

Abstract
Cytochrome P450 BM-3 is a bacterial enzyme with sequence similarity to mammalian P450s that catalyzes the hydroxylation of fatty acids with high efficiency. Enzyme–substrate binding and dynamics has been an important topic of study for cytochromes P450 because most of the crystal structures of substrate-bound structures show the complex in an inactive state. We have determined a new crystal structure for cytochrome P450 BM-3 in complex with N-palmitoylglycine (NPG), which unexpectedly showed a direct bidentate ion pair between NPG and arginine 47 (R47). We further explored the role of R47, the only charged residue in the binding pocket in cytochrome P450 BM-3, through mutagenesis and crystallographic studies. The mutations of R47 to glutamine (R47Q), glutamic acid (R47E), and lysine (R47K) were designed to investigate the role of its charge in binding and catalysis. The oppositely charged R47E mutation had the greatest effect on activity and binding. The crystal structure of R47E BMP shows that the glutamic acid side chain is blocking the entrance to the binding pocket, accounting for NPG’s low binding affinity and charge repulsion. For R47Q and R47K BM-3, the mutations caused only a slight change in kcat and a large change in Km and Kd, which suggests that R47 mostly is involved in binding and that our crystal structure, 4KPA, represents an initial binding step in the P450 cycle.
Graphical abstract
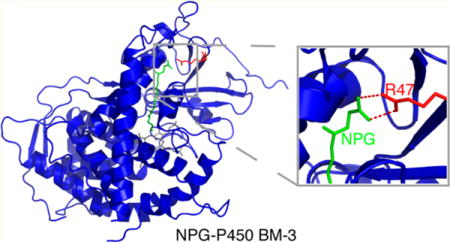
Cytochromes P450 are a class of heme-dependent enzymes that perform the stereospecific oxidation of substrates, metabolize drugs and other xenobiotics, and synthesize steroids and vitamins. Because the active site of cytochrome P450 can incorporate substrates of varying size, predicting the range of chemistry of this enzyme is an important topic of study. However, cryogenic crystal structures of numerous P450 isoforms show that the substrate is positioned too far away from the heme to be a catalytically relevant binding mode.1–4 Therefore, solving new crystal structures, identifying important amino acids in the binding pocket, and understanding the active site’s structure and dynamics remain crucial areas of research for drug discovery, protein engineering, and insight into the biological function of BM-3.
A bacterial model widely used to study mammalian P450s, cytochrome P450 BM-3 from Bacillus megaterium, was identified by Fulco and has structural similarity to the corresponding CYP4A mammalian fatty acid hydroxylases.5,6 BM-3’s exact function is not known, but it hydroxylates fatty acids with high efficiency in the presence of reduced nicotinamide adenine dinucleotide phosphate (NADPH) and oxygen. BM-3 with arachidonate as a substrate has the highest catalytic activity determined for a P450 mono-oxygenase (17 000 min−1).7 The efficient activity can be explained by BM-3 having an attached reductase domain and a high efficiency of intramolecular electron transfer. The heme domain of BM-3 (residues 1–472) is a 55 kDa protein and is referred to as BMP. In this Article, we use the full-length enzyme for biochemical activity studies and the heme domain for crystallization.
Before a crystal structure of cytochrome P450 BM-3 was ever published, researchers speculated that in addition to a hydrophobic pocket there was a charged residue in the binding pocket because P450 BM-3 is inactive to n-hydrocarbons and fatty methyl esters but reactive to fatty alcohols, acids, and amides.8 When the crystal structure of P450 BMP was solved, it showed that the binding site has a long hydrophobic channel that is 8–10 Å in diameter and the beginning of the channel contained the only charged residue, arginine 47 (R47),9 as shown in Figure 1. The role of R47 has been proposed and demonstrated by mutagenesis studies to be involved in the initial binding of the substrate, steering the substrate into the active site,10,11 and the positioning and stabilizing of the substrate in the active site.7,12–14 Protein engineering of P450 BM-3 has become a popular area of study,15–26 changing the charge at position 47 has been shown to change the substrate specificity. For example, the R47E BM-3 mutant showed an increase in activity for positively charged trimethylammonium (TMA) compounds.27 In addition, the mutations R47Q and R47S BM-3 have shown selectivity for acyl homoserine lactone over acyl homoserine substrates.28 Overall, R47 is a commonly mutated residue to allow for the catalysis of non-natural substrates29–40 and shorter substrates because the carboxylate binding site is eliminated.26
Figure 1.
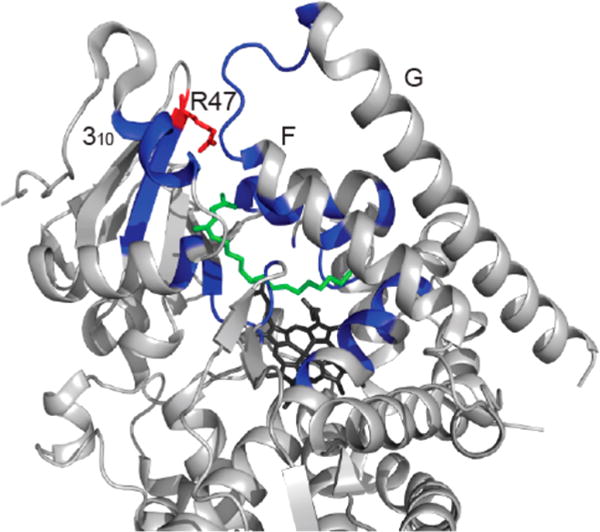
Structure of cytochrome P450 BMP with NPG bound. The F and G loops form one side of the substrate entrance channel, whereas the other side of the channel is formed by the 310 helix (residues 16–20) and the R47 β-sheet. The binding pocket and access channel are colored in blue, NPG, green, heme, black, and R47, red. This figure shows that arginine is in a key position to interact with substrates at the entrance of the binding pocket. This structure was prepared in PyMOL41 with PDB file 1JPZ.11
The charge at position 47 has been explored by reversing the charge with a R47E BM-3 mutant, which could not catalyze the oxidation of large substrates such as arachidonic acid (AA) and eicosapentanoic acid (EPA) that both have 20 carbons.13 However, the hydroxylation and epoxidation of AA and EPA were catalyzed by R47A and R47G BM-3 at a reduced rate.7,13 For smaller substrates such as laurate, myristate, and palmitate, mutant R47E BM-3 was able to perform catalysis at a decreased rate,13,27 which can be explained by hydrophobic interactions that also play a role in guiding the ligand into the binding pocket. Mutations replacing R47 with a neutral amino acid, R47Q or R47S, resulted in a decreased binding affinity for N-myristoyl-L-methionine. In addition, changes to the charge on N-acyl amino acid substrates showed that BM-3-mediated catalysis of N-palmitoyl-L-glutamine and N-palmitoyl-L-gutamic acid was reduced compared to that of N-palmitoylglycine (NPG).42
Although there is mutagenesis evidence for R47’s role in binding and catalysis, the majority of the previous crystal structures of BMP have not shown substrates interacting with R47. In PDB entries 1JPZ,11 1ZOA,43 and 1ZO9,42 the carboxylate carbon (C1) of NPG is hydrogen bonded to Q73 and A74, and the amide carbonyl (C3) is hydrogen bonded to Y51. This is also seen in the crystal structure of N-palmitoyl-L-methionine bound to BM-3.42 For smaller substrates such as BMP with palmitoleic acid (1FAG,14 1SMJ,44 and 3EKD45) and palmitic acid (2UWH46), the carboxyl group (C1) of the substrate is hydrogen bonded to Y51. Only the crystal structure of a laboratory-evolved octane mono-oxygenase, a P450 BM-3 enzyme with 11 mutations, in complex with NPG (3CBD) showed that R47 is hydrogen bonded to only one of the C1 oxygens in NPG.18
We further investigated the role of R47 through a systematic study to clarify the role of electrostatics and steric bulk as key factors at position 47. Mutations were made to neutral glutamine (R47Q), negatively charged glutamic acid (R47E), and positively charged but smaller lysine (R47K), and their binding and kinetics were monitored with NPG (Figure 2) as the substrate. The largest effect was seen with R47E BM-3; therefore, we solved a crystal structure of R47E BMP to see if there are any structural limitations to NPG binding or if it is purely a consequence of charge repulsion.
Figure 2.
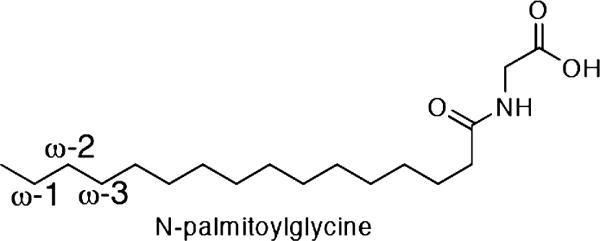
Structure of N-palmitoylglycine (NPG). NPG is used as a substrate because of its increased water solubility and tight binding to BM-3. P450 BM-3 in the presence of NADPH and O2 catalyzes the hydroxylation of fatty acids, such as NPG, at positions ω-1, ω-2, and ω-3.
MATERIALS AND METHODS
Crystallization, Data Collection, and Structure Determination
Cytochrome P450 BMP and R47E BMP were overexpressed in E. coli BL21(DE3) and purified as previously described.47 After purification, cytochrome P450 was concentrated to either 25 or 12.5 mg/mL in 50 mM potassium phosphate pH 7.4. If the protein was stored at −80 °C, then 10% glycerol was added, and the protein was divided into aliquots of 30 μL. Crystals were grown with the hanging-drop method at 4 °C with 1:1 aliquots of the protein and reservoir solutions. The protein solution contained either 12.5 or 25 mg/ mL of stock BMP with either 1 or 1.1 equiv of NPG, and the reservoir solution contained 12–21% (w/v) PEG 3350, 150–225 mM MgCl2, and 100 mM 2-(N-morpholino)ethanesulfonic acid (MES) pH 6. Crystals with high concentrations of PEG grew within 24 h and those with lower concentrations of PEG usually grew within 2 days. Diffraction-quality crystals were obtained by microseeding a drop of 16 mg/mL of BMP in 50 mM potassium phosphate pH 7.4, 1 equiv of NPG, 150 mM MgCl2, 50 mM MES pH 6.0, 10% glycerol, and 13% PEG 3350. The structure of cytochrome P450 BMP with NPG was determined at a 2.0 Å resolution (Table 1).
Table 1.
Data Collection and Refinement
| crystal structure | BMP NPG complex | BMP R47E mutant |
|---|---|---|
| maximum resolution (Å) | 2.0 | 2.1 |
| space group | P21212 | P21 |
| unit cell parameters (angstroms, degrees) | a = 188.7, b = 59.32, c = 56.24 |
a = 58.9, b = 152.7, c = 62.0 β = 94.4 |
| no. of observations | 241 183 | 272 722 |
| redundancy | 5.9 (3.0) | 4.3 (4.4) |
| I/σI | 11.4 (2.1) | 21.9 (5.8) |
| Rmerge (%) | 11.6 (37.3) | 8.5 (20.5) |
| resolution range used for refinement | 30.0–2.0 | 29.4–2.1 |
| no. of reflections | 38 044 | 63 247 |
| completeness (%) | 87.2 | 99.5 |
| R factor (%) | 19.4 | 18.8 |
| free R factor (%) | 23.0 | 23.2 |
| rms deviation in bond lengths (angstroms) | 0.017 | 0.008 |
| rms deviation in bond angles (degrees) | 1.7 | 1.2 |
R47E BMP crystals were grown by microseeding a drop of 24 mg/mL of protein in 50 mM potassium phosphate pH 7.4, 1 equiv of NPG, 150 mM MgCl2, 100 mM MES pH 6.0, and 14% PEG 3350. After crystals were formed, 15% glycerol was added for cryoprotection. The structure of R47E BMP in the presence of NPG was determined at a 2.1 Å resolution (Table 1).
X-ray diffraction data were collected at the X29A beamline of the National Synchrotron Light Source (NSLS). Diffraction images were processed with the HKL package.48 The structure was solved by molecular replacement with the COMO49 program using the structure of BMP with NPG, 1JPZ,11 as the search model. The model was refined with the CNS50 program and manually rebuilt using the O51 program.
Mutagenesis Methods
The site-directed mutagenesis of R47E, R47Q, and R47K BM-3 and BMP were performed using designed primers from Invitrogen (Table S1). The sense and antisense primers were designed with the QuikChange primer-design program available on Agilent’s Web site (http://www.genomics.agilent.com). The mutagenesis was performed using the QuikChange Lightning Site-Directed Mutagenesis Kit from Agilent, and the protocol was from the handbook. All sequencing was performed at Columbia University’s Protein Core Facility (New York, NY). The successfully mutated plasmids were transformed into BL21 DE3 cells for protein expression. The mutants were expressed and purified by the same method as that for the wild-type protein.47
Characterization of R47E, R47Q, and R47K BM-3
NPG was synthesized from a known protocol.52 For the determination of spin state change with temperature, optical absorption spectra of enzyme/NPG mixtures at temperatures between −3 and 37 °C were taken under the same conditions used previously.53 Thirty equiv of NPG (129 μM) were added to a solution of BM-3 (2.15 μM) in 500 μL of 50 mM MOPS pH 7.4 and 30% glycerol at room temperature. The solution then was cooled to −3 °C and equilibrated for 30 min before a measurement was taken, and the temperature was increased for the next measurement. The percentage of high spin was calculated using the equations stated by Jovanovic et al.54
To determine the dissociation constant (Kd), approximately 0.5 μM of protein was added to 500 μL of 50 mM potassium phosphate monobasic pH 7.4 in a cuvette. NPG (stock of 0.05 mM) was titrated into the solution in 1 μL aliquots, and the sample was equilibrated for 1 min before the optical absorption data were collected. The data were fitted using a biomolecular dissociation equation11 with Igor Pro55 where the absorbance at 393 nm was subtracted from the absorbance at 418 nm to maximize the change in absorbance due to binding.
For the determination of the catalytic constant (kcat), various concentrations of NPG (0–250 μM) were added to 0.25 μM of BM-3 in 50 mM potassium phosphate pH 7.4 in a cuvette, and the mixture was incubated for 5 min at room temperature. The enzymatic reaction was started by the addition of 200 μM NADPH, and the reaction was monitored by the consumption of NADPH at 340 nm. The rate was determined using the slope of the first 30 s of the absorbance data, ε340 for NADPH (6200 M−1 cm−1), and the path length (b = 1 cm). We plotted the velocity divided by the enzyme concentration against the concentration of NADPH to determine kcat and Km using Igor Pro.55
RESULTS
Crystal Structure of NPG with P450 BMP
The crystal structure of P450 BMP in complex with NPG has been determined at a 2.0 Å resolution. Our structure, 4KPA, was compared to PDB entry 1JPZ,11 which is the first NPG–BMP complex that was solved. The Cα atoms of 4PKA and 1JPZ were aligned in PyMOL41 and showed an rmsd of 0.34 Å41 (Figure 3A). In fact, the two crystals were grown under very similar conditions. The 1JPZ crystal was grown from 12.5 mg/mL of protein with 12% PEG 3350 and 1.1 equiv of NPG, and our crystal was grown from 16 mg/mL of protein with 10% glycerol, 13% PEG 3350, and 1 equiv of NPG. Even though the differences in the crystallization conditions between the two crystal structures were small, the two crystals are in different space groups, and in the unit cell 1JPZ is a dimer whereas our structure is a monomer. Previous structures of BMP have been solved in both space groups.9,42,44–46,56–65
Figure 3.
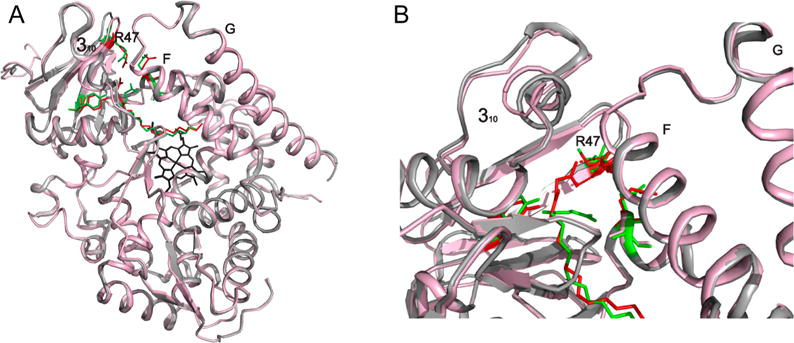
Comparison of 4KPA and 1JPZ. (A) Cα alignment of 1JPZ (gray and green) and 4KPA (red and pink) with an rmsd of 0.34 Å showing that the overall fold is the same. (B) Interaction of NPG with R47 in 4KPA, which is in a different conformation than it is in 1JPZ. The structures were prepared in PyMOL.41
The largest difference between 4KPA and 1JPZ is that NPG is in a different conformation (Figure 3B). Our structure shows R47 interacting with the carboxylate oxygens of NPG in a direct bidentate ion pair at distances of 2.9 and 3.1 Å, which has not been observed in previous crystal structures.11,18,42,43 Figure 4 shows the comparison of 4KPA with 1JPZ and 3CBD, with all of them having a different conformation of the carboxylate part of the ligand. In 1JPZ, the carboxylate carbon (C1) of NPG is hydrogen bonded to Q73 and A74, and the amide carbonyl (C3) is hydrogen bonded to Y51. The crystal structure of a laboratory-evolved octane mono-oxygenase, a P450 BM-3 enzyme with 11 mutations, in complex with NPG (3CBD)18 shows that R47 is hydrogen bonded to only one of the C1 oxygens in NPG at 3.1 Å, wherease the other one is at 4.1 Å. In both 4KPA and 3CBD, the C3 oxygen of NPG is too far away (5 Å) to be hydrogen bonded to Y51 in comparison to a distance of 2.6 Å in 1JPZ.
Figure 4.
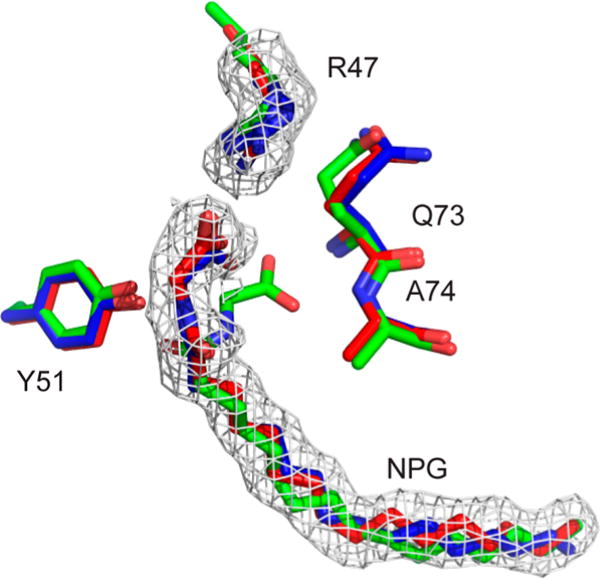
Hydrogen-bonding networks of NPG bound to BM-3. The comparison of the hydrogen-bonding networks between NPG and BMP from the crystal structures is as follows: 1JPZ (green), 3CBD (blue), 4KPA (red), and the omit Fo-Fc maps (gray). In 1JPZ, the carboxylate (C1) of NPG is close enough to hydrogen bond to the backbone of Q73 and A74, and the amide carbonyl (C3) is hydrogen bonded to Y51. However, in our structure, 4KPA, C1 of NPG is in a direct bidentate ion-pair interaction with R47, and it does not appear that C3 is hydrogen bonding to any residue. In 3CBD, only one of the oxygens of carboxylate (C1) is close enough to hydrogen bond to R47. The structures were prepared in PyMOL.41
Spin State
The heme in P450 BM-3 undergoes a spin state change from a low-spin resting state in which water is coordinated as the sixth ligand, to a high-spin state in which the water is displaced upon ligand binding. The wild-type enzyme with NPG has a temperature-dependent spin state, which is proposed to represent a change between the proximal and distal conformation of the ligand.53 As shown in Figure 5A, at 37 °C the wild-type enzyme has a high population of heme in the high-spin state (393 nm). As the temperature decreases, the population in the low-spin state increases (418 nm), suggesting that the substrate is in a distant alternative binding mode.47
Figure 5.
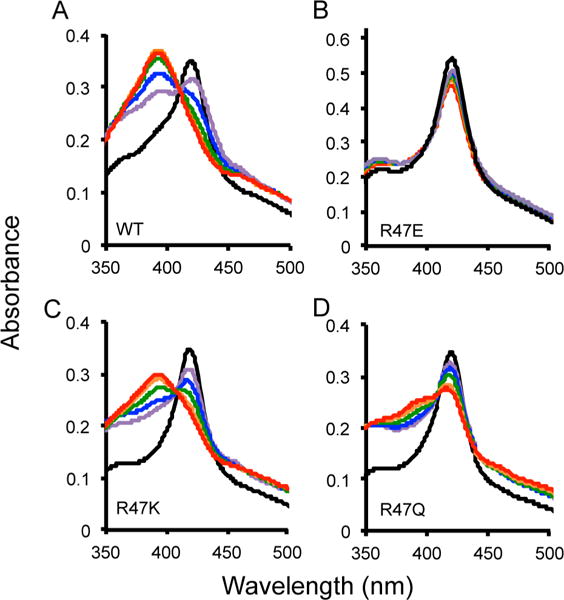
Spin state temperature dependence for BM-3 wild type (A), R47E (B), R47K (C), and R47Q (D). The optical absorbance spectra with a 30-fold excess of NPG was collected at temperatures of 37 (red), 27 (orange), 17 (green), 7 (blue), and −3 °C (purple). The resting state of the enzyme without NPG is shown in black at room temperature. A temperature-dependent change in the spin state of the heme iron was observed with excess NPG because the absorbance at 418 nm (low spin) decreased and at 393 nm (high spin) increased as the temperature increased for wild-type, R47K, and R47Q. There was no temperature-dependent spin state change observed for R47E BM-3 with NPG bound. The percentage of the population in high spin at 37 °C is 97% for the wild-type, 80% for R47K, 50% for R47Q, and 8% for R47E.
The temperature dependence of the spin state for the mutants was conducted with a 30-fold excess of NPG to BM-3, and optical spectroscopy was performed at the indicated temperatures. For R47E BM-3 with NPG at 37 °C, we only observed a small population of the heme iron in the high-spin state (∼8%) and no temperature-dependent spin state change, as shown in Figure 5B. In the case of NPG bound to R47K BM-3 at 37 °C, the heme iron is 80% in the high-spin state (Figure 5C). As shown in Figure 5D, for NPG bound to R47Q BM-3 the heme iron is 50% in the high-spin state at 37 °C. By contrast, the heme iron in wild-type BM-3 is 97% in the high-spin state. Both R47K and R47Q have a temperature-dependent spin state. These results show that charge along with temperature plays a role in correctly positioning the substrate for catalysis because the wild-type and R47K BM-3 with NPG have a higher percentage in the high-spin state, possibly indicating a proximal catalytically preferred conformation. For R47Q and R47E BM-3, there is no specific substrate interaction with residue 47, causing the substrate to occupy other binding modes and have a lower percentage in the high-spin state.
Ligand Binding
The Kd of NPG was determined for wild-type, R47K, R47Q, and R47E BM-3 (Supporting Information Figures S1–S4 and Table 2). The Kd that we determined for wild-type BM-3 with NPG is 0.37 ± 0.05 μM, which is comparable to the previous literature value.11 For R47K BM-3 with NPG, the Kd is 1.2 ± 0.1 μM, which is 3-fold weaker than that of the wild-type. For R47Q with NPG, the Kd is 7.0 ± 0.7 μM, which is 20-fold weaker than that of the wild-type. This is expected because the substrate no longer has attractive forces with residue 47. For R47E BM-3, a Kd was not determined because type I binding was not observed. As seen in Figures 5 and S2, with a 30-fold excess of ligand to protein concentration there is only a peak at 418 nm. It is unclear as to whether the ligand binds in a nonproductive (distant) conformation where it is unable to cause a change in the spin state or whether the charge repulsion is too strong and it is not binding at all.
Table 2.
Spectral Dissociation Constant, Percentage of High Spin, and Enzymatic Data for P450 BM-3 and Mutants for the Metabolism of NPGa
| enzyme | Kd (μM) | high spin (%) | Km (μM) | kcat (min−1) | kcat/Km (min−1 μM −1) |
|---|---|---|---|---|---|
| wild type | 0.37 ± 0.05 | 80 | 5 ± 3 | 1500 ± 140 | 282 |
| R47E | ND | 14 | 90 ± 24 | 42 ± 8 | 0.47 |
| R47K | 1.2 ± 0.1 | 70 | 55 ± 17 | 1400 ± 140 | 25 |
| R47Q | 7.0 ± 0.7 | 45 | 410 ± 160 | 1320 ± 210 | 3.2 |
Kd, kcat, Km, and kcat/Km and the percentage of high spin for NPG with wild-type, R47E, R47K, and R47Q BM-3 were determined at room temperature and pH 7.4. The Kd for R47E was not determined (ND) because type-1 binding was not observed. A small amount of catalysis occurred for R47E BM-3 because the value for NADPH without ligand was determined to be 5 ± 4 min−1. For the R47K and R47Q mutations, both Kd and Km increased, showing that R47 is important for binding.
Enzyme Activity
The Km, kcat, and Km/kcat values for the hydroxylation of NPG were determined for wild-type, R47K, R47Q, and R47E BM-3 (Supporting Information Figures S5–S8 and Table 2). The mutations significantly decreased the efficiency of the enzyme (kcat/Km) compared to the wild-type. The activity of R47E with NPG was 30-fold weaker compared to wild-type, and the Km increased. For R47Q and R47K, less activity was observed than for the wild-type, and both the Kd and Km increased, showing that R47 is important for binding. However, there is an order of magnitude difference between the Km and Kd for the wild-type, R47K, and R47Q, which may be a result of the ligand accessing different noncatalytically active conformations or it may indicate that binding is not the rate-limiting step. However, the kcat for R47Q and R47K are within the error of the wild-type, indicating that the mutation does not affect kcat but only binding.
Crystal Structure of R47E BMP
To see if the R47E mutation caused any structural changes preventing NPG from binding to BM-3, we determined a crystal structure of R47E BMP at a 2.1 Å resolution (Table 1). Crystals of R47E BMP were grown in the presence of 1 equiv of NPG, but in the crystal structure no NPG is bound. From the crystal structure, an explanation for NPG’s weak or nonexistent binding is that residue E47 is blocking access to the substrate channel and the electron density of E47 indicates that there may be multiple conformations. In comparison to our crystal structure of NPG-BMP, the largest differences are seen in the F and G helices, which form one side of the substrate channel, and, on the other side of the channel, the 310 helix and the R47 β-sheet. As seen in previous substrate–BMP complexes as well as in our structure, the two sides of the channel are closer together in the substrate-bound structure. There also are changes in the B′, C, D, H, and I helices, as shown in Figure 6A,B. Because no substrate is present, R47E BMP more closely resembles substrate-free structure 1BU79 shown in Figure 6C,D.
Figure 6.
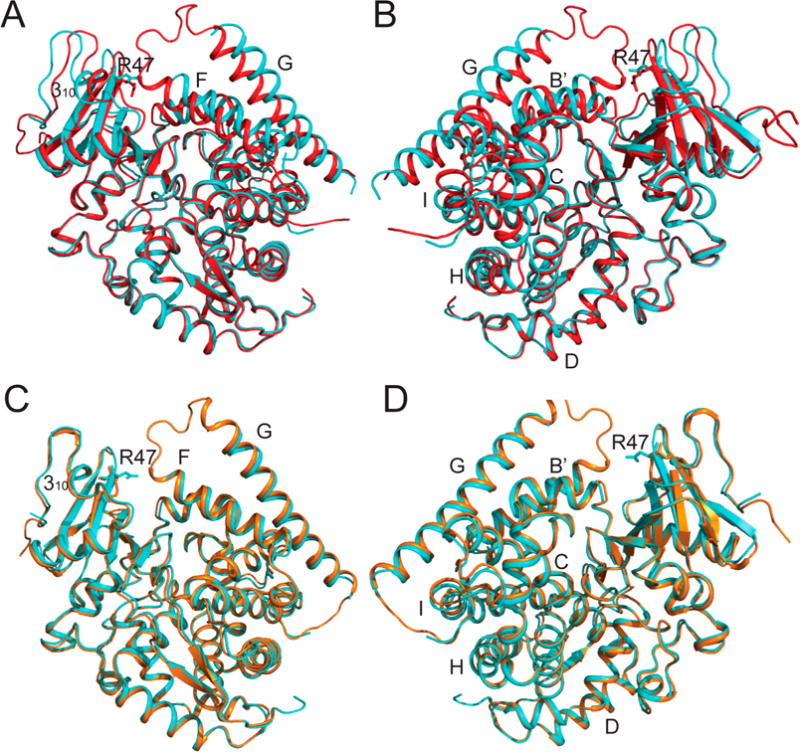
Crystal structure of R47E BMP. The structure of R47E BMP (4KPB, cyan) is compared to our crystal structure of NPG bound to BMP (4KPA, red) and to wild-type BMP (1BU7, orange). (A) Comparison of NPG–BMP with R47E BMP. (B) The same structure from A rotated 180°. The largest differences in structure are the F, G, and 310 helices as well as residue 47, which forms the entrance to the substrate-binding channel and is known to close upon substrate binding. (C) Comparison of R47E BMP with wild-type BMP free enzyme. (D) The same structure from C rotated 180°. R47E BMP more closely resembles substrate-free structure 1BU7.9 These two structures are very similar with the exception of E47’s position, which appears to be blocking the entrance of the channel, and the electron density of E47 indicates that there may be multiple confirmations. The structures were prepared in PyMOL.41
DISCUSSION
In all of the crystal structures published for NPG complexed with BMP,11,18,42,43 including our structure, NPG is still too far away from the heme iron to be in the catalytically relevant binding mode in comparison to that of camphor in P450cam. Our crystal structure with a direct bidentate ion-pair interaction between NPG and R47 therefore represents an initial binding step in the mechanistic reaction of cytochrome P450. This is supported by random expulsion molecular dynamics studies that showed arginine interacting with NPG in a bidentate ionpair interaction.66,67 By comparing the conformation of NPG bound to BMP in our crystal structure to other crystal structures, the different conformations of NPG provide insight into the dynamics of the initial binding in the carboxylate region of the ligand.
The mobility of NPG has been shown through previous studies. Roberts et al. showed by paramagnetic relaxation techniques that the substrate moved 6 Å upon reduction.68,69 Harris and Jovanovic et al., through solid-state NMR experiments, showed a non-Curie temperature dependence of the chemical shift of the terminal methyl of NPG, indicating a change in the ligand conformation.47,53,54 In addition, replica exchange molecular dynamics (REMD) trajectories70 were also used to determine the order parameters for NPG bound to BM-3. The results showed that the order parameters for the crystallographic conformation were higher than those for the predicted room-temperature structure, which is in agreement with the solid-state NMR experimentally determined order parameters.71 All of these experiments indicate that the substrate moves; however, there is no information on how it repositions or structural evidence of the new binding site close to the heme. The mobility of the ligand is not unique to BM-3 and was probably first seen through kinetic isotope studies of the demethylation of anisole.72
Our mutagenesis studies along with previous studies7,10,12,13,27 showed that R47 is essential for the binding of NPG and possibly other large substrates either during encounter complex formation and/or for positioning the substrate correctly for catalysis. For the R47A mutant with AA, an increased rate of epoxidation and a decreased rate of hydroxylation compared to that of the wild type was observed. Because the rate of epoxidation increased, the absence of R47 allowed the substrate to penetrate further into the binding pocket, allowing chemistry to occur more favorably at C15 compared to C18, which supports that R47 is involved in substrate positioning. In addition, for EPA the rate of catalysis increased in forming epoxidation product 17,18-EETA.13 This may also be due to the fact that for R47A there is more room in the binding pocket.
In our mutagenesis studies, R47 is essential for the binding of NPG and the positioning for catalysis, as indicated by the decrease in the percentage of the heme in the high-spin state at 37 °C and the decrease in catalytic efficiency for the R47Q, R47E, and R47K BM-3 mutants. The largest effect was seen for NPG bound to R47E BM-3 where the substrate was not bound or was bound in a noncatalytic mode, as shown by our optical absorption data, such that catalysis was greatly diminished. This is in agreement with previous mutations of R47E BM-3 that showed diminished activity for large substrates such as AA and EPA.13 On the basis of our R47E BMP crystal structure, it appears that NPG does not bind under stoichiometric substrate concentrations because of the charge repulsion and position of E47. For R47Q, an increase in binding affinity was also seen previously.28 However, the kcat determined was within the error for the wild-type at a large concentration of ligand, showing that hydrophobic interactions also play a role in guiding the ligand into the binding pocket. For R47K, there was also an increase in Km and Kd, causing a decrease in the catalytic efficiency of the enzyme. Thus, not only a positive charge is necessary but also the size of the residue at position 47 is crucial in maintaining catalytic efficiency.
Other P450 enzymes show important arginine–substrate interactions. For example, R108 in CYP2C973 is proposed to play a similar role as that of R47 in BM-3. CYP2C9 is found in the human liver and is involved in metabolizing commonly prescribed drugs such as S-warfarin, phenytoin, and many other nonsteroidal anti-inflammatory drugs.74 In the crystal structure of flurbiprofen with CYP2C9, the substrate is in a presumably catalytically active conformation and is interacting with R108, as shown in Figure 7.73 In comparing CYP2C9 R108 to BM-3 R47, the positioning of the arginine residue is dependent on the size of the substrate for that particular P450 and on the position of hydroxylation. In cytochrome P450BSβ, which hydroxylates fatty acids at the α or β position of the carboxylate headgroup, an arginine is in the I helix near the heme.75 The crystal structure shows the substrate interacting with R242, and mutations of this arginine residue have shown a loss in activity.76 However, because the arginine is located near the heme it is also proposed to be involved in the mechanism.75
Figure 7.
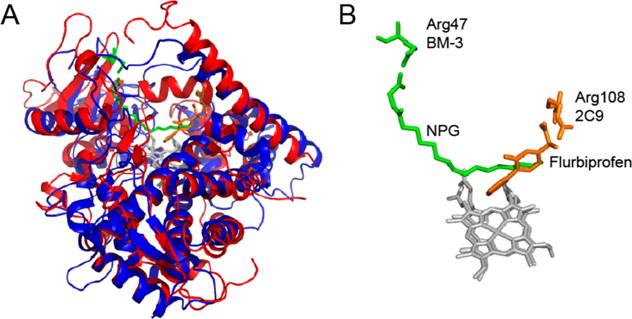
Comparison of the position of arginine in NPG bound to BMP in 4KPA (4KPA, red and green) and flurbiprofen bound to CYP2C9 (1R90, blue and orange). Both proteins have the same overall fold (A); however, the arginine residues in the binding pocket are in different locations to accommodate the difference in the size of the substrates (B). The structures were prepared in PyMOL.41
A comparison of the sequence of BM-3 with those of fatty acid ω-hydroxylases shows that Y51 is conserved whereas R47 is not.77 Previous crystal structures of BM-3 show ligand interactions with Y51.11,14,42–46 The mutation of Y51 to alanine along with double mutant R47A/Y51A has been shown to decrease the rate of activity and increase the binding constants for lauric, palmitoleic, and arachidonic acid.12 Analysis of the products for AA showed that ω-1 hydroxylation occurred for Y51A and R47A/Y51A, which is not a wild-type or R47A product. For double mutant R47A/Y51A and R47A, a larger percentage of epoxidation product 14,15-EET was produced compared to the wild type, which was not true for Y51A.12 Therefore, R47 may be more involved in the initial binding, as shown with our mutagenesis and crystallographic studies, preventing large substrates from penetrating too far into the binding pocket, whereas Y51 may be more involved in the positioning of the substrate at the time of catalysis. Although mutations of Y51F showed only a slight decrease in activity, the effect of the Y51A mutation may be a result of the difference in the size and hydrophobicity and not necessarily the hydroxyl group.7 Together, R47 and Y51 are important for the monooxygenation with high regioselectivity to correctly position some substrates. The arginine–tyrosine pair is not unique to cytochrome P450 BM-3. An arginine–tyrosine pair is used to stabilize the binding of lactate in L-lactate cytochrome c oxidoreductase from Saccharomyces cerevisiae.78 Similarly, flavocytochrome b2 from Hansenula anomala and L-mandelate dehydrogenase from Rhodotorula graminis have an important arginine–tyrosine pair, and spinach glycolate oxidase also has an arginine–serine pair.79
CONCLUSIONS
Our crystal structure of NPG–BMP in combination with previous crystal structures provide insight into the binding of ligands to cytochrome P450 BM-3. We clarified the role of electrostatics by using amino acids of similar steric bulk to arginine, and we showed that not only a positive charge but also arginine is necessary to achieve the same catalytic efficiency. In addition, all three mutants caused the Km and Kd to increase, showing that R47 is important for the binding of substrates.
Supplementary Material
Acknowledgments
Funding
NIH Biophysics Training Grant GM08281
ABBREVIATIONS
- P450 BM-3
cytochrome P450 from Bacillus megaterium
- P450 BMP
heme domain of cytochrome P450
- NADPH
reduced nicotinamide adenosine dinucleotide phosphate
- AA
arach-idonic acid
- EPA
eicosapentanoic acid
Footnotes
Table of primers used to construct the mutants, spectroscopy data for the Kd and the corresponding fits, and the curve fits for the enzymatic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates and structure factors have been deposited in the Protein Data Bank under accession nos. 4KPA for the BMP NPG complex and 4KPB for BMP R47E mutant.
Notes
The authors declare no competing financial interest.
References
- 1.Schoch GA, Yano JK, Wester MR, Griffin KJ, Stout CD, Johnson EF. Structure of human microsomal cytochrome P4502C8 - Evidence for a peripheral fatty acid binding site. J Biol Chem. 2004;279:9497–9503. doi: 10.1074/jbc.M312516200. [DOI] [PubMed] [Google Scholar]
- 2.Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 3.Williams PA, Cosme J, Ward A, Angova HC, Vinkovic DM, Jhoti H. Crystal structure of human cytochrome P4502C9 with bound warfarin. Nature. 2003;424:464–468. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 4.Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, Johnson EF. The structure of human cytochrome P4502C9 complexed with flurbiprofen at 2.0-angstrom resolution. J Biol Chem. 2004;279:35630–35637. doi: 10.1074/jbc.M405427200. [DOI] [PubMed] [Google Scholar]
- 5.Narhi LO, Fulco AJ. Phenobarbital induction of a soluble cytochrome P-450-dependent fatty acid monooxygenase in Bacillus megaterium. J Biol Chem. 1982;257:2147–2150. [PubMed] [Google Scholar]
- 6.Narhi LO, Fulco AJ. Characterization of a catalytically self-sufficient 119,000-dalton cytochrome P-450 mono-oxygenase induced by barbiturates in Bacillus megaterium. J Biol Chem. 1986;261:7160–7169. [PubMed] [Google Scholar]
- 7.Noble MA, Miles CS, Chapman SK, Lysek DA, Mackay AC, Reid GA, Hanzlik RP, Munro AW. Roles of key active-site residues in flavocytochrome P450 BM3. Biochem J. 1999;339:371–379. [PMC free article] [PubMed] [Google Scholar]
- 8.Miura Y, Fulco AJ. Omega-1, omega-2 and omega-3 hydroxylation of long-chain fatty acids, amides and alcohols by a soluble enzyme system from Bacillus megaterium. Biochim Biophys Acta. 1975;388:305–317. doi: 10.1016/0005-2760(75)90089-2. [DOI] [PubMed] [Google Scholar]
- 9.Sevrioukova IF, Li H, Zhang H, Peterson JA, Poulos TL. Structure of a cytochrome P450-redox partner electron-transfer complex. Proc Natl Acad Sci USA. 1999;96:1863–1868. doi: 10.1073/pnas.96.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ost TWB, Miles CS, Murdoch J, Cheung YF, Reid GA, Chapman SK, Munro AW. Rational re-design of the substrate binding site of flavocytochrome P450BM3. FEBS Lett. 2000;486:173–177. doi: 10.1016/s0014-5793(00)02267-5. [DOI] [PubMed] [Google Scholar]
- 11.Haines DC, Tomchick DR, Machius M, Peterson JA. Pivotal role of water in the mechanism of P450BM-3. Biochemistry. 2001;40:13456–13465. doi: 10.1021/bi011197q. [DOI] [PubMed] [Google Scholar]
- 12.Cowart LA, Falck JR, Capdevila JH. Structural determinants of active site binding affinity and metabolism by cytochrome P450BM-3. Arch Biochem Biophys. 2001;387:117–124. doi: 10.1006/abbi.2000.2246. [DOI] [PubMed] [Google Scholar]
- 13.Graham-Lorence S, Truan G, Peterson JA, Falck JR, Wei S, Helvig C, Capdevila JH. An active site substitution, F87V, converts cytochrome P450BM-3 into a regio- and stereo-selective (14S,15R)-arachidonic acid epoxygenase. J Biol Chem. 1997;272:1127–1135. doi: 10.1074/jbc.272.2.1127. [DOI] [PubMed] [Google Scholar]
- 14.Li HY, Poulos TL. The structure of the cytochrome P450 BM-3 haem domain complexed with the fatty acid substrate, palmitoleic acid. Nat Struct Biol. 1997;4:140–146. doi: 10.1038/nsb0297-140. [DOI] [PubMed] [Google Scholar]
- 15.Otey CR, Bandara G, Lalonde J, Takahashi K, Arnold FH. Preparation of human metabolites of propranolol using laboratory-evolved bacterial cytochromes P450. Biotechnol Bioeng. 2006;93:494–499. doi: 10.1002/bit.20744. [DOI] [PubMed] [Google Scholar]
- 16.Kubo T, Peters MW, Meinhold P, Arnold FH. Enantioselective epoxidation of terminal alkenes to (R)- and (S)-epoxides by engineered cytochromes P450BM-3. Chem—Eur J. 2006;12:1216–1220. doi: 10.1002/chem.200500584. [DOI] [PubMed] [Google Scholar]
- 17.Landwehr M, Hochrein L, Otey CR, Kasrayan A, Backvall JE, Arnold FH. Enantioselective alpha-hydroxylation of 2-arylacetic acid derivatives and buspirone catalyzed by engineered cytochrome P450BM-3. J Am Chem Soc. 2006;128:6058–6059. doi: 10.1021/ja061261x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fasan R, Meharenna YT, Snow CD, Poulos TL, Arnold FH. Evolutionary history of a specialized P450 propane monooxygenase. J Mol Biol. 2008;383:1069–1080. doi: 10.1016/j.jmb.2008.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong TS, Arnold FH, Schwaneberg U. Laboratory evolution of cytochrome P450BM-3 monooxygenase for organic cosolvents. Biotechnol Bioeng. 2004;85:351–358. doi: 10.1002/bit.10896. [DOI] [PubMed] [Google Scholar]
- 20.Warman AJ, Roitel O, Neeli R, Girvan HM, Seward HE, Murray SA, McLean KJ, Joyce MG, Toogood H, Holt RA, Leys D, Scrutton NS, Munro AW. Flavocytochrome P450BM3: an update on structure and mechanism of a biotechnologically important enzyme. Biochem Soc Trans. 2005;33:747–753. doi: 10.1042/BST0330747. [DOI] [PubMed] [Google Scholar]
- 21.Jung ST, Lauchli R, Arnold FH. Cytochrome P450: taming a wild type enzyme. Curr Opin Biotechnol. 2011;22:809–817. doi: 10.1016/j.copbio.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farinas ET, Bulter T, Arnold FH. Directed enzyme evolution. Curr Opin Biotechnol. 2001;12:545–551. doi: 10.1016/s0958-1669(01)00261-0. [DOI] [PubMed] [Google Scholar]
- 23.Li QS, Ogawa J, Schmid RD, Shimizu S. Engineering cytochrome P450BM-3 for oxidation of polycyclic aromatic hydrocarbons. Appl Environ Microbiol. 2001;67:5735–5739. doi: 10.1128/AEM.67.12.5735-5739.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peters MW, Meinhold P, Glieder A, Arnold FH. Regio- and enantioselective alkane hydroxylation with engineered cytochromes P450BM-3. J Am Chem Soc. 2003;125:13442–13450. doi: 10.1021/ja0303790. [DOI] [PubMed] [Google Scholar]
- 25.Appel D, Lutz-Wahl S, Fischer P, Schwaneberg U, Schmid RD. A P450 BM-3 mutant hydroxylates alkanes, cycloalkanes, arenes and heteroarenes. J Biotechnol. 2001;88:167–171. doi: 10.1016/s0168-1656(01)00249-8. [DOI] [PubMed] [Google Scholar]
- 26.Li QS, Schwaneberg U, Fischer M, Schmitt J, Pleiss J, Lutz-Wahl S, Schmid RD. Rational evolution of a medium chain-specific cytochrome P-450 BM-3 variant. Biochim Biophys Acta. 2001;1545:114–121. doi: 10.1016/s0167-4838(00)00268-5. [DOI] [PubMed] [Google Scholar]
- 27.Oliver CF, Modi S, Primrose WU, Lian LY, Roberts GCK. Engineering the substrate specificity of Bacillus megaterium cytochrome P-450 BM3: hydroxylation of alkyl trimethylammonium compounds. Biochem J. 1997;327:537–544. doi: 10.1042/bj3270537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chowdhary PK, Stewart L, Lopez C, Haines DC. A single mutation in P450 BM-3 enhances acyl homoserine lactone: Acyl homoserine substrate binding selectivity nearly 250-fold. J Biotechnol. 2008;135:374–376. doi: 10.1016/j.jbiotec.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Whitehouse CJ, Yang W, Yorke JA, Rowlatt BC, Strong AJ, Blanford CF, Bell SG, Bartlam M, Wong LL, Rao Z. Structural basis for the properties of two single-site proline mutants of CYP102A1 (P450BM3) ChemBioChem. 2010;11:2549–2556. doi: 10.1002/cbic.201000421. [DOI] [PubMed] [Google Scholar]
- 30.Otey CR, Landwehr M, Endelman JB, Hiraga K, Bloom JD, Arnold FH. Structure-guided recombination creates an artificial family of cytochromes P450. PLoS Biol. 2006;4:e112-1–e112-10. doi: 10.1371/journal.pbio.0040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Vugt-Lussenburg BM, Damsten MC, Maasdijk DM, Vermeulen NP, Commandeur JN. Heterotropic and homotropic cooperativity by a drug-metabolising mutant of cytochrome P450 BM3. Biochem Biophys Res Commun. 2006;346:810–818. doi: 10.1016/j.bbrc.2006.05.179. [DOI] [PubMed] [Google Scholar]
- 32.Carmichael AB, Wong LL. Protein engineering of Bacillus megaterium CYP102. The oxidation of polycyclic aromatic hydrocarbons. Eur J Biochem. 2001;268:3117–3125. doi: 10.1046/j.1432-1327.2001.02212.x. [DOI] [PubMed] [Google Scholar]
- 33.Noble MA, Quaroni L, Chumanov GD, Turner KL, Chapman SK, Hanzlik RP, Munro AW. Imidazolyl carboxylic acids as mechanistic probes of flavocytochrome P-450 BM3. Biochemistry. 1998;37:15799–15807. doi: 10.1021/bi980462d. [DOI] [PubMed] [Google Scholar]
- 34.Urlacher VB, Makhsumkhanov A, Schmid RD. Biotransformation of beta-ionone by engineered cytochrome P450 BM-3. Appl Microbiol Biotechnol. 2006;70:53–59. doi: 10.1007/s00253-005-0028-4. [DOI] [PubMed] [Google Scholar]
- 35.Sawayama AM, Chen MM, Kulanthaivel P, Kuo MS, Hemmerle H, Arnold FH. A panel of cytochrome P450 BM3 variants to produce drug metabolites and diversify lead compounds. Chem — Eur J. 2009;15:11723–11729. doi: 10.1002/chem.200900643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sowden RJ, Yasmin S, Rees NH, Bell SG, Wong LL. Biotransformation of the sesquiterpene (+)-valencene by cytochrome P450cam and P450BM-3. Org Biomol Chem. 2005;3:57–64. doi: 10.1039/b413068e. [DOI] [PubMed] [Google Scholar]
- 37.Lussenburg BM, Babel LC, Vermeulen NP, Commandeur JN. Evaluation of alkoxyresorufins as fluorescent substrates for cytochrome P450 BM3 and site-directed mutants. Anal Biochem. 2005;341:148–155. doi: 10.1016/j.ab.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 38.Whitehouse CJ, Bell SG, Tufton HG, Kenny RJ, Ogilvie LC, Wong LL. Evolved CYP102A1 (P450 BM3) variants oxidise a range of non-natural substrates and offer new selectivity options. Chem Commun (Cambridge, UK) 2008:966–968. doi: 10.1039/b718124h. [DOI] [PubMed] [Google Scholar]
- 39.Lewis JC, Bastian S, Bennett CS, Fu Y, Mitsuda Y, Chen MM, Greenberg WA, Wong CH, Arnold FH. Chemoenzymatic elaboration of monosaccharides using engineered cytochrome P450 BM3 demethylases. Proc Natl Acad Sci USA. 2009;106:16550–16555. doi: 10.1073/pnas.0908954106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitehouse CJ, Bell SG, Wong LL. P450(BM3) (CYP102A1): connecting the dots. Chem Soc Rev. 2012;41:1218–1260. doi: 10.1039/c1cs15192d. [DOI] [PubMed] [Google Scholar]
- 41.DeLano WL. The PyMOL Molecular Graphics System. Schrödinger, LLC; San Carlos, CA: 2002. [Google Scholar]
- 42.Hegde A, Haines DC, Bondlela M, Chen B, Schaffer N, Tomchick DR, Machius M, Nguyen H, Chowdhary PK, Stewart L, Lopez C, Peterson JA. Interactions of substrates at the surface of P450s can greatly enhance substrate potency. Biochemistry. 2007;46:14010–14017. doi: 10.1021/bi701667m. [DOI] [PubMed] [Google Scholar]
- 43.Haines DC, Hegde A, Chen B, Zhao W, Bondlela M, Humphreys JM, Mullin DA, Tomchick DR, Machius M, Peterson JA. A single active-site mutation of P450BM-3 dramatically enhances substrate binding and rate of product formation. Biochemistry. 2011;50:8333–8341. doi: 10.1021/bi201099j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joyce MG, Girvan HM, Munro AW, Leys D. A single mutation in cytochrome P450 BM3 induces the conformational rearrangement seen upon substrate binding in the wild-type enzyme. J Biol Chem. 2004;279:23287–23293. doi: 10.1074/jbc.M401717200. [DOI] [PubMed] [Google Scholar]
- 45.Girvan HM, Toogood HS, Littleford RE, Seward HE, Smith WE, Ekanem IS, Leys D, Cheesman MR, Munro AW. Novel haem co-ordination variants of flavocytochrome P450 BM3. Biochem J. 2009;417:65–76. doi: 10.1042/BJ20081133. [DOI] [PubMed] [Google Scholar]
- 46.Huang WC, Westlake AC, Marechal JD, Joyce MG, Moody PC, Roberts GC. Filling a hole in cytochrome P450 BM3 improves substrate binding and catalytic efficiency. J Mol Biol. 2007;373:633–651. doi: 10.1016/j.jmb.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 47.Jovanovic T, McDermott AE. Observation of ligand binding to cytochrome P450-BM-3 by means of solid-state NMR spectroscopy. J Am Chem Soc. 2005;127:13816–13821. doi: 10.1021/ja0438314. [DOI] [PubMed] [Google Scholar]
- 48.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 49.Tong L. Combined molecular replacement. Acta Crystallogr Sect A. 1996;52:782–784. doi: 10.1107/s0907444901006783. [DOI] [PubMed] [Google Scholar]
- 50.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr Sect D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 51.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr, Sect A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 52.Lapidot Y, Rappopor S, Wolman Y. Use of esters of N-hydroxysuccinimide in synthesis of N-acylamino acids. J Lipid Res. 1967;8:142–145. [PubMed] [Google Scholar]
- 53.Jovanovic T, Farid R, Friesner RA, McDermott AE. Thermal equilibrium of high- and low-spin forms of cytochrome P450 BM-3: repositioning of the substrate? J Am Chem Soc. 2005;127:13548–13552. doi: 10.1021/ja0524604. [DOI] [PubMed] [Google Scholar]
- 54.Jovanovic T, Harris M, McDermott AE. Cytochrome P450 BM-3 in complex with its substrate: Temperature-dependent spin state equilibria in the oxidized and reduced states. Appl Magn Reson. 2007;31:411–429. [Google Scholar]
- 55.Igor Pro. WaveMetrics, Inc; Lake Oswego, OR: 2008. (Version 6.03A2). [Google Scholar]
- 56.Kuper J, Wong TS, Roccatano D, Wilmanns M, Schwaneberg U. Understanding a mechanism of organic cosolvent inactivation in heme monooxygenase P450 BM-3. J Am Chem Soc. 2007;129:5786–5787. doi: 10.1021/ja067036x. [DOI] [PubMed] [Google Scholar]
- 57.Haines DC, Chen B, Tomchick DR, Bondlela M, Hegde A, Machius M, Peterson JA. Crystal structure of inhibitor-bound P450BM-3 reveals open conformation of substrate access channel. Biochemistry. 2008;47:3662–3670. doi: 10.1021/bi7023964. [DOI] [PubMed] [Google Scholar]
- 58.Yeom H, Sligar SG, Li H, Poulos TL, Fulco AJ. The role of Thr268 in oxygen activation of cytochrome P450BM-3. Biochemistry. 1995;34:14733–14740. doi: 10.1021/bi00045a014. [DOI] [PubMed] [Google Scholar]
- 59.Ost TWB, Munro AW, Mowat CG, Taylor PR, Pesseguiero A, Fulco AJ, Cho AK, Cheesman MA, Walkinshaw MD, Chapman SK. Structural and spectroscopic analysis of the F393H mutant of flavocytochrome p450 BM3. Biochemistry. 2001;40:13430–13438. doi: 10.1021/bi010717e. [DOI] [PubMed] [Google Scholar]
- 60.Ost TW, Clark J, Mowat CG, Miles CS, Walkinshaw MD, Reid GA, Chapman SK, Daff S. Oxygen activation and electron transfer in flavocytochrome P450 BM3. J Am Chem Soc. 2003;125:15010–15020. doi: 10.1021/ja035731o. [DOI] [PubMed] [Google Scholar]
- 61.Clark JP, Miles CS, Mowat CG, Walkinshaw MD, Reid GA, Daff SN, Chapman SK. The role of Thr268 and Phe393 in cytochrome P450 BM3. J Inorg Biochem. 2006;100:1075–1090. doi: 10.1016/j.jinorgbio.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 62.Li H, Poulos TL. Modeling protein-substrate interactions in the heme domain of cytochrome P450(BM-3) Acta Crystallogr Sect D. 1995;51:21–32. doi: 10.1107/S0907444994009194. [DOI] [PubMed] [Google Scholar]
- 63.Girvan HM, Seward HE, Toogood HS, Cheesman MR, Leys D, Munro AW. Structural and spectroscopic characterization of P450 BM3 mutants with unprecedented P450 heme iron ligand sets. New heme ligation states influence conformational equilibria in P450 BM3. J Biol Chem. 2007;282:564–572. doi: 10.1074/jbc.M607949200. [DOI] [PubMed] [Google Scholar]
- 64.Whitehouse CJ, Bell SG, Yang W, Yorke JA, Blanford CF, Strong AJ, Morse EJ, Bartlam M, Rao Z, Wong LL. A highly active single-mutation variant of P450BM3 (CYP102A1) ChemBioChem. 2009;10:1654–1656. doi: 10.1002/cbic.200900279. [DOI] [PubMed] [Google Scholar]
- 65.Ravichandran KG, Boddupalli SS, Hasemann CA, Peterson JA, Deisenhofer J. Crystal-structure of hemoprotein domain of P450bm-3, a prototype for microsomal P450s. Science. 1993;261:731–736. doi: 10.1126/science.8342039. [DOI] [PubMed] [Google Scholar]
- 66.Winn PJ, Ludemann SK, Gauges R, Lounnas V, Wade RC. Comparison of the dynamics of substrate access channels in three cytochrome P450s reveals different opening mechanisms and a novel functional role for a buried arginine. Proc Natl Acad Sci USA. 2002;99:5361–5366. doi: 10.1073/pnas.082522999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ludemann SK, Lounnas V, Wade RC. How do substrates enter and products exit the buried active site of cytochrome P450cam? 1. Random expulsion molecular dynamics investigation of ligand access channels and mechanisms. J Mol Biol. 2000;303:797–811. doi: 10.1006/jmbi.2000.4154. [DOI] [PubMed] [Google Scholar]
- 68.Modi S, Sutcliffe MJ, Primrose WU, Lian LY, Roberts GC. The catalytic mechanism of cytochrome P450 BM3 involves a 6 Å movement of the bound substrate on reduction. Nat Struct Biol. 1996;3:414–417. doi: 10.1038/nsb0596-414. [DOI] [PubMed] [Google Scholar]
- 69.Modi S, Primrose WU, Boyle JM, Gibson CF, Lian LY, Roberts GC. NMR studies of substrate binding to cytochrome P450 BM3: comparisons to cytochrome P450 cam. Biochemistry. 1995;34:8982–8988. doi: 10.1021/bi00028a006. [DOI] [PubMed] [Google Scholar]
- 70.Ravindranathan KP, Gallicchio E, McDermott AE, Levy RM. Conformational dynamics of substrate in the active site of cytochrome P450 BM-3/NPG complex: insights from NMR order parameters. J Am Chem Soc. 2007;129:474–475. doi: 10.1021/ja0672371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jovanovic T. PhD Thesis. Columbia University; New York, NY: 2005. [Google Scholar]
- 72.Smith JRL, Sleath PR. Model systems for cytochrome-P450 dependent mono-oxygenases 0.2 Kinetic isotope effects for the oxidative demethylation of anisole and [Me-H-2(3)]-labeled anisole by cytochrome-P450 dependent mono-oxygenases and model systems. J Chem Soc Perkin Trans. 1983;2:621–628. [Google Scholar]
- 73.Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, Johnson EF. The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0-Å resolution. J Biol Chem. 2004;279:35630–35637. doi: 10.1074/jbc.M405427200. [DOI] [PubMed] [Google Scholar]
- 74.Miners JO, Birkett DJ. Cytochrome P4502C9: an enzyme of major importance in human drug metabolism. Br J Clin Pharmacol. 1998;45:525–538. doi: 10.1046/j.1365-2125.1998.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee DS, Yamada A, Sugimoto H, Matsunaga I, Ogura H, Ichihara K, Adachi S, Park SY, Shiro Y. Substrate recognition and molecular mechanism of fatty acid hydroxylation by cytochrome P450 from Bacillus subtilis. Crystallographic, spectroscopic, and mutational studies. J Biol Chem. 2003;278:9761–9767. doi: 10.1074/jbc.M211575200. [DOI] [PubMed] [Google Scholar]
- 76.Matsunaga I, Ueda A, Sumimoto T, Ichihara K, Ayata M, Ogura H. Site-directed mutagenesis of the putative distal helix of peroxygenase cytochrome P450. Arch Biochem Biophys. 2001;394:45–53. doi: 10.1006/abbi.2001.2512. [DOI] [PubMed] [Google Scholar]
- 77.Li H, Poulos TL. Fatty acid metabolism, conformational change, and electron transfer in cytochrome P-450(BM-3) Biochim Biophys Acta. 1999;1441:141–149. doi: 10.1016/s1388-1981(99)00161-4. [DOI] [PubMed] [Google Scholar]
- 78.Xia ZX, Mathews FS. Molecular structure of flavocytochrome b2 at 2.4 Å resolution. J Mol Biol. 1990;212:837–863. doi: 10.1016/0022-2836(90)90240-M. [DOI] [PubMed] [Google Scholar]
- 79.Stenberg K, Clausen T, Lindqvist Y, Macheroux P. Involvement of Tyr24 and Trp108 in substrate binding and substrate specificity of glycolate oxidase. Eur J Biochem. 1995;228:408–416. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.