

Abstract
Background
IL33 plays an important role in development of experimental asthma.
Objective
To study the role of the IL33 receptor (ST2) in persistence of asthma in a mouse model.
Methods
We studied allergen-induced experimental asthma in ST2 knockout (KO) and wild-type control mice. We measured airway hyperresponsiveness (AHR) by flexivent, inflammatory indices by ELISA, histology and real-time PCR, and ILC2s in lung single cell preparations by flow cytometry.
Results
The AHR level was elevated in allergen-treated ST2 KO mice and was comparable to that from allergen-treated WT controls. Peribronchial and perivascular inflammation and mucus production were largely similar in both groups. Persistence of experimental asthma in ST2 KO mice was associated with an increase in the level of TSLP, IL9 and IL13 but not IL5 in bronchoalveolar lavage (BAL). ST2 deletion expectedly caused a reduction in IL13+ CD4 T cells, Foxp3+ Tregs and IL5+ ILC2s. Unexpectedly, ST2 deletion led to an overall increase in ILCs (CD45+lin-CD25+ cells), IL13+ ILC2s, the emergence of a TSLP-R+ IL9+ ILC2 population and an increase in intraepithelial mast cells in the lung. An anti-TSLP antibody abrogated AHR, inflammation and mucus production in allergen-treated ST2 KO mice. It also caused a reduction in ILCs, ILC2s, and IL9+ and IL13+ ILC2s in the lung.
Conclusions
Genetic deletion of the IL33 receptor paradoxically increases TSLP production, which stimulates the emergence of IL9+ and IL13+ ILC2s and mast cells, and leads to the development of chronic experimental asthma. An anti-TSLP antibody abrogates all pathologic features of asthma in this model.
Keywords: Asthma, type 2 innate lymphoid cells, IL33 receptor, thymic stromal lymphopoietin, interleukin-9, mast cells
Introduction
ILC2s represent an important group of type 2 immune cells (reviewed in ref. 1 and 2). They are regulated by many growth factors. IL33 is an important growth factor for ILC2s, especially lung ILC2s (1, 2). IL33 promotes their differentiation, maturation and type 2 cytokine expression. IL33 signals through its receptor ST2, also known as T1/ST2L (3). ST2 signaling is critically important for IL5 induction in Th2 cells and is required for Th2 memory (4). ST2 signaling regulates many other cells including Tregs (5), dendritic cells (6), mast cells (7), basophils (8), eosinophils (9) and macrophages (10). By regulating so many immune cells, the IL33/ST2 axis plays a seminal role in host defense and allergic and chronic inflammatory diseases (reviewed in ref. 11).
The role of ST2 was previously investigated in various models of experimental asthma (12–17). Some studies reported a complete or substantial resolution of airway hyperreactivity, inflammation, goblet cell hyperplasia and other features of asthma (12–15). A few studies observed persistence of experimental asthma in ST2 knockout mice (16, 17). A complete resolution of experimental asthma and lung inflammation was observed only when all three type 2 inducing epithelial cytokines—IL25, IL33 and TSLP or their receptors were blocked (17). Interestingly, most of these studies were performed in acute asthma models or in a model of chronic allergen exposure where the outcomes were measured within 1–3 days of the last allergen exposure. We developed a mouse model where experimental asthma persists for more than 6 months after the last allergen exposure (18, 19). The persistence of experimental asthma in this model was dependent on ILC2s but not T cells, although the latter contributed to the magnitude of airway inflammation. We asked if ST2 played an essential role in this chronic asthma model.
Materials and Methods
The protocol for this study was approved by the National Jewish Health IACUC. We obtained ST2 KO mice from Dr. Andrew McKenzie, (Cambridge, UK) (12). These mice were backcrossed to BALB/C mice for more than eight generations. We used BALB/C mice as wild-type controls (Jackson Laboratory).
Mucosal sensitization of mice
The allergen extract used in this experiment was a mixture of 3 allergens: dust mite (Dermatophagoides farinae), ragweed (Ambrosia artemislifolia), and Aspergillus fumigatus (Greer Laboratories). We used the following protein concentrations of the allergen extracts: D farina: 5 μg (LPS content: 3–35 EU by means of LAL assay); ragweed: 50 μg (LPS content: 5 EU); Aspergillus species: 5 μg (LPS content: 0.1 EU). The allergen dose was based on the results of a survey of previous publications (a total of 7 publications) indicating successful sensitization and elicitation of allergic inflammation in the lungs (18). This allergen mixture was delivered intranasally in 15μL aliquots in saline. Chronic experimental asthma was developed by intranasal delivery of the triple allergen mixture twice a week for six consecutive weeks in female mice 8–12 weeks of age as described previously (18, 19). A timeline of manipulations and interventions for the chronic asthma protocol with allergens is shown in Figure 1A.
Figure 1.

A: A schematic presentation of the mouse model of chronic asthma. B: Airway hyperreactivity (lung resistance) to inhaled methacholine as measured by Flexivent. WT: wild-type controls; KO: ST2 knockout; A: allergen exposed; S: saline exposed;*P<0.05, N=6 per group. C&D: Comparison of airway inflammation (H&E staining in B) and mucus (PAS staining in pink in C) production between WT and ST2 KO mice (representative of 5 mice per group). E, F & G: Morphometric quantification of peribronchial (E), perivascular (F) inflammation and goblet cell hyperplasia (G) in WT and ST2 KO mice.
Antibody, pharmacological and genetic interventions
For TSLP blockade experiments an anti-TSLP antibody (#MAB555 from R&D, Inc., 20μg/20μL in saline intranasally) was delivered for 3 consecutive days, 3 days prior to analysis in week 10. An isotype rat IgG2a (#MAB006) antibody was used as a control.
Airway hyperreactivity measurement
Measurement methodologies have been explained in depth elsewhere (18). Briefly, mice were anesthetized with ketamine (180 mg/kg), xylazine (9 mg/kg), and acepromazine (4 mg/kg). After the loss of foot-pad pinch reflex, a tracheotomy was performed and the mouse was attached via an 18 gauge cannula to a small-animal ventilator with a computer-controlled piston (Flexivent Fx; Scireq). After performing initial calibrations (cylinder pressure channel and nebulizer calibration) we conducted dynamic tube calibration and used a default program called QuickPrime 3 v7 for measurement of airway resistance in response to methacholine. This program utilizes prime perturbations, which are a family of complex forced oscillation perturbations at a frequency above and below the subject’s ventilation frequency (1–20.5 Hz). The amplitude of the oscillatory signal is pre-set to a volume that is slightly smaller than the subject’s tidal volume (0.2 ml). Volume and pressure signal are recorded during a measurement and the flow signal is derived from the volume. The foregoing allows calculation of Newtonian resistance, tissue damping, tissue elastance and hysteresivity. Resistance measurements were taken to establish the baseline for the total lung resistance and at each methacholine dose. Group averages were expressed as fold increase over the baseline resistance (mean ± SEM).
Histology and Immunofluorescence Staining
Paraffin embedded lungs were sectioned and stained with hematoxylin and eosin (H&E) for morphometric analysis, PAS for mucin staining, Mason’s trichrome for collagen deposition, and toluidine blue for mast cell staining. Sections for immunofluorescence staining were permeabilized with 0.01% saponin in PBS, blocked with 2% BSA, and stained with a primary antibody against mouse TSLP (rabbit polyclonal; abcam#ab115700) and visualized with an Alexa-594 conjugated secondary antibody as described previously (19) and counterstained with DAPI for nuclear staining.
Images were acquired on a Nikon Eclipse TE2000-U microscope using 20x dry lenses at room temperature through a Diagnostics Instruments camera model #4.2 using Spot software 5.0. H&E sections were mounted using Permount medium. Images were adjusted for brightness and contrast to improve viewing.
Lung digestions
Lungs were perfused with saline and single cell suspensions of lung cells were acquired by mechanical mincing of the lungs followed by digestion at 37°C for 45 minutes in RPMI with 10% FBS, 1% penicillin/streptomycin and collagenase Type I (1mg/mL) (Worthington # LS004197) as described previously (19). Cell suspensions were agitated at room temperature for 10 minutes in RPMI with 100U/mL DNAse I prior to filtration through 40μm filters and red blood cell lysis. Single cell suspensions were subsequently cultured in RPMI with 10% FBS, 1% penicillin/ streptomycin at 37°C in CO2 incubator overnight. Next day cells were used for flow cytometric staining and analysis.
Flow cytometric analyses of ILCs and T cells
We stained live cells first for cell surface receptors. Then we fixed the cells with 4% paraformaldehyde for 15 min at 4° C, washed them with the staining buffer (PBS+ 1% BSA) and then incubated with the permeabilization buffer (PBS pH 7.4 + 1% BSA + 0.1% saponin) at 4° C for 20 min. After centrifugation, we resuspended the cells in permeabilization buffer and incubated with antibodies against the intracellular proteins (cytokines and transcription factors) at 4° C for 30 min. We washed the cells twice with the permeabilization buffer and resuspended in the staining buffer and kept at 4° C until flow analysis. For intercellular cytokines, we added monensin (2 μM) to the cells and cultured for 4 hrs before staining. Most of the fluorophore-conjugated antibodies were purchased from Biolegend (San Diego, CA) and some from eBioscience or R&D, Inc., unless otherwise stated. Mouse lung single cell suspension was blocked with an anti-FcR blocking reagent (Miltenyi Biotec; # 130-092-575) before staining. ILC cells were stained with FITC-labelled anti-CD45.2 (clone 104), pacific blue-labeled lineage marker antibodies (CD3, Ly-6G/Ly-6C, CD11b, CD45R/B220, TER-119/Erythroid cells with added antibodies against NK1.1 and FcεRIα), PerCP-Cy5.5-conjugated anti-CD25 (eBioscience; clone PC61.5), allophycocyanin- labeled anti-IL9 and anti-IL5 (TRFK5), PE-Cy7 labeled anti-IL13 (eBioscience; clone eBio13A). ILC2 cells were initially characterized with the addition of brilliant violet 605 conjugated anti-KLRG1 (MAFA; clone: 2F1/KLRG1), brilliant violet 510 conjugated anti-ICOS (CD278; clone: C398.4), PE conjugated anti-TSLPR (R&D # FAB5461P), APC conjugated anti-IL17RB (IL-25R; R&D # FAB10402A) followed by pacific blue-labeled lineage, FITC conjugated anti-CD45.2 and PerCP-Cy5.5 conjugated anti-CD25 antibody. PE/Cy7 conjugated anti-IL13 antibody and PE conjugated anti-Foxp3 & PerCP-Cy5.5 conjugated anti-CD25 antibody followed by APC conjugated anti-CD4 and FITC conjugated anti-CD45.2 was used for T cells and Foxp3+ Treg analysis. We routinely performed 5–7 color flow cytometry. Since we studied more than 7 target molecules, we split the isolated lung cells into two or more samples (depending on the need). We used fluorophore-conjugated antibodies against CD45.2, the lineage cocktail, and CD25 to detect ILCs. Then we treated cells with 4 additional color-matched antibodies against ILC2 markers—KLRG1, TSLPR, ICOS, IL17RB, IL7Rα, GATA3, IL5, IL-9 and IL-13 as appropriate for the experimental question. For controls, we used unstained cells and cells stained with an isotype control antibody. We performed simultaneous flow cytometry for FMO (fluorescence minus one). We used the following equipment: LSRII and LSRFortessa.
Real time PCR
Total RNA was isolated from frozen tissues using Trizol (Invitrogen), and cDNA was synthesized using the Verso-cDNA synthesis kit (Thermo Scientific # AB-1453/B) according to manufacturer’s instructions as described previously (19). Gene specific PCR products were amplified using the qPCR SYBRGreen Rox mix (Thermo Scientific # AB-4162/B) and primers outlined in online repository table E1. Primers were designed using the Applied Biosystems 7000 Sequence Detection System software. The levels of target gene expression were normalized to 18S ribosomal RNA expression using the 2−ΔΔCt method.
ELISA for cytokines
TSLP (ELISA kit from eBioscience;#88-7490-88), IL9(ELISA kit from eBiscience;#88-8092-88) and IL13 (ELISA kit from R&D;#DY413-05) in the bronchoalveolar lavage fluid were measured by ELISA as per manufacturer’s instruction.
Statistical Analyses
Statistical analyses were performed, and data figures were prepared using the software GraphPad Prism, v6 (San Diego, CA).
Results
ST2 is not essential for persistence of airway hyperreactivity in chronic experimental asthma
We examined the effect of germ-line deletion/knockout (KO) of the IL33 receptor ST2 on chronic experimental asthma in a mouse model. Mice were intranasally exposed to a combination of 3 allergens (A: dust mites, ragweed and Aspergillus sp.) or saline (S) twice a week for 6 weeks, rested for 3 weeks and then subjected to measurements of airway hyperreactivity, inflammation and tissue remodeling (Figure 1A). Allergen-exposed wild-type (WT) and KO mice showed a significant increase in airway resistance in response to methacholine when compared to saline-exposed mice (Figure 1B). There was no significant difference in airway hyperreactivity between WT and KO mice. The total number of cells in bronchoalveolar lavage (BAL) was significantly elevated in allergen-treated WT mice as compared to saline treated mice (online repository figure E1A–C). Allergen-treated ST2 KO mice had higher numbers of total BAL cells as compared to allergen-treated WT mice. Allergen-treated WT mice had elevated neutrophils and eosinophils, which were absent in ST2 KO mice. Lung histology showed reduced levels of peribronchial and perivascular inflammation (Fig 1C) in ST2 KO mice. However, the level of mucus-producing airway goblet cell hyperplasia (Fig 1D) and airway remodeling/collagen deposition (online repository figure E1 D&E) were similar in allergen-treated ST2 KO and wild-type mice. The foregoing findings indicate that many features of chronic experimental asthma, especially airway hyperreactivity and mucus production, persist in the absence of ST2.
IL9+ and IL13+ ILC2s are elevated in ST2 KO mice
Next we examined the effect of ST2 deletion on ILC2s in the lung digest from the study animals. The gating strategy for detection of ILC2s and ILC2 subpopulations is shown in online repository figure E2A and E2B. The frequency of dead cells in gated live CD45+ cells was ~7% in the lung digest from both WT and KO mice (online repository figure E2A). We identified ILCs as CD45+Lin-CD25+ cells. Over 80% of these cells were positive for IL7Ra. A majority of these cells expressed surface markers of ILC2 (KLRG1, TSLPR) and a smaller fraction expressed GATA3 and type 2 cytokines. The total ILC (CD45+Lin-CD25+) number was increased in chronic experimental asthma as compared to saline control (Figure 2A), which confirmed our previous report (19). Unexpectedly, the total ILC number was even higher in ST2 KO mice as compared to WT controls in the chronic asthma model. This increase was observed not only with total ILCs but also with ILC2s carrying specific markers—KLRG1 (Figure 2B), TSLPR (Figure 2C) and ICOS but not IL17RB (IL25R) (Online repository figure E3A & B). Next, we examined the expression of type 2 cytokines by ILC2s. ST2 KO mice had higher number of IL13+ and IL9+ ILC2s as compared to WT mice (Figure 2D & E). Interestingly, they had lower numbers of IL5+ ILC2s as compared to WT mice (Figure 2F). Nonetheless, ST2 KO mice had elevated number of IL5+ ILC2s when compared with saline treated mice.
Figure 2.

Flow cytometric enumeration of ILCs. Released lung cells obtained from WT and ST2 KO mice following collagenase digestion were stained with a combination of labeled antibodies, detected by flow cytometry and analyzed by FlowJo. The absolute number of various receptor (A–C) positive and cytokine (D–F) positive ILCs present in the lung digest is shown. P values are shown on the top of the dot plots. Each dot/symbol represents a mouse.
We previously reported that this chronic asthma model was associated with heightened numbers of CD4 T cells and CD4+ nTregs (18), and that CD4 T cells contributed to the magnitude of airway inflammation (19). We examined the role of ST2 in this model. As expected, the number of Th2 cells as measured by IL13+CD4 T cells and the number of CD25+FoxP3+ nTregs was elevated in the chronic asthma model (Figure 3A & B). ST2 deletion did not have any effect on the number of these cell types in chronic experimental asthma. Interestingly, the number of IL13+ CD4 T cells and CD25+FoxP3+ nTregs were higher in saline-treated ST2 knockout as compared to saline-treated wild-type mice.
Figure 3.

A& B: Enumeration of IL13+ CD4 T cells (Th2 cells) and FoxP3+ CD25+ Tregs in the lung digest as measured by flow cytometry. C–E: Select cytokine levels in bronchoalveolar lavage fluid from WT and ST2 KO mice. Cytokines—IL13 (C), IL9 (D) and TSLP (E) were assayed by ELISA. Each symbol represents a mouse.
IL9, IL13 and TSLP are elevated in the airways from ST2 KO mice
In order to establish the relevance of elevated type 2 cytokine+ ILC2s, we measured type 2 cytokine mRNA in the lung and protein in the BAL. IL13 and IL9 were elevated in the lung (online repository figure E4 A & B) and in BAL from the chronic asthma model (Figure 3C & D). Their levels were even higher in ST2 KO mice, which was in agreement with heightened IL13+ and IL9+ ILC2s in this group. The magnitude of elevation of IL9 in BAL from ST2 KO mice was remarkable. Even the saline-treated ST2 KO mice showed a higher baseline level of IL9. Amongst the epithelial cytokines that control tissue ILC2s, TSLP was previously shown to regulate Th9 differentiation and IL9 expression. We examined the expression of TSLP in the lung and BAL. The mRNA and protein for TSLP were elevated in both WT and ST2 KO mice from the chronic asthma model (Online repository figure E4C and Figure 3E). The levels paralleled IL9 concentration in BAL, i.e. the TSLP level was higher in ST2 knockout mice as compared to WT mice. Interestingly, saline-treated ST2 KO mice had increased TSLP in BAL suggesting that ST2 negatively regulated TSLP expression. We examined the source of TSLP in the lung. TSLP was primarily produced by airway epithelial cells (online repository figure E5). In accordance with the TSLP level in BAL, TSLP expression was higher in ST2 KO mice.
We also examined the expression of mRNA for IL33, which was elevated in the chronic asthma model (online repository figure E4D). Interestingly, IL33 mRNA was even higher in ST2 KO mice suggesting the existence of a negative feedback inhibition in WT mice, whose loss caused an increase in IL33 expression. IL17A has been shown to be involved in some mouse models of experimental asthma (20). We observed increased expression of mRNA for IL17A and IL17F in the chronic asthma model. ST2 KO did not affect mRNA for IL17A but reduced mRNA for IL17F (online repository figure E4E&F).
Increased expression of intraepithelial mast cells and mucus in ST2 KO mice
IL9 is known to induce mast cell hyperplasia (21–23). Both IL9 and IL13 stimulate mucus production (21). We examined the expression of mRNA for the mast cell protease 1 (MCP1) and the mucus genes MUC5AC and MUC5B. The expression of mRNA for MCP1 and MUC5AC but not MUC5B was increased in ST2 KO mice as compared to WT controls in the chronic asthma model (Figure 4 A–C). The result of mucus staining in ST2 KO mice is in agreement with the MUC5AC data. We previously reported that chronic experimental asthma was associated with increased intraepithelial mast cells (18). To validate the result of increased MCP1 mRNA expression, we examined intraepithelial mast cells. ST2 KO mice from the chronic asthma model expressed higher number of intraepithelial mast cells as compared to WT mice (Figure 4 D&E). The results in aggregate suggest that increased TSLP production in ST2 KO mice led to a cascade of events involving induction of IL9+ and IL13+ ILC2s and intraepithelial mast cells.
Figure 4.

A–C: The expression of mRNA for the mast cell protease MCP1 (A), and the mucin genes MUC5AC (B) and MUC5B (C) in the lung tissue. The expression of mRNA was measured by real-time PCR and the data expressed in reference to the expression level of 18S ribosomal RNA. D: A representative image of toluidine positive intraepithelial mast cells in the lung from a ST2 KO mouse with chronic experimental asthma. E: Comparison of intraepithelial mast cells between allergen-treated WT and ST2 KO mice. The results were shown as the number of mast cells per mm of basement membrane (BM).
An anti-TSLP antibody induces resolution of experimental asthma in ST2 KO mice
To determine if TSLP played a critical role in persistence of experimental asthma in ST2 KO mice, we treated mice with an anti-TSLP antibody or isotype control antibody in week 10, 3 days before outcome measurements (Figure 5A). The anti-TSLP antibody significantly (p<0.001) inhibited airway hyperreactivity, tissue inflammation and mucus production as compared to the isotype control antibody (Figure 5B–D).
Figure 5.

A: A schematic presentation of the timing of anti-TSLP intervention. B: Comparison of airway hyperreactivity (lung resistance) to inhaled methacholine between anti-TSLP and isotype antibody-treated ST2 KO mice (*P<0.05, N=5 per group). C&D: Comparison of airway inflammation (H&E staining in C) and mucus (PAS staining in pink in D) production between anti-TSLP and isotype antibody treated ST2 KO mice (representative of 5 mice per group). E, F & G: Morphometric quantification of peribronchial (E), perivascular (F) inflammation and goblet cell hyperplasia (G) in ST2 KO mice treated with anti-TSLP and isotype control antibody.
The TSLP blockade abrogates the induction of IL9+ and IL13+ ILC2s and the expression of IL9 and IL13 in the airways
We examined the effect of the anti-TSLP antibody on ILC populations in the lung. The blockade of TSLP reduced the number of total ILCs (CD45+ Lin-and CD25+), KLRG1+ ILC2s, TSLPR+ILC2s, IL9+ILC2s and IL13+ILC2s (Figure 6A–E). As anticipated, this treatment also reduced the expression of TSLP, IL9 and IL13level in BAL (Figure 7 A–C).
Figure 6.

A–D: Effect of anti-TSLP antibody on ILCs and cytokine+ ILCs in the lungs. Released lung cells from the sensitized ST2 KO mice were analyzed by flow cytometry for ILCs (A–C) and IL9+ (D) and IL13+ (E) ILCs.
Figure 7.
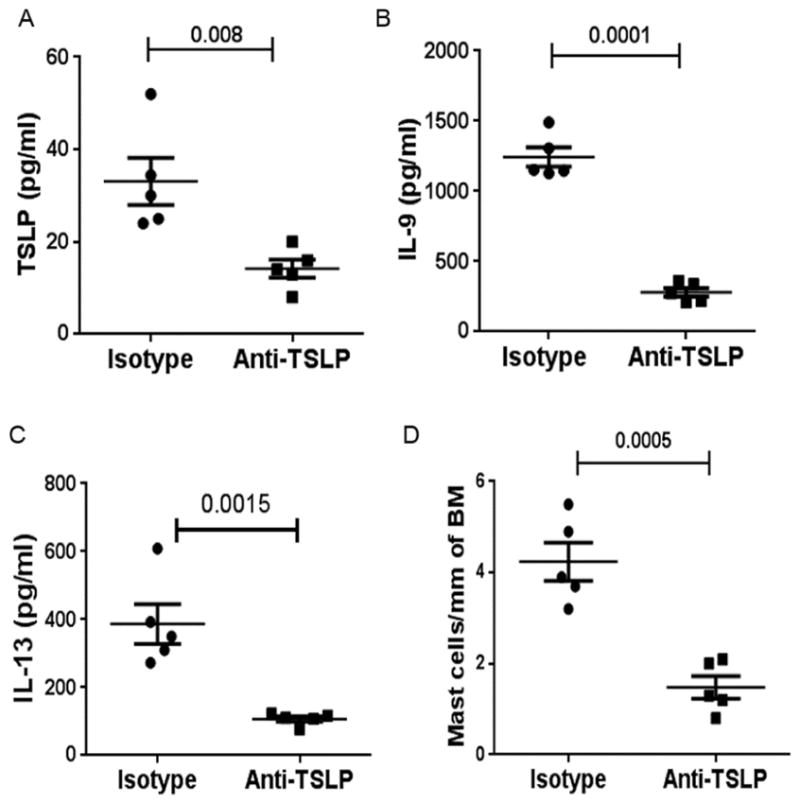
A & B: Levels of TSLP and IL9 in the BAL fluid from sensitized ST2 KO mice after anti-TSLP treatment. Each symbol represents a mouse. C: Toluidine-positive intraepithelial mast cells in ST2 KO mice after treatment with TSLP. The results were shown as the number of mast cells per mm of basement membrane (BM).
Next, we studied the effect of the anti-TSLP antibody on mast cells. The MCP1 mRNA level was significantly reduced in anti-TSLP treated mice (online repository figure E6A). There was a corresponding reduction in intraepithelial mast cell number in anti-TSLP treated mice (Figure 7D). PAS staining of the lung tissue showed an inhibition of mucus production after anti-TSLP treatment (Figure 5D). An analysis of expression of mRNA for MUC5AC confirmed this data and showed a significant inhibition of its expression after TSLP treatment (online repository figure E6B).
Discussion
The role of IL33 and its receptor ST2 has previously been studied in mouse models of experimental asthma. ST2 KO mice produced variable results depending on the mouse model used. ST2 KO showed inhibition of acute asthma in some models (12–15) but not in others (16, 17). We developed a chronic model where experimental asthma persists in the absence of allergen challenge for longer than 6 months. In a different model of experimental asthma where sensitized mice were intratracheally challenged with Aspergillus fumigatus conidia, airway pathology and hyperreactivity persisted up to day 44 (24, 25). We believe that these chronic models more closely mimic human asthma since the latter persists without regard to allergen exposure. In this paper we showed that chronic experimental asthma persisted in ST2 KO mice. Airway hyperreactivity and mucus production persisted unabated. The absence of ST2 did reduce the magnitude of airway inflammation. We showed that in the absence of ST2 signaling there was a paradoxical increase in TSLP expression in airway epithelial cells, which led to an increase in IL9+ and IL13+ ILC2s in the airways and an increase in mast cells. We believe that the foregoing contributed to the persistence of experimental asthma. Inhibition TSLP completely abrogated chronic experimental asthma suggesting that TSLP played an essential role in development of chronic experimental asthma in ST2 KO mice.
Previously an IL33-independent and IL25-regulated subtype of ILC2s was reported by Huang et al (26). These IL25-dependent ST2- ILC2s were equally effective in mounting a type 2 immune response against parasitic infection. A TSLP-induced subtype of ILC2, that functioned independent of IL25 and IL33, was initially identified in the skin (27). This TSLP-dependent ILC2 population induced atopic dermatitis-like lesions in mice. A TSLP-dependent and IL33-independent ILC2 population was also identified in the mouse lung following infection with respiratory syncytial virus (28). The foregoing studies suggest that TSLP-dependent ILC2s can independently induce a type 2 inflammatory phenotype in the mouse model.
Many cells produce IL33 and TSLP but the primary source for these cytokines in the lung is airway epithelial cells (reviewed in ref. 29). The endogenous mechanism of IL33 and TSLP production is less known. Our results suggest that IL33 signaling through ST2 triggers a negative feedback inhibition of IL33. In the absence of ST2 this negative feedback is lost. Consequently, IL33 production is increased in ST2 KO mice. We believe that TSLP and IL33 are co-regulated. Increased expression of IL33 also leads to heightened production of TSLP in ST2 KO mice. Co-regulation of IL33 and TSLP has previously been reported. Many environmental agents simultaneously induce IL33 and TSLP (30). Previous studies demonstrated a positive regulation of TSLP by IL33. IL33 induced TSLP in human nasal epithelial cells in vitro (30) and in mouse airways in vivo (31). Since we observed increased TSLP and IL33 in ST2 KO mice, we speculate that this co-regulation was the result of activation of a common signaling/transcriptional mechanism(s) not involving ST2.
We examined the mechanism by which TSLP promotes persistence of experimental asthma in ST2 KO mice. TSLP increased the number of IL9+ and IL13+ but not IL5+ ILC2s. The reduction of IL5+ ILC2s suggests that the IL33-ST2 axis was critical for IL5+ ILC2s. The TSLP blockade reduced the number of IL9+ ILC2s and the expression of IL9 protein in the lung. The result suggests that TSLP was important for IL9 induction in ST2 KO mice. TSLP was previously shown to induce Th9 and increase IL9 production (32, 33). TSLP induced STAT5, which directly bound to the IL9 promoter and facilitated its transcription (32). An anti-IL9 antibody abolished airway inflammation associated with TSLP overexpression. Based upon these studies IL9 is considered a critical effector molecule for TSLP-induced airway inflammation. IL9 was previously shown to induce experimental asthma in mice (34). The effect of IL9 was considered to be mediated by IL13. An allergen challenge induced IL9 in the airway tissue from asthmatic patients suggesting an association with human asthma (35). The effect of IL9 on goblet cell function and mucus production is complex. IL9 did not directly induce goblet cell proliferation or mucus production from human airway epithelial cells (36). The effect of IL9 on mucus production in mice was mixed. IL9 deficiency was associated with defective goblet cell hyperplasia and mucus production in parasite-induced pulmonary granuloma in mice (22). On the other hand, IL9 was redundant for mucus production as well as airway inflammation and hyperreactivity in an allergen-induced asthma model (37). These studies suggest that the effect of IL9 on allergic inflammation varies depending upon the experimental model. ILC2-derived IL9 was previously shown to exert an autocrine effect and amplified IL13 production, which sustained inflammation by inducing IL33 and TSLP production by airway epithelial cells (38). Thus, IL9 could have contributed to sustenance of inflammation in ST2 KO mice.
TSLP deficient mice had impaired airway inflammation and mucus production in an allergen-induced asthma model (39, 40). Conversely, lung-specific overexpression of TSLP caused airway hyperreactivity, type 2 inflammation, goblet cell hyperplasia and subepithelial fibrosis. The foregoing findings suggest an important role for TSLP in multiple aspects of asthma pathology including mucus production. We observed increased mucus production and heightened expression of mRNA for MUC5AC in ST2 KO mice. The anti-TSLP antibody abrogated mucus production. We believe that TSLP directly induced mucus production in ST2 KO mice.
IL9 is a known mast cell growth factor (21). It stimulated mast cell development from stem cells in vitro. IL9 overexpression caused mast cell hyperplasia in vivo (22, 23). Interestingly, IL9 overexpression caused an increase in intraepithelial mast cells in the airways (21). IL9 genetic deletion was associated with an inability to increase the number of mast cells in the airways (22). We observed increased intraepithelial mast cells in ST2 KO mice. This increase was abrogated by the anti-TSLP intervention. We believe that the anti-TSLP-induced reduction in IL9 led to a decline in mast cells and resolution of experimental asthma.
As noted above ST2 KO mice produced variable results for experimental asthma (12–17). Most of the previously described models involved an acute form of experimental asthma, which is different from our model. Our model is closer to that described by Iijima et al (13) and Vannella et al (13). Both studies applied chronic exposure to a single allergen (17) or multiple allergens (13). However, the foregoing studies analyzed experimental outcomes 24 hours after the last allergen exposure, which is different from our model as we analyzed outcomes 3 weeks after the last allergen exposure. The Vannella study outcomes are similar to that of our study. The Iijima study did report a significant reduction in airway eosinophils and hyperreactivity. It should be noted that although it declined statistically, airway hyperreactivity remained highly elevated in ST2 KO mice in the Iijima study. ST2 KO mice had a significant reduction in IL5+ ILC2s. In accordance, BAL eosinophil count was also reduced in ST2 KO mice, which is similar to the Iijima study. This is also in line with previous publications that demonstrated a pivotal role for the IL33/ST2 axis in generation of IL5+ memory Th2 cells (41) and production of IL5 in allergic inflammation (42). The lack of IL5+ but not IL13+ ILC2s in ST2 KO mice also suggests that production of IL5 and IL13 can be discordant under certain circumstances. This is not uncommon in the clinical setting as eosinophilia is not always associated with asthma and vice versa. The effect of ST2 deletion had a differential effect on BAL leukocytes and tissue inflammation scores. ST2 KO mice had reduced eosinophils and neutrophils in BAL but increased total cells and macrophages. The increase in BAL macrophages is likely due to heightened levels of TSLP as the later has been shown to stimulate extramedullary hematopoiesis and macrophage differentiation (43). This mixed effect of ST2 deletion on BAL leukocytes mirrored the effect on the airway tissue, which showed inhibition of perivascular but not peribronchial inflammation. Our results in aggregate suggest that the IL33/ST2 pathway is important for eosinophilic inflammation but redundant for airway hyperreactivity and mucus production. The combined action of IL33 and TSLP is essential for maintenance of airway hyperreactivity and goblet cell metaplasia during the chronic phase of experimental asthma. Since ST2 deletion has a mixed outcome in acute asthma, additional work is needed to more fully define how the pathogenesis of allergic lung disease differs between acute and chronic contexts.
Supplementary Material
Key Messages.
Experimental asthma persists in IL33R knockout mice due to increased TSLP, IL9+ and IL13+ ILC2s and mast cells.
An anti-TSLP intervention is likely to benefit asthmatic patients, who fail to respond to an anti-IL33R therapy.
Acknowledgments
Funding support: The work was supported by NIH grants RO1 AI091614, AI102943 and HL126895.
Abbreviations
- A
allergen
- AHR
airway hyperreactivity
- BAL
bronchoalveolar lavage
- ILC
innate lymphoid cell
- KO
knockout
- S
saline
- ST2
suppressor of tumorigenicity 2 (also serum stimulation 2)
- TSLP
thymic stromal lymphopoietin
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morita H, Moro K, Koyasu S. Innate lymphoid cells in allergic and nonallergic inflammation. J Allergy Clin Immunol. 2016;138:1253–1264. doi: 10.1016/j.jaci.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. 2016;17:765–74. doi: 10.1038/ni.3489. [DOI] [PubMed] [Google Scholar]
- 3.Chackerian AA, Oldham ER, Murphy EE, Schmitz J, Pflanz S, Kastelein RA. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J Immunol. 2007;179:2551–5. doi: 10.4049/jimmunol.179.4.2551. [DOI] [PubMed] [Google Scholar]
- 4.Endo Y, Hirahara K, Iinuma T, Shinoda K, Tumes DJ, Asou HK, et al. The interleukin-33-p38 kinase axis confers memory T helper 2 cell pathogenicity in the airway. Immunity. 2015;42:294–308. doi: 10.1016/j.immuni.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 5.Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513:564–8. doi: 10.1038/nature13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, et al. IL-33 is an unconventional Alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J Immunol. 2014;193:4010–20. doi: 10.4049/jimmunol.1400481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol. 2007;179:2051–4. doi: 10.4049/jimmunol.179.4.2051. [DOI] [PubMed] [Google Scholar]
- 8.Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20:791–800. doi: 10.1093/intimm/dxn037. [DOI] [PubMed] [Google Scholar]
- 9.Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol. 2008;121:1484–90. doi: 10.1016/j.jaci.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183:6469–77. doi: 10.4049/jimmunol.0901575. [DOI] [PubMed] [Google Scholar]
- 11.Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016;16:676–689. doi: 10.1038/nri.2016.95. [DOI] [PubMed] [Google Scholar]
- 12.Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, McKenzie AN. T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J Exp Med. 2000;191:1069–76. doi: 10.1084/jem.191.6.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iijima K, Kobayashi T, Hara K, Kephart GM, Ziegler SF, McKenzie AN, et al. IL-33 and thymic stromal lymphopoietin mediate immune pathology in response to chronic airborne allergen exposure. J Immunol. 2014;193:1549–59. doi: 10.4049/jimmunol.1302984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saglani S, Lui S, Ullmann N, Campbell GA, Sherburn RT, Mathie SA, Denney L, Bossley CJ, Oates T, Walker SA, Bush A, Lloyd CM. IL-33 promotes airway remodeling in pediatric patients with severe steroid-resistant asthma. J Allergy Clin Immunol. 2013;132:676–685. e13. doi: 10.1016/j.jaci.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zoltowska AM, Lei Y, Fuchs B, Rask C, Adner M, Nilsson GP. The interleukin-33 receptor ST2 is important for the development of peripheral airway hyperresponsiveness and inflammation in a house dust mite mouse model of asthma. Clin Exp Allergy. 2016;46:479–90. doi: 10.1111/cea.12683. [DOI] [PubMed] [Google Scholar]
- 16.Hoshino K, Kashiwamura S, Kuribayashi K, Kodama T, Tsujimura T, Nakanishi K, et al. The absence of interleukin 1 receptor-related T1/ST2 does not affect T helper cell type 2 development and its effector function. J Exp Med. 1999;190:1541–8. doi: 10.1084/jem.190.10.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vannella KM, Ramalingam TR, Borthwick LA, Barron L, Hart KM, Thompson RW, et al. Combinatorial targeting of TSLP, IL-25, and IL-33 in type 2 cytokine-driven inflammation and fibrosis. Sci Transl Med. 2016;8:337ra65. doi: 10.1126/scitranslmed.aaf1938. [DOI] [PubMed] [Google Scholar]
- 18.Goplen N, Karim MZ, Liang Q, Gorska MM, Rozario S, Guo L, et al. Combined sensitization of mice to extracts of dust mite, ragweed, and Aspergillus species breaks through tolerance and establishes chronic features of asthma. J Allergy Clin Immunol. 2009;123:925–32. e11. doi: 10.1016/j.jaci.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christianson CA, Goplen NP, Zafar I, Irvin C, Good JT, Jr, Rollins DR, et al. Persistence of asthma requires multiple feedback circuits involving type 2 innate lymphoid cells and IL-33. J Allergy Clin Immunol. 2015;136:59–68. e14. doi: 10.1016/j.jaci.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 2008;181(6):4089–97. doi: 10.4049/jimmunol.181.6.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Godfraind C, Louahed J, Faulkner H, Vink A, Warnier G, Grencis R, et al. Intraepithelial infiltration by mast cells with both connective tissue-type and mucosal-type characteristics in gut, trachea, and kidneys of IL-9 transgenic mice. J Immunol. 1998;160:3989–96. [PubMed] [Google Scholar]
- 22.Townsend JM, Fallon GP, Matthews JD, Smith P, Jolin EH, McKenzie NA. IL-9-deficient mice establish fundamental roles for IL-9 in pulmonary mastocytosis and goblet cell hyperplasia but not T cell development. Immunity. 2000;13:573–83. doi: 10.1016/s1074-7613(00)00056-x. [DOI] [PubMed] [Google Scholar]
- 23.Kearley J, Erjefalt JS, Andersson C, Benjamin E, Jones CP, Robichaud A, et al. IL-9 governs allergen-induced mast cell numbers in the lung and chronic remodeling of the airways. Am J Respir Crit Care Med. 2011;183:865–75. doi: 10.1164/rccm.200909-1462OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blease K, Jakubzick C, Westwick J, Lukacs N, Kunkel SL, Hogaboam CM. Therapeutic effect of IL-13 immunoneutralization during chronic experimental fungal asthma. J Immunol. 2001;166:5219–5224. doi: 10.4049/jimmunol.166.8.5219. [DOI] [PubMed] [Google Scholar]
- 25.Blease K, Schuh JM, Jakubzick C, Lukacs NW, Kunkel SL, Joshi BH, Puri RK, Kaplan MH, Hogaboam CM. Stat6-deficient mice develop airway hyperresponsiveness and peribronchial fibrosis during chronic fungal asthma. American Journal of Pathology. 2002;160:481–490. doi: 10.1016/S0002-9440(10)64867-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential 'inflammatory' type 2 innate lymphoid cells. Nat Immunol. 2015;16:161–9. doi: 10.1038/ni.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, et al. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 2013;5:170ra16. doi: 10.1126/scitranslmed.3005374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stier MT, Bloodworth MH, Toki S, Newcomb DC, Goleniewska K, Boyd KL, et al. Respiratory syncytial virus infection activates IL-13-producing group 2 innate lymphoid cells through thymic stromal lymphopoietin. J Allergy Clin Immunol. 2016;138:814–824. e11. doi: 10.1016/j.jaci.2016.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammad H, Lambrecht BN. Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity. 2015;43:29–40. doi: 10.1016/j.immuni.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 30.Liao B, Cao PP, Zeng M, Zhen Z, Wang H, Zhang YN, et al. Interaction of thymic stromal lymphopoietin, IL-33, and their receptors in epithelial cells in eosinophilic chronic rhinosinusitis with nasal polyps. Allergy. 2015;70:1169–80. doi: 10.1111/all.12667. [DOI] [PubMed] [Google Scholar]
- 31.Kabata H, Moro K, Fukunaga K, Suzuki Y, Miyata J, Masaki K, et al. Thymic stromal lymphopoietin induces corticosteroid resistance in natural helper cells during airway inflammation. Nat Commun. 2013;4:2675. doi: 10.1038/ncomms3675. [DOI] [PubMed] [Google Scholar]
- 32.Yao W, Zhang Y, Jabeen R, Nguyen ET, Wilkes DS, Tepper RS, et al. Interleukin-9 is required for allergic airway inflammation mediated by the cytokine TSLP. Immunity. 2013;38:360–72. doi: 10.1016/j.immuni.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Froidure A, Shen C, Gras D, Van Snick J, Chanez P, Pilette C. Myeloid dendritic cells are primed in allergic asthma for thymic stromal lymphopoietin-mediated induction of Th2 and Th9 responses. Allergy. 2014;69:1068–76. doi: 10.1111/all.12435. [DOI] [PubMed] [Google Scholar]
- 34.Temann UA, Laouar Y, Eynon EE, Homer R, Flavell RA. IL9 leads to airway inflammation by inducing IL13 expression in airway epithelial cells. Int Immunol. 2007;19:1–10. doi: 10.1093/intimm/dxl117. [DOI] [PubMed] [Google Scholar]
- 35.Erpenbeck VJ, Hohlfeld JM, Volkmann B, Hagenberg A, Geldmacher H, Braun A, et al. Segmental allergen challenge in patients with atopic asthma leads to increased IL-9 expression in bronchoalveolar lavage fluid lymphocytes. J Allergy Clin Immunol. 2003;111:1319–27. doi: 10.1067/mai.2003.1485. [DOI] [PubMed] [Google Scholar]
- 36.Parker JC, Thavagnanam S, Skibinski G, Lyons J, Bell J, Heaney LG, et al. Chronic IL9 and IL-13 exposure leads to an altered differentiation of ciliated cells in a well-differentiated paediatric bronchial epithelial cell model. PLoS One. 2013;8:e61023. doi: 10.1371/journal.pone.0061023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McMillan SJ, Bishop B, Townsend MJ, McKenzie AN, Lloyd CM. The absence of interleukin 9 does not affect the development of allergen-induced pulmonary inflammation nor airway hyperreactivity. J Exp Med. 2002;195:51–7. doi: 10.1084/jem.20011732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohapatra A, Van Dyken SJ, Schneider C, Nussbaum JC, Liang HE, Locksley RM. Group 2 innate lymphoid cells utilize the IRF4-IL-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal Immunol. 2016;9:275–86. doi: 10.1038/mi.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou B, Comeau MR, De Smedt T, Liggitt HD, Dahl ME, Lewis DB, et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. 2005;6:1047–53. doi: 10.1038/ni1247. [DOI] [PubMed] [Google Scholar]
- 40.Yadava K, Massacand J, Mosconi I, Nicod LP, Harris NL, Marsland BJ. Thymic stromal lymphopoietin plays divergent roles in murine models of atopic and nonatopic airway inflammation. Allergy. 2014;69:1333–42. doi: 10.1111/all.12469. [DOI] [PubMed] [Google Scholar]
- 41.Endo Y, Hirahara K, Iinuma T, Shinoda K, Tumes DJ, Asou HK, et al. The interleukin-33-p38 kinase axis confers memory T helper 2 cell pathogenicity in the airway. Immunity. 2015;42:294–308. doi: 10.1016/j.immuni.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 42.Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. 2008;181:4780–90. doi: 10.4049/jimmunol.181.7.4780. [DOI] [PubMed] [Google Scholar]
- 43.Siracusa MC, Saenz SA, Wojno ED, Kim BS, Osborne LC, Ziegler CG, Benitez AJ, et al. Thymic stromal lymphopoietin-mediated extramedullary hematopoiesis promotes allergic inflammation. Immunity. 2013;39:1158–70. doi: 10.1016/j.immuni.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.