

Abstract
Parkinson’s disease (PD) is a complex multifactorial disorder where environmental factors interact with genetic susceptibility. Accumulating evidence suggests that mitochondria have a central role in the progression of neurodegeneration in sporadic and/or genetic forms of PD. We previously reported that exposure to a secondary metabolite from the soil bacterium, Streptomyces venezuelae, results in age- and dose-dependent dopaminergic (DA) neurodegeneration in Caenorhabditis elegans and human SH-SY5Y neurons. Initial characterization of this environmental factor indicated that neurodegeneration occurs through a combination of oxidative stress, mitochondrial complex I impairment, and proteostatic disruption. Here we present extended evidence to elucidate the interaction between this bacterial metabolite and mitochondrial dysfunction in the development of DA neurodegeneration. We demonstrate that it causes a time-dependent increase in mitochondrial fragmentation through concomitant changes in the gene expression of mitochondrial fission and fusion components. In particular, the outer mitochondrial membrane fission and fusion genes, drp-1 (a dynamin-related GTPase) and fzo-1 (a mitofusin homolog), are up- and down-regulated, respectively. Additionally, eat-3, an inner mitochondrial membrane fusion component, an OPA1 homolog, is also down regulated. These changes are associated with a metabolite-induced decline in mitochondrial membrane potential and enhanced DA neurodegeneration that is dependent on PINK-1 function. Genetic analysis also indicates an association between the cell death pathway and drp-1 following S. ven exposure. Metabolite-induced neurotoxicity can be suppressed by DA-neuron-specific RNAi knockdown of eat-3. AMPK activation by 5-amino-4-imidazole carboxamide riboside (AICAR) ameliorated metabolite- or PINK-1-induced neurotoxicity; however, it enhanced neurotoxicity under normal conditions. These studies underscore the critical role of mitochondrial dynamics in DA neurodegeneration. Moreover, given the largely undefined environmental components of PD etiology, these results highlight a response to an environmental factor that defines distinct mechanisms underlying a potential contributor to the progressive DA neurodegeneration observed in PD.
Introduction
Neurons are reliant on mitochondrial function due to their high-energy demands. As a result, this organelle is dynamic and fission and fusion cycles occur regularly. Mitochondria also change size or number in response to environmental cues, which can affect cellular integrity. Mitochondrial network maintenance is primarily regulated by GTPases (Drp1, Opa1) and the mitofusins (Mfn1/Mfn2). Together, these fission and fusion gene products, located on mitochondrial membranes, regulate organelle dynamics while a fission/fusion imbalance can lead to an accumulation of ultrastructural defects and eventually cell death.
Mitochondrial dysfunction is associated with Parkinson’s disease (PD) through both genetic and environmental routes. For example, mitochondrial toxicity can occur via the PD-linked recessive mutations PARK2 (Parkin) and PARK6 (PINK1)1–5. However, only 5–10% of PD cases are linked to genetic susceptibilities6, 7. Thus, environmental factors alone, or in combination genetic susceptibility, are thought to increase the risk of developing PD8, 9.
Previously, we identified a novel environmental contributor to dopaminergic (DA) neurodegeneration from Streptomyces venezuelae (S. ven). These common soil bacteria produce a secondary metabolite that causes age- and dose-dependent DA neurodegeneration in Caenorhabditis elegans (C. elegans) and cultured human DA neurons. The neurodegenerative activity, which is amphipathic, is stable and will retain neurotoxic activity after 30 min of boiling8. S. ven metabolite also increases oxidative stress, inhibits mitochondrial complex I, and reduces ATP production8, 10, 11. Moreover, we discovered that the cellular response to this bacterial product is epistatically regulated by pink-1 loss-of function, suggesting that it causes DA neuronal cell death through mitotoxicity11.
Here we report that S. ven metabolite exposure is associated with an increase in mitochondrial fragmentation. There are also concomitant gene expression changes whereby drp-1 gene expression is increased following exposure to the metabolite while fzo-1 and eat-3 gene expression (C. elegans mitofusin and OPA1, respectively) are decreased. Bacterial metabolite exposure is also associated with DA neurodegeneration through increased pink-1-dependent drp-1 fission activity and eat-3 resistance to pink-1 mutant- and metabolite-induced neurotoxicity. Genes within the cell death pathway also interact with drp-1 following metabolite exposure. These results demonstrate the critical response mitochondria exhibit when challenged with an environmental stressor, leading to neurodegeneration, and provide mechanistic details underlying susceptibility to neuronal cell death.
Results
S. venezuelae metabolite exposure causes mitochondrial fragmentation
As previously described8, S. ven is grown in liquid media, and then the small secondary product (MW < 300) is separated through column chromatography and extracted with dichloromethane. The S. ven extract fraction is collected through rotary evaporation to remove the solvent. The fraction is resuspended in ethyl acetate (EtAc) for experimentation. Hereafter, this compound will be referred to as the metabolite. EtAc is used as a negative solvent control in all experiments and does not cause any significant neurodegeneration8, 10, 11.
Mitochondrial toxicants often cause an imbalance between mitochondrial fission and fusion9. We therefore wanted to determine if S. ven exposure could also disrupt mitochondrial fission/fusion. We initially conducted a time course exposure with 5-, 7- and 9-day old animals. Because the body-wall muscle cells of C. elegans contain numerous mitochondria, it is an ideal system to investigate mitochondrial morphology. Using a reporter strain, we expressed a mitochondrial outer membrane (OMM)protein-fused to mRFP (Pmyo-3::TOM20::mRFP) and observed that metabolite addition significantly enhanced mitochondrial fragmentation. This was characterized by disordered and small circular-shaped mitochondria compared to solvent control at days 5, 7, and 9 post-hatching (Fig. 1a, b). We noted an increase of mitochondrial fragmentation in aging nematodes from either solvent or S. ven treatment; however, morphological abnormalities were significantly accelerated in metabolite-treated animals compared to solvent controls (Fig. 1a), indicating that mitochondrial morphology changes occur following metabolite exposure in a time-dependent manner.
Fig. 1. The S. venezuelae (S. ven) metabolite causes perturbations of mitochondrial fission and fusion, resulting in time-dependent mitochondrial fragmentation in C. elegans.
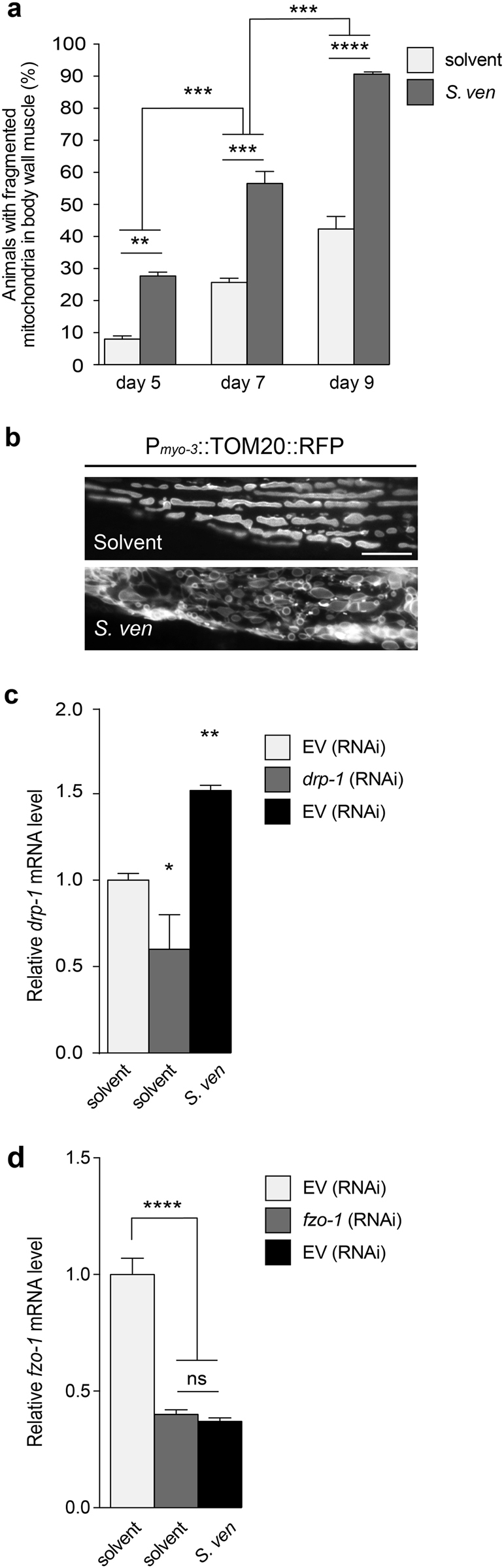
a Quantification of mitochondrial fragmentation as indicated at young adult (5-day old), middle (7-day old), and older aged (9-day old) nematodes in response to metabolite exposure using Pmyo-3:TOM20:mRFP. S. ven metabolite significantly enhanced mitochondrial fragmentation in aging nematodes. Data represented as mean ± S.E.M.; n = 30 animals per replicate, three independent replicates; two-way ANOVA with Tukey’s post hoc test for multiple comparisons. **P < 0.01, ***P < 0.001, and ****P < 0.0001. b TOM20::mRFP images of the representative mitochondrial morphology following exposure of solvent or metabolite at day 5 post-hatching. The metabolite causes disordered and small donut-shaped morphology in the mitochondrial outer membrane. The scale bar is 20 μm. c, d Animals exposed to solvent (EtAc) or S. ven metabolite were assessed for drp-1 and fzo-1 mRNA levels by qRT-PCR. Metabolite exposure significantly increased drp-1 expression and reduced fzo-1 expression levels compared to empty vector (EV) solvent control. Relative mRNA expression levels were normalized to EV control. Data represented as mean ± S.E.M.; three replicates comprising at least 100 animals each; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. *P < 0.05, **P < 0.01, and ****P < 0.0001.
Mitochondrial morphology reflects a balance between mitochondrial fission and fusion7, 12, 13. Fission removes damaged components through the OMM dynamin-related GTPase, Drp1 (C. elegans DRP-1) while fusion, via the conserved GTPases, Mfn1/Mfn2 (C. elegans FZO-1), compensates by forming long and interconnected networks. Because mitochondrial fission and fusion are primarily mediated by DRP-1 and FZO-1, respectively on the OMM, in response to stressors14–16, mRNA expression of drp-1 and fzo-1 was examined by qRT-PCR following exposure of the metabolite. Metabolite exposure significantly increased the expression levels of drp-1 mRNA and reduced fzo-1 expression (Fig. 1c,d). RNAi knockdown of drp-1 or fzo-1 served as a control. Consistent with previous observations in human cells that mitochondrial oxidative stress causes mitochondrial fragmentation through an imbalance in Drp1 and Mfn217, we concluded that metabolite exposure induces a similar imbalance of fission and fusion through changes in drp-1 and fzo-1 gene expression in the OMM, resulting in mitochondrial fragmentation.
We quantitated morphological changes using two different mitochondrial marker strains; Pmyo-3::mitoGFP was used to visualize the mitochondrial matrix and Pmyo-3:: TOM20::mRFP for the OMM in body-wall muscle cells. In combination with RNAi depletion of OMM and inner mitochondrial membrane (IMM) fission/fusion components, these transgenic animals were used to investigate epistatic relationships. In both transgenic lines, more than half the muscle cells displayed normal tubular structures when exposed to solvent control in the EV RNAi cells (Fig. 2a, b). In notable contrast, exposure to S. ven metabolite resulted in a significant shift to a distinctive, circular-shaped morphology characteristic of mitochondrial fragmentation (Fig. 2a, b). RNAi knockdown of the OMM fission gene product drp-1 increased rates of fusion (interconnected and tubular) compared to EV RNAi (Fig. 2a, b). Following metabolite exposure, a significant shift back to mitochondrial fragmentation was observed, indicating an epistatic interaction with the metabolite. Knockdown of fis-1, which has been shown to colocalize with DRP1 at the OMM18, also caused significant fusion; this phenotype could be reverted back to fragmentation with the addition of metabolite (Fig. 2a, b). RNAi knockdown of the fusion gene products fzo-1 (OMM) and eat-3 (IMM) resulted in a significant enhancement in mitochondrial fragmentation compared to EV control. Following treatment with metabolite, we did not observe an additional increase in fragmentation because fragmentation was already at maximum (Fig. 2a, b).
Fig. 2.

The effect of S. ven metabolite-mediated fission and fusion on the mitochondrial matrix (a) and mitochondrial outer membrane (b) in C. elegans body-wall muscles. Transgenic nematodes expressing Pmyo-3::mitoGFP were analyzed to monitor mitochondrial matrix in response to each treatment, and mitochondrial outer membrane was detected with Pmyo-3::TOM20::mRFP strain. Representative mitoGFP and TOM20 images (left panel in a and b) and quantification of the different mitochondrial morphologies observed (right panel with graphical stacks in a and b, Y axis represents distribution of mitochondrial morphology phenotypes in worm populations in percentage) are shown. Compared with solvent treatment, populations of RNAi-treated fission gene-associated drp-1 and fis-1 animals exhibited significantly more mitochondrial fragmentation following metabolite exposure, characterized by disordered and small circularly shaped mitochondria at day 8 post-hatching. Animals reduced for the fusion regulators, fzo-1 and eat-3, displayed mitochondrial fragmentation following exposure to metabolite in a manner similar to solvent-treated animals, respectively. Arrows indicate representative areas of mitochondrial fusion. These data are presented as mean ± S.E.M.; n = 30 animals per replicate, two independent replicates; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. *P < 0.05, **P < 0.01. The scale bar is 20 μm.
Metabolite-mediated effects on mitochondrial function
Abnormal mitochondrial fission/fusion is associated with mitochondrial membrane potential (ΔΨm) collapse15, 19. To explore the hypothesis that the metabolite alters ΔΨm, we measured the relative mitochondrial uptake of the fluorescent dye tetramethylrhodamine ethyl ester (TMRE) in live nematodes following S. ven exposure. This lipophilic cation dye has been used previously in C. elegans as an indicator of mitochondrial activity20–22. We used RNAi knockdown of the familial recessive PD gene, pink-1, as a positive control. Animals exposed to metabolite or pink-1 (RNAi) displayed significant decreases in TMRE fluorescence in comparison with solvent-treated EV control group at day 8 (Fig. 3a, b).
Fig. 3. The effect of S. ven metabolite-mediated fission and fusion on mitochondrial function.
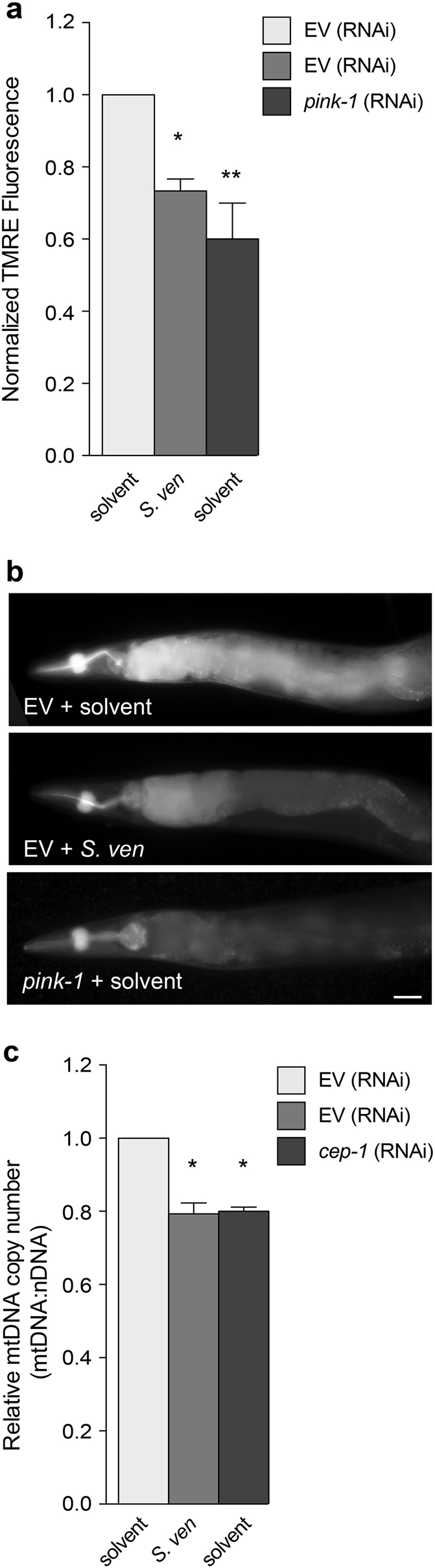
a Relative mitochondrial uptake of the fluorescent dye tetramethylrhodamine ethyl ester (TMRE) was assessed for the mitochondrial membrane potential (ΔΨm) of C. elegans. Animals (wild-type N2) exposed to S. venezuelae metabolite had a significantly lower ΔΨm then solvent-treated EV control animals at day 8 post-hatching. RNAi knockdown of pink-1 was used as a positive control. Relative TMRE fluorescent intensity was normalized to EV solvent control. These data are presented as mean ± S.E.M.; n = 30 animals per replicate, three independent replicates; one-way ANOVA with Dunnett’s post hoc test for multiple comparisons. *P < 0.05, **P < 0.01. b Representative images of TMRE-stained nematodes following treatment with solvent and metabolite, respectively. The scale bar is 5 μm. c Quantitation of mitochondrial DNA (mtDNA) copy number by qRT-PCR in N2 animals. The metabolite caused a relative decrease of mtDNA copy number in comparison to EV solvent control. cep-1 (RNAi) was included as a positive control. Data represented as mean ± S.E.M.; three biological and three technical replicates; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. *P < 0.05
Given the observed perturbations of fission/fusion by metabolite, it is possible that mitochondrial DNA (mtDNA) becomes unstable and accumulates mutations as a consequence of defective mitochondrial fusion. Thus, we assessed mtDNA levels in response to the metabolite. Because mtDNA is more sensitive than nuclear DNA (nDNA) to environmental stress exposures23, the mtDNA:nDNA ratio was determined in N2 wild-type worms treated with metabolite by qRT-PCR. As a positive control, we depleted the C. elegans homolog of p53 (cep-1) by RNAi, which causes sensitivity to mtDNA damage as previously described24. Compared to solvent treatment, metabolite-treated animals displayed a significant decrease in the mtDNA:nDNA ratio in comparison with solvent-only animals (Fig. 3c). Taken together, these results suggest that a metabolite-induced imbalance of fission/fusion exacerbates mitochondrial toxicity.
Down regulation of eat-3 suppresses neurotoxicity caused by the metabolite
Considering the severe mitochondrial morphological changes induced by S. ven exposure, we sought to evaluate how modulating fission/fusion impacts DA neurodegeneration. Our analysis was performed through RNAi knockdown of mitochondrial fission/fusion components following exposure of metabolite in an RNAi strain that selectively enables RNAi only in DA neurons25, 26. Consistent with the effect on mitochondrial morphology observed in body-wall muscle cells, RNAi depletion of fission (drp-1 and fis-1) and fusion (fzo-1 and eat-3) genes resulted in significant DA neurodegeneration in comparison with solvent-only EV control at day 9 post-hatching (Fig. 4a). When metabolite was added, it enhanced degeneration in EV RNAi conditions, but it did not result in further degenerative changes in drp-1, fis-1 or fzo-1 RNAi knockdown conditions (Fig. 4a, b). However, a combination of eat-3 (RNAi) and metabolite exposure resulted in resistance to DA neurotoxicity (Fig. 4a, b). A previously reported study suggested that the OMM fission component drp-1 acts upstream of eat-3, an IMM fusion component, and that mutual compensation occurs for physiological defects27. Since we observed that metabolite increases drp-1 gene expression levels (Fig. 1c), it is possible that depletion of eat-3 could be compensated by metabolite-induced drp-1, or vice versa, providing resistance to DA neurodegeneration. To explain the interaction between eat-3 and metabolite-induced drp-1, we examined the effect of eat-3 (RNAi) on DA neurodegeneration in a drp-1(tm1108) null mutant background crossed to the DA neuron-sensitive RNAi strain. We hypothesized that if metabolite-induced drp-1 interacts with eat-3 (RNAi), then it would not exhibit the resistance phenotype under the drp-1 null mutant background. Because mitochondrial fission/fusion components are interdependent, we also knocked down fis-1 and fzo-1 in the drp-1 mutant background. RNAi depletion of fis-1 or fzo-1 resulted in severe neurodegeneration in the drp-1 mutant background; solvent and the addition of metabolite did not cause further neurodegeneration (Fig. 4c). This indicated that drp-1 acts as an upstream component for fission/fusion as it pertains to DA neurodegeneration. Interestingly, a combination of metabolite and eat-3 (RNAi) in the drp-1 null background no longer displayed neuroprotection against the metabolite, indicating that there is epistatic regulation between eat-3 depletion and metabolite-induced drp-1 activity (Fig. 4a vs. c). We also monitored eat-3 transcriptional activity by qRT-PCR to determine the interaction between drp-1 and eat-3. When drp-1 was depleted by RNAi, the eat-3 gene expression levels were significantly higher than when drp-1 was activated by metabolite treatment (Fig. 4d). These results provide evidence that response to metabolite involves drp-1 fission activity, which genetically interacts with eat-3 in a compensatory manner.
Fig. 4. Down-regulation of eat-3 suppresses neurotoxicity caused by the metabolite.

a, b RNAi knockdown of fission (drp-1 and fis-1) and fusion (fzo-1 and eat-3) components with solvent caused DA neurodegeneration at day 9 post-hatching. With metabolite exposure, RNAi treated with drp-1, fis-1, or fzo-1 animals did not exhibit enhanced DA neurotoxicity compared to the corresponding solvent control. In contrast, a combination of S. ven metabolite and RNAi targeting eat-3 resulted in significant resistance to neurotoxicity. The scale bar is 10 μm. These data are presented as mean ± S.E.M.; n = 30 animals per replicate, three independent replicates; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. *P < 0.05, **P < 0.01. Representative images of the six anterior dopaminergic neurons are shown in (b), with degenerating or missing neurons marked by arrows and normal neurons indicated by arrowheads. c An RNAi-sensitive strain with GFP expression in DA neurons was crossed to drp-1(tm1108) mutant animals. Homozygosity was confirmed by PCR. Loss-of-function drp-1(tm1108) exacerbated DA neurotoxicity compared with GFP only solvent control at day 9 post-hatching. RNAi knockdown of eat-3 did not result in resistance of neurotoxicity as observed in Fig. 4a. Data are represented as mean ± S.E.M; n = 30 animals per replicate, three independent replicates; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. *P < 0.05, *P < 0.0001. d eat-3 mRNA levels were measured by qRT-PCR in animals treated with solvent (EtAc), S. ven metabolite, drp-1 (RNAi), or eat-3 (RNAi). The animals treated with metabolite showed significant lower eat-3 mRNA levels than when drp-1 was depleted by RNAi. Data represented as mean ± S.E.M.; three replicates comprises at least 100 animals each; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. ****P < 0.0001.
DRP-1-dependent metabolite induction of the cell death pathway
To explore neurodegeneration within the context mitochondrial fission we turned to CED-9, a protein involved in both proapoptotic and antiapoptotic activities in C. elegans. More specifically, CED-9, an ortholog of BCL2-like protein, has been reported to interact with DRP-1 to promote mitochondrial fission28. Since RNAi knockdown of CED-9 led to lethality, we examined EGL-1, an upstream activator of the cell death pathway, which is specifically required to regulate CED-928, 29. RNAi knockdown egl-1 enhanced DA neurodegeneration in comparison with EV solvent control; however, metabolite addition significantly attenuated neurotoxicity (Fig. 5a). We also introduced the drp-1(tm1108) mutant background, which caused severe neurotoxicity; here, the addition of metabolite did not attenuate neurodegeneration in egl-1 RNAi (Fig. 5a). We then examined ced-9 expression level by qRT-PCR (Fig. 5b). We observed that it was significantly increased following S. ven exposure, however, under egl-1 or drp-1 RNAi depletion there was no significant change of ced-9 expression with S. ven (Fig. 5b). These data suggest a possible interaction between CED-9 and DRP-1 following metabolite induction.
Fig. 5. Exposure to S. ven metabolite is associated with cell death.

a Reduction of egl-1 (RNAi) caused neurodegeneration in comparison with EV solvent control; however, addition of metabolite showed significant reduction of neurotoxicity. Under the drp-1(tm1108) mutant background, the animals exhibited severe neurodegeneration, and the addition of metabolite did not cause further neurodegeneration in EV solvent control or egl-1 RNAi. These data are presented as mean ± S.E.M.; n = 30 animals per replicate, three independent replicates; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. **P < 0.01. b ced-9 mRNA levels were measured by qRT-PCR. Metabolite exposure resulted in a significant increase in ced-9 transcriptional activity compared to EV solvent control, however, there was no significant change in ced-9 expression level when egl-1 or drp-1 was depleted (RNAi). Data are represented as mean ± S.E.M; three replicates comprised of at least 100 animals each; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. ****P < 0.0001. c, d A dopaminergic (DA) neuron RNAi-sensitive strain with GFP expression in DA neurons was crossed to strains harboring mutant alleles for pink-1(tm1779) or pdr-1(gk448). Homozygosity was confirmed by PCR. Loss of either pink-1(tm1779) (c) or pdr-1(gk448) (d) function enhanced DA neurodegeneration with or without metabolite compared to GFP only solvent control at day 9 post-hatching. RNAi knockdown of eat-3 resulted in significant resistance to both pink-1 (c) and pdr-1-induced (d) neurotoxicity. Data presented as mean ± S.E.M.; n = 30 animals per replicate, three independent replicates; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Metabolite induces PINK-1/PDR-1-dependent fission
Mitochondrial fission is also associated with the PINK1/Parkin pathway30, 31. These proteins generally function together to regulate mitochondrial homeostasis32–34 in part by promoting DRP1-dependent mitochondrial fission30, 31. We hypothesized that if metabolite-induced fission is independent of PINK-1, it would suppress the pink-1 mutant phenotype. Conversely, if dependent on PINK-1 function, metabolite addition would not enhance neurodegeneration when pink-1 was depleted. We assessed these predictions in null mutant alleles of pink-1(tm1779) and pdr-1(gk448) (corresponding to human PINK1 and Parkin, respectively), which were crossed into the DA-neuron-specific RNAi strain. Consistent with prior reports32, 35, loss of pink-1 enhanced DA neurodegeneration compared to GFP only animals (solvent comparison) (Fig. 5c). Metabolite addition did not enhance neurotoxicity from pink-1 loss (Fig. 5c). RNAi knockdown of drp-1 in the pink-1 loss-of-function background did not further enhance DA neurodegeneration, indicating that metabolite-induced pink-1 neurodegeneration is dependent on DRP-1 fission. We also found that reduction of eat-3 (RNAi) significantly suppressed pink-1-induced neurodegeneration with or without metabolite exposure (Fig. 5c). Similar results were obtained under the pdr-1(gk448) mutant background (Fig. 5d). These data suggest that down regulation of eat-3 is neuroprotective in pink-1/pdr-1 mutant conditions.
AICAR-mediated AMPK activation rescues metabolite and pink-1-induced dopaminergic neurodegeneration
We previously reported that the metabolite can decrease ATP production10. Since neurotoxicity from either metabolite and/or the pink-1 mutant was reduced by eat-3 depletion, we next wanted to determine whether down regulation of eat-3 acts as a compensatory mechanism to maintain adequate ATP levels. We performed an ex vivo ATP assay on pink-1(tm1779) mutant nematodes treated with eat-3 RNAi with metabolite. ATP levels were significantly decreased in metabolite-treated control animals (EV), pink-1 mutant and eat-3 RNAi knockdown animals (Fig. 6a). However, down regulation of eat-3 did not exhibit an effect on ATP levels (Fig. 6a).
Fig. 6. AICAR-mediated AMPK activation rescues metabolite-induced dopaminergic neurodegeneration.

a The effect of eat-3 RNAi on ATP levels in pink-1(tm1779) mutant animals after exposure of solvent (EtAc) or metabolite. ATP content was measured in young adult nematodes using a luciferase-based assay. Values are mean ± S.E.M.; n = 3 independent samples with 100 worms in each; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. ****P < 0.0001. b Reduction of either aak-2 or mff-1 enhanced DA neurotoxicity with or without metabolite in comparison with EV solvent control. Data represented as mean ± S.E.M.; n = 30 animals per replicate, three independent replicates; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. **P < 0.005. c A timeline representing an experimental paradigm depicting the relative length of S. ven metabolite exposure and AICAR treatment. The abbreviations L1–L4 are the larval stages of C. elegans, and the ‘young adult’ designations represent days post-hatching. F1 animals were treated with either RNAi and/or S. ven metabolite from hatching to the day of analysis; neurodegeneration assays were performed at day 9. Animals were treated with 1 mM AICAR from hatching until day 4. d Animals expressing only GFP in the DA neurons treated with AICAR (alone) displayed enhanced DA neurodegeneration compared to solvent controls; however, metabolite-induced neurotoxicity was reduced by AICAR treatment. Furthermore, AICAR rescued neurotoxicity caused by pink-1(tm1779) in comparison to non-AICAR treated (solvent) animals with pink-1 mutation. AICAR did not rescue DA neuron cell death from the combined stress of metabolite and pink-1. Data are presented as mean ± S.E.M.; n = 30 animals per replicate, three independent replicates; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. *P < 0.05, ***P < 0.001. e AICAR-mediated AMPK activation was sufficient to protect neurodegeneration caused by eat-3 deficiency (RNAi) alone or with metabolite, however, it could not reduce neurotoxicity in the absence of pink-1 function (tm1779) and eat-3 RNAi together. Data represented as mean ± S.E.M.; n = 30 animals per replicate, three independent replicates; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. *P < 0.05, **P < 0.01. f AICAR-mediated AMPK activation failed to attenuate neurotoxicity from drp-1 (RNAi) depletion in the GFP only or pink-1(tm1779) mutant background; however, AICAR suppressed neurotoxicity in the pink-1 mutant background in combination with metabolite exposure. These data are presented as mean ± S.E.M.; n = 30 animals per replicate, three independent replicates; one-way ANOVA with Tukey’s post hoc test for multiple comparisons. *P < 0.05.
AMP-activated protein kinase (AMPK) is activated by ATP depletion from mitochondrial stress and enhances mitochondrial biogenesis to supplant energy deficiency16, 36. Moreover, AMPK is neuroprotective against the mitochondria complex I inhibitors 1-methyl-4-phenylpyridinium (MPP+) and rotenone36, 37. Therefore, we investigated whether depletion of the C. elegans homolog of AMPK, aak-2, and an AMPK substrate and DRP-1 receptor, MFF-1, caused DA neurodegeneration in response to metabolite. Knockdown of both gene products resulted in significant DA neurodegeneration while the addition of metabolite did not further enhance neurodegeneration, indicating this process is modulated by the metabolite (Fig. 6b). We next examined the activation of AMPK by a pharmacological method to determine if we could ameliorate metabolite-induced neurotoxicity. Animals were treated with 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), an activator of AMPK (Fig. 6c), and analyzed for DA neurodegeneration. AICAR treatment caused significant DA neurodegeneration under normal conditions (GFP only with solvent). Interestingly, AICAR attenuated neurotoxicity when animals were exposed to metabolite (Fig. 6d). We further examined whether AMPK activation would suppress neurodegeneration from pink-1 loss-of-function. Compared with non-AICAR treated pink-1(tm1779) animals, AICAR treatment was significantly neuroprotective in solvent-only animals (Fig. 6d). However, AICAR could not attenuate neurotoxicity from the combination of pink-1 loss-of-function and metabolite (Fig. 6d).
We further delved into the effect of AMPK activation under RNAi knockdown of eat-3 or drp-1 (Fig. 6e,f, respectively). We observed that AICAR-mediated AMPK activation was sufficient to protect against eat-3 deficiency (GFP only) but it failed to suppress neurotoxicity in the pink-1 mutant background (Fig. 6e). Conversely, AICAR-mediated AMPK activation did not protect against drp-1 deficiency in the GFP only background, but it was able to suppress neurotoxicity in the pink-1 mutant background in combination with the metabolite (Fig. 6f).
Discussion
Perturbations in mitochondrial fission/fusion have been reported as critical mechanisms during neurodegeneration33, 38, 39. Although genetic risk factors are associated with this process, the interaction between an environmental contributor and mitochondrial dynamics remains poorly understood. In an effort to uncover potentially more prevalent sources of environmental influence on neurodegeneration, we previously identified a metabolite from the common soil bacterium, S. venezuelae, that induces DA neuron cell death by targeting mitochondria8, 10, 11. Here, we extend these studies by examining mitochondrial dynamics and mechanistically discerning genetic modulators altering its neurotoxicity in vivo.
We initially determined that the metabolite enhances mitochondrial fragmentation in a time-dependent manner (Fig. 1a). We were curious to know if the mitotoxic properties of S. ven could be distinguished from other mitochondrial complex I inhibitors. This was performed by thoroughly examining mitochondrial fission/fusion changes following metabolite exposure in combination with RNAi knockdown of mitochondrial fission or fusion gene products in C. elegans body-wall muscle cells and DA neurons. We discovered that the mechanism of action underlying S. ven neurotoxicity is distinct from that of either rotenone or MPP+.
S. ven exposure increases mitochondrial fragmentation (Figs. 1, 2) while rotenone decreases fission and induces fusion40–42. Likewise, drp-1 gene expression increases following S. ven treatment (Fig. 1c) but it decreases following rotenone treatment41. Similar to S. ven, MPP+ increases mitochondrial fragmentation and increases DRP1 levels43, 44. However, genetic inactivation of drp-1 in SH-SY5Y cells blocks MPP+ mitochondrial fragmentation43 whereas fragmentation still occurs in C. elegans when drp-1 is knocked down following treatment with S. ven (Fig. 2). While all three of these neurotoxic substances inhibit mitochondrial complex I, their mitotoxic mechanisms of action are clearly distinguishable.
We have previously reported that the metabolite upregulates two pathways associated with mitochondrial dysfunction, the mitochondrial unfolded protein response pathway (UPRmt) and intracellular ROS accumulation10. The UPRmt can be activated by a mitonuclear protein imbalance that shifts the relative nDNA and mtDNA levels45. Following metabolite exposure, a mtDNA:nDNA imbalance was identified (Fig. 3c) and we have shown that the UPRmt was upregulated in response to metabolite10. The UPRmt signaling pathway regulates > 900 gene products including nDNA-encoded mitochondrial molecular chaperones, the mitochondrial protease CLPP, and DRP-146.
While ROS can cause toxicity and alter mitochondrial function at higher levels, it has also been shown, at lower levels, to be beneficial to C. elegans where it extends lifespan47. Likewise, lifespan extension has been also noted with reduction of mitochondrial electron transport chain components48. Such differences are interesting and may reflect tissue-specific distinctions in exposure to acute and chronic stressors. ROS overproduction both enhances DRP-1 and reduces FZO-1 activities, resulting in mitochondrial fragmentation;17 likewise, S. ven increases drp-1 gene expression and decreases fzo-1 gene expression (Fig. 1).
The impact of increased drp-1 gene expression following S. ven exposure was examined in more detail in our study. Notably, eat-3 RNAi attenuated metabolite-induced neurodegeneration, while other combinations of S. ven and fission/fusion genes did not alter levels of neurodegeneration. We hypothesized that, in the presence of metabolite-induced stress, the absence of eat-3 led to an increase in mitochondrial fission. Indeed, through an epistasis analysis we determined that eat-3 was no longer neuroprotective against S. ven in the drp-1 null mutant background (Fig. 4c), indicating a genetic interaction between drp-1 and eat-3. The interaction between drp-1 and eat-3 was also examined by monitoring eat-3 transcriptional activity. When drp-1 was depleted by RNAi, eat-3 levels were significantly higher than when drp-1 was activated by metabolite (Fig. 4d). These results provide evidence that the metabolite induces drp-1-mediated fission activity, which genetically interacts with eat-3 in a compensatory manner.
It has been reported that low ΔΨm in mitochondria serves as a trigger for PINK-1 accumulation on OMM to signal the degradation of damaged organelles through autophagy5, 30, 49, 50. This process requires DRP1 fission to generate small mitochondrial particles for efficient organelle degradation5, 30, 49, 50. We previously reported that the metabolite-induced PINK-1-dependent autophagy10, 11. Here, metabolite-induced drp-1 function is dependent on PINK-1 (Fig. 5c). Therefore, it is possible that metabolite-induced ROS could enhance drp-1 gene expression directly and lower ΔΨm caused by metabolite exposure (Fig. 3a,b), which would also induce PINK-1-dependent DRP-1 fission for removal of damaged mitochondria through autophagy (Fig. 7).
Fig. 7. Proposed mechanism of altered mitochondrial homeostasis caused by S.
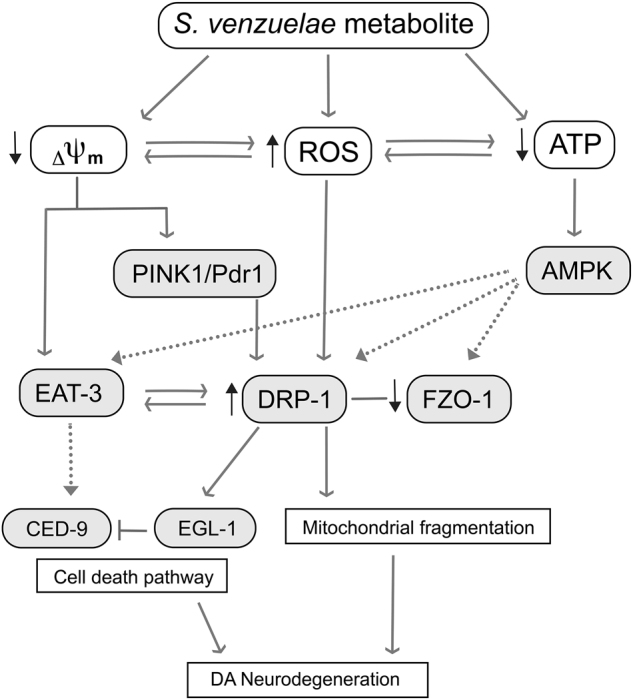
ven metabolite-induced toxicity. This experimental model illustrates our current understanding of the effect of S. ven metabolite on mitochondrial homeostatic mechanisms in the development of neurodegeneration. metabolite-induced ROS gradually impairs mitochondria through mitochondrial complex I damage, energy deprivation, and eventually, the loss of ΔΨm. As ΔΨm is decreased, C. elegans PINK-1 and PDR-1 would be mobilized to the OMM, which would then recruit the OMM fission factor, DRP-1, to the OMM to degrade the damaged organelle, resulting in mitochondrial fragmentation. Moreover, proteolytic cleavage of EAT-3 by the decline in ΔΨm and increased DRP-1 could lead to neuron cell death. In our genetic studies, the reduction of eat-3 (RNAi) attenuated neurotoxicity induced by metabolite or in the pink-1 mutant background, demonstrating that eat-3 depletion (RNAi) might counteract cell death and have a role as an antiapoptotic factor when the cell death pathway is activated by DRP-1. Moreover, we observed a genetic interaction between drp-1 and egl-1, a component of the cell death pathway as well as increased mRNA expression of ced-9 in the presence of metabolite. Another DRP-1-related response observed involves C. elegans AAK-2 (AMPK), which is a candidate signaling molecule and known regulator of mitochondrial biogenesis. The AMPK pathway converges with the gene expression of mitochondrial fission and fusion genes, such as drp-1 and eat-3, representing a prospective mechanism of response to S. ven exposure.
It was previously reported that DRP-1 recruitment to mitochondria is promoted by EGL-1 and CED-9 in C. elegans28, 51. Exposure to metabolite causes an increase in ced-9 gene expression (Fig. 5b), and neurotoxicity from disrupting egl-1 was reduced by metabolite (Fig. 5a). This observation suggests a possible interaction between PINK-1/PDR-1 and the apoptotic pathway. PINK-1 has been shown as mediator of cytochrome c release, which is a key component for initiating caspase acitivity52. Collapse of ΔΨm by metabolite could trigger PINK-1 accumulation on OMM, gathering DRP-1 for fission while it induces the cell death pathway.
Previously, it was shown that an increase in DRP-1 activity occurred in response to mitochondrial dysfunction through AMPK, a key regulator of cellular energy metabolism16, 53. AMPK can promote mitochondrial division and is activated in response to mitochondrial complex inhibitors, such as rotenone and antimycin A16. AMPK suppressed the DA neurotoxicity associated with metabolite exposure in N2 wildtype, eat-3 (RNAi), and pink-1; drp-1(RNAi) animals (Fig. 6). These data indicate that AMPK has an important role underlying the mechanism of metabolite-induced DA neurodegeneration. Although further work will be required to elucidate how the metabolite triggers mitochondrial dysfunction, modulation of AMPK activity, an established effector of mitochondrial biogenesis, is a potential means by which changes in drp-1 and eat-3 gene transcription levels are regulated in response to metabolite (Fig. 7).
In considering the etiological understanding of PD, there remains an urgent need to define putative gene-by-environment interactions that underlie neurodegeneration, well beyond current explanations. Our data provide evidence that a bacterial source of an environmental toxin causes DA neuron cell death through mitotoxicity and that established genetic susceptibility factors for PD, such as pink-1 mutation, exacerbate neurodegeneration. Chronic exposure of the S. ven metabolite causes alterations in mitochondrial fission/fusion, resulting in neurodegeneration. This study advances our understanding of the mitochondrial homeostatic mechanism in response to an environmental stressor that induces neuronal cell death and, with the identification of AMPK as a neuroprotective agent, provides an avenue for subsequent investigation of environmental exposures and potentially targeted modulation of their impact on neurodegeneration.
Materials and Methods
C. elegans strains
Nematodes were maintained through well-established procedures54. The following strains were provided by the CGC: SJ4103 [zcls14(Pmyo-3::mitoGFP)], PS6192 [syls243(Pmyo-3::TOM20::mRFP + unc-119( + ) + pBS Sk + )], and CU6372 [drp-1(tm1108) IV]. Strain pink-1(tm1779) was provided by the Mitani laboratory through the National BioResource Project of the MEXT, Japan. Other strains included UA202 [vtIs7(Pdat-1::GFP); sid-1
(pk3321); baIn33(Pdat-1::sid-1, Pmyo-2::mCherry)], UA312 [vtIs7(Pdat-1::GFP); drp-1(tm1108); baIn33(Pdat-1::sid-1, Pmyo-2::mCherry), sid-1(pk3321)], UA313 [vtIs7(Pdat-1::GFP); pink-1(tm1779); sid-1(pk3321); baIn33(Pdat-1::sid-1, Pmyo-2::mCherry)], UA316 [vtIs7(Pdat-1::GFP); pdr-1(gk448); sid-1(pk3321); baIn33(Pdat-1::sid-1, Pmyo-2::mCherry)].
Construction of drp-1 (tm1108), pink-1 (tm1779), and pdr-1 (gk448) RNAi strains
The mutant animals were crossed into strain UA202 [sid-1(pk3321); baIn33(Pdat-1::sid-1, Pmyo-2::mCherry); (Pdat-1::GFP)] to generate UA312, UA313, and UA316, respectively. The sid-1(pk3321) point mutation was detected using the following primers: forward CTTCTTGTATACTGAACGACG and reverse GCACAGTTATCAGATTTGCA. PCR samples were digested with the restriction enzyme ApoI for 2 h at 50 °C. The products were examined by agarose gel electrophoresis. Total PCR product is 874 bp; sid-1(pk3321) cut by ApoI restriction enzyme generated two fragments: 523 bp and 350 bp. After crossing mutant strains containing these alleles, the molecular lesions were probed using the following primers:
drp-1 (tm1108) Forward: CCAGACTTCGATGCCGTG
drp-1 (tm1108) Reverse: CCTCCGCATAGCTCAGTT G
pink-1(tm1779) Forward: GTTACAAGGCGAGCCTGAAAG
pink-1(tm1779) Reverse: GAAGCCTCGGGCTTATTAAGG
pdr-1(gk448) Forward: CACTTACGCAAGTGCTTCTTCG
pdr-1(gk448) Reverse: GTACGTGAGTTAGAGCTGC
Isolation and extraction of S. venezuelae metabolite
S. venezuelae metabolite was generated as previously described10, 11. Briefly, spores from S. venezuelae (ARS NRRL ISP-5230) were rehydrated at a density of 1 × 108 in 5 L SYZ media and were grown at 30 °C in a shaker for 3 weeks. For harvesting samples, cell debris was removed by centrifugation at 10,000 × g for 10 min and supernatants were sequentially passed through eight PES filter membranes with the following range of pore sizes: 11, 6, 2.7, 1.7, 1.2, 0.7, 0.45, and 0.22 μm. The conditioned media was extracted with an equal volume of dimethylchloride for 3 times using a separatory funnel. The dimethylchloride organic layers were collected, dried, and resuspended at a 1 × concentration, which is equivalent to 1 μl of metabolite resuspended in 1 ml EtAc.
S. venezuelae metabolite treatment
For the assays in this study, 20 × concentration of metabolite was used [2 μl of the concentrated stock solution was reconstituted in 100 μl EtAc] or EtAc (solvent control) was added to the surface of bacterial lawn on nematode growth medium (NGM) Petri plates (60 mm diameter) and allowed to dry. Animals were transferred to freshly made plates every other day and were exposed to the metabolite or solvent from hatching until the day of analysis, which was performed with the investigator blinded to sample identities. It should be noted that 20 × is the standard concentration used when analyzing GFP-only DA neurons. When our lab introduces a proteostasis stressor such as α-synuclein to DA neurons a 5 × concentration is appropriate11.
AICAR assay
AICAR (Adipogen, San Diego, CA, USA) was added directly to agar media at a final concentration of 1 mM as previously described55. Nematodes were exposed to AICAR from hatching to day 4, then were transferred to corresponding treatment plates without AICAR, as described in Fig. 6c. Dopaminergic neurodegeneration assays were then performed with the investigator blinded to sample identities using normal assay conditions.
RNAi treatments
RNAi feeding constructs were obtained from the C. elegans Ahringer library56. RNAi feeding clones were inoculated in LB culture with 100 μg/ml ampicillin, and grown overnight at 37 °C, shaking. Small (35 mm diameter) NGM plates containing IPTG-ampicillin (1 mM IPTG, 100μg/ml ampicillin) were seeded with 250 μl of RNAi bacteria culture and allowed to dry. RNAi feeding clones and double-stranded RNA was induced overnight at 20 °C. S. venezuelae metabolite solution [2 μl (20 × ) of the concentrated stock solution is reconstituted in 100 μl EtAc] or EtAc (solvent control) alone was spread onto corresponding RNAi plates and allowed to dry. Adult hermaphrodites were placed onto corresponding plates and allowed to lay eggs for 5 h to synchronize the F1 progeny.
Analysis of dopaminergic neurodegeneration
C. elegans dopaminergic neurons were analyzed for degeneration as previously described57. Briefly, on the day of analysis, which was performed with the investigator blinded to sample identities, the six anterior dopaminergic neurons [four CEP (cephalic) and two ADE (anterior deirid)] were examined in 30 randomly selected worms, repeated three times, for a total of 90 animals. Each animal was considered normal when there was a full complement of six anterior DA neurons. However, if a worm displayed any degenerative phenotype, such as a missing dendritic process, cell body loss, or a blebbing neuronal process, it was scored as degenerating. An average of total percentage of worms with normal neurons was reported in the study. A one-way ANOVA and Tukey’s post hoc test were used for statistical analysis.
Analysis of mitochondrial morphology in body-wall muscle cells
SJ4103 and PS6192 were utilized and scored as described previously11, 12. The mitochondrial matrix reporter was composed of the myo-3 promoter ligated to a mitochondrial leader sequence fused to GFP (referred to as “mitoGFP”)12, 58. Adult hermaphrodites were allowed to lay eggs on corresponding treatment plates for 5 h at 20 °C and were then removed. Animals were treated with 20 × concentrations of metabolite until day of analysis, which was performed with the investigator blinded to sample identities. Mitochondrial morphology in nematodes was measured by examining a consistent region near the vulva. In all cases, between five to eight muscle cells were examined. Mitochondrial morphology was considered normal when the majority of the cells within a single worm had ordered, tubular-shaped mitochondria. Fragmented cells consisted of disorganized circular forms of mitochondria and fused mitochondria had elongated and connected formations12. We examined 30 worms and repeated the analysis three times, so that 90 animals were analyzed. An average percentage of worms with each mitochondrial phenotype was reported in the study. A one-way ANOVA and Tukey’s post hoc test were used for statistical analysis.
Analysis of mitochondrial membrane potential (ΔΨm)
Tetramethylrhodamine ethyl ester (TMRE; Biotium, Fremont, CA, USA) is a lipophilic cation that accumulate into the mitochondrial matrix of live nematodes based on its ΔΨm20, 21, 59. C. elegans takes up TMRE from culture medium and incorporate it broadly into its tissues. Synchronized N2 (Bristol) animals were exposed to RNAi bacteria with metabolite in EtAc or EtAc alone until day 8 post-hatching. Worms were then placed on NGM plates containing 0.1μM TMRE for 18 h and washed with M9 before analysis, which was performed with the investigator blinded to sample identities. The live animals were photographed using a Nikon Eclipse E800 epifluorescence microscope at 10 × magnification. Relative uptake of the fluorescent dye was measured using MetaMorph software (Molecular Devices, Sunnyvale, CA, USA). When analyzing each animal, three 65 × 65 μm boxes were placed from most anterior portion of the intestine toward the vulva. Three independent replicates were conducted for each treatment with 30 animals per replicates for a total of 90 animals. A one-way ANOVA and Tukey’s post hoc were used for statistical analysis.
ATP measurements
The ATP measurement assay was conducted as described previously60, with minor modifications. Animals were fed bacterial clones expressing EV or eat-3 (RNAi) with metabolite in EtAc or EtAc alone from hatching until the analysis. The assay was performed with the investigator blinded to sample identities. Briefly, 100 age-synchronized young adult worms were collected in M9 buffer and washed three times. Worm pellets were treated with three freeze-thaw cycles and boiled for 15 min to releases ATP and destroy ATPase activity. Samples were then spun at 4 °C at 11000 g for 10 min. ATP contents were measured using an ATP determination kit (Molecular Probes, Eugene, OR, USA). Three independent samples were obtained for each condition and each sample was measured in duplicate using a Biotek Synergy H1 microplate reader. For normalization, protein levels from the same preparation were determined using a BCA assay (Thermo Scientific, Rockford, IL, USA).
Quantitative Real-Time PCR
Total RNA was isolated from 100 L4 worms (N2) from each independent sample using TRI reagent (Molecular Research Center). cDNA was synthesized with iScript Reverse Transcription Supermix for qRT-PCR (Bio-Rad, Hercules, CA, USA), as previously described11. qRT-PCR was performed with IQ-SYBR Green Supermix (Bio-Rad) with the Bio-Rad CFX96 Real-Time System. Each reaction contained: 7.5 μl of the IQSYBR Green Supermix, 200 nM of forward and reverse primers, and 0.3 μl cDNA, to a final volume of 15 μl. Expression levels were normalized to three reference genes (cdc-42, tba-1, and snb-1) and were calculated using qBasePLUS version 2.6 (Biogazelle, Gent, Belgium) for determining reference target stability. This study was performed using three technical replicates with three independent biological replicates. Each primer pair was confirmed for at least 90-110% efficiency in a standard curve on N2 cDNA. The following primers were used for the assays;
drp-1 Forward: GAAGACGGTCAAATGGAACAC
drp-1 Reverse: GCACGGCATCGAAGTCTGT
fzo-1 Forward: GTGCTGCCGATAATGAACCAC
fzo-1 Reverse: TTCCCGCTGTTCAGAACTAAC
eat-3 Forward: CGACATCTGCTCAAACTTCGAT
eat-3 Reverse: CCAAGACCCATTTGAATCGAAC
ced-9 Forward: CCATCACCGAGTAGGCAG
ced-9 Reverse: CGACCACAAATCCCTCGATA
snb-1 Forward: CCGGATAAGACCATCTTGACG
snb-1 Reverse: GACGACTTCATCAACCTGAGC
tba-1 Forward: GTACACTCCACTGCTCTGCTGACAAG
tba-1 Reverse: CTCTGTACAAGAGGCAAACAGCCATG
cdc-42 Forward: CTGCTGGACAGGAAGATTACG
cdc-42 Reverse: CTCGGACATTCTCGAATGAAG
Genomic DNA isolation and mitochondrial DNA copy number determination
C. elegans genomic DNA was isolated as previously described61, with minor modification. Briefly, N2 animals at day 8 post-hatching, exposed to RNAi conditions, were washed with M9 buffer to remove bacteria. Then, 6 worms from each condition were collected in 25 μl of worm lysis buffer pre-aliquoted into PCR tubes, and frozen in liquid nitrogen immediately for 5 min. The standard thermal cycler program used in this analysis was 60 min at 65 °C, 15 min at 95 °C, and hold at 8 °C. Samples were prepared in triplicate per condition. Using the lysed worm samples, the relative copy number of mtDNA to ncDNA was measured by qRT-PCR as described previously, which is specific for C. elegans DNA61. For each sample, three biological and three technical replicates were averaged from a single real-time qPCR run. A one-way ANOVA with Tukey’s post hoc test were used for multiple comparisons.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism software (Graphpad Software, Inc., La Jolla, CA, USA) and the appropriate statistical test and post hoc tests were applied as described. Data were expressed as the mean ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 were considered statistically significant.
Acknowledgements
Special thanks to Laura Berkowitz for her invaluable assistance, as well as all members of the Caldwell laboratory for their collegiality and collaborative nature. Some strains were provided by the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). RJP received support from a University of Alabama College of Arts & Sciences Undergraduate Creative Activity and Research Award.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by C. Munoz-Pinedo
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Subramaniam SR, Vergnes L, Franich NR, Reue K, Chesselet MF. Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol. Dis. 2014;70:204–213. doi: 10.1016/j.nbd.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kamp F, et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010;29:3571–3589. doi: 10.1038/emboj.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakamura K, et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011;286:20710–20726. doi: 10.1074/jbc.M110.213538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poole AC, et al. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl Acad. Sci. USA. 2008;105:638–643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb. Perspect. Biol. 2012;4:a011338–8. doi: 10.1101/cshperspect.a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu CT. A pivotal role for PINK1 and autophagy in mitochondrial quality control: implications for Parkinson disease. Hum. Mol. Genet. 2010;19:R28–R37. doi: 10.1093/hmg/ddq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luz AL, et al. Deficiencies in mitochondrial dynamics sensitize Caenorhabditis elegans to arsenite and other mitochondrial toxicants by reducing mitochondrial adaptability. Toxicology. 2017;387:1–14. doi: 10.1016/j.tox.2017.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caldwell KA, et al. Investigating bacterial sources of toxicity as an environmental contributor to dopaminergic neurodegeneration. PLoS ONE. 2009;4:e7227–10. doi: 10.1371/journal.pone.0007227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Richardson RJ. The risk of Parkinson’s disease with exposure to pesticides, farming, well water, and rural living. Neurology. 1998;50:1346–1350. doi: 10.1212/WNL.50.5.1346. [DOI] [PubMed] [Google Scholar]
- 10.Ray A, Martinez BA, Berkowitz LA, Caldwell GA, Caldwell KA. Mitochondrial dysfunction, oxidative stress, and neurodegeneration elicited by a bacterial metabolite in a C. elegans Parkinson’s model. Cell Death Dis. 2014;5:e984. doi: 10.1038/cddis.2013.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez BA, Kim H, Ray A, Caldwell GA, Caldwell KA. A bacterial metabolite induces glutathione-tractable proteostatic damage, proteasomal disturbances, and PINK1-dependent autophagy in C. elegans. Cell Death Dis. 2015;6:e1908–13. doi: 10.1038/cddis.2015.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Labrousse AM, Zappaterra MD, Rube DA, Van Der Bliek AMC. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol. Cell. 1999;4:815–826. doi: 10.1016/S1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- 13.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 14.van der Bliek AM. Fussy mitochondria fuse in response to stress. EMBO J. 2009;28:1533–1534. doi: 10.1038/emboj.2009.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishihara N, Jofuku A, Eura Y, Mihara K. Regulation of mitochondrial morphology by membrane potential, and DRP1-dependent division and FZO1-dependent fusion reaction in mammalian cells. Biochim. Biophys. Acta. 2003;301:891–898. doi: 10.1016/s0006-291x(03)00050-0. [DOI] [PubMed] [Google Scholar]
- 16.Toyama EQ, et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. doi: 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu S, Zhou F, Zhang Z, Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission–fusion proteins. FEBS J. 2011;278:941–954. doi: 10.1111/j.1742-4658.2011.08010.x. [DOI] [PubMed] [Google Scholar]
- 18.Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J. Cell Biol. 2000;151:367–380. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev. Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 20.Yoneda T. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 2004;117:4055–4066. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- 21.Zuryn S, Kuang J, Ebert P. Mitochondrial modulation of phosphine toxicity and resistance in Caenorhabditis elegans. Toxicol. Sci. 2008;102:179–186. doi: 10.1093/toxsci/kfm278. [DOI] [PubMed] [Google Scholar]
- 22.Narendra DP, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298–21. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bess AS, Crocker TL, Ryde IT, Meyer JN. Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans. Nucleic Acids Res. 2012;40:7916–7931. doi: 10.1093/nar/gks532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahn JM, Eom HJ, Yang X, Meyer JN, Choi J. Comparative toxicity of silver nanoparticles on oxidative stress and DNA damage in the nematode, Caenorhabditis elegans. Chemosphere. 2014;108:343–352. doi: 10.1016/j.chemosphere.2014.01.078. [DOI] [PubMed] [Google Scholar]
- 25.Calixto A, Chelur D, Topalidou I, Chen X, Chalfie M. Enhanced neuronal RNAi in C. elegans using SID-1. Nat. Methods. 2010;7:554–559. doi: 10.1038/nmeth.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harrington AJ, Yacoubian TA, Slone SR, Caldwell KA, Caldwell GA. Functional analysis of VPS41-mediated neuroprotection in Caenorhabditis elegans and mammalian models of Parkinson’s disease. J. Neurosci. 2012;32:2142–2153. doi: 10.1523/JNEUROSCI.2606-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanazawa T, et al. The C. elegans Opa1 Homologue EAT-3 is essential for resistance to free radicals. PLoS Genet. 2008;4:e1000022–12. doi: 10.1371/journal.pgen.1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu Y, Rolland SG, Conradt B. A molecular switch that governs mitochondrial fusion and fission mediated by the BCL2-like protein CED-9 of Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 2011;108:E813–E822. doi: 10.1073/pnas.1103218108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conradt B, Horvitz HR. The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell. 1998;93:519–529. doi: 10.1016/S0092-8674(00)81182-4. [DOI] [PubMed] [Google Scholar]
- 30.Buhlman L, et al. Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. BBA—Mol. Cell Res. 2014;1843:2012–2026. doi: 10.1016/j.bbamcr.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 31.Sandebring A, et al. Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS ONE. 2009;4:e5701. doi: 10.1371/journal.pone.0005701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pickrell AM, Youle RJ. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itoh K, Nakamura K, Iijima M, Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013;23:64–71. doi: 10.1016/j.tcb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Lett. Nat. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 35.Gandhi S, et al. Dopamine induced neurodegeneration in a PINK1 model of Parkinson’s disease. PLoS ONE. 2012;7:e37564–11. doi: 10.1371/journal.pone.0037564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi JS, Park C, Jeong JW. AMP-activated protein kinase is activated in Parkinson’s disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem Biophys. Res Commun. 2010;391:147–151. doi: 10.1016/j.bbrc.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 37.Wu Y, et al. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. NSG. 2011;19:163–174. doi: 10.1159/000328516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao J, et al. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants. 2017;6:25–19. doi: 10.3390/antiox6020025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seo AY, et al. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J. Cell Sci. 2010;123:2533–2542. doi: 10.1242/jcs.070490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arnold B, Cassady SJ, VanLaar VS, Berman SB. Integrating multiple aspects of mitochondrial dynamics in neurons: age-related differences and dynamic changes in a chronic rotenone model. Neurobiol. Dis. 2011;41:189–200. doi: 10.1016/j.nbd.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peng K, et al. The interaction of mitochondrial biogenesis and fission/fusion mediated by PGC-1α regulates rotenone-induced dopaminergic neurotoxicity. Mol. Neurobiol. 2017;54:3783–3797. doi: 10.1007/s12035-016-9944-9. [DOI] [PubMed] [Google Scholar]
- 42.Koopman WJH, et al. Inhibition of complex I of the electron transport chain causes O2-. -mediated mitochondrial outgrowth. Am. J. Physiol., Cell Physiol. 2005;288:C1440–C1450. doi: 10.1152/ajpcell.00607.2004. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, et al. DLP1-dependent mitochondrial fragmentation mediates 1-methyl-4-phenylpyridinium toxicity in neurons: implications for Parkinson’s disease. Aging Cell. 2011;10:807–823. doi: 10.1111/j.1474-9726.2011.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bajpai P, et al. Metabolism of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine by mitochondrion-targeted cytochrome P450 2D6: implications in Parkinson disease. J. Biol. Chem. 2013;288:4436–4451. doi: 10.1074/jbc.M112.402123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Houtkooper RH, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Lett. Nat. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt) Mol. Cell. 2015;58:123–133. doi: 10.1016/j.molcel.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increases HIF-1 activity. Curr. Biol. 2010;20:2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dillin A, et al. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- 49.Lutz AK, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 2009;284:22938–22951. doi: 10.1074/jbc.M109.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pryde KR, Smith HL, Chau KY, Schapira AHV. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 2016;213:163–171. doi: 10.1083/jcb.201509003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jagasia R, Grote P, Westermann B, Conradt B. DRP-1 mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–760. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- 52.Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev. Genet. 2016;43:95–118. doi: 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang C, Youle R. Cell biology: Form follows function for mitochondria. Lett. Nat. 2016;530:288–289. doi: 10.1038/530288a. [DOI] [PubMed] [Google Scholar]
- 54.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gioran A, Nicotera P, Bano D. Impaired mitochondrial respiration promotes dendritic branching via the AMPK signaling pathway. Cell Death Dis. 2014;5:e1175. doi: 10.1038/cddis.2014.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kamath RS, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Lett. Nat. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 57.Hamamichi S, et al. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. USA. 2008;105:728–733. doi: 10.1073/pnas.0711018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174:229–239. doi: 10.1534/genetics.106.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loew LM, Tuft RA, Carrington W, Fay FS. Imaging in five dimensions: time-dependent membrane potentials in individual mitochondria. Biophys. J. 1993;65:2396–2407. doi: 10.1016/S0006-3495(93)81318-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000556–14. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rooney JP, et al. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol. Biol. 2015;1241:23–38. doi: 10.1007/978-1-4939-1875-1_3. [DOI] [PMC free article] [PubMed] [Google Scholar]