MDMi in the present study were derived from frozen PBMCs; this means that large numbers of samples may be collected and banked for use in large-scale studies, such as the eQTL experiment presented here that are done in a single batch, minimizing the effect of experimental variation. In addition, the use of a well-defined set of recombinant cytokines to generate MDMi further reduces variability (compared to approaches involving astrocyte-conditioned media) and enhances reproducibility by being standardized, which is ideal for high-throughput screening and translational genetic studies.
In terms of understanding the functional outcomes of GWAS SNPs, it is now clear that they must be examined in the correct cell type and context. Due to the limitations of acquiring human microglia in sufficient quantity from large numbers of human subjects (as is needed for genetic association studies) and a lack of a cell line that adequately mimics human microglia, we propose that the MDMi system could fill that void. In addition to genetic studies, an application of the MDMi system may be in drug screening, in which a biochemical or functional change can be monitored, when microglia are thought to be the target cell type. Additional potential applications of MDMi in clinical research include the exploration of various unknown biological behaviors of human microglia in neurodegenerative disorders. Enhancing the MDMI approach by co-culturing these cells with human neurons and astrocytes would bring us closer to a so-called “brain in a dish” system and would allow us to model innate immune dysfunction in a more complex system where the target cell is interacting with other relevant cells. Finally, given that the differentiation protocol takes just 10 days, this system has the potential to contribute to the personalization of therapies in which MDMi derived from a patient’s monocytes can be rapidly characterized to individualize drug selection and the management of a neurodegenerative or other disease (71). In conclusion, we propose an in vitro translational tool for generating microglia-like cells quickly and easily from adult blood, and for exploring microglia function and dysfunction on a large scale.
Materials and Methods
Detailed Materials and Methods are provided in Supplementary Materials.
Study Design
The overall aim of the study was to characterize and refine a translational research tool whereby microglia-like cells are generated from human peripheral blood monocytes in the presence of polarizing cytokines. We refer to these cells as monocyte-derived microglia-like cells (MDMi). The objective of the first portion of the study was to determine whether MDMi express known microglia genes and have a functional phenotype that is similar to microglia. To do this, MDMi were derived from the peripheral venous blood of healthy control volunteers from the PhenoGenetic Project. This is a living tissue bank that consists of healthy subjects who are re-contactable and can therefore be recalled based on their genotype. 1,753 healthy subjects >18 years of age have been recruited from the general population of Boston. Subjects are free of chronic inflammatory, infectious and metabolic diseases and are of diverse ethnicities (29% are non-Caucasian) and are 62.7% women. Median age is 24. The objective of the second portion of the study was to extend our understanding of the genetic modulation of gene expression in our in vitro model of microglia. To do this we associated MDMi RNA expression with SNPs. We used this model system to perform an expression quantitative trait locus (eQTL) study examining the expression of 94 genes in loci associated with increased risk of developing AD, MS or PD in MDMi from 94 young healthy individuals from the PhenoGenetic Project and compared their expression to microarray data from monocytes in 211 individuals generated in an earlier study (13). For the eQTL analysis in monocytes (N = 211), which included only Causasians, the median age was 25 and 56.9% were women. For the eQTL analysis in MDMi (N = 95), also exclusively Caucasian, the median age was 29 and 67.4% were women. Informed consent was obtained from all human subjects. All blood draws and data analyses were done in compliance with protocols approved by the Institutional Review Boards of each institution.
Induction of Monocyte-Derived Microglia-like Cells (MDMi) and Monocyte-Derived Macrophages (MDM)
Peripheral blood mononuclear cells (PBMCs) were separated by Lymphoprep gradient centrifugation (StemCell Technologies). PBMCs were frozen at a concentration of 1–3 × 107 cells ml–1 in 10% DMSO (Sigma-Aldrich)/90% fetal bovine serum (vol/vol, Corning). Prior to each study, aliquots of frozen PBMCs from the PhenoGenetic cohort were thawed and washed in 10 ml PBS. Monocytes were positively selected from whole PBMCs using anti-CD14+ microbeads (Miltenyi Biotech) and plated at the following densities per well: 1 × 105 cells (96-well plate), 3 × 105 cells (24-well plate), 1 × 106 cells (6-well plate). To induce the differentiation of MDMi, monocytes were incubated in serum-free conditions using RPMI-1640 Glutamax (Life Technologies) with 1% penicillin/streptomycin (Lonza) and 2.5 μg/ml Fungizone (Life Technologies) and a mixture of the following human recombinant cytokines: M-CSF (10 ng/ml; Biolegend 574806), GM-CSF (10 ng/ml; R&D Systems 215-GM-010/CF), NGF-β (10 ng/ml; R&D Systems 256-GF-100), CCL2 (100 ng/ml; Biolegend 571404) and IL-34 (100 ng/ml; R&D Systems 5265-IL-010/CF) at standard humidified culture conditions (37°C, 5% CO2) for up to 15 days. Monocyte-derived macrophages were generated by incubating monocytes with 20 ng/ml M-CSF in RMPI plus 10% fetal bovine serum (FBS). The cells were used for experiments and characterization at different time points as indicated. For the 30-day culture, both MDM and MDMi cells were moved on day 15 to unconditioned RPMI media until day 30.
Statistical Analyses
Statistical analyses for Fig. 1A, Fig. 1D, and Fig. 5B–D were performed using Prism 5 (GraphPad Software). Comparisons of protein and mRNA expression across groups were analyzed by Student’s t-test or one-way ANOVA as appropriate and significant differences were deconvoluted using Tukey’s multiple comparison test. Probability values of <0.05 were considered to represent statistically significant differences.
Supplementary Material
Fig. S1. MDMi express more P2RY12 and TMEM119 compared to monocytes.
Fig. S2. The pro-apoptotic gene BAX is increased in monocytes and MDM compared to MDMi.
Fig. S3. Many SNPs have differential eQTLs between monocytes and MDMi.
Table S1. Cis-eQTLs in MDMi
Table S2. LD pruned list of cis-eQTLs in MDMi
Acknowledgments
We thank the participants of PGP for their time and specimens that they contributed. This work was supported by the US National Institutes of Health grants R01 AG043617, R01 NS089674, R01 AG048015, and Alzheimer’s Association grant DVT-14-321148. K.J.R, P.L.D. and E.M.B. designed and implemented the study and wrote the manuscript. K.J.R. conducted the experiments with technical assistance from K.P, M.F, M.C, P.A.W, A.McH, M.O, B.J.K, G.C, J.I, and W.E. C.C.W, J.M.R. and J.X. performed statistical analyses and assisted with the interpretation of results. P.L.D and N.E.C. coordinated the collection of blood from PGP. E.M.B is a consultant for Pfizer. P.L.D is a consultant for Sanofi Genzyme, Roche and Celgene. J.I. has editorial activities with the Journal of Neuroimmunology and is a consultant with Biogen.
References
- 1.Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, Morris MC, Evans DA, Johnson K, Sperling RA, Schneider JA, Bennett DA, De Jager PL. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nature neuroscience. 2013;16:848–850. doi: 10.1038/nn.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malik M, Simpson JF, Parikh I, Wilfred BR, Fardo DW, Nelson PT, Estus S. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:13320–13325. doi: 10.1523/JNEUROSCI.1224-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guerreiro RJ, Lohmann E, Bras JM, Gibbs JR, Rohrer JD, Gurunlian N, Dursun B, Bilgic B, Hanagasi H, Gurvit H, Emre M, Singleton A, Hardy J. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA neurology. 2013;70:78–84. doi: 10.1001/jamaneurol.2013.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J. TREM2 variants in Alzheimer’s disease. The New England journal of medicine. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K. Variant of TREM2 associated with the risk of Alzheimer’s disease. The New England journal of medicine. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P, Baloh RH, Ravits J, Simpson E, Appel SH, Pestronk A, Goate AM, Miller TM, Cruchaga C, Harms MB. TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA neurology. 2014;71:449–453. doi: 10.1001/jamaneurol.2013.6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, Hatanpaa KJ, Lomen-Hoerth C, Kertesz A, Bigio EH, Lippa C, Josephs KA, Knopman DS, White CL, 3rd, Caselli R, Mackenzie IR, Miller BL, Boczarska-Jedynak M, Opala G, Krygowska-Wajs A, Barcikowska M, Younkin SG, Petersen RC, Ertekin-Taner N, Uitti RJ, Meschia JF, Boylan KB, Boeve BF, Graff-Radford NR, Wszolek ZK, Dickson DW, Rademakers R, Ross OA. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Molecular neurodegeneration. 2013;8:19. doi: 10.1186/1750-1326-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu G, Liu Y, Jiang Q, Jiang Y, Feng R, Zhang L, Chen Z, Li K, Liu J. Convergent Genetic and Expression Datasets Highlight TREM2 in Parkinson’s Disease Susceptibility. Molecular neurobiology. 2015 doi: 10.1007/s12035-015-9416-7. [DOI] [PubMed] [Google Scholar]
- 10.Lee MN, Ye C, Villani AC, Raj T, Li W, Eisenhaure TM, Imboywa SH, Chipendo PI, Ran FA, Slowikowski K, Ward LD, Raddassi K, McCabe C, Lee MH, Frohlich IY, Hafler DA, Kellis M, Raychaudhuri S, Zhang F, Stranger BE, Benoist CO, De Jager PL, Regev A, Hacohen N. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science (New York, NY) 2014;343:1246980. doi: 10.1126/science.1246980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ye CJ, Feng T, Kwon HK, Raj T, Wilson MT, Asinovski N, McCabe C, Lee MH, Frohlich I, Paik HI, Zaitlen N, Hacohen N, Stranger B, De Jager P, Mathis D, Regev A, Benoist C. Intersection of population variation and autoimmunity genetics in human T cell activation. Science (New York, NY) 2014;345:1254665. doi: 10.1126/science.1254665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, Jostins L, Plant K, Andrews R, McGee C, Knight JC. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science (New York, NY) 2014;343:1246949. doi: 10.1126/science.1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raj T, Rothamel K, Mostafavi S, Ye C, Lee MN, Replogle JM, Feng T, Lee M, Asinovski N, Frohlich I, Imboywa S, Von Korff A, Okada Y, Patsopoulos NA, Davis S, McCabe C, Paik HI, Srivastava GP, Raychaudhuri S, Hafler DA, Koller D, Regev A, Hacohen N, Mathis D, Benoist C, Stranger BE, De Jager PL. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science (New York, NY) 2014;344:519–523. doi: 10.1126/science.1249547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pollard JW. Trophic macrophages in development and disease. Nature reviews Immunology. 2009;9:259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vinet J, Weering HR, Heinrich A, Kalin RE, Wegner A, Brouwer N, Heppner FL, Rooijen N, Boddeke HW, Biber K. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. Journal of neuroinflammation. 2012;9:27. doi: 10.1186/1742-2094-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Priller J, Flugel A, Wehner T, Boentert M, Haas CA, Prinz M, Fernandez-Klett F, Prass K, Bechmann I, de Boer BA, Frotscher M, Kreutzberg GW, Persons DA, Dirnagl U. Targeting gene-modified hematopoietic cells to the central nervous system: use of green fluorescent protein uncovers microglial engraftment. Nature medicine. 2001;7:1356–1361. doi: 10.1038/nm1201-1356. [DOI] [PubMed] [Google Scholar]
- 18.Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, Schwartz M. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:11784–11789. doi: 10.1073/pnas.0604681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC, Vinters HV. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. European journal of clinical investigation. 2002;32:360–371. doi: 10.1046/j.1365-2362.2002.00994.x. [DOI] [PubMed] [Google Scholar]
- 20.Rektor I, Goldemund D, Sheardova K, Rektorova I, Michalkova Z, Dufek M. Vascular pathology in patients with idiopathic Parkinson’s disease. Parkinsonism & related disorders. 2009;15:24–29. doi: 10.1016/j.parkreldis.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 21.Song IU, Kim YD, Cho HJ, Chung SW. The effects of silent cerebral ischemic lesions on the prognosis of idiopathic Parkinson’s disease. Parkinsonism & related disorders. 2013;19:761–763. doi: 10.1016/j.parkreldis.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 22.Cramer SP, Simonsen H, Frederiksen JL, Rostrup E, Larsson HB. Abnormal blood-brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. NeuroImage Clinical. 2014;4:182–189. doi: 10.1016/j.nicl.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullivan SE, Young-Pearse TL. Induced pluripotent stem cells as a discovery tool for Alzheimers disease. Brain research. 2015 doi: 10.1016/j.brainres.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang PW, Haidet-Phillips AM, Pham JT, Lee Y, Huo Y, Tienari PJ, Maragakis NJ, Sattler R, Rothstein JD. Generation of GFAP::GFP astrocyte reporter lines from human adult fibroblast-derived iPS cells using zinc-finger nuclease technology. Glia. 2016;64:63–75. doi: 10.1002/glia.22903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao MC, Muratore CR, Gierahn TM, Sullivan SE, Srikanth P, De Jager PL, Love JC, Young-Pearse TL. Single-Cell Detection of Secreted Abeta and sAPPalpha from Human IPSC-Derived Neurons and Astrocytes. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2016;36:1730–1746. doi: 10.1523/JNEUROSCI.2735-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, Corcoran S, Bakiasi G, Tsai LH, Aubourg P, Ransohoff RM, Jaenisch R. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nature medicine. 2016;22:1358–1367. doi: 10.1038/nm.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Etemad S, Zamin RM, Ruitenberg MJ, Filgueira L. A novel in vitro human microglia model: characterization of human monocyte-derived microglia. Journal of neuroscience methods. 2012;209:79–89. doi: 10.1016/j.jneumeth.2012.05.025. [DOI] [PubMed] [Google Scholar]
- 28.Leone C, Le Pavec G, Meme W, Porcheray F, Samah B, Dormont D, Gras G. Characterization of human monocyte-derived microglia-like cells. Glia. 2006;54:183–192. doi: 10.1002/glia.20372. [DOI] [PubMed] [Google Scholar]
- 29.Ohgidani M, Kato TA, Setoyama D, Sagata N, Hashimoto R, Shigenobu K, Yoshida T, Hayakawa K, Shimokawa N, Miura D, Utsumi H, Kanba S. Direct induction of ramified microglia-like cells from human monocytes: dynamic microglial dysfunction in Nasu-Hakola disease. Scientific reports. 2014;4:4957. doi: 10.1038/srep04957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noto D, Sakuma H, Takahashi K, Saika R, Saga R, Yamada M, Yamamura T, Miyake S. Development of a culture system to induce microglia-like cells from haematopoietic cells. Neuropathology and applied neurobiology. 2014;40:697–713. doi: 10.1111/nan.12086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sievers J, Parwaresch R, Wottge HU. Blood monocytes and spleen macrophages differentiate into microglia-like cells on monolayers of astrocytes: morphology. Glia. 1994;12:245–258. doi: 10.1002/glia.440120402. [DOI] [PubMed] [Google Scholar]
- 32.Sellgren CM, Sheridan SD, Gracias J, Xuan D, Fu T, Perlis RH. Patient-specific models of microglia-mediated engulfment of synapses and neural progenitors. Molecular psychiatry. 2017;22:170–177. doi: 10.1038/mp.2016.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (New York, NY) 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Simone R, Ambrosini E, Carnevale D, Ajmone-Cat MA, Minghetti L. NGF promotes microglial migration through the activation of its high affinity receptor: modulation by TGF-beta. Journal of neuroimmunology. 2007;190:53–60. doi: 10.1016/j.jneuroim.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 35.El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nature medicine. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 36.Esen N, Kielian T. Effects of low dose GM-CSF on microglial inflammatory profiles to diverse pathogen-associated molecular patterns (PAMPs) Journal of neuroinflammation. 2007;4:10. doi: 10.1186/1742-2094-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schilling T, Nitsch R, Heinemann U, Haas D, Eder C. Astrocyte-released cytokines induce ramification and outward K+ channel expression in microglia via distinct signalling pathways. The European journal of neuroscience. 2001;14:463–473. doi: 10.1046/j.0953-816x.2001.01661.x. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, Barrow AD, Diamond MS, Colonna M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nature immunology. 2012;13:753–760. doi: 10.1038/ni.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, Duncan JA, 3rd, Cheshier SH, Shuer LM, Chang EF, Grant GA, Gephart MG, Barres BA. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron. 2016;89:37–53. doi: 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, Weissman IL, Chang EF, Li G, Grant GA, Hayden Gephart MG, Barres BA. New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E1738–1746. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Durafourt BA, Moore CS, Zammit DA, Johnson TA, Zaguia F, Guiot MC, Bar-Or A, Antel JP. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. 2012;60:717–727. doi: 10.1002/glia.22298. [DOI] [PubMed] [Google Scholar]
- 42.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi SH, Reitz C, Pasquier F, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letenneur L, Moron FJ, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fievet N, Huentelman MW, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuiness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossu P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Deniz Naranjo MC, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannefelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O’Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH, Jr, Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RF, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JS, Boerwinkle E, Riemenschneider M, Boada M, Hiltuenen M, Martin ER, Schmidt R, Rujescu D, Wang LS, Dartigues JF, Mayeux R, Tzourio C, Hofman A, Nothen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nature genetics. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Replogle JM, Chan G, White CC, Raj T, Winn PA, Evans DA, Sperling RA, Chibnik LB, Bradshaw EM, Schneider JA, Bennett DA, De Jager PL. A TREM1 variant alters the accumulation of Alzheimer-related amyloid pathology. Annals of neurology. 2015;77:469–477. doi: 10.1002/ana.24337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, Ikram MA, Ioannidis JP, Hadjigeorgiou GM, Bis JC, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L, Eriksson N, Foroud T, Singleton AB. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nature genetics. 2014;46:989–993. doi: 10.1038/ng.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, Cotsapas C, Shah TS, Spencer C, Booth D, Goris A, Oturai A, Saarela J, Fontaine B, Hemmer B, Martin C, Zipp F, D’Alfonso S, Martinelli-Boneschi F, Taylor B, Harbo HF, Kockum I, Hillert J, Olsson T, Ban M, Oksenberg JR, Hintzen R, Barcellos LF, Agliardi C, Alfredsson L, Alizadeh M, Anderson C, Andrews R, Sondergaard HB, Baker A, Band G, Baranzini SE, Barizzone N, Barrett J, Bellenguez C, Bergamaschi L, Bernardinelli L, Berthele A, Biberacher V, Binder TM, Blackburn H, Bomfim IL, Brambilla P, Broadley S, Brochet B, Brundin L, Buck D, Butzkueven H, Caillier SJ, Camu W, Carpentier W, Cavalla P, Celius EG, Coman I, Comi G, Corrado L, Cosemans L, Cournu-Rebeix I, Cree BA, Cusi D, Damotte V, Defer G, Delgado SR, Deloukas P, di Sapio A, Dilthey AT, Donnelly P, Dubois B, Duddy M, Edkins S, Elovaara I, Esposito F, Evangelou N, Fiddes B, Field J, Franke A, Freeman C, Frohlich IY, Galimberti D, Gieger C, Gourraud PA, Graetz C, Graham A, Grummel V, Guaschino C, Hadjixenofontos A, Hakonarson H, Halfpenny C, Hall G, Hall P, Hamsten A, Harley J, Harrower T, Hawkins C, Hellenthal G, Hillier C, Hobart J, Hoshi M, Hunt SE, Jagodic M, Jelcic I, Jochim A, Kendall B, Kermode A, Kilpatrick T, Koivisto K, Konidari I, Korn T, Kronsbein H, Langford C, Larsson M, Lathrop M, Lebrun-Frenay C, Lechner-Scott J, Lee MH, Leone MA, Leppa V, Liberatore G, Lie BA, Lill CM, Linden M, Link J, Luessi F, Lycke J, Macciardi F, Mannisto S, Manrique CP, Martin R, Martinelli V, Mason D, Mazibrada G, McCabe C, Mero IL, Mescheriakova J, Moutsianas L, Myhr KM, Nagels G, Nicholas R, Nilsson P, Piehl F, Pirinen M, Price SE, Quach H, Reunanen M, Robberecht W, Robertson NP, Rodegher M, Rog D, Salvetti M, Schnetz-Boutaud NC, Sellebjerg F, Selter RC, Schaefer C, Shaunak S, Shen L, Shields S, Siffrin V, Slee M, Sorensen PS, Sorosina M, Sospedra M, Spurkland A, Strange A, Sundqvist E, Thijs V, Thorpe J, Ticca A, Tienari P, van Duijn C, Visser EM, Vucic S, Westerlind H, Wiley JS, Wilkins A, Wilson JF, Winkelmann J, Zajicek J, Zindler E, Haines JL, Pericak-Vance MA, Ivinson AJ, Stewart G, Hafler D, Hauser SL, Compston A, McVean G, De Jager P, Sawcer SJ, McCauley JL. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nature genetics. 2013;45:1353–1360. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Healy DG, O’Sullivan SS, Falchi M, Bonifati V, Durr A, Bressman S, Aasly A, Zabetian CP, Goldwurm S, Ferreira JJ, Tolosa E, Kay DM, Klein C, Williams DR, Marras C, Lang AE, Wszolek ZK, Berciano J, Schapira AHV, Lynch T, Bhatia KP, Gasser T, Lees AJ, Wood NW. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS genetics. 2014;10:e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, Goate AM. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PloS one. 2012;7:e50976. doi: 10.1371/journal.pone.0050976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hernandez-Caselles T, Martinez-Esparza M, Perez-Oliva AB, Quintanilla-Cecconi AM, Garcia-Alonso A, Alvarez-Lopez DM, Garcia-Penarrubia P. A study of CD33 (SIGLEC-3) antigen expression and function on activated human T and NK cells: two isoforms of CD33 are generated by alternative splicing. Journal of leukocyte biology. 2006;79:46–58. doi: 10.1189/jlb.0205096. [DOI] [PubMed] [Google Scholar]
- 50.Fiala M, Cribbs DH, Rosenthal M, Bernard G. Phagocytosis of amyloid-beta and inflammation: two faces of innate immunity in Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2007;11:457–463. doi: 10.3233/jad-2007-11406. [DOI] [PubMed] [Google Scholar]
- 51.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nature neuroscience. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bialas AR, Stevens B. TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nature neuroscience. 2013;16:1773–1782. doi: 10.1038/nn.3560. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Fourgeaud L, Traves PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, Callaway P, Zagorska A, Rothlin CV, Nimmerjahn A, Lemke G. TAM receptors regulate multiple features of microglial physiology. Nature. 2016;532:240–244. doi: 10.1038/nature17630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Monif M, Reid CA, Powell KL, Smart ML, Williams DA. The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:3781–3791. doi: 10.1523/JNEUROSCI.5512-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nature medicine. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O’Banion MK. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. The Journal of clinical investigation. 2007;117:1595–1604. doi: 10.1172/JCI31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, Kidd G, Zorlu MM, Sun N, Hu W, Liu L, Lee JC, Taylor SE, Uehlein L, Dixon D, Gu J, Floruta CM, Zhu M, Charo IF, Weiner HL, Ransohoff RM. Differential roles of microglia and monocytes in the inflamed central nervous system. The Journal of experimental medicine. 2014;211:1533–1549. doi: 10.1084/jem.20132477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nature neuroscience. 2011;14:1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 59.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 60.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 61.Thevenet J, Pescini Gobert R, Hooft van Huijsduijnen R, Wiessner C, Sagot YJ. Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PloS one. 2011;6:e21519. doi: 10.1371/journal.pone.0021519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, Cowell RM, West AB. LRRK2 inhibition attenuates microglial inflammatory responses. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:1602–1611. doi: 10.1523/JNEUROSCI.5601-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raj T, Ryan KJ, Replogle JM, Chibnik LB, Rosenkrantz L, Tang A, Rothamel K, Stranger BE, Bennett DA, Evans DA, De Jager PL, Bradshaw EM. CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Human molecular genetics. 2014;23:2729–2736. doi: 10.1093/hmg/ddt666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nature genetics. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mao YF, Guo ZY, Pu JL, Chen YX, Zhang BR. Association of CD33 and MS4A cluster variants with Alzheimer’s disease in East Asian populations. Neuroscience letters. 2015;609:235–239. doi: 10.1016/j.neulet.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 66.Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, Valladares O, Lin CF, Larson EB, Graff-Radford NR, Evans D, De Jager PL, Crane PK, Buxbaum JD, Murrell JR, Raj T, Ertekin-Taner N, Logue M, Baldwin CT, Green RC, Barnes LL, Cantwell LB, Fallin MD, Go RCP, Griffith P, Obisesan TO, Manly JJ, Lunetta KL, Kamboh MI, Lopez OL, Bennett DA, Hendrie H, Hall KS, Goate AM, Byrd GS, Kukull WA, Foroud TM, Haines JL, Farrer LA, Pericak-Vance MA, Schellenberg GD, Mayeux R. Variants in the ATP-Binding Cassette Transporter (ABCA7), Apolipoprotein E ε4, and the Risk of Late-Onset Alzheimer Disease in African Americans. JAMA: the journal of the American Medical Association. 2013;309:1483–1492. doi: 10.1001/jama.2013.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carrasquillo MM, Belbin O, Hunter TA, Ma L, Bisceglio GD, Zou F, Crook JE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, Passmore P, Morgan K, Younkin SG. Replication of EPHA1 and CD33 associations with late-onset Alzheimer’s disease: a multi-centre case-control study. Molecular neurodegeneration. 2011;6:54. doi: 10.1186/1750-1326-6-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chan G, White CC, Winn PA, Cimpean M, Replogle JM, Glick LR, Cuerdon NE, Ryan KJ, Johnson KA, Schneider JA, Bennett DA, Chibnik LB, Sperling RA, Bradshaw EM, De Jager PL. CD33 modulates TREM2: convergence of Alzheimer loci. Nature neuroscience. 2015;18:1556–1558. doi: 10.1038/nn.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sachamitr P, Hackett S, Fairchild PJ. Induced pluripotent stem cells: challenges and opportunities for cancer immunotherapy. doi: 10.3389/fimmu.2014.00176. [DOI] [PMC free article] [PubMed]
- 70.Haw RT, Tong CK, Yew A, Lee HC, Phillips JB, Vidyadaran S. A three-dimensional collagen construct to model lipopolysaccharide-induced activation of BV2 microglia. Journal of neuroinflammation. 2014;11:134. doi: 10.1186/1742-2094-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ohgidani M, Kato TA, Kanba S. Introducing directly induced microglia-like (iMG) cells from fresh human monocytes: a novel translational research tool for psychiatric disorders. Frontiers in cellular neuroscience. 2015;9:184. doi: 10.3389/fncel.2015.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, Macmanes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, Leduc RD, Friedman N, Regev A. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature protocols. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Leidi M, Gotti E, Bologna L, Miranda E, Rimoldi M, Sica A, Roncalli M, Palumbo GA, Introna M, Golay J. M2 macrophages phagocytose rituximab-opsonized leukemic targets more efficiently than m1 cells in vitro. Journal of immunology (Baltimore, Md: 1950) 2009;182:4415–4422. doi: 10.4049/jimmunol.0713732. [DOI] [PubMed] [Google Scholar]
- 75.Lim AS, Srivastava GP, Yu L, Chibnik LB, Xu J, Buchman AS, Schneider JA, Myers AJ, Bennett DA, De Jager PL. 24-hour rhythms of DNA methylation and their relation with rhythms of RNA expression in the human dorsolateral prefrontal cortex. PLoS genetics. 2014;10:e1004792. doi: 10.1371/journal.pgen.1004792. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 76.Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the rush Memory and Aging Project. Current Alzheimer research. 2012;9:646–663. doi: 10.2174/156720512801322663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS. Overview and findings from the religious orders study. Current Alzheimer research. 2012;9:628–645. doi: 10.2174/156720512801322573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics (Oxford, England) 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. MDMi express more P2RY12 and TMEM119 compared to monocytes.
Fig. S2. The pro-apoptotic gene BAX is increased in monocytes and MDM compared to MDMi.
Fig. S3. Many SNPs have differential eQTLs between monocytes and MDMi.
Table S1. Cis-eQTLs in MDMi
Table S2. LD pruned list of cis-eQTLs in MDMi