

Abstract
Thymosin β4 (Tβ4), a ubiquitous peptide, regulates several cellular processes that include cell morphology, wound healing and inflammatory response. Administration of exogenous Tβ4 is protective in diabetic nephropathy and in a unilateral ureteral obstruction model. However, the role of endogenous Tβ4 in health and disease conditions remains unclear. To elucidate the pathophysiological role of endogenous Tβ4 in hypertension, we examined angiotensin-II (Ang-II)-induced renal and cardiac damage in Tβ4 knockout (Tβ4 KO) mice. Tβ4 KO and wild-type (WT) C57BL/6 mice were infused continuously for six weeks with either vehicle or Ang-II (980 ng/kg/min). At baseline, Tβ4 deficiency did not affect renal and cardiac function. Systolic blood pressure (SBP) in the Ang-II group was similar in WT and Tβ4 KO mice (WT Ang-II 179.25±10.11 mmHg; Tβ4 KO Ang-II 169.81±6.54 mmHg). Despite the similar SBP following Ang-II infusion, Tβ4-deficient mice had dramatically increased albuminuria and decreased nephrin expression in the kidney (P<0.005). In the heart of Tβ4 KO mice, Ang-II reduced ejection fraction (EF) and shortening fraction (EF: WT Ang-II 77.95±1.03%; Tβ4 KO Ang-II 62.58±3.25%; P<0.005), which was accompanied by cardiac hypertrophy and left ventricular dilatation. Additionally, renal and cardiac infiltration of CD68 macrophages, intercellular adhesion molecule-1 (ICAM-1) and total collagen content were increased after Ang-II infusion in Tβ4 KO mice (P<0.005). Overall, our data indicate that endogenous Tβ4 is crucial in preventing tissue injury from Ang-II-induced hypertension. This study gives new insights into the protective role of endogenous Tβ4 in hypertensive end-organ damage.
Keywords: Thymosin β4, Angiotensin-II, Inflammation, Fibrosis, Ac-SDKP
Introduction
The development of hypertensive end-organ damage results from the interaction between humoral and mechanical stimuli1. In angiotensin-II (Ang-II)-induced hypertension, tissue damage is dependent on the magnitude of blood pressure, but some of Ang-II actions are independent of its pressure related effects2. Ang-II-induced hypertension is often characterized by increased inflammation and fibrosis in the heart and kidney1. Although a low level of circulating Ang-II is required for maintaining basal vascular tone, salt and water homeostasis; increased Ang-II in chronic hypertension stimulates the expression of proinflammatory and profibrotic gene products and thereby promotes renal and cardiac damage3.
Thymosin β4 (Tβ4) is a 43 amino acid short peptide that was originally described to regulate lymphocyte maturation in the thymus4, and subsequently, Tβ4 was identified as a primary G-actin sequestering peptide5. Tβ4 is highly expressed in platelets, macrophages and wound fluids6, 7. Tβ4 is also found in other tissues of the body, including heart and kidney8. Our group recently demonstrated that Tβ4 is enzymatically hydrolyzed to release the anti-inflammatory product N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP)9. Ac-SDKP treatment in murine models of cardiovascular diseases has shown beneficial organ effects10, 11. Similar to Ac-SDKP, Tβ4 also ameliorates tissue injury associated with heart failure, kidney diseases, stroke, lung diseases and dry eye, and these protective effects are in part due to the role of Tβ4 in regulating cell movement, angiogenesis, wound healing, inflammation and fibrosis12–16. Despite such evidence of exogenously infused Tβ4 in organ protection, the functional role of endogenous Tβ4 in the heart and kidney in normal and disease conditions remains unclear. To elucidate the pathophysiological role of endogenous Tβ4 in hypertension, we evaluated Ang-II-induced renal and cardiac damage in Tβ4 KO mice. Here, we aimed to address the effect of Ang-II infusion in WT mice that normally express Tβ4 and in mice lacking endogenous Tβ4 on 1) systolic blood pressure, 2) renal and cardiac function, 3) renal and cardiac inflammation and 4) renal and cardiac fibrosis. These data will facilitate our understanding on the role of endogenous Tβ4 in the pathophysiology of hypertension and related organ damage.
Methods
The data, analytic methods, and study materials that support the findings of this study are available from the corresponding author on reasonable request.
All the experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Henry Ford Hospital and were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Male C57BL/6J rats (Jackson Laboratories, Sacramento, CA) and male Tβ4 KO mice (on C57BL/6 background) at 8-10 weeks old were randomly divided and subcutaneously infused with either vehicle (0.01 N acetic acid saline solution) or Ang-II (980 ng/kg/min; Bachem, Bubendorf, Switzerland) for 6 weeks using an osmotic minipump (Alzet, Cupertino, CA, USA). The key experimental procedures included the following measurements: systolic blood pressure via a non-invasive tail-cuff method; albuminuria by ELISA; Doppler echocardiography on awake mice to assess cardiac function; immunohistochemistry on heart and kidney sections to determine infiltration of CD68+ macrophages and total collagen content (picrosirius red staining); determination of protein expression by immunoblotting; and Ac-SDKP measurement in tissues by ELISA. A detailed methods section is available in the data supplement for all these procedures.
Statistical analyses
A nonparametric two-sample Wilcoxon test with a Fliqner-Policello correction for unequal variances was used to compare contrasts of interest in all the data. Significance was determined using Hochberg’s method to adjust for multiple testing. The adjustment was performed on groups of similar tests. A significant difference was considered at P-values less than 0.01. Analysis was generated using SAS/STAT software, Version 9.3 of the SAS System for Windows (Copyright SAS Institute Inc., Cary, NC)
Results
Effect of Ang-II infusion on systolic blood pressure, body weight, and kidney weight in WT and Tβ4 KO mice
At baseline, systolic blood pressure (SBP) was similar in WT and Tβ4 KO mice (Fig. 1). Ang-II infusion increased the SBP in both strains, compared with that of the vehicle group (P<0.01). SBP at any week was not significantly different between the two strains (Fig. 1A). Body weight (b.w.) and kidney weight was similar in WT and Tβ4 KO mice in the vehicle group (Table S1). As expected, under continuous Ang-II infusion for six weeks, the b.w. of WT mice decreased moderately, whereas a significant reduction in b.w. was observed in Tβ4 KO mice, compared with that of the vehicle group. Furthermore, under Ang-II infusion, the kidney weight to tibia length ratio was significantly higher in Tβ4 KO mice than in WT mice (Table S1).
Figure 1.
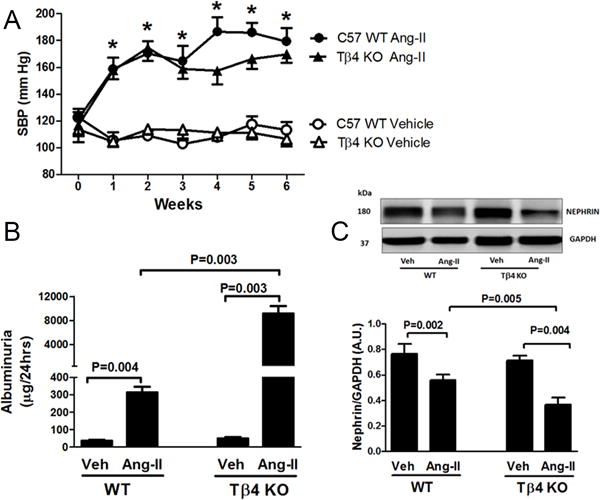
Effect of Angiotensin-II (Ang-II) on the systolic blood pressure (SBP), albuminuria and renal nephrin expression in Tβ4 KO mice. (A) In the vehicle (Veh) treatment, no difference in SBP was observed between WT (open circle) and Tβ4 KO mice (open triangle). Ang-II increased SBP in both WT (closed circle) and Tβ4 KO (closed triangle) mice, with no significant difference observed at any week compared with that in the vehicle group. (B) Ang-II moderately increased albumin excretion in WT mice; however, albuminuria increased dramatically in Ang-II-infused Tβ4 KO mice. (C) Top figure: At baseline, renal nephrin expression was similar in WT and Tβ4 KO mice; Ang-II significantly decreased nephrin expression in Tβ4 KO mice compared with that in the WT. Bottom figure shows the quantitative data. All data are expressed as the mean ± SEM, n = 6-10 in each group. *P<0.01 Veh vs. Ang-II. Arbitrary Unit (A.U.).
Effect of Ang-II infusion on albuminuria and nephrin expression in the kidney of WT and Tβ4 KO mice
Basal urinary albumin excretion was similar in WT and Tβ4 KO mice (Fig. 1B). Ang-II infusion moderately increased albuminuria in WT mice; however, the increase was dramatic in Tβ4-deficient mice (Fig. 1B). We also measured nephrin expression in the kidney, an important protein forming the renal filtration barrier. Whereas basal nephrin expression in WT and Tβ4 KO mice remained largely unchanged, Ang-II infusion in Tβ4 KO mice caused a significant reduction in nephrin expression, compared with that in WT mice (Fig. 1C).
Effect of Ang-II infusion on renal and cardiac macrophages and ICAM-1 expression in WT and Tβ4 KO mice
At baseline, no difference was observed in renal and cardiac infiltration of CD68 positive macrophages between WT and Tβ4 KO mice (Fig. 2A–B; Fig. 5A–B). With Ang-II infusion, renal and cardiac infiltration of CD68 macrophages increased significantly in Tβ4 KO mice, compared with that in WT mice (Fig. 2A–B; Fig. 5A–B). These changes were associated with increased expression of the intercellular adhesion molecule-1 (ICAM-1), a pro-inflammatory protein, in the heart and kidney of Ang-II-infused Tβ4 KO mice (Fig. 2C; Fig. 5C).
Figure 2.
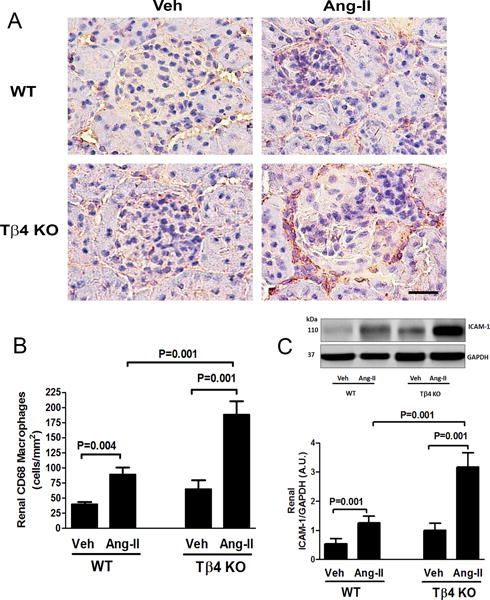
Effect of Ang-II on CD68 macrophage infiltration and intercellular adhesion molecule-1 (ICAM-1) protein expression in the kidney of Tβ4 KO mice. (A) Representative images of renal macrophage infiltration using anti-CD68 antibody. Positive signals were detected as reddish-brown staining in the cytoplasm. Shown are images captured under a 20× microscope objective. Scale bar = 25 μM. In Ang-II infusion, renal infiltration of CD68 positive macrophages increased significantly in Tβ4 KO mice, compared with that in the WT. (B) Quantitative data for Figure 2A. (C) Top figure: At the basal level, no significant difference was observed in renal ICAM-1 protein expression; however, Ang-II infusion significantly increased ICAM-1 expression in Tβ4 KO mice compared with that in the WT. Bottom figure shows the quantitative data. All data are expressed as the mean ± SEM, n = 6-10 in each group. Vehicle (Veh), Angiotensin-II (Ang-II), Arbitrary Unit (A.U.).
Figure 5.
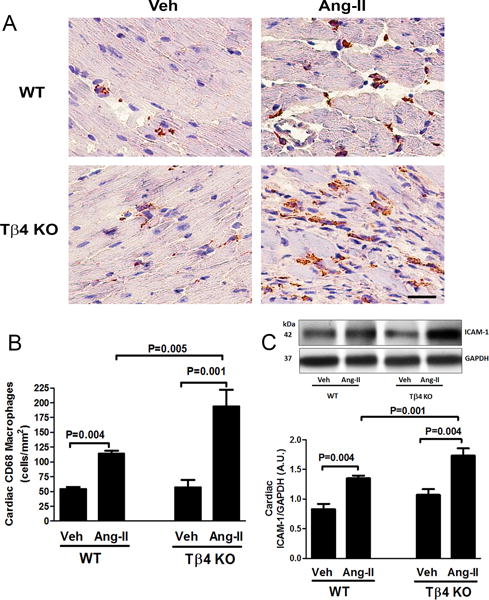
Effect of Ang-II on cardiac macrophage infiltration and intercellular adhesion molecule-1 (ICAM-1) protein expression in the heart of Tβ4 KO mice. (A) Representative images of cardiac macrophage infiltration using anti-CD68 antibody. Positive signals were detected as reddish-brown staining in the cytoplasm. Shown are images captured under a 20× microscope objective. Scale bar = 25 μM. In Ang-II infusion, cardiac infiltration of CD68 positive macrophages increased significantly in Tβ4 KO mice, compared with that in the WT. (B) Quantitative data for Figure 5A. (C) Top figure: At baseline, no significant difference was observed in cardiac ICAM-1 expression; Ang-II infusion significantly increased ICAM-1 expression in the heart of Tβ4 KO mice, compared with that in WT mice. Bottom figure shows the quantitative data. All data are expressed as the mean ± SEM, n = 6-10 in each group.
Effect of Ang-II infusion on renal and cardiac fibrosis and α-smooth muscle actin (α-SMA) expression in WT and Tβ4 KO mice
Total collagen content in heart and kidney was quantified by PSR staining (histology) and by hydroxyproline assay (biochemical method), and we found collagen was similar in WT and Tβ4 KO mice under the basal condition (Fig. 3A–C; Fig. 6A–C). Under Ang-II infusion, renal and cardiac collagen content was significantly higher in Tβ4 KO mice than that in WT mice (Fig. 3A–C; Fig. 6A–C). These changes were associated with increased expression of pro-fibrotic α-SMA in the heart and kidney of Ang-II-infused Tβ4 KO mice (Fig. 3D; Fig. 6D).
Figure 3.
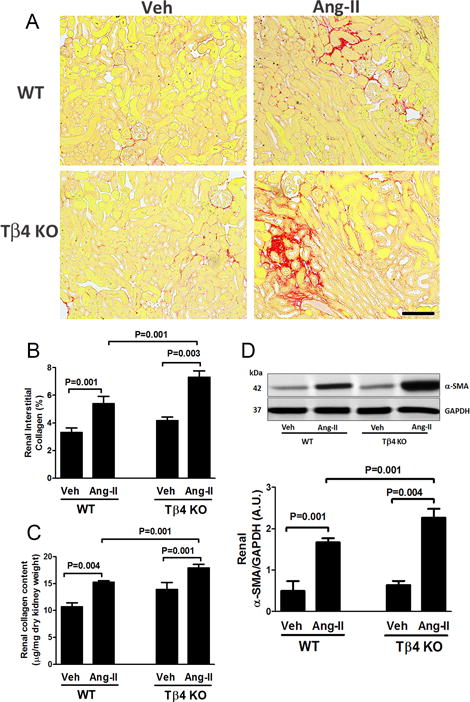
Effect of Ang-II on renal fibrosis and α-smooth muscle actin (α-SMA) protein expression in the kidney of Tβ4 KO mice. (A) Representative images of renal interstitial fibrosis. Red color indicates total collagen content by picrosirius red staining (PSR). Images were captured using a 20× microscope objective. Scale bar = 100 μM. In Ang-II infusion, total renal collagen content increased significantly in Tβ4 KO mice, compared with that in the WT. (B) Quantitative data for Figure 3A. (C) Total renal collagen content was further quantified by a different method utilizing a biochemical hydroxyproline assay. (D) Top figure: At the basal level, no significant difference was observed in renal α-SMA expression; Ang-II infusion significantly increased α-SMA expression in Tβ4 KO mice, compared with that in the WT. Bottom figure shows the quantitative data. All data are expressed as the mean ± SEM, n = 6-10 in each group.
Figure 6.
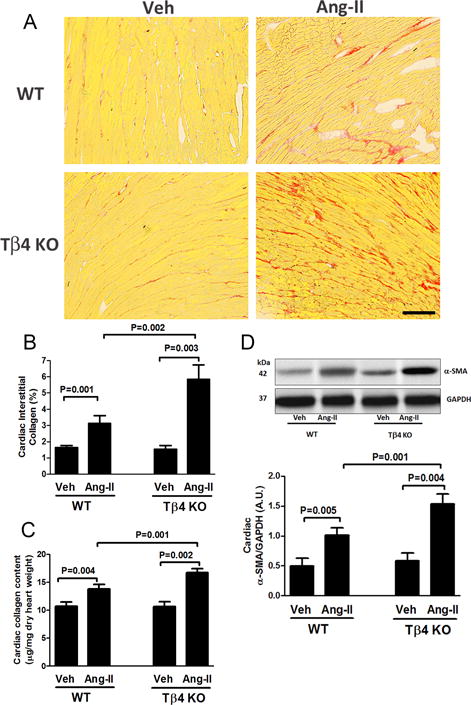
Effect of Ang-II on cardiac fibrosis and α-smooth muscle actin (α-SMA) protein expression in the heart of Tβ4 KO mice. (A) Representative images of cardiac interstitial fibrosis. Red color indicates total collagen content by picrosirius red staining. Images were captured using a 20× microscope objective. Scale bar = 100 μM. In Ang-II infusion, total cardiac collagen content increased significantly in Tβ4 KO mice, compared with that in the WT. (B) Quantitative data for Figure 6A. (C) Total cardiac collagen content was further quantified by a biochemical hydroxyproline assay. (D) Top figure: At baseline, no significant difference in cardiac α-SMA expression was observed; Ang-II infusion significantly increased α-SMA expression in the heart of Tβ4 KO mice, compared with that in WT mice. Bottom figure shows the quantitative data. All data are expressed as the mean ± SEM, n = 6-10 in each group.
Effect of Ang-II infusion on cardiac hypertrophy and cardiac function in WT and Tβ4 KO mice
In the vehicle group, cardiac hypertrophy (heart weight to tibia length ratio) was similar in WT and Tβ4 KO mice (Fig. S1). With Ang-II infusion, cardiac hypertrophy increased significantly in Tβ4 KO mice, compared with that in WT mice (Fig. S1). Additionally, we found ejection fraction and shortening fraction were significantly reduced in Tβ4 KO mice (Fig. 4A–B), indicating reduced cardiac function. Posterior wall thickness increased in both strains in the Ang-II infusion group (Fig. 4C). We also noted that Ang-II-infused Tβ4 KO mice showed a markedly enlarged left ventricular chamber, compared with that of WT mice (Fig. 4D). Increase in left ventricular chamber dimensions indicated eccentric hypertrophy in Ang-II-infused Tβ4 KO mice, in contrast to concentric hypertrophy in Ang-II-infused C57BL/6 WT mice.
Figure 4.
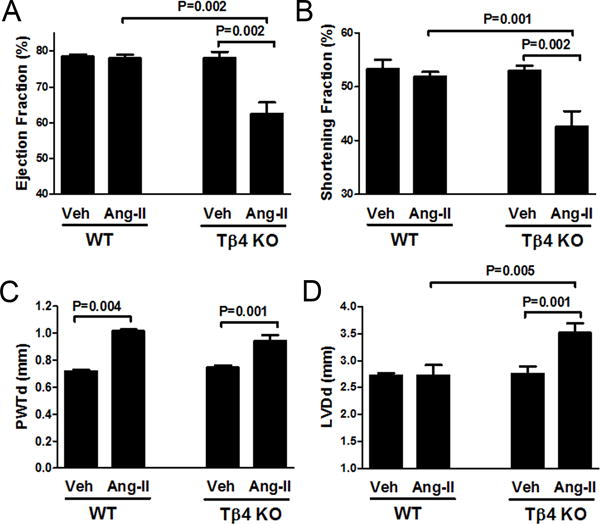
Effect of Ang-II infusion on cardiac function in Tβ4 KO mice. (A & B) Ang-II infusion reduced ejection and shortening fraction in Tβ4 KO mice compared with that in the WT. (C) Ang-II infusion caused a similar increase in posterior wall thickness-in diastole (PWTd) in both strains. (D) Enlarged left ventricular dimension-in diastole (LVDd) was observed in Ang-II-infused Tβ4 KO mice. All data are expressed as the mean ± SEM, n = 6-10 in each group.
Effect of Ang-II infusion on the mortality rate in WT and Tβ4 KO mice
No mortality was observed in WT mice in either vehicle or Ang-II-infusion group or even in Tβ4 KO mice in the vehicle group during the protocol (Fig. S2). However, the mortality rate increased significantly in Ang-II-infused Tβ4 KO mice (~40%, Fig. S2).
Ac-SDKP content in the heart and kidney tissue of WT and Tβ4 KO mice
We further determined the Ac-SDKP content in the heart and kidney tissue of WT and Tβ4 KO mice. Although Ac-SDKP content remained detectable in the kidney and heart of Tβ4 KO mice, the amount was markedly lower than that of WT mice (Fig. S3).
Discussion
In this study, we found Tβ4 deficiency did not alter heart and kidney function in the basal condition. However, in an experimental model of Ang-II-induced hypertension, renal and cardiac injury was exacerbated in Tβ4 KO mice. We found Ang-II infusion increased the systolic blood pressure (SBP) to a similar extent in WT and Tβ4 KO mice. Despite a similar increase in SBP following Ang-II infusion, inflammation and fibrosis in the heart and kidney were significantly higher in Tβ4 KO mice than in WT mice, accompanied by increased cardiac and renal damage. Our study provides novel insights into the protective role of endogenous Tβ4 in hypertensive end-organ damage.
Tβ4 is a primary G-actin sequestering peptide and is implicated in regulating cell movement and cell migration via mechanism(s) involving actin binding5. In addition to a role in regulating cell movement, several reports demonstrate non-traditional roles of Tβ4 in disease states17, 18. In cardiovascular diseases, our group and others have shown organ protective effects of exogenously administered Tβ4, and these effects were due to its anti-inflammatory and anti-fibrotic nature12, 13, 19. Despite reports that show beneficial effects of exogenous Tβ4, the role of endogenous Tβ4 in health and disease conditions has remained unclear. In this study, we evaluated the heart and kidney injury in Tβ4 KO mice following Ang-II infusion.
Whether the deleterious organ effects in Ang-II-infused Tβ4 KO mice were due to loss of Tβ4 ability to bind G-actin is intriguing. Cell structure is tightly regulated by precise actin polymerization, and any major alterations in actin-cytoskeletal organization can lead to organ dysfunction20, 21. At baseline, we found no changes in renal and cardiac function in Tβ4 KO mice, suggesting that binding of Tβ4 to G-actin is either not required or alternate compensatory mechanisms(s) exist in the absence of Tβ4 to maintain normal renal and cardiac function. Vasilopoulou et al. support this hypothesis in a recent study22 in which they found that in the basal condition, mice lacking Tβ4 did not show changes in glomerular morphology or podocyte architecture, processes that require correct organization of actin-cytoskeletal structure22. Moreover, expression of another actin binding protein cofilin was significantly up-regulated in the kidney of Tβ4 KO mice, which might partly compensate for the loss of Tβ422. However, because renal and cardiac damage was exacerbated in Tβ4 KO mice after Ang-II stimuli, whereas no damage was associated in the vehicle group, we concluded that in the presence of injurious stimuli, compensatory mechanism(s) in Tβ4 deficiency might not be sufficient to prevent the subsequent tissue injury; thus, endogenous Tβ4 is rendered crucial in preventing hypertensive end-organ damage.
We initially aimed to assess SBP of WT and Tβ4 KO mice with vehicle and Ang-II infusion. Our results for the vehicle group revealed no differences in SBP between WT and Tβ4 KO mice. Infusion of Ang-II increased SBP similarly in both strains. Though, we noticed a lower SBP response after Ang-II in Tβ4 KO mice at week 4, 5 and 6, however it did not reach statistical significance, compared to the Ang-II infused WT. Mortality observed after Ang-II infusion in Tβ4 KO mice starting at week 3 (Online Supplement) may not be the likely factor for the lower SBP response in Tβ4 KO mice, but rather could be related either with the variability of the tail-cuff method or due to the attenuation of Ang-II chronic effects in Tβ4 KO mice. Nonetheless, despite a similar increase in SBP, cardiac and renal injury were much higher in Ang-II-infused Tβ4 KO mice. These data indicated that Tβ4 deficiency, without affecting the blood pressure, increased the susceptibility to Ang-II-induced tissue injury.
A progressive increase in albuminuria is an indicator of declining kidney function23. Excessive protein filtration across the glomeruli stimulates the proliferation and expansion of the mesangial cells and activates proximal tubular cells to synthesize excess collagen and fibronectin, which causes renal fibrosis and damage24. We found at the basal condition that albuminuria was similar in both strains. Ang-II infusion in WT mice caused a moderate but significant increase in albuminuria; however, albumin excretion increased massively in Tβ4 KO mice, indicating Tβ4 deficiency increased renal damage. Nephrin protein maintains integrity of the renal filtration barrier by forming tight junctions between podocytes25. Decrease in nephrin expression in the kidney is accompanied by albuminuria26. Mutation in the nephrin gene causes a rare form of congenital nephrotic syndrome, affecting primarily infants and is characterized by massive proteinuria27. Notably, normally high expression of Tβ4 occurs in the glomeruli in which nephrin is localized22. In Ang-II infusion, nephrin expression was significantly reduced in Tβ4-deficient mice, and this indicates that loss of nephrin was one of the key mechanism(s) for massive albuminuria in Ang-II-infused Tβ4 KO mice.
To identify whether inflammatory cells were associated with advanced renal damage in Ang-II-infused Tβ4 KO mice, histological and biochemical analyses were conducted. Inflammation is an early event in tissue injury associated with Ang-II hypertension and is often accompanied by an increase in expression of endothelial cell-surface adhesion proteins, such as ICAM-128. Increased ICAM-1 expression then promotes tissue infiltration of immune cells including macrophages, which contributes to increased inflammation29. High expression of Tβ4 is also found in macrophages in which the precise function is not yet clear6. Kannan L. et al. reported that macrophages secrete Tβ4 when stimulated with lipopolysaccharide30. Released Tβ4 has been suggested to counterbalance the detrimental effects of inflammation to restore physiological homeostasis. In Ang-II infusion, we found expression of ICAM-1 in the kidney increased significantly in Tβ4 deficiency, which was associated with increased renal infiltration of macrophages. Loss of Tβ4 secretion from macrophages in Tβ4 KO mice might partially explain the suppressed counter-regulatory mechanism, resulting in further inflammation. We also observed increased renal fibrosis in Tβ4-deficient mice, which might represent a secondary process following increased inflammation.
We assessed cardiac damage in Tβ4 KO mice in the present study. As mentioned earlier, SBP was similar in WT and Tβ4 KO mice in the Ang-II group, but cardiac hypertrophy was significantly higher in Tβ4-deficient mice than in WT mice. Additionally, we noticed enlarged left ventricular (LV) chamber in Ang-II-infused Tβ4 KO mice, which is indicative of eccentric hypertrophy, in contrast to concentric hypertrophy in WT mice. Multiple mechanisms may be involved in LV dilatation; however, the loss of Tβ4 ability to bind G-actin and subsequent effects on actin-cytoskeletal organization in cardiomyocytes of Tβ4-deficient mice may likely to contribute in reduced contractility, which could in turn explain the reduced ejection fraction and LV dilation. These data again suggested that the compensatory mechanism in Tβ4 deficiency might not sustain overwhelming cardiac insults caused by injurious Ang-II. Cardiac inflammation in pathological conditions, such as in myocarditis, is often accompanied by dilated cardiomyopathy and may result in heart failure31. Increased cardiac inflammation in Ang-II-infused Tβ4 KO mice might also be a contributing factor in promoting LV dilation and reduction in ejection fraction. Additionally, we found that the organ detrimental effects of Ang-II infusion were associated with significantly high mortality in Tβ4 KO mice.
In the present study, the determination of Ac-SDKP content in the heart and kidney of Tβ4 KO mice was another intriguing factor. Tβ4 is the primary source of the anti-inflammatory peptide Ac-SDKP32. Ac-SDKP is released from Tβ4 after sequential hydrolysis by meprin-α and prolyl-oligopeptidase9. Our group previously reported the beneficial effects of Ac-SDKP in hypertension and cardiovascular diseases10, 11, 33. In the present study, we found Ac-SDKP content was not completely absent but was significantly reduced in the heart and kidney of Tβ4 KO mice (Online Supplement). Thus, deleterious organ effects observed in Ang-II-infused Tβ4 KO mice might be mediated in part because of reduced tissue Ac-SDKP content. Our laboratory has previously reported that Ang-II infusion per se did not alter Ac-SDKP content in the heart and kidney, whereas exogenous Ac-SDKP treatment increased Ac-SDKP concentration and ameliorated end-organ damage in Ang-II challenge34, 35. Initially, we did not expect Ac-SDKP to be present in the tissue of Tβ4 KO mice. However, evidence suggest that thymosin-beta family comprise of 15 homologous variants found in the vertebrates and invertebrates, but most mammalian cells normally express two variants simultaneously, one at higher (usually Tβ4) and second at lower concentration (for e.g. Tβ15)36. Interestingly, Tβ15 also carries Acetyl-SDKP at its N-terminal end and our preliminary study (data not published) indicate that Tβ15 could be compensatory in the absence of Tβ4, explaining part of the Ac-SDKP found in the tissue of Tβ4 KO mice in the present study. Further investigation will be required to delineate the exact role played by Ac-SDKP in Tβ4 KO mice in heart and kidney diseases.
In conclusion, Tβ4 deficiency in the normal condition did not cause changes in renal or cardiac function; however, under Ang-II infusion, Tβ4 deficiency was associated with increased tissue inflammation and fibrosis, thereby causing exacerbated renal and cardiac injury. Overall, our data provide new insights into the role of endogenous Tβ4 in hypertensive end-organ damage.
Supplementary Material
Perspectives.
Hypertension exerts deleterious effects on vital organs including the heart and kidney. Our current data showed that endogenous Tβ4 is important in ameliorating inflammation and fibrosis associated with hypertension, and Tβ4 deficiency resulted in exacerbated organ damage. Our study will facilitate the understanding of the role of endogenous Tβ4 in the pathophysiology of hypertension, and its modulation could provide novel therapeutic targets in the treatment of hypertension and cardiovascular diseases.
Novelty and Significance.
-
What is new
This study describes the crucial role of endogenous Tβ4 in preventing angiotensin-II-induced renal and cardiac damage. Tβ4 deficiency resulted in loss of protection and exacerbated organ damage in Ang-II hypertension.
-
What is relevant
Endogenous Tβ4 involvement in preventing Ang-II-induced organ damage could provide a new mechanism and may explain part of the variability in organ damage observed in humans with hypertension.
-
Summary
Loss of endogenous Tβ4 is detrimental in Ang-II-induced renal and cardiac damage.
Acknowledgments
We thank Gulser Gurocak, Xiangguo Dai and Carl Polomoski for their technical support.
Source(s) of Funding
This work was supported by the National Institute of Health Grant P01HL028982 (O.A.C.).
Footnotes
Disclosures
No conflicts of interest, financial or otherwise, are declared by the author(s).
References
- 1.McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res. 2015;116:1022–1033. doi: 10.1161/CIRCRESAHA.116.303697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mervaala E, Muller DN, Schmidt F, Park JK, Gross V, Bader M, Breu V, Ganten D, Haller H, Luft FC. Blood pressure-independent effects in rats with human renin and angiotensinogen genes. Hypertension. 2000;35:587–594. doi: 10.1161/01.hyp.35.2.587. [DOI] [PubMed] [Google Scholar]
- 3.Navar LG. Physiology: Hemodynamics, endothelial function, renin-angiotensin-aldosterone system, sympathetic nervous system. J Am Soc Hypertens. 2014;8:519–524. doi: 10.1016/j.jash.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Low TL, Hu SK, Goldstein AL. Complete amino acid sequence of bovine thymosin beta 4: A thymic hormone that induces terminal deoxynucleotidyl transferase activity in thymocyte populations. Proc Natl Acad Sci U S A. 1981;78:1162–1166. doi: 10.1073/pnas.78.2.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Safer D, Elzinga M, Nachmias VT. Thymosin beta 4 and fx, an actin-sequestering peptide, are indistinguishable. J Biol Chem. 1991;266:4029–4032. [PubMed] [Google Scholar]
- 6.Paulussen M, Landuyt B, Schoofs L, Luyten W, Arckens L. Thymosin beta 4 mrna and peptide expression in phagocytic cells of different mouse tissues. Peptides. 2009;30:1822–1832. doi: 10.1016/j.peptides.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 7.Sosne G, Qiu P, Kurpakus-Wheater M. Thymosin beta 4: A novel corneal wound healing and anti-inflammatory agent. Clin Ophthalmol. 2007;1:201–207. [PMC free article] [PubMed] [Google Scholar]
- 8.Pradelles P, Frobert Y, Creminon C, Ivonine H, Frindel E. Distribution of a negative regulator of haematopoietic stem cell proliferation (acsdkp) and thymosin beta 4 in mouse tissues. FEBS Lett. 1991;289:171–175. doi: 10.1016/0014-5793(91)81062-d. [DOI] [PubMed] [Google Scholar]
- 9.Kumar N, Nakagawa P, Janic B, Romero CA, Worou ME, Monu SR, Peterson EL, Shaw J, Valeriote F, Ongeri EM, Niyitegeka JM, Rhaleb NE, Carretero OA. The anti-inflammatory peptide ac-sdkp is released from thymosin-beta4 by renal meprin-alpha and prolyl oligopeptidase. Am J Physiol Renal Physiol. 2016;310:F1026–1034. doi: 10.1152/ajprenal.00562.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng H, Carretero OA, Liao TD, Peterson EL, Rhaleb NE. Role of n-acetyl-seryl-aspartyl-lysyl-proline in the antifibrotic and anti-inflammatory effects of the angiotensin-converting enzyme inhibitor captopril in hypertension. Hypertension. 2007;49:695–703. doi: 10.1161/01.HYP.0000258406.66954.4f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang F, Yang XP, Liu YH, Xu J, Cingolani O, Rhaleb NE, Carretero OA. Ac-sdkp reverses inflammation and fibrosis in rats with heart failure after myocardial infarction. Hypertension. 2004;43:229–236. doi: 10.1161/01.HYP.0000107777.91185.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuo Y, Chun B, Potthoff SA, Kazi N, Brolin TJ, Orhan D, Yang HC, Ma LJ, Kon V, Myohanen T, Rhaleb NE, Carretero OA, Fogo AB. Thymosin beta4 and its degradation product, ac-sdkp, are novel reparative factors in renal fibrosis. Kidney Int. 2013;84:1166–1175. doi: 10.1038/ki.2013.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans MA, Smart N, Dube KN, Bollini S, Clark JE, Evans HG, Taams LS, Richardson R, Levesque M, Martin P, Mills K, Riegler J, Price AN, Lythgoe MF, Riley PR. Thymosin beta4-sulfoxide attenuates inflammatory cell infiltration and promotes cardiac wound healing. Nat Commun. 2013;4:2081. doi: 10.1038/ncomms3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris DC, Chopp M, Zhang L, Lu M, Zhang ZG. Thymosin beta4 improves functional neurological outcome in a rat model of embolic stroke. Neuroscience. 2010;169:674–682. doi: 10.1016/j.neuroscience.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sosne G, Dunn SP, Kim C. Thymosin beta4 significantly improves signs and symptoms of severe dry eye in a phase 2 randomized trial. Cornea. 2015;34:491–496. doi: 10.1097/ICO.0000000000000379. [DOI] [PubMed] [Google Scholar]
- 16.Conte E, Iemmolo M, Fagone E, Gili E, Fruciano M, Genovese T, Esposito E, Cuzzocrea S, Vancheri C. Thymosin beta4 reduces il-17-producing cells and il-17 expression, and protects lungs from damage in bleomycin-treated mice. Immunobiology. 2014;219:425–431. doi: 10.1016/j.imbio.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Goldstein AL, Hannappel E, Sosne G, Kleinman HK. Thymosin beta4: A multi-functional regenerative peptide. Basic properties and clinical applications. Expert Opin Biol Ther. 2012;12:37–51. doi: 10.1517/14712598.2012.634793. [DOI] [PubMed] [Google Scholar]
- 18.Bollini S, Riley PR, Smart N. Thymosin beta4: Multiple functions in protection, repair and regeneration of the mammalian heart. Expert Opin Biol Ther. 2015;15(Suppl 1):S163–174. doi: 10.1517/14712598.2015.1022526. [DOI] [PubMed] [Google Scholar]
- 19.Peng H, Xu J, Yang XP, Dai X, Peterson EL, Carretero OA, Rhaleb NE. Thymosin-beta4 prevents cardiac rupture and improves cardiac function in mice with myocardial infarction. Am J Physiol Heart Circ Physiol. 2014;307:H741–751. doi: 10.1152/ajpheart.00129.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desouza M, Gunning PW, Stehn JR. The actin cytoskeleton as a sensor and mediator of apoptosis. Bioarchitecture. 2012;2:75–87. doi: 10.4161/bioa.20975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol. 2010;177:512–524. doi: 10.2353/ajpath.2010.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vasilopoulou E, Kolatsi-Joannou M, Lindenmeyer MT, White KE, Robson MG, Cohen CD, Sebire NJ, Riley PR, Winyard PJ, Long DA. Loss of endogenous thymosin beta4 accelerates glomerular disease. Kidney Int. 2016;90:1056–1070. doi: 10.1016/j.kint.2016.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Remuzzi G, Ruggenenti P, Benigni A. Understanding the nature of renal disease progression. Kidney Int. 1997;51:2–15. doi: 10.1038/ki.1997.2. [DOI] [PubMed] [Google Scholar]
- 24.Zoja C, Morigi M, Remuzzi G. Proteinuria and phenotypic change of proximal tubular cells. J Am Soc Nephrol. 2003;14(Suppl 1):S36–41. doi: 10.1097/01.asn.0000068626.23485.e0. [DOI] [PubMed] [Google Scholar]
- 25.Fukasawa H, Bornheimer S, Kudlicka K, Farquhar MG. Slit diaphragms contain tight junction proteins. J Am Soc Nephrol. 2009;20:1491–1503. doi: 10.1681/ASN.2008101117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G, Camussi G. Nephrin expression is reduced in human diabetic nephropathy: Evidence for a distinct role for glycated albumin and angiotensin ii. Diabetes. 2003;52:1023–1030. doi: 10.2337/diabetes.52.4.1023. [DOI] [PubMed] [Google Scholar]
- 27.Jalanko H. Congenital nephrotic syndrome. Pediatric nephrology (Berlin, Germany) 2009;24:2121–2128. doi: 10.1007/s00467-007-0633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harrison DG, Marvar PJ, Titze JM. Vascular inflammatory cells in hypertension. Front Physiol. 2012;3:128. doi: 10.3389/fphys.2012.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brake DK, Smith EO, Mersmann H, Smith CW, Robker RL. Icam-1 expression in adipose tissue: Effects of diet-induced obesity in mice. Am J Physiol Cell Physiol. 2006;291:C1232–1239. doi: 10.1152/ajpcell.00008.2006. [DOI] [PubMed] [Google Scholar]
- 30.Kannan L, Rath NC, Liyanage R, Lay JO., Jr Effect of toll-like receptor activation on thymosin beta-4 production by chicken macrophages. Mol Cell Biochem. 2010;344:55–63. doi: 10.1007/s11010-010-0528-0. [DOI] [PubMed] [Google Scholar]
- 31.Cooper LTJ. Myocarditis. New England Journal of Medicine. 2009;360:1526–1538. doi: 10.1056/NEJMra0800028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cavasin MA, Rhaleb NE, Yang XP, Carretero OA. Prolyl oligopeptidase is involved in release of the antifibrotic peptide ac-sdkp. Hypertension. 2004;43:1140–1145. doi: 10.1161/01.HYP.0000126172.01673.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin CX, Rhaleb NE, Yang XP, Liao TD, D’Ambrosio MA, Carretero OA. Prevention of aortic fibrosis by n-acetyl-seryl-aspartyl-lysyl-proline in angiotensin ii-induced hypertension. Am J Physiol Heart Circ Physiol. 2008;295:H1253–H1261. doi: 10.1152/ajpheart.00481.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cavasin MA, Liao TD, Yang XP, Yang JJ, Carretero OA. Decreased endogenous levels of ac-sdkp promote organ fibrosis. Hypertension. 2007;50:130–136. doi: 10.1161/HYPERTENSIONAHA.106.084103. [DOI] [PubMed] [Google Scholar]
- 35.Peng H, Carretero OA, Vuljaj N, Liao TD, Motivala A, Peterson EL, Rhaleb NE. Angiotensin-converting enzyme inhibitors: A new mechanism of action. Circulation. 2005;112:2436–2445. doi: 10.1161/CIRCULATIONAHA.104.528695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mannherz HG, Hannappel E. The beta-thymosins: Intracellular and extracellular activities of a versatile actin binding protein family. Cell Motil Cytoskeleton. 2009;66:839–851. doi: 10.1002/cm.20371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.