

Abstract
Aim: Non-alcoholic steatohepatitis (NASH) increases cardiovascular risk regardless of risk factors in metabolic syndrome. However, the intermediary factors between NASH and vascular disease are still unknown because a suitable animal model has never been established. The stroke-prone (SP) spontaneously hypertensive rat, SHRSP5/Dmcr, simultaneously develops hypertension, acute arterial lipid deposits in mesenteric arteries, and NASH when feed with a high-fat and high-cholesterol (HFC) diet. We investigated whether SHRSP5/Dmcr affected with NASH aggravates the cardiac or vascular dysfunction.
Method: Wister Kyoto and SHRSP5/Dmcr rats were divided into 4 groups of 5 rats each, and fed with a SP or HFC diet. After 8 weeks of HFC or SP diet feeding, glucose and insulin resistance, echocardiography, blood biochemistry, histopathological staining, and endothelial function in aorta were evaluated.
Results: We demonstrate that SHRSP5/Dmcr rats fed with a HFC diet presented with cardiac and vascular dysfunction caused by cardiac fibrosis, endothelial dysfunction, and left ventricular diastolic dysfunction, in association with NASH and hypertension. These cardiac and vascular dysfunctions were aggravated and not associated with the presence of hypertension, glucose metabolism disorder, and/or obesity.
Conclusions: SHRSP5/Dmcr rats may be a suitable animal model for elucidating the organ interaction between NASH and cardiac or vascular dysfunction.
Keywords: Atherosclerosis, Non-alcoholic steatohepatitis, SHRSP5/Dmcr, Endothelial function, High-fat, High-cholesterol diets
Introduction
Arteriosclerotic lesions induce cerebrovascular disease or ischemic heart disease either through the process of endothelial dysfunction; intimal lipid accumulation caused by macrophage infiltration or smooth muscle cells; atherosclerotic plaque formation; and/or vascular occlusion or thrombus formation1). The effective prevention, treatment, and care of vascular disease are still critical issues. However, research has been hampered by the fact that the majority of experimental rodents exhibit resistance to atherosclerotic lesions2). Therefore, transgenic mice without the apolipoprotein-E or low-density lipoprotein (LDL) receptor have been developed for the research of the arteriosclerotic mechanism3). Meanwhile, the development of an alternative arteriosclerosis model similar to the human pathology, which is characterized by dietary intake without genetic modification, is aspired to in order to clarify the metabolic syndrome.
The stroke-prone (SP) spontaneously hypertensive rat, SHRSP5/Dmcr, formerly called the arteriolipidosis-prone rat, simultaneously develops hypertension, acute arterial lipid deposits in mesenteric arteries, and non-alcoholic steatohepatitis (NASH) upon exposure to high-fat and high-cholesterol (HFC) diets4, 5). NASH has been observed as a comorbidity of the metabolic syndrome phenotype, and is closely related to insulin resistance or oxidative stress4, 6). Recent data suggests that NASH is linked to increased cardiovascular risk, independent of the broad spectrum of risk factors in metabolic syndrome7–10). Furthermore, retrospective and prospective studies have provided evidence for a strong relationships between NASH and the subclinical manifestation of atherosclerosis, as represented by increased intima-media thickness, endothelial dysfunction, arterial stiffness, impaired left ventricular (LV) function, and coronary calcification10–15). As commonly recognized in previous studies, compared with patients with simple fat liver, chronic inflammation in those with NASH aggravates arteriosclerosis, thereby increasing cardiovascular risks during the process of non-alcoholic fatty liver disease (NAFLD) progression11). However, the intermediary factors between NASH and vascular disease are still unknown (except for insulin resistance and oxidative stress) because no suitable animal model has been established to date.
Aim
The SHRSP5/Dmcr rat model shows a human NASH-like phenotype with severe hepatic fibrosis, hypertension, and lipid deposition in the mesenteric arteries within a short period of 8 weeks of being fed with an HFC diet4, 16). Most research on the SHRSP5/Dmcr rat model focuses on NASH4, 16–25). There are no reports on the relationship between NASH and cardiac and vascular function. Therefore, we first investigated whether NASH aggravates cardiac or vascular dysfunction in SHRSP5/Dmcr rats.
Materials and Methods
Animals and Feeding Diets
Nine-week-old male rats of Wister Kyoto (WKY) (n = 10) and SHRSP5/Dmcr (n = 10) strains were supplied from the Disease Model Cooperative Research Association (Kyoto, Japan). SHRSP5/Dmcr rats are established as parallel lines from outbred WKY rats19, 23). In this study, the animals were maintained in a temperature-controlled (24°C ± 2°C) and humidity-controlled (55% ± 5%) facility with a 12-h light/dark cycle, and allowed to consume a water and SP diet (20.8% crude protein, 4.8% crude lipid, 3.2% crude fiber, 5.0% crude ash, 8.0% moisture, and 58.2% carbohydrate) ad libitum. The HFC diet (a mixture of a 68% SP diet, 25% palm oil, 5.0% cholesterol, and 2.0% cholic acid), in order to induce NASH, was obtained from Funabashi Farm (Chiba, Japan)4). At 10 weeks of age, the WKY and SHRSP5/Dmcr rats were divided into 4 groups of 5 rats each, and fed with a SP or HFC diet as follows: WKY + SP diet (CONT group), WKY + HFC diet (CONT + HFC group), SHRSP5/Dmcr + SP diet (SHRSP5 group), and SHRSP5/Dmcr + HFC diet (SHRSP5 + HFC group), which induces NASH. Body weight and food intake were measured weekly. Systolic blood pressure (SBP), diastolic blood pressure (DBP), mean aortic pressure (MAP=(SBP-DBP) / 3 + DBP), and heart rate (HR) were measured biweekly in conscious animals by tail-cuff plethysmography (BP-98A, Softron, Tokyo, Japan).
All animal experiments were carried out in strict accordance with the recommendations of the standards relating to the Care and Management of Laboratory Animals and Relief of Pain (2006) published by the Japanese ministry of environment. The protocol was approved by the Animal Experiment Committee of Okayama University (approval nos. OKU-2016222 and OKU-2016223).
Glucose- and Insulin-Resistance
At 16 weeks of age, rats were orally administered glucose (2 g/kg) by gavage following an overnight fast. Tail blood was sampled before and at 15, 30, 60, and 120 min after the provision of glucose to measure blood glucose levels. Insulin (0.75 U/kg) was intraperitoneally injected into animals following a 6-h fast. Blood glucose levels were determined using a glucose analyzer (Glutest Neo Super, Sanwa Kagaku Kenkyusho, Nagoya, Japan). Blood glucose responses during the oral glucose tolerance test (OGTT) and insulin tolerance test (ITT) were estimated by the area under the curve (AUC) using the trapezoidal method26, 27).
Echocardiographic Analysis
All rats were intraperitoneally anesthetized using ketamine (50 mg/kg) and xylazine (10 mg/kg), and M-mode LV echograms and Doppler mitral flow were measured at 17 weeks of age. The echocardiography was performed with a 12-MHz transducer (Xario PLT-1202S, SSA-680A, Toshiba Medical Systems, Tochigi, Japan). Anterior and posterior wall thickness and LV internal dimensions were measured at end-diastole and end-systole by using a modification of the American Society for Echocardiography leading edge method on the basis of at least 3 consecutive cardiac cycles on the M-mode tracings28–31). The basic parameters for the LV echogram, LV end-diastolic (LVDd) and end-systolic (LVDs) dimensions, as well as the thickness of the interventricular septum (IVST) and LV posterior wall (LVPWT), were measured. LV systolic function was evaluated by LV fractional shortening (%FS), LV ejection fraction (EF), isovolumic contraction time (ICT), and cardiac output (CO). LVEF and CO were calculated using the Teichholz formula. End diastolic volume (EDV), end systolic volume (ESV), and CO were calculated as follows: EDV = 7 × LVDd3 / (2.4 + LVDd), ESV = 7 × LVDs3 / (2.4 + LVDs), and CO = HR × (EDV − ESV). The HR was recorded during echocardiograhy. LV diastolic function was evaluated by isovolumic relaxation time (IRT) and deceleration time (DcT). The total ejection isovolumic index (TEI index), incorporating both systolic and diastolic time intervals for expressing systolic and diastolic ventricular function (thereby presenting the total cardiac function), was calculated as follows: TEI index = (ICT + IRT) / ejection time27).
Dissection and Blood Analysis
At 18 weeks of age, blood was collected from the right carotid artery of rats that had been deprived of food overnight. Rats were then anesthetized by an intraperitoneal injection of pentobarbital sodium (50 mg/kg). Blood was centrifuged at 1,400 g for 10 min at 4°C, and the resultant serum supernatants were maintained at −80°C until analysis. Aspartate aminotransferase (AST), alanine aminotransferase (ALT), high-density lipoprotein (HDL) cholesterol, LDL cholesterol, triglycerides, and free fatty acids (FFAs) were measured with routine laboratory methods (SRL Inc., Tokyo, Japan). All rats were killed by euthanasia, and the heart, liver, aorta, and lung were removed and weighted. Tibial length and body weight were used to correct each organ's weight. The mesenteric artery was separated from the mesenteric adipose tissue for oil red O staining2). Hearts and liver were sectioned for histopathological staining, and the aorta was cut into 2 mm rings for evaluating the endothelial function.
Histopathological and Oil red O Staining
LVs and livers were fixed in 10% formalin for 48 h, embedded in paraffin and sectioned for histology. Transverse sections (5 µm) were stained with standard hematoxylin and eosin (HE) staining, and LVs and livers were stained with Picro-sirius red (PSR) staining and Masson-trichrome staining, respectively, to evaluate fibrosis. All images were obtained by an all-in-one fluorescence microscope (BZ-X700, KEYENCE, Osaka, Japan). Areas of the myocardial fiber and fibrosis were analyzed by a digital imaging software (Image J, NIH, ver. 1.47). To evaluate LV hypertrophy, 5 myocardial fibers stained with HE were randomly selected, and the shape of the fibers was traced to measure the area of the fibers. Interstitial fibrosis was evaluated using PSR staining. The fibrosis (stained red) and myocardial fiber (stained yellow) were automatically separated using the chromatic threshold, and the total area of interstitial fibrosis, except for perivascular fibrosis, in the microscopic field was calculated. The area of interstitial fibrosis in each group was evaluated as a relative value to the CONT group. Similarly, the area of perivascular fibrosis was calculated, and interstitial fibrosis and myocardial fibers were excluded from the analysis. The perivascular fibrosis area was corrected by the coronary perimeter area. Areas of interstitial and perivascular fibrosis were randomly selected across 5 microscopic fields (× 100), and average interstitial and perivascular fibrosis areas were calculated.
To evaluate macrophage infiltration into the myocardium, we performed immunostaining for the monocyte-macrophage maker CD68 with the paraffin-embedded section (3 µm). Endogenous peroxidase activity was blocked by exposure of the sections to methanol containing 0.3% H2O2. Sections were first incubated at 4°C overnight with mouse monoclonal antibodies to CD68 (clone ED1, BIO-RAD, Hercules, CA) and then for 30 min with Histofine Simple Stain Rat MAX PO (Nichirei Biosciences, Tokyo Japan)27). The number of immunoreactive myocardial interstitial macrophages per unit area (mm2) was calculated as previously described27).
Oil red O staining for the mesenteric artery was performed as described in previous reports2, 32). The mesenteric artery was carefully isolated from the intestine, and then fixed with 10% formalin for 10 min. Following washing with distilled water, the tissue was immersed in 50% isopropanol for 5 min, with oil red O stain solution (O0625, Sigma-aldrich, Tokyo, Japan) for 10 min, and with 50% isopropanol for 8 min.
Endothelial Function
The endothelial function of the aorta of each rat was measured at the end of the experimental period. The assay was performed following the method of Hosoo et al.33). The isolated aorta was cut into rings and placed in cold Krebs-Henseleit buffer (119 mM NaCl, 4.7 mM KCl, 1.1 mM KH2PO4, 1.2 mM MgSO4, and 25 mM NaHCO3, pH 7.4). The rings were mounted at a resting tension of 1.5 g in a 5 ml organ bath (UFER UC-05A Micro-easy Magnus system, Kishimoto Medical Instruments, Kyoto, Japan) containing warmed (30°C) and oxygenated (O2: CO2, 19: 1) Krebs-Henseleit buffer. After a 60 min equilibration at this tension, vasoconstrictor prostaglandin F2alpha (PGF2α, 0.1 µM) was added to the rings. When a constriction plateau was reached, acetylcholine (ACh, 10−1010−4 M) was then applied to the constricted rings to obtain the concentration-response curves. The vasorelaxant effect was expressed as the percentage relaxation of PGF2α-induced constriction. At the end of the evaluation of endothelial function using ACh, the rings were completely dilated by the addition of excess vasorelaxant (100 µM papaverine) to confirm that the mesothelium functioned properly.
Quantitative RT-PCR Analysis
Total RNA was extracted from LV tissues (10 mg), and treated with an RNeasy Mini Kit (Qiagen, Hilden, Germany). Extracted total RNA (1000 ng) was subjected to reverse transcription using the PrimerScript RT Reagent Kit (Takara, Shiga, Japan). Quantitative PCR analysis was performed using SYBR Mix Ex Taq II (Talara, Shiga, Japan) and a StepOne-Plus Realtime PCR system (Applied Biosystems, Massachusetts, USA)34–36). The specific primers for cDNAs are as follows: interleukin-6 (IL-6; forward: 5′–GTCAACTCCATCTGCCCTTCAG–3′ and reverse: 5′–GGCAGTGGCTGTCAACAACAT–3′, GenBank Accession No. NM_012589), tumor necrosis factor α (TNF-α; forward: 5′–ACTGAACTTCGGGGTGATTG–3′ and reverse: 5′–CTTGGTGGTTTGCTACGAC–3′, GenBank Accession No. NM_012675.3), atrial natriuretic peptide (ANP; forward: 5′–AGCGGACTAGGCTGCAACAG–3′ and reverse: 5′–CTGCAGCTCCAGGAGGGTAT–3′, GenBank Accession No. NM_012612.2), RAS-related C3 botulinum toxin substrate 1 (Rac-1; forward: 5′–ATCCGAGCCGTTCTCTGTCC–3′ and reverse: 5′–GCGAAGACTCCGCCATTTTC–3′, GenBank Accession No. NM_134366.1), toll-like receptor 4 (TLR4: forward: 5′–CAGGGCACAAGGAAGTAGCA–3′ and reverse: 5′–GTTCTCACTGGGCCTTAGCC–3′, GenBank Accession No. NM_019178.1) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; forward: 5′-TCAAGAAGGTGGTGAAGCAG–3′ and reverse: 5′–AGGTGGAAGAATGGGAGTTG–3′, GenBank Accession No. NM_017008.4). GAPDH mRNA was used as an internal standard.
Statistical Analysis
Relevant data is presented as means ± standard error (SE). Differences among the 4 groups at 18 weeks of age were assessed by one-way factorial ANOVA and, if a significant difference was detected, intergroup comparisons were performed with Fisher's multiple-comparison test. P-values of < 0.05 were considered indicative of statistical significance.
Results
Physiological Data
Body weight (Fig. 1A) gradually increased in the 4 groups until 16 weeks of age. SHRSP5/Dmcr rats are generally smaller than WKY rats, according to previous studies16, 23). The present data set showed similar results as those of the previous studies at the 9–16 weeks of age. At 16 weeks of age, the HFC diet reduced the body weight at a similar level in CONT and SHRSP5 groups. Weight loss is considered to be related to food intake. There were no significant differences between the 4 groups in food intake (Fig. 1B) at 9 weeks of age, before feeding of the HFC diets. However, food intake for the HFC diet was decreased during 1016 weeks of age. SBP (Fig. 1C) was decreased in the SHRSP5 + HFC group (143.6 ± 1.7 mmHg) relative to the SHRSP5 group (173.8 ± 7.4 mmHg) at 17 weeks of age. The HFC diet had no influence on SBP in the CONT groups. The time course of MAP, which is calculated from SBP and DBP, showed the same tendency as that of SBP (Supplemental Fig. 1). HR (Fig. 1D) showed no significant differences among the 4 groups.
Fig. 1.
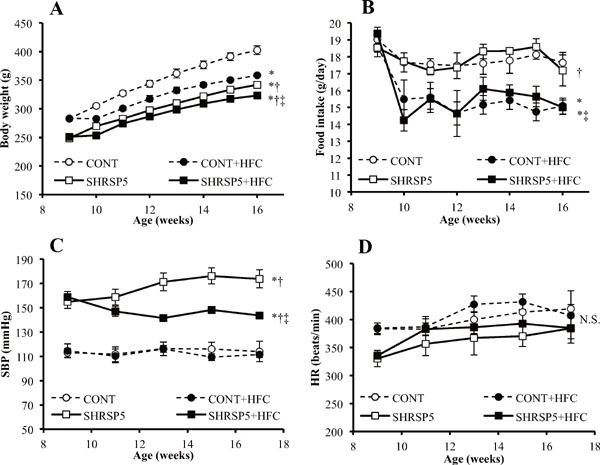
Changes in body weight (A), food intake (B), systolic blood pressure (SBP; C), and heart rate (HR; D) at 916 weeks of age
The following 4 groups were used: CONT group; WKY rats were fed a stroke-prone (SP) diet, CONT + HFC group; WKY rats were fed a high-fat and high-cholesterol (HFC) diet, SHRSP5 group; SHRSP5/Dmcr rats were fed an SP diet, and SHRSP5 + HFC group; SHRSP5/Dmcr rats were fed an HFC diet, which induce the non-alcoholic steatohepatitis (NASH). Data are shown as means ± standard error (SE); n = 5 in each group. *P < 0.05 vs. the CONT group, †P < 0.05 vs. the CONT + HFC group, ‡P < 0.05 vs. the SHRSP5 group.
Supplemental Fig. 1.
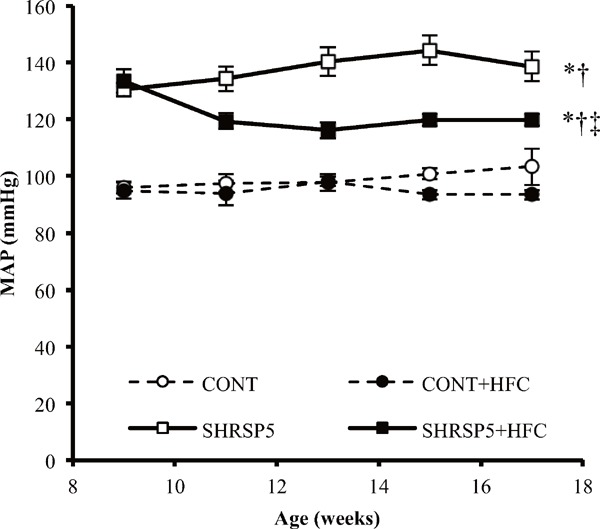
Change in mean aortic pressure (MAP) from 9 to 17 weeks of age
All data are shown as mean ± standard error (SE); n = 5 in each group. *P < 0.05 vs. the CONT group, †P < 0.05 vs. the CONT + HFC group, ‡P < 0.05 vs. the SHRSP5 group.
Pathological Condition in the Liver
Macroscopic findings of the liver (Fig. 2A) were obviously different among the 4 groups. The color tone of the organs obtained from rats from HFC diet groups was more whitish than that from SP diet groups. Moreover, the weight of livers obtained from rats from HFC diet groups was significantly larger than that from SP diet groups (Fig. 2E). The CONT + HFC group showed only hepatic steatosis without hepatocyte ballooning and fibrosis (Fig. 2B). In contrast, the SHRSP5 + HFC group showed hepatocyte ballooning and severe fibrosis in addition to hepatic steatosis at 18 weeks of age (Fig. 2B), and their hepatic function, AST, and ALT were the worst among the 4 groups (Fig. 2C and D). The HFC diet did not aggravate hepatic function in the CONT group. SHRSP5/Dmcr rats fed the SP diet presented as normal for fibrosis and hepatic function.
Fig. 2.
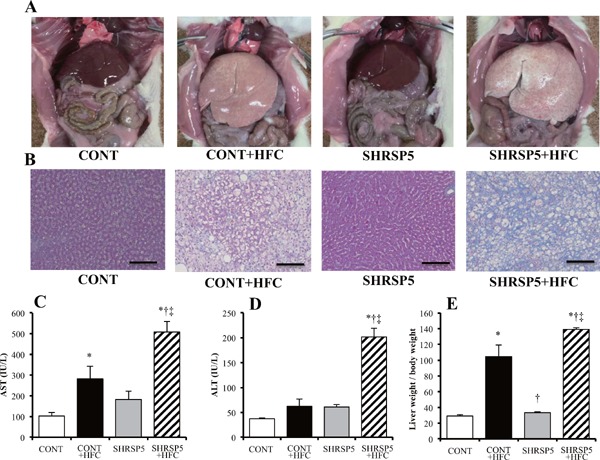
Pathological liver conditions in the 4 groups at 18 weeks of age
A: Macroscopic findings of the liver in the abdominal cavity. B: Masson trichrome staining for hepatic fibrosis. Scale bars = 100 µm. C and D: Hepatic function in blood biochemistry assays. The serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were measured. E: Liver weight corrected by body weight. All data are shown as means ± standard error (SE); n = 5 in each group. *P < 0.05 vs. the CONT group, †P < 0.05 vs. the CONT + HFC group, ‡P < 0.05 vs. the SHRSP5 group.
LV Hypertrophy, Fibrosis, and Inflammation
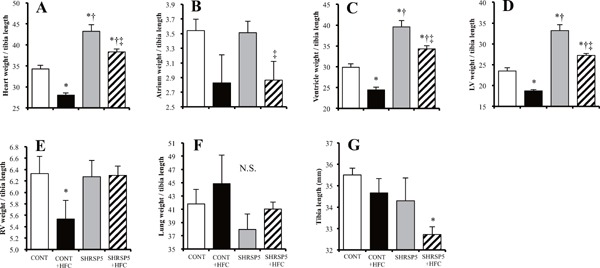
Heart, ventricle, LV, and right ventricle (RV) weights corrected by BW were increased in both of the SHRSP5/Dmcr rat groups fed the SP and HFC diets, as compared with those of the WKY rats (Fig. 3A, CE). Myocyte cross-sectional areas and ANP mRNA levels in the SHRSP5 + HFC group showed the same tendency as LV weight (Fig. 3D H, and I). Therefore, SHRSP5 and SHRSP5 + HFC groups showed LV hypertrophy. The HFC diet aggravated lung weight and RV hypertrophy (Fig. 3E, F). No significant differences were observed among the CONT groups. The weight of each individual heart section (heart, atrium, ventricle, RV, and LV; corrected by tibial length) showed LV hypertrophy in the SHRSP5 + HFC group (Supplemental Fig. 2D). This result shows the same tendency as BW correction (Figs. 3AF). However, a part of the results, especially for the WKY + HFC group, was different. The LV weight/tibial length in the WKY + HFC group was significantly lower/shorter than that in the WKY group (Supplemental Fig. 2D), although the SBP values in the WKY + HFC and WKY groups were almost the same (Fig. 1C).
Fig. 3.
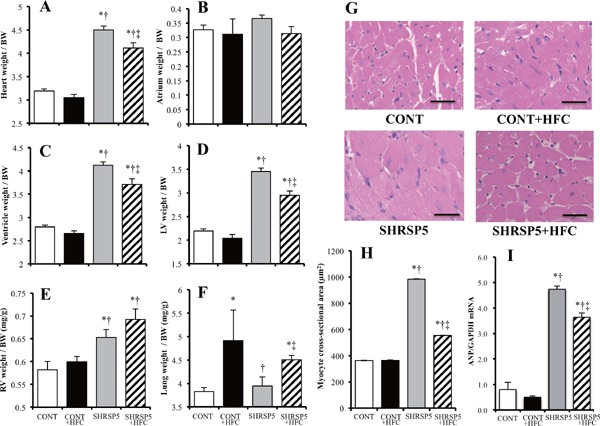
Evaluation of left-ventricle (LV) hypertrophy in the 4 groups at 18 weeks of age
AF: Organ weights corrected by body weight (BW) for heart (A), atrium (B), ventricle (C), LV (D), right-ventricle (RV) (E), and lung (F). G: Hematoxylin and eosin (HE) staining of cross sections of the LV myocardium. Scale bars = 100 µm. H: Cross-sectional areas of cardiac myocytes calculated by those in (G). I: Quantitative RT-PCR analysis of atrial natriuretic peptide (ANP). The amount of mRNA was normalized to that of GAPDH mRNA and then expressed relative to the mean value of the CONT group. All data are shown as means ± standard error (SE); n = 5 in each group. *P < 0.05 vs. the CONT group, †P < 0.05 vs. the CONT + HFC group, ‡P < 0.05 vs. the SHRSP5 group.
Supplemental Fig. 2.
Evaluation of left ventricular (LV) hypertrophy in the 4 groups at 18 weeks of age
AF: Organ weights (heart (A), atrium (B), ventricle (C), LV (D), right ventricle (RV) (E), and lung (F)) corrected by tibial length (G). All data are shown as mean ± standard error (SE); n = 5 in each group. *P < 0.05 vs. the CONT group, †P < 0.05 vs. the CONT + HFC group, ‡P < 0.05 vs. the SHRSP5 group.
Perivascular fibrosis and interstitial fibrosis found in the SHRSP5 + HFC group were the worst among the 4 groups (Fig. 4AD), whereas LV hypertrophy was ameliorated in this group (Fig. 3D, G, H). SHRSP5/Dmcr rats fed the SP diet, which have a phenotype of hypertension, showed less fibrosis compared than the CONT group. LV myocardium inflammation caused by macrophage accumulation was confirmed in the SHRSP5 + HFC group (Figs. 5A, B); in addition, mRNA levels of inflammatory cytokines such as TNF-α and IL-6 were upregulated (Figs. 5C, D). These results indicate that cardiac fibrosis was not caused by only hypertension but also by inflammation. Inflammation is derived from NASH.
Fig. 4.
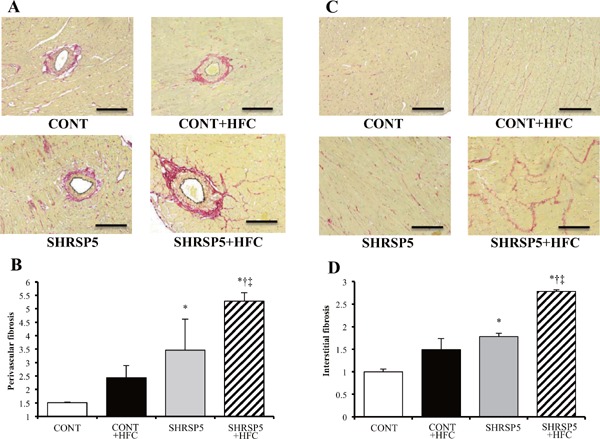
Evaluation of cardiac fibrosis in the 4 groups at 18 weeks of age
AD: Collagen deposition as revealed by picro-sirius red (PSR) staining in perivascular (A) and interstitial (C) regions of the LV myocardium. The perivascular fibrosis area was corrected by the area of the perimeter (B), and interstitial fibrosis was expressed relative to the CONT group (D). Scale bars = 100 µm. All data are shown as means ± SE; n = 5 in each group. *P < 0.05 vs. the CONT group, †P < 0.05 vs. the CONT + HFC group, ‡P < 0.05 vs. the SHRSP5 group.
Fig. 5.
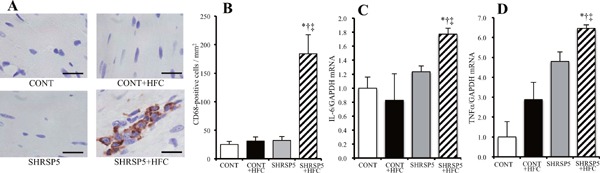
Macrophage infiltration and inflammation-related gene expression in the LV of rats from the 4 groups at 18 weeks of age
A: Immunohistochemical staining with antibodies of the monocyte-macrophage marker CD68. Scale bars = 20 µm. B: Density of CD68-positive cells per unit area (mm2) determined from sections similar to those shown in A. C, D: Quantitative RT-PCR analysis of interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) in the LV. The amount of mRNA was normalized to that of GAPDH mRNA and then expressed relative to the mean value for the CONT group. All data are shown as mean ± standard error (SE); n = 5 in each group. *P < 0.05 vs. the CONT group, †P < 0.05 vs. the CONT + HFC group, ‡P < 0.05 vs. the SHRSP5 group.
LV Systolic and Diastolic Function
LV systolic function evaluated by %FS and LVEF did not differ significantly among the 4 groups (Table 1). CO in the SHRSP5 + HFC group was lower than that in the SHRSP5 group. In contrast, LV diastolic function evaluated by IRT and DcT showed significantly differed between WKY rats and SHRSP5/Dmcr rats. Particularly, the SHRSP5 + HFC group affected with NASH showed aggravated LV diastolic function, as compared with the SHRSP5 group. The TEI index, which reflects total LV function, showed the same tendency of LV diastolic function.
Table 1. Ultrasonographic parameters in LV systolic and diastolic function in 4 experimental groups at 18 weeks of age.
| Parameters | CONT | CONT + HFC | SHRSP5 | SHRSP5 + HFC | |
|---|---|---|---|---|---|
| Systolic function | LV fractional shortening, % | 36.7 ± 3.1 | 37.7 ± 2.5 | 44.5 ± 6.1 | 36.3 ± 3.8 |
| LV ejection fraction, % | 64.4 ± 4.0 | 65.8 ± 3.1 | 72.6 ± 6.6 | 63.8 ± 5.0 | |
| Isovolumic contraction time, ms | 21.6 ± 3.9 | 33.8 ± 2.3 | 31.6 ± 4.7 | 40.2 ± 1.6*‡ | |
| Cardiac output, mL/min | 49.1 ± 5.0 | 47.7 ± 4.3 | 41.3 ± 3.8 | 28.7 ± 1.7*†‡ | |
| Diastolic function | Isovolumic relaxation time, ms | 30.8 ± 2.1 | 31.4 ± 1.3 | 57.6 ± 5.4*† | 77.3 ± 6.3*†‡ |
| Deceleration time, ms | 42.4 ± 3.4 | 35.8 ± 2.0 | 53.0 ± 0.9*† | 67.8 ± 2.9*†‡ | |
| Total LV function | Total ejection isovolume index | 0.61 ± 0.08 | 0.71 ± 0.12 | 1.36 ± 0.17*† | 2.01 ± 0.22*†‡ |
All data are means ± SE; n = 5 in each group.
P < 0.05 vs. the CONT group,
P < 0.05 vs. the CONT + HFC group,
P < 0.05 vs. the SHRSP5 group.
Mesenteric Lipid Deposition and Aortic Endothelial Function
Many lipid depositions were observed in SHRSP5/Dmcr rats fed the HFC diet (Fig. 6). The CONT + HFC and SHRSP5 groups showed less lipid depositions than the SHRSP5 + HFC group. The CONT group showed no lipid deposition at all. The isolated aorta and coronary artery were not stained with oil red O.
Fig. 6.
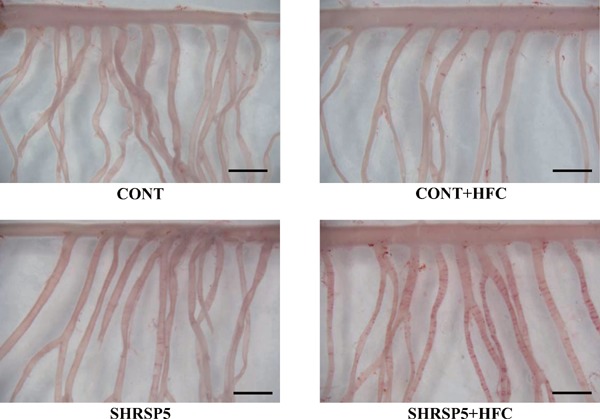
Lipid deposition in the branches of the mesenteric artery in the 4 groups at 18 weeks of age
Isolated mesenteric arteries were stained with oil red O to visualize ring-shaped lipid deposition. Scale bar = 2 mm.
Endothelial function in SHRSP5/Dmcr rats was significantly impaired by consumption of the HFC diet; the percentage of maximum contraction was only 40%, even at the 10−4 M ACh concentration (Fig. 7). Endothelial function in the CONT group was normal and was not affected by the HFC diet. In SHRSP5/Dmcr rats fed the SP diet endothelial function was damaged compared with the CONT rats.
Fig. 7.
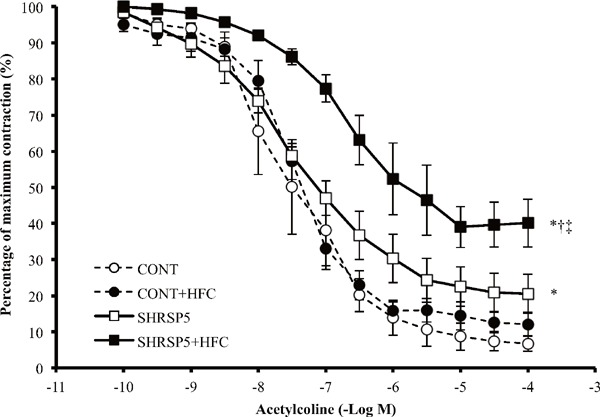
Endothelium-dependent relaxation curve induced by acetylcholine (ACh) of aortic rings
Statistical analysis for the 4 groups was performed at the ACh 10−4M. All data are shown as means ± standard error (SE); n = 5 in each group. *P < 0.05 vs. the CONT group, †P < 0.05 vs. the CONT + HFC group, ‡P < 0.05 vs. the SHRSP5 group.
Lipid- and Glucose-Metabolism
HDL-cholesterol in the SHRSP5 group was lower than that in the CONT group (Table 2). HFC diets increased HDL-cholesterol in both CONT and SHRSP5 groups. Furthermore, LDL-cholesterol showed the same tendency as HDL-cholesterol. Thus, the HFC diets increased both HDL- and LDL-cholesterol; however, LDL-cholesterol was increased more strongly than HDL-cholesterol by consumption of the HFC diets (see LDL/HDL cholesterol ratio in Table 2). Triglycerides and FFAs showed no significant differences among the 4 groups. Glucose-metabolism evaluated by OGTT, ITT, and fasting serum glucose did not show significant differences between groups.
Table 2. Lipid- and glucose-metabolism of blood analysis in 4 experimental groups at 18 weeks of age.
| Parameters | CONT | CONT + HFC | SHRSP5 | SHRSP5 + HFC | |
|---|---|---|---|---|---|
| Lipid-metabolism | HDL-cholesterol, mg/dL | 25.0 ± 0.8 | 37.0 ± 2.3* | 16.8 ± 0.8*† | 28.8 ± 0.9†‡ |
| LDL-cholesterol, mg/dL | 14.0 ± 0.9 | 64.7 ± 23.5* | 5.4 ± 0.5*† | 104.6 ± 13.8*†‡ | |
| Triglyceride, mg/dL | 29.2 ± 1.1 | 52.3 ± 18.4 | 42.0 ± 5.8 | 32.8 ± 4.1 | |
| Free fatty acids, µEq/L | 567.8 ± 25.2 | 421.7 ± 55.8 | 689.4 ± 120.5 | 518.8 ± 73.3 | |
| LDL- / HDL- cholesterol ratio | 0.56 ± 0.03 | 1.81 ± 0.75* | 0.32 ± 0.02† | 3.70 ± 0.60*†‡ | |
| Glucose-metabolism | Area under curve in OGTT, mg/dL*min*1000 | 17.3 ± 0.7 | 17.1 ± 0.7 | 18.0 ± 1.1 | 17.3 ± 1.6 |
| Area under curve in ITT, mg/dL*min*1000 | 5.2 ± 0.7 | 5.6 ± 0.2 | 4.9 ± 0.3 | 5.0 ± 0.4 | |
| Fasting serum glucose, mg/dL | 162.4 ± 6.0 | 147.3 ± 1.5 | 166.0 ± 12.5 | 125.0 ± 5.1 | |
All data are means ± SE; n = 5 in each group.
P < 0.05 vs. the CONT group,
P < 0.05 vs. the CONT + HFC group,
P < 0.05 vs. the SHRSP5 group.
HDL; high-density lipoprotein, LDL; low-density lipoprotein, OGTT; oral glucose tolerance test, and ITT; insulin tolerance test.
Discussion
The SHRSP5/Dmcr rat model, which has congenital hypertension, induces the NASH, lipid deposition of mesenteric artery and hyperlipidemia via feeding of the HFC diet4, 16–21, 25). In the present study, we reveal that a SHRSP5/Dmcr rat model fed a HFC diet presented with cardiac and vascular dysfunctions such as: (1) cardiac fibrosis, (2) endothelial function, and (3) LV diastolic function. It is interesting to note that these cardiac and vascular dysfunctions were unrelated to hypertension level and/or glucose-metabolism (see Fig. 1C and Table 2). These results demonstrate that the NASH disease interposes to cardiac and vascular dysfunction. To our knowledge, our study is the first to show that SHRSP5/Dmcr rats may be a suitable animal model for elucidating the organ interaction between NASH and cardiac or vascular dysfunction.
Characteristics of SHRSP5/Dmcr Rat Model
SHRSP5/Dmcr is a newly established substrain of SHRSP. HFC diet-fed SHRSP5/Dmcr rats were first reported in 2012 as a novel rat model for developing hepatic lesions similar to human NASH16). Hepatic steatosis and lobular inflammation are present with gradually increasing severity from 2 weeks after the introduction of the HFC diet. Partial hepatic fibrosis is also detected at 6 weeks, and is spread over the entire region of the liver with more severe bridging formation by 16 weeks16). In the present study, SHRSP5/Dmcr rats were fed an HFC diet for 8 weeks. Severe hepatic fibrosis was observed only in SHRSP5 + HFC group, in addition to hepatic hypertrophy, hypohepatia, and high LDL cholesterol (Fig. 2AE and Table 2). These results are consistent with previous studies, in which SHRSP5/Dmcr rats were induced with NASH by being fed an HFC diet4, 17, 21). Importantly, WKY rats fed an HFC diet also showed hepatic hypertrophy, weak hypohepatia, and high LDL cholesterol. Horai et al. reported that the scores of steatosis and lobular inflammation in WKY rats are as same as those in SHRSP5/Dmcr; however, the fibrosis stage score was very low at 8 weeks of HFC diet feedings16). Therefore, WKY rats fed an HFC diet might develop simple fatty liver, but not NASH.
NAFLD/NASH patients mostly exhibit weight gain, insulin resistance, and elevated triglyceride or total cholesterol levels in the blood. In the present study, the body weight decreased (Fig. 1A). In addition, glucose-metabolism indicators such as OGTT, ITT, and fasting serum glucose and partial lipidmetabolism factors, such as triglycerides and FFAs, were normal (Table 2). This glycolipid amelioration is attributed to the cholic acid contained in the HFC diet. Cholic acid induces liver toxicity, increases the ratio of chenodeoxycholic acid, which is even more toxic, and decreases the expression levels of bile acid transporters, which play an important role in the pathogenesis of NASH18). On the other hand, the presence of cholic acid increases energy expenditure by activating brown adipose tissue, thereby ameliorating glucose tolerance, insulin resistance, triglycerides, and obesity37, 38). Insulin resistance is well known to be involved in the many pathways for arteriosclerosis, hypertension, hyperlipidemia, and diabetes27).
Previous studies have reported that the SBP in SHRSP5/Dmcr rats fed an HFC diet was lower than that in SHRSP5/Dmcr rats fed a SP diet4, 16). However, the mechanism underlying the decrease in blood pressure was not discussed in these previous studies. Blood pressure, especially MAP, is determined based on CO and peripheral vascular resistance. Although the time course of MAP showed the same trend as that of SBP, the CO in the SHRSP5 + HFC group was lower than that in the SHRSP5 group (Table 1). This result indicates that the decrease in SBP in the SHRSP5 + HFC group may be caused by CO. Endothelial dysfunction in the SHRSP5 + HFC group (Fig. 7) may be related to peripheral vascular resistance. However, this issue was not investigated in the present study. Hepatic function may be another possible cause of the decrease in blood pressure. It is well known that chronic or severe liver diseases induce a low blood pressure39). We conclude that SHRSP5/Dmcr rats fed an HFC diet are a suitable model for NASH research. However, we should appreciate the distinct characteristics of this animal model.
NASH and Cardiac or Vascular Dysfunction
The second notable characteristic of the SHRSP5/Dmcr model in this study is the lipid deposition on the mesenteric artery, which is incidental to NASH and hypertension. NAFLD/NASH patients have been associated with a higher prevalence of early asymptomatic cardiovascular alterations, such as increased carotid intima-media thickness and atherosclerosis10, 12, 13), coronary calcification10, 14), altered LV geometry and diastolic dysfunction15), and endothelial dysfunction11). These observations indicate that NAFLD/NASH has some influence on cardiac and/or vascular dysfunction.
Endothelial dysfunction is one of the earliest markers of atherosclerosis. Recent cohort studies have shown an association between NAFLD/NASH and endothelial dysfunction, assessed by brachial artery flow mediated dilatation40–42). In fact, endothelial function in SHRSP5/Dmcr rats fed the HFC diet was aggravated, as compared with SHRSP5/Dmcr rats fed the SP diet, who were not affected with NASH (Fig. 7). In the hypertensive state, mechanical stress induced by hemodynamic forces like shear stress and stretch force is one of the most important factors that contributes to endothelial dysfunction/injury43). Interestingly, endothelial dysfunction was observed even though blood pressure was decreased (Fig. 1C), glucose-metabolism was normal (Table 2), and there were no indicators of obesity (Fig. 1A) in the SHRSP5 + HFC group. In addition, WKY rats fed the HFC diet, who presented with simple fatty liver, demonstrated normal levels with regards to endothelial function (Fig. 7). Simple fatty liver disease develops into NASH via oxidative stress11). The activation of oxidative stress may also aggravate endothelial dysfunction. Nakano et al. reported that a lectin-like oxidized LDL receptor-1 in a SHRSP rat model induced the activation of nicotinamide adenine dinucleotide phosphate oxidase, which led to oxidative stress in the vessel wall43).
NAFLD/NASH is associated with LV diastolic dysfunction, alterations in energy metabolism, and the disturbance of cardiac rhythm15, 44–49). Goland et al. found that patients with NAFLD, in the absence of morbid obesity, hypertension and/or diabetes, had early features of LV diastolic dysfunction and mildly altered LV geometry44). Fallo et al., in a cohort study of never-treated essential hypertensive patients with NAFLD, reported that NAFLD was significantly associated with insulin resistance and abnormalities of LV diastolic function, suggesting a concomitant increase of metabolic and cardiac risk46). These case-control studies concluded that significant changes in cardiac structure and function are evident in NAFLD/NASH without accompanying overt metabolic changes or cardiac disease11). As the common distinctive feature between humans and the SHRSP5/Dmcr rat model in the present study, NASH significantly aggravated LV diastolic function (Table 1). Diastolic heart failure is caused by impaired relaxation (e.g., aging, ischemia, or cardiomyopathy), reduced compliance (e.g., hypertrophy, myocardial fibrosis or restrictive cardiomyopathy) and/or pericardial constriction27). We demonstrated that SHRSP5/Dmcr rats fed a HFC diet presented with LV and myocardial hypertrophy (Fig. 3D, G and H) and severe myocardial fibrosis (Fig. 4AD). Activation of oxidative stress may be involved in myocardial fibrosis and endothelial dysfunction in the SHRSP5/Dmcr rat model.
Hyperlipidemia is one of the interesting signatures of SHRSP5/Dmcr rats fed HFC diets. Myocardial fibrosis is caused by various factors, such as hypertension, oxidative stress, and inflammation. SHRSP5/Dmcr rats fed HFC diets exhibit considerable hypertension (SBP over 140 mmHg) compared with WKY rats. A previous study has shown that NASH aggravates oxidative stress and inflammation50). In fact, cardiac oxidative stress (Rac-1 mRNA level; Supplemental Fig. 3A) and inflammation (IL-6, TNF-α mRNA levels) were upregulated in the SHRSP5 + HFC group (Fig. 5). Weixin et al. demonstrated that hyperlipidemia aggravated cardiac inflammation, fibrosis, hypertrophy, and apoptosis via TLR4 and epidermal growth factor receptor on the myocardium51). Among the 4 groups in our study, the SHRSP5 + HFC group showed the highest TLR4 mRNA levels (Supplemental Fig. 3B). Hyperlipidemia may promote cardiac fibrosis in this model. However, further studies are needed to consolidate this finding. Xiaofang et al. demonstrated that serum IL-6 and TNF-α levels in the SHRSP5 + HFC group were much higher than those in the SHRSP5 group17). Those results support the assumption that systemic inflammation due to liver damage affects cardiac dysfunction. The HFC diet increased LDL cholesterol levels in both WKY and SHRSP5 rats, because it contains 25% palm oil and 5% cholesterol. In a recent NASH study, large VLDL1, which contains rich TG, and small dense LDL are remarkable keywords. Fujita et al. demonstrated that mRNA levels of microsomal triacylglyceride-transfer protein in an NASH liver were lower than that in an NAFLD liver, and that TG-rich VLDL1 and small dense LDL levels increased only in NASH patients52). Kitamori et al. reported that HFC diets increased hepatic TG and cholesterol in the SHRSP5/Dmcr rat4). Therefore, TG-rich livers in SHRSP5 + HFC rats may produce copious amounts of VLDL1 and small dense LDL. Small dense LDL readily converts to oxidative LDL, and promotes systemic inflammation.
Supplemental Fig. 3.
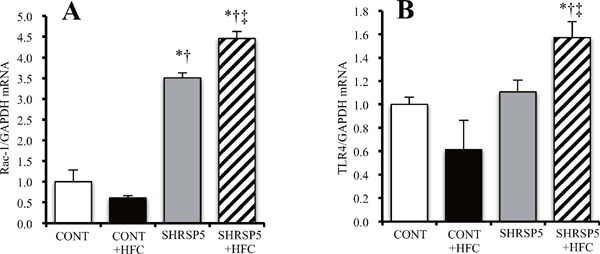
A quantitative reverse transcription–polymerase chain reaction analysis of RAS-related C3 botulinum toxin substrate 1 (Rac-1) (A) and Toll-like receptor 4 (TLR4) (B) in the left ventricle of the 4 groups at 18 weeks of age
The amount of mRNA was normalized to that of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA and then expressed relative to the mean value of the CONT group. All data are shown as mean ± standard error (SE); n = 5 in each group. *P < 0.05 vs. the CONT group, †P < 0.05 vs. the CONT + HFC group, ‡P < 0.05 vs. the SHRSP5 group.
Although outcomes in the SHRSP5/Dmcr rats were as those expected in a animal model that is a carrier of ischemic heart disease, indicators of LV systolic function, such as LV %FS and LV EF, were normal (Table 1), and lipid deposition in the coronary arteries was not be observed. Ikeda et al. administered the nitric oxide synthesis inhibitor NG-nitro-L-arginine to a SHRSP/Izm rat model, which did not present with NASH, and established a myocardial infarction-induced hypercholesterolaemic model53). Thus, administeration of with NG-nitro-L-arginine to SHRSP5/Dmcr rats may lead to an induction of ischemic heart disease in association with NASH and this model can be used for future studies.
Conclusions
The SHRSP5/Dmcr rat model, which has congenital hypertension, induces NASH, lipid deposition in the mesenteric arteries, and hyperlipidemia upon being fed an HFC diet. We demonstrated that, in addition, the SHRSP5/Dmcr rat model fed with a HFC diet presents with cardiac and vascular dysfunction, such as cardiac fibrosis, endothelial function, and LV diastolic function. These cardiac and vascular dysfunctions were unrelated to hypertension level, glucose-metabolism, and/or the degree of obesity. SHRSP5/Dmcr rats may be a suitable animal model for elucidating the organ interaction between NAFLD/NASH and cardiac or vascular dysfunction.
Acknowledgment
This work was supported by a JSPS Grant-in-Aid for Young Scientists (B) Grant Number 15K19178 to S. Watanabe.
Grant Support
This work was supported by a Japan Society for the Promotion of Science Grant-in-Aid for Young Scientists (B), Grant Number 15K19178 to S. Watanabe.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1). Bentzon JF, Otsuka F, Virmani R, Falk E: Mechanisms of plaque formation and rupture. Circ Res, 2014; 114: 1852-1866 [DOI] [PubMed] [Google Scholar]
- 2). Kunimasa K, Yoshitomi H, Miura C, Mori H, Tsuchikura S, Ikeda K, Gao M, Yamori Y, Mori M: High susceptibility of obese hypertensive SHRSP.Z-Lepr(fa) /IzmDmcr rats to lipid deposition in the mesenteric artery. Clin Exp Pharmacol Physiol, 2010; 37: 1102-1104 [DOI] [PubMed] [Google Scholar]
- 3). Gordts PL, Bartelt A, Nilsson SK, Annaert W, Christoffersen C, Nielsen LB, Heeren J, Roebroek AJ: Impaired LDL receptor-related protein 1 translocation correlates with improved dyslipidemia and atherosclerosis in apoEdeficient mice. PLoS One, 2012; 7: e38330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4). Kitamori K, Naito H, Tamada H, Kobayashi M, Miyazawa D, Yasui Y, Sonoda K, Tsuchikura S, Yasui N, Ikeda K, Moriya T, Yamori Y, Nakajima T: Development of novel rat model for high-fat and high-cholesterol dietinduced steatohepatitis and severe fibrosis progression in SHRSP5/Dmcr. Environ Health Prev Med, 2012; 17: 173-182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5). Yamori Y, Kitamura Y, Nara Y, Iritani N: Mechanism of hypercholesterolemia in arteriolipidosis-prone rats (ALR). Jpn Circ J, 1981; 45: 1068-1073 [DOI] [PubMed] [Google Scholar]
- 6). Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S, Forlani G, Melchionda N: Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes, 2001; 50: 1844-1850 [DOI] [PubMed] [Google Scholar]
- 7). Sookoian S, Pirola CJ: Non-alcoholic fatty liver disease is strongly associated with carotid atherosclerosis: a systematic review. J Hepatol, 2008; 49: 600-607 [DOI] [PubMed] [Google Scholar]
- 8). Targher G, Day CP, Bonora E: Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med, 2010; 363: 1341-1350 [DOI] [PubMed] [Google Scholar]
- 9). Targher G, Bertolini L, Poli F, Rodella S, Scala L, Tessari R, Zenari L, Falezza G: Nonalcoholic fatty liver disease and risk of future cardiovascular events among type 2 diabetic patients. Diabetes, 2005; 54: 3541-3546 [DOI] [PubMed] [Google Scholar]
- 10). Oni ET, Agatston AS, Blaha MJ, Fialkow J, Cury R, Sposito A, Erbel R, Blankstein R, Feldman T, Al-Mallah MH, Santos RD, Budoff MJ, Nasir K: A systematic review: burden and severity of subclinical cardiovascular disease among those with nonalcoholic fatty liver; should we care? Atherosclerosis, 2013; 230: 258-267 [DOI] [PubMed] [Google Scholar]
- 11). Fargion S, Porzio M, Fracanzani AL: Nonalcoholic fatty liver disease and vascular disease: state-of-the-art. World J Gastroenterol, 2014; 20: 13306-13324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12). Brea A, Puzo J: Non-alcoholic fatty liver disease and cardiovascular risk. Int J Cardiol, 2013; 167: 1109-1117 [DOI] [PubMed] [Google Scholar]
- 13). Fracanzani AL, Burdick L, Raselli S, Pedotti P, Grigore L, Santorelli G, Valenti L, Maraschi A, Catapano A, Fargion S: Carotid artery intima-media thickness in nonalcoholic fatty liver disease. Am J Med, 2008; 121: 72-78 [DOI] [PubMed] [Google Scholar]
- 14). Kim D, Choi SY, Park EH, Lee W, Kang JH, Kim W, Kim YJ, Yoon JH, Jeong SH, Lee DH, Lee HS, Larson J, Therneau TM, Kim WR: Nonalcoholic fatty liver disease is associated with coronary artery calcification. Hepatology, 2012; 56: 605-613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15). Bonapace S, Perseghin G, Molon G, Canali G, Bertolini L, Zoppini G, Barbieri E, Targher G: Nonalcoholic fatty liver disease is associated with left ventricular diastolic dysfunction in patients with type 2 diabetes. Diabetes Care, 2012; 35: 389-395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16). Horai Y, Utsumi H, Ono Y, Kishimoto T, Ono Y, Fukunari A: Pathological characterization and morphometric analysis of hepatic lesions in SHRSP5/Dmcr, an experimental non-alcoholic steatohepatitis model, induced by high-fat and high-cholesterol diet. Int J Exp Pathol, 2016; 97: 75-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17). Jia X, Naito H, Yetti H, Tamada H, Kitamori K, Hayashi Y, Yamagishi N, Wang D, Yanagiba Y, Ito Y, Wang J, Tanaka N, Ikeda K, Yamori Y, Nakajima T: The modulation of hepatic adenosine triphosphate and inflammation by eicosapentaenoic acid during severe fibrotic progression in the SHRSP5/Dmcr rat model. Life Sci, 2012; 90: 934-943 [DOI] [PubMed] [Google Scholar]
- 18). Jia X, Suzuki Y, Naito H, Yetti H, Kitamori K, Hayashi Y, Kaneko R, Nomura M, Yamori Y, Zaitsu K, Kato M, Ishii A, Nakajima T: A possible role of chenodeoxycholic acid and glycine-conjugated bile acids in fibrotic steatohepatitis in a dietary rat model. Dig Dis Sci, 2014; 59: 1490-1501 [DOI] [PubMed] [Google Scholar]
- 19). Kozaki Y, Umetsu R, Mizukami Y, Yamamura A, Kitamori K, Tsuchikura S, Ikeda K, Yamori Y: Peripheral gene expression profile of mechanical hyperalgesia induced by repeated cold stress in SHRSP5/Dmcr rats. J Physiol Sci, 2015; 65: 417-425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20). Moriya T, Kitamori K, Naito H, Yanagiba Y, Ito Y, Yamagishi N, Tamada H, Jia X, Tsuchikura S, Ikeda K, Yamori Y, Nakajima T: Simultaneous changes in high-fat and high-cholesterol diet-induced steatohepatitis and severe fibrosis and those underlying molecular mechanisms in novel SHRSP5/Dmcr rat. Environ Health Prev Med, 2012; 17: 444-456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21). Naito H, Jia X, Yetti H, Yanagiba Y, Tamada H, Kitamori K, Hayashi Y, Wang D, Kato M, Ishii A, Nakajima T: Importance of detoxifying enzymes in differentiating fibrotic development between SHRSP5/Dmcr and SHRSP rats. Environ Health Prev Med, 2016; 21: 368-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22). Nakajima T, Naito H: [Mechanism Analysis and Prevention of Pathogenesis of Nonalcoholic Steatohepatitis]. Nihon eiseigaku zasshi Japanese journal of hygiene, 2015; 70: 197-204 [DOI] [PubMed] [Google Scholar]
- 23). Sweet JG, Chan SL, Cipolla MJ: Effect of hypertension and carotid occlusion on brain parenchymal arteriole structure and reactivity. Journal of applied physiology (Bethesda, Md: 1985), 2015; 119: 817-823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24). Tamada H, Naito H, Kitamori K, Hayashi Y, Yamagishi N, Kato M, Nakajima T: Efficacy of Dietary Lipid Control in Healing High-Fat and High-Cholesterol Diet-Induced Fibrotic Steatohepatitis in Rats. PLoS One, 2016; 11: e0145939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25). Yetti H, Naito H, Jia X, Shindo M, Taki H, Tamada H, Kitamori K, Hayashi Y, Ikeda K, Yamori Y, Nakajima T: High-fat-cholesterol diet mainly induced necrosis in fibrotic steatohepatitis rat by suppressing caspase activity. Life Sci, 2013; 93: 673-680 [DOI] [PubMed] [Google Scholar]
- 26). Wei J, Lin Y, Li Y, Ying C, Chen J, Song L, Zhou Z, Lv Z, Xia W, Chen X, Xu S: Perinatal exposure to bisphenol A at reference dose predisposes offspring to metabolic syndrome in adult rats on a high-fat diet. Endocrinology, 2011; 152: 3049-3061 [DOI] [PubMed] [Google Scholar]
- 27). Matsuura N, Nagasawa K, Minagawa Y, Ito S, Sano Y, Yamada Y, Hattori T, Watanabe S, Murohara T, Nagata K: Restraint stress exacerbates cardiac and adipose tissue pathology via beta-adrenergic signaling in rats with metabolic syndrome. Am J Physiol Heart Circ Physiol, 2015; 308: H1275-1286 [DOI] [PubMed] [Google Scholar]
- 28). Ono K, Masuyama T, Yamamoto K, Doi R, Sakata Y, Nishikawa N, Mano T, Kuzuya T, Takeda H, Hori M: Echo doppler assessment of left ventricular function in rats with hypertensive hypertrophy. J Am Soc Echocardiogr, 2002; 15: 109-117 [DOI] [PubMed] [Google Scholar]
- 29). Tada Y, Kagota S, Matsumoto M, Naito Y, Shibata H, Nejime N, Tsujino T, Koshiba M, Masuyama T, Shinozuka K: Characterization of cardiac size and function in SHRSP.Z-Lepr(fa)/IzmDmcr rats, a new animal model of metabolic syndrome. Biol Pharm Bull, 2010; 33: 1971-1976 [DOI] [PubMed] [Google Scholar]
- 30). Sahn DJ, DeMaria A, Kisslo J, Weyman A: Recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements. Circulation, 1978; 58: 1072-1083 [DOI] [PubMed] [Google Scholar]
- 31). Iwamoto M, Hirohata S, Ogawa H, Ohtsuki T, Shinohata R, Miyoshi T, Hatipoglu FO, Kusachi S, Yamamoto K, Ninomiya Y: Connective tissue growth factor induction in a pressure-overloaded heart ameliorated by the angiotensin II type 1 receptor blocker olmesartan. Hypertens Res, 2010; 33: 1305-1311 [DOI] [PubMed] [Google Scholar]
- 32). Kumar S, Chen M, Li Y, Wong FH, Thiam CW, Hossain MZ, Poh KK, Hirohata S, Ogawa H, Angeli V, Ge R: Loss of ADAMTS4 reduces high fat diet-induced atherosclerosis and enhances plaque stability in ApoE(-/-) mice. Sci Rep, 2016; 6: 31130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33). Hosoo S, Koyama M, Kato M, Hirata T, Yamaguchi Y, Yamasaki H, Wada A, Wada K, Nishibe S, Nakamura K: The Restorative Effects of Eucommia ulmoides Oliver Leaf Extract on Vascular Function in Spontaneously Hypertensive Rats. Molecules (Basel, Switzerland), 2015; 20: 21971-21981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34). Yaykasli KO, Oohashi T, Hirohata S, Hatipoglu OF, Inagawa K, Demircan K, Ninomiya Y: ADAMTS9 activation by interleukin 1 beta via NFATc1 in OUMS-27 chondrosarcoma cells and in human chondrocytes. Mol Cell Biochem, 2009; 323: 69-79 [DOI] [PubMed] [Google Scholar]
- 35). Cilek MZ, Hirohata S, Faruk Hatipoglu O, Ogawa H, Miyoshi T, Inagaki J, Ohtsuki T, Harada H, Kamikawa S, Kusachi S, Ninomiya Y: AHR, a novel acute hypoxiaresponse sequence, drives reporter gene expression under hypoxia in vitro and in vivo. Cell Biol Int, 2011; 35: 1-8 [DOI] [PubMed] [Google Scholar]
- 36). Demircan K, Gunduz E, Gunduz M, Beder LB, Hirohata S, Nagatsuka H, Cengiz B, Cilek MZ, Yamanaka N, Shimizu K, Ninomiya Y: Increased mRNA expression of ADAMTS metalloproteinases in metastatic foci of head and neck cancer. Head Neck, 2009; 31: 793-801 [DOI] [PubMed] [Google Scholar]
- 37). Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J: Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest, 2004; 113: 1408-1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38). Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, Schoonjans K, Bianco AC, Auwerx J: Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature, 2006; 439: 484-489 [DOI] [PubMed] [Google Scholar]
- 39). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis, 2004; 43: S1-290 [PubMed] [Google Scholar]
- 40). Villanova N, Moscatiello S, Ramilli S, Bugianesi E, Magalotti D, Vanni E, Zoli M, Marchesini G: Endothelial dysfunction and cardiovascular risk profile in nonalcoholic fatty liver disease. Hepatology, 2005; 42: 473-480 [DOI] [PubMed] [Google Scholar]
- 41). Senturk O, Kocaman O, Hulagu S, Sahin T, Aygun C, Konduk T, Celebi A: Endothelial dysfunction in Turkish patients with non-alcoholic fatty liver disease. Intern Med J, 2008; 38: 183-189 [DOI] [PubMed] [Google Scholar]
- 42). Sciacqua A, Perticone M, Miceli S, Laino I, Tassone EJ, Grembiale RD, Andreozzi F, Sesti G, Perticone F: Endothelial dysfunction and non-alcoholic liver steatosis in hypertensive patients. Nutr Metab Cardiovasc Dis, 2011; 21: 485-491 [DOI] [PubMed] [Google Scholar]
- 43). Nakano A, Inoue N, Sato Y, Nishimichi N, Takikawa K, Fujita Y, Kakino A, Otsui K, Yamaguchi S, Matsuda H, Sawamura T: LOX-1 mediates vascular lipid retention under hypertensive state. J Hypertens, 2010; 28: 1273-1280 [DOI] [PubMed] [Google Scholar]
- 44). Goland S, Shimoni S, Zornitzki T, Knobler H, Azoulai O, Lutaty G, Melzer E, Orr A, Caspi A, Malnick S: Cardiac abnormalities as a new manifestation of nonalcoholic fatty liver disease: echocardiographic and tissue Doppler imaging assessment. J Clin Gastroenterol, 2006; 40: 949-955 [DOI] [PubMed] [Google Scholar]
- 45). Lautamaki R, Borra R, Iozzo P, Komu M, Lehtimaki T, Salmi M, Jalkanen S, Airaksinen KE, Knuuti J, Parkkola R, Nuutila P: Liver steatosis coexists with myocardial insulin resistance and coronary dysfunction in patients with type 2 diabetes. Am J Physiol Endocrinol Metab, 2006; 291: E282-290 [DOI] [PubMed] [Google Scholar]
- 46). Fallo F, Dalla Pozza A, Sonino N, Lupia M, Tona F, Federspil G, Ermani M, Catena C, Soardo G, Di Piazza L, Bernardi S, Bertolotto M, Pinamonti B, Fabris B, Sechi LA: Non-alcoholic fatty liver disease is associated with left ventricular diastolic dysfunction in essential hypertension. Nutr Metab Cardiovasc Dis, 2009; 19: 646-653 [DOI] [PubMed] [Google Scholar]
- 47). Perseghin G, Lattuada G, De Cobelli F, Esposito A, Belloni E, Ntali G, Ragogna F, Canu T, Scifo P, Del Maschio A, Luzi L: Increased mediastinal fat and impaired left ventricular energy metabolism in young men with newly found fatty liver. Hepatology, 2008; 47: 51-58 [DOI] [PubMed] [Google Scholar]
- 48). Hallsworth K, Hollingsworth KG, Thoma C, Jakovljevic D, MacGowan GA, Anstee QM, Taylor R, Day CP, Trenell MI: Cardiac structure and function are altered in adults with non-alcoholic fatty liver disease. J Hepatol, 2013; 58: 757-762 [DOI] [PubMed] [Google Scholar]
- 49). Targher G, Valbusa F, Bonapace S, Bertolini L, Zenari L, Rodella S, Zoppini G, Mantovani W, Barbieri E, Byrne CD: Non-alcoholic fatty liver disease is associated with an increased incidence of atrial fibrillation in patients with type 2 diabetes. PLoS One, 2013; 8: e57183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50). Reid AE: Nonalcoholic steatohepatitis. Gastroenterology, 2001; 121: 710-723 [DOI] [PubMed] [Google Scholar]
- 51). Li W, Fang Q, Zhong P, Chen L, Wang L, Zhang Y, Wang J, Li X, Wang Y, Wang J, Liang G: EGFR Inhibition Blocks Palmitic Acid-induced inflammation in cardiomyocytes and Prevents Hyperlipidemia-induced Cardiac Injury in Mice. Sci Rep, 2016; 6: 24580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52). Fujita K, Nozaki Y, Wada K, Yoneda M, Fujimoto Y, Fujitake M, Endo H, Takahashi H, Inamori M, Kobayashi N, Kirikoshi H, Kubota K, Saito S, Nakajima A: Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis. Hepatology, 2009; 50: 772-780 [DOI] [PubMed] [Google Scholar]
- 53). Ikeda K, Nara Y, Tagami M, Yamori Y: Nitric oxide deficiency induces myocardial infarction in hypercholesterolaemic stroke-prone spontaneously hypertensive rats. Clin Exp Pharmacol Physiol, 1997; 24: 344-348 [DOI] [PubMed] [Google Scholar]