

Abstract
Affective disorders such as major depression and bipolar disorder are among the most prevalent forms of mental illness and their etiologies involve complex interactions between genetic and environmental risk factors. Over the past ten years, several genome wide association studies (GWAS) have identified CACNA1C as one of the strongest genetic risk factors for the development of affective disorders. However, its role in disease pathogenesis is still largely unknown. Vulnerability to affective disorders also involves diverse environmental risk factors such as perinatal insults, childhood maltreatment, and other adverse pathophysiological or psychosocial life events. At the cellular level, such environmental influences may activate oxidative stress pathways, thereby altering neuronal plasticity and function. Mitochondria are the key organelles of energy metabolism and, further, highly important for the adaptation to oxidative stress. Accordingly, multiple lines of evidence including post-mortem brain and neuro-imaging studies suggest that psychiatric disorders are accompanied by mitochondrial dysfunction. In this study, we investigated the effects of Cacna1c downregulation in combination with glutamate-induced oxidative stress on mitochondrial function, Ca2+ homeostasis, and cell viability in mouse hippocampal HT22 cells. We found that the siRNA-mediated knockdown of Cacna1c preserved mitochondrial morphology, mitochondrial membrane potential, and ATP levels after glutamate treatment. Further, Cacna1c silencing inhibited excessive mitochondrial reactive oxygen species formation and calcium influx, and protected the HT22 cells from oxidative cell death. Overall, our findings suggest that the GWAS-confirmed psychiatric risk gene CACNA1C plays a major role in oxidative stress pathways with particular impact on mitochondrial integrity and function.
Introduction
Affective disorders such as major depression (MDD) and bipolar disorder (BD) are among the most prevalent forms of mental illness and affect more than 350 million people worldwide1. Their etiologies involve complex interactions between genetic and environmental risk factors2,3. The pronounced heritability of affective disorders suggests that genetic influences play a key role in disease pathogenesis4. In fact, the heritability ranges from 40–50% in MDD to 80–85% in BD, and family studies indicate for MDD a two- to threefold and for BD a nearly tenfold increase in lifetime risk among first-degree relatives5,6. During the past decade, several genome wide association studies (GWAS) have identified CACNA1C as one of the strongest genetic risk factors for the development of affective disorders7–9. It has been shown that the main single nucleotide polymorphism (SNP) rs1006737 is associated with increased mRNA expression of CACNA1C and enhanced calcium signaling10–12. CACNA1C codes for the pore-forming α1C subunit of CaV1.2, the major L-type voltage-gated calcium channel in the brain. Brain CaV1.2 channels couple Ca2+ influx to neuronal gene transcription and play an important role in synaptic plasticity, dendritic development, and cell survival13,14. However, the underlying cellular and molecular mechanisms explaining how this genetic risk variant contributes to the pathophysiology of neuropsychiatric disorders remain largely unknown15,16.
Vulnerability to affective disorders is not only based on genetic predisposition, but also involves diverse environmental risk factors such as intrauterine infections of the embryo, perinatal brain injuries attributed to birth complications, childhood maltreatment, migration, and other adverse pathophysiological and psychosocial life events17–19. There is accumulating evidence that such psychosocial stressors activate oxidative stress pathways, thereby disrupting the oxidant-antioxidant balance within the brain and causing oxidative damage to DNA, mitochondria, lipids, and proteins. Mitochondrial impairment, in turn, can accelerate the production of reactive oxygen species (ROS), which further modifies biological macromolecules and alters cellular functions20. In the brain, the accumulation of oxidative damage may result in loss of neuronal plasticity and function, and ultimately in apoptotic cell death, processes that have also been implicated as underlying mechanisms in the neuropathology of affective disorders21,22.
Mitochondria are the key organelles of energy metabolism and, thus, are highly important for adaptation to oxidative stress, calcium homeostasis, and regulation of cellular viability and function. Accordingly, numerous findings from neuro-imaging, post-mortem brain analyses, GWAS, and studies on peripheral biomarkers suggest that psychiatric disorders are accompanied by mitochondrial dysfunction23,24. These investigations revealed reduced complex I activity, increased ROS levels, decreased ATP production, downregulated expression of anti-apoptotic proteins, and upregulated expression of pro-apoptotic proteins in patients suffering from affective disorders or schizophrenia, all indicating impaired mitochondrial function.
In the present study, neuronal HT22 cells were used to elucidate whether the psychiatric risk gene Cacna1c affects oxidative stress-mediated disruption of mitochondrial function and, thereby, neuronal cell homeostasis. The applied mouse hippocampal HT22 cell line allows for the targeted manipulation of gene expression and serves as a well-established model system for glutamate-induced oxidative stress with significant mitochondrial impairment25,26. The specific aim of this study was to investigate the effects of reduced Cacna1c gene expression in combination with oxidative stress on mitochondrial function, Ca2+ homeostasis, and cell viability.
Results
Downregulation of Cacna1c preserved mitochondrial morphology and function independent of glutathione depletion in glutamate-challenged HT22 cells
In order to study the effects of Cacna1c depletion on mitochondrial parameters, we first validated the siRNA-mediated knockdown at the level of mRNA and protein. As shown in Fig. 1a, Cacna1c siRNA significantly reduced mRNA expression by 75% compared to control and siScr. In line with this finding, the immunoblot revealed a pronounced downregulation of CaV1.2 protein levels in siRNA-transfected HT22 cells (Fig. 1b). A comparable knockdown was also achieved using another Cacna1c siRNA sequence (siCacna1c 3; Supplementary Fig. S1).
Fig. 1. siRNA-mediated knockdown of Cacna1c prevented lipid peroxidation, but not glutathione depletion following glutamate exposure.

a Cacna1c mRNA levels were analyzed 24 h after siRNA transfection with 20 and 40 nM. Gapdh served as internal control. b Protein samples were collected 48 h after transfection with 40 nM siRNA and the CaV1.2 expression levels were then identified by Western blot. Vinculin was used as loading control. c Total glutathione levels were calculated from three replicates per condition after 0, 2, 4, and 6 h of glutamate treatment (10 mM). Data are provided as mean + SD. d, e Lipid peroxidation in HT22 cells was determined using BODIPY staining after an 8-h incubation with 9 mM glutamate. The dot plots show representative replicates and the bar graph summarizes the associated experiment where three replicates per sample are shown as percentage of cells in the upper right quarter (mean + SD; 10,000 cells per replicate). Ctrl control, siScr scrambled siRNA, siCacna1c Cacna1c siRNA, Glut glutamate. ***p < 0.001; ns (not significant) compared to glutamate-treated ctrl (ANOVA, Scheffé’s-test)
To investigate the effects of Cacna1c gene silencing on the mechanisms of glutamate-induced oxidative stress, we next analyzed glutathione levels (GSH) and lipid peroxidation in HT22 cells exposed to glutamate. Total GSH were determined 2, 4, and 6 h after the onset of glutamate treatment (Fig. 1c). Loss of GSH marks the initial step in oxidative glutamate toxicity and in fact, we observed a steady decline in GSH concentration over time27. Notably, there were no significant differences in GSH depletion between Cacna1c siRNA-transfected HT22 cells and controls. Glutathione depletion leads to the accumulation of lipid peroxides, which was evaluated here 8 h post-glutamate treatment using the fluorescent dye BODIPY followed by flow cytometric analysis26. The results revealed that the initial rise in lipid peroxidation upon glutamate exposure was largely prevented in HT22 cells transfected with Cacna1c siRNA, indicating a partial action upstream of mitochondria in the considered oxidative stress cascade (Fig. 1d,e).
Then, we focused on specific parameters of mitochondria, since these organelles play a crucial role in the employed paradigm of oxidative stress28. As displayed in Fig. 2a, we found a pronounced increase in mitochondrial fragmentation in the control and siScr-transfected cells after glutamate treatment. In contrast, Cacna1c siRNA-transfected cells mostly maintained their tubular, elongated morphology despite the glutamate challenge. The mitochondrial morphology was quantified by means of a classification into three different categories25. Briefly, cells containing healthy mitochondria with an elongated, tubule-like structure were defined as Category 1; while dying cells show strongly fragmented mitochondria accumulating around the nucleus and were classified as Category 3. Based on this classification system, the systematic quantification confirmed that Cacna1c gene silencing preserved mitochondrial morphology at Category 1 also after glutamate exposure (Fig. 2b). To substantiate these results, we additionally evaluated the expression of mitochondrial fission and fusion proteins, as well as mitochondrial biogenesis and mitophagy markers (Supplementary Fig. S2). While the total levels of mitofusin 2 (Mfn2), a protein that regulates the fusion of the outer mitochondrial membrane, were not significantly altered, we observed changes in the expression of the mitochondrial fission proteins Drp1 and Fis1. During mitochondrial fission, cytosolic dynamin-related protein 1 (Drp1) is recruited to the mitochondrial outer membrane where it interacts with mitochondrial fission protein 1 (Fis1) leading to constriction and fragmentation of the organelles. Upon glutamate challenge, the levels of both Drp1 and Fis1 were increased in the control conditions, whereas in the siCacna1c-transfected cells their levels remained unchanged. In addition to the rather moderate differences in the actual levels of the related fission and fusion proteins, it is likely that mitochondrial fragmentation is also regulated by modifications in their activity. The peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC1α) is a transcription factor that regulates genes involved in mitochondrial biogenesis. In glutamate-treated HT22 cells, Cacna1c knockdown led to elevated PGC1α levels, contrary to controls showing slightly decreased levels. Sequestosome 1 (p62) is involved in the recruitment of autophagosomal membranes to the mitochondria and interacts with the microtubule-associated protein light chain 3 isoform B (LC3B). During autophagy LC3B-I (16 kDa) is converted to LC3B-II (14 kDa) through lipidation. The expression of p62 and LC3B-II was increased after 16 h of glutamate treatment in Cacna1c siRNA-transfected and control cells alike. However, silencing of Cacna1c induced already a considerable increase in basal levels of both p62 and LC3B-II compared to controls. This finding is consistent with earlier studies stating that decreases in cytosolic Ca2+ can induce autophagy29. Our results indicate that Cacna1c downregulation stimulates mitochondrial biogenesis under oxidative stress without affecting mitophagic processes, thereby promoting the turnover of mitochondria and preventing the accumulation of dysfunctional mitochondria in HT22 cells.
Fig. 2. Cacna1c gene silencing preserved the morphology of healthy mitochondria in glutamate-challenged HT22 cells.
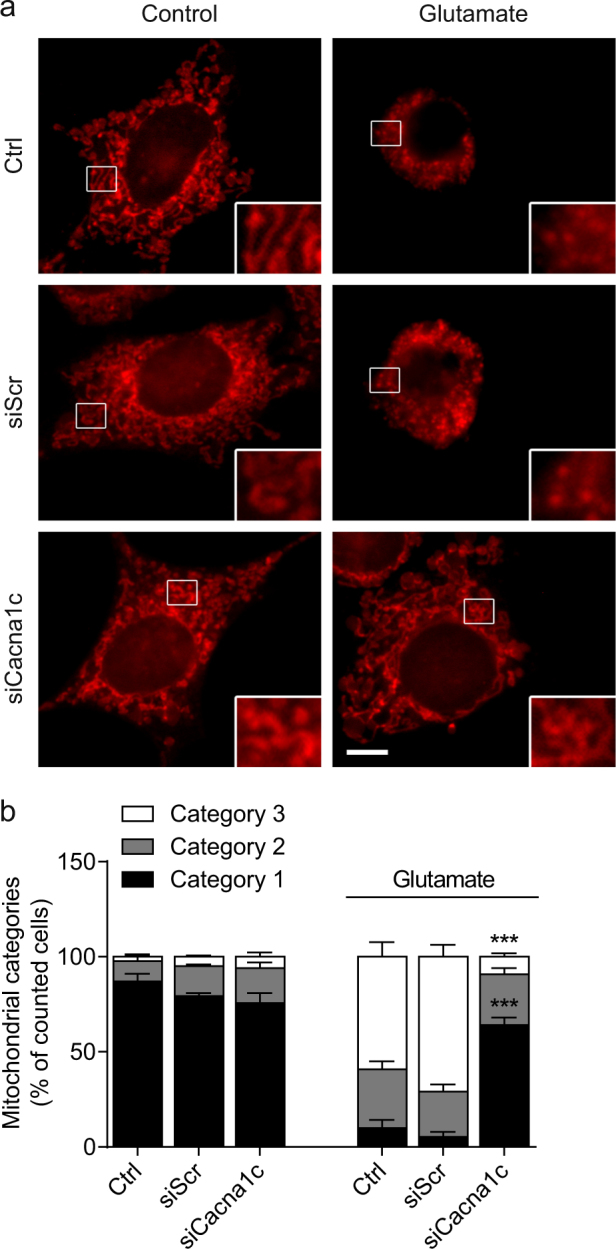
a Changes in mitochondrial morphology were visualized after 15 h of glutamate exposure using MitoTracker Deep Red. Epifluorescence images of representative HT22 cells are shown with details enlarged by 300% at the bottom right. Scale bar 10 µm. b Five hundred cells per condition were classified into three categories of mitochondrial fragmentation (Category 1: elongated; Category 2: intermediate; Category 3: fragmented) and quantified as percentage of counted cells (n = 3, mean + SD). ***p < 0.001 compared to glutamate-treated ctrl (ANOVA, Scheffé’s-test)
At the functional level, we next determined the concentration of cellular adenosine triphosphate (ATP), which provides the energetic basis for neuronal plasticity, viability, and function. In line with the previous findings on mitochondrial morphology, the downregulation of Cacna1c blocked the loss of ATP levels in response to glutamate (Fig. 3a). To further confirm the protective effects of Cacna1c knockdown in this model of oxytosis, the oxygen consumption rate (OCR) was analyzed as a measure of mitochondrial respiration. As illustrated by the OCR graphs shown in Fig. 3b, the basal respiration, represented by the OCR before injection of oligomycin (Oligo), was considerably higher in Cacna1c siRNA-transfected cells compared to controls after glutamate treatment. In line with this observation, the maximal mitochondrial respiration, detected after FCCP injection, was also significantly higher in glutamate-exposed cells with Cacna1c depletion versus controls. Here, the mitochondrial uncoupler FCCP mimics an increased energy demand as, for example, present in times of stress and, thereby, allows for the evaluation of the respiratory reserve. Our findings imply that mitochondrial dysfunction attributed to oxidative glutamate toxicity leads to decreased stress adaptation, which is prevented by a knockdown of Cacna1c. Besides the effects of Cacna1c downregulation after glutamate challenge, differences in the OCR between untreated conditions and Cacna1c siRNA-transfected cells can be observed, which may be functionally relevant. Compared to untreated controls, silencing of Cacna1c led to a lower basal and maximal OCR without a compensatory increase in glycolysis, as measured by the extracellular acidification rate (ECAR; Supplementary Fig. 3a). Thus, consistent with the slightly decreased basal ATP levels, depletion of Cacna1c resulted in a reduced metabolic potential, suggesting in particular a constitutively inhibited respiratory chain (Fig. 3a and Supplementary Fig. 3b). Reportedly, the activity of the electron transport complex IV is disinhibited by increases in cytosolic calcium concentration ([Ca2+]i) and is thereby presumably dependent on Cacna1c function. Consequently, Cacna1c knockdown may act as a form of preconditioning that prevents glutamate toxicity via complex IV inhibition and concurrent decrease in mitochondrial ROS production30. As described previously, the mitochondrial electron transport chain is the major source of ROS in this paradigm of glutamate-induced stress. Therefore, mitochondrial superoxide levels were detected by MitoSOX Red staining27. Fig. 3c demonstrates that mitochondrial ROS formation was significantly elevated in glutamate-challenged HT22 cells, but not in the Cacna1c siRNA-transfected cells. This protective effect was confirmed by another siRNA sequence (siCacna1c 3; comparable 62% reduction of CaV1.2 protein levels versus control), whereas a further siRNA (siCacna1c 2; only 21% reduction of CaV1.2 protein levels compared to controls) did not provide sufficient knockdown to affect mitochondrial ROS levels in glutamate-treated cells (Supplementary Fig. S4a). These results suggest that the functional effects of Cacna1c siRNA transfection strongly correlate with the extent of reduction in mRNA and protein levels (Supplementary Fig. S1). Another common feature of apoptotic and metabolically stressed cells is the collapse of the mitochondrial membrane potential (ΔΨm)31. According to Fig. 3d, the breakdown of ΔΨm in response to glutamate is indicated by a loss in TMRE fluorescence and this was visibly attenuated in Cacna1c-silenced cells. Furthermore, Cacna1c siRNA-transfected HT22 cells exhibited significantly less ΔѰm hyperpolarization in the surviving cell population after glutamate damage (Supplementary Fig. S5). A lower ΔѰm results from a decreased [Ca2+]i and is accompanied by less mitochondrial superoxide formation (Fig. 3c)32. These results further substantiate our findings from the OCR measurements and assist in elucidating the protective mechanisms of Cacna1c downregulation.
Fig. 3. Downregulation of Cacna1c gene expression mediated protection of mitochondrial function against glutamate-induced oxidative stress.

a A luciferase-based assay was used to determine ATP levels in glutamate-exposed (8 mM) HT22 cells. Values from eight replicate wells per condition are displayed as mean + SD. The luminescence under control conditions is set as 100%. b After 16 h of glutamate challenge, the oxygen consumption rate (OCR) was analyzed with a Seahorse XFe96 Analyzer. Data of 3–7 replicate wells per condition are given as mean ± SD. Oligo oligomycin, FCCP carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone, Rot rotenone, AA antimycin A. c Mitochondrial superoxide formation was investigated by flow cytometry using the fluorescent dye MitoSOX. HT22 cells were treated with 9 mM glutamate for 18 h. d Flow cytometric analysis of the mitochondrial membrane potential was realized via TMRE staining after 18 h of glutamate treatment. CCCP (carbonyl cyanide 3-chlorophenylhydrazone, 50 µM) is a mitochondrial membrane depolarizer and serves as positive control. c, d Each bar chart depicts one representative experiment with three replicates per sample (mean + SD; 10,000 cells per replicate). ***p < 0.001; **p < 0.01 compared to glutamate-treated ctrl (ANOVA, Scheffé’s-test)
Importantly, the detrimental mitochondrial Ca2+ overload constitutes a central event during programmed cell death and is tightly coupled to excessive mitochondrial ROS production33. Here, we assessed mitochondrial calcium concentrations using the fluorescent dye rhodamine-2. The fluorescence intensity of rhodamine-2 increases upon binding calcium and the quantification of gated cells displays that the glutamate-induced rise in mitochondrial calcium was fully blocked in Cacna1c siRNA-transfected cells compared to the controls (Fig. 4a,b). This effect of Cacna1c siRNA could be attributed to lower levels of CaV1.2 in the cell membrane, which may contribute to a reduced influx of exogenous Ca2+, thereby positively influencing the deregulated calcium homeostasis in this model of oxidative stress34. This conclusion was further supported by the effects of the dihydropyridine (DHP) calcium channel blocker nimodipine, which protected HT22 cells against glutamate-induced mitochondrial calcium overload at a concentration of 100 µM similar to the effects achieved by Cacna1c knockdown (Supplementary Fig. S6a). To further investigate the mechanisms linking Cacna1c downregulation to mitochondrial calcium load we next assessed the cellular localization of CaV1.2 in HT22 cells (Supplementary Fig. S7a). Consistent with existing evidence describing a plasma membrane localization of voltage-gated calcium channels, we found that CaV1.2 was mostly present in the total lysate and to a lesser extent in the cytosolic fraction. As expected, CaV1.2 was not appreciably expressed in the mitochondrial fraction rather excluding a direct effect of Cacna1c gene silencing on mitochondrial calcium concentration. Therefore, we then evaluated if Cacna1c knockdown indirectly affects the expression of the mitochondrial calcium uniporter (MCU; Supplementary Fig. S7b,c). The MCU is an inner mitochondrial membrane transport protein and essential for the regulation of calcium uptake. Under control conditions, oxidative glutamate toxicity caused an increase in MCU levels after 16 h. This glutamate-induced rise in MCU expression was significantly abolished in both Cacna1c siRNA-transfected and nimodipine-treated cells, thereby correlating well with our findings from the Rhod-2 calcium measurements. Furthermore, these results are in line with a previous study showing that knockdown of MCU in mouse neurons reduces NMDA-induced increases in mitochondrial calcium resulting in resistance to excitotoxicity35. Overall, these results demonstrate that both Cacna1c gene silencing and pharmacological inhibition of L-type calcium channels (LTCCs) provided a consistent protection of different mitochondrial parameters in paradigms of oxidative glutamate neurotoxicity.
Fig. 4. Depletion of Cacna1c attenuated the glutamate-induced rise in mitochondrial calcium concentration.
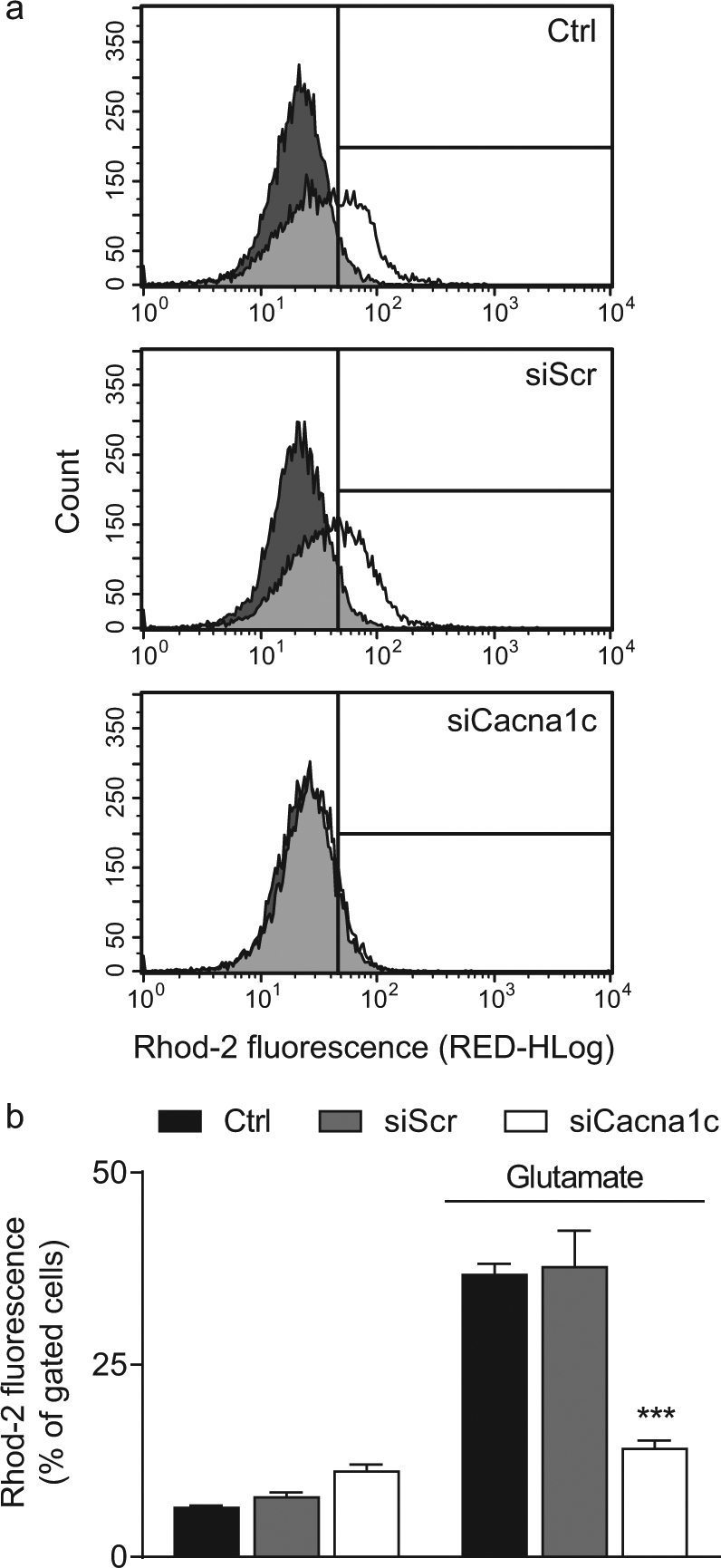
a The mitochondria-specific dye dihydrorhod-2 AM was used for the flow cytometric calcium measurements. The dark grey graphs represent the untreated controls, the white graphs the glutamate-treated samples, and the light grey areas the overlay of both graphs. b Here, the right shift in Rhod-2 fluorescence in response to 11 mM glutamate was quantified. The bar graph shows one representative experiment with three replicates per sample (mean + SD; 10,000 cells per replicate). ***p < 0.001 compared to glutamate-treated ctrl (ANOVA, Scheffé’s-test)
Knockdown of Cacna1c mediated protection against glutamate-induced cell death
The positive effects of Cacna1c depletion in the applied model of oxidative stress were also reflected at the level of cell morphology and viability. Representative images of glutamate-treated controls showed characteristic features of dying cells, which were shrinking, rounding up, and detaching from the culture dish. In contrast, healthy and viable cells appeared elongated and well attached to the well bottom, as detected in the untreated control and glutamate-exposed siCacna1c conditions (Fig. 5a).
Fig. 5. Cacna1c silencing protected HT22 cells from glutamate-induced cell death.

a Representative phase-contrast images of glutamate-treated HT22 cells illustrate changes in cell morphology. Scale bar 25 µm. b A representative real time cell impedance measurement over 30 h after the treatment with 8 mM glutamate is shown. Eight replicate wells per condition are displayed as mean ± SD. c Before cell viability was determined by MTT assay, HT22 cells were challenged with 8 mM glutamate for 16 h. Values from 8 wells per condition are presented as mean + SD. The absorbance under control conditions is set as 100%. d The bar graph shows the percentage of annexin V and AV/PI positive cells. One representative experiment with three replicates per sample is depicted (mean + SD; 10,000 cells per replicate). e In addition, the dot plots exemplify the subdivision into AV (lower right quarter) and AV/PI positive cells (upper right quarter). AV annexin V, PI propidium iodide. ***p < 0.001 compared to glutamate-treated ctrl (ANOVA, Scheffé’s-test)
Finally, we investigated cell proliferation and viability in real time via continuous electrical impedance readout and found that, compared to controls, Cacna1c siRNA-transfected cells showed a significantly lower proliferation rate under basal conditions (Fig. 5b). As cell proliferation is tightly coupled to energy metabolism, this effect may be due to the before mentioned siCacna1c-mediated inhibition of the mitochondrial electron transport chain36. However, Cacna1c silencing mediated persistent protection of HT22 cells from glutamate toxicity. On the contrary, in the glutamate-treated controls a fast decrease in the normalized cell index was observed after 12 to 15 h of glutamate administration, indicating cell death. In line with this result from real time impedance measurements, the protective effect of the Cacna1c siRNA was also detected in the MTT reduction assay after 16 h of glutamate exposure (Fig. 5c). Both findings were independently confirmed by an additional siRNA sequence (siCacna1c 3; Supplementary Fig. S4b,c). Accordingly, flow cytometric measurements using annexin V and propidium iodide (PI) provided concurrent results. The two dyes stain early apoptotic and late apoptotic/necrotic cells respectively (Fig. 5d,e). The quantification presented in Fig. 5d indicates a significantly enhanced resistance against glutamate-induced cell death in Cacna1c siRNA-transfected cells. Supporting these results, nimodipine (100 µM) also protected the cells from glutamate toxicity as shown by the MTT assay and annexin V/PI staining (Supplementary Fig. S6b,c).
Discussion
In the present study, we found that both siRNA-mediated downregulation and pharmacological inhibition of Cacna1c in neuronal HT22 cells mediated substantial protective effects on lipid peroxidation, mitochondrial integrity and function, and cell viability in a model of glutamate-induced cell death. These results suggest that, in paradigms of oxidative stress, Cacna1c is significantly involved in mechanisms of neuronal damage.
Our findings may also be of potential relevance in human diseases, since hippocampal and cortical atrophy, which are closely associated with abnormalities in neuronal plasticity and reduced resilience to cellular stress, have been frequently reported in patients with psychiatric disorders and early-life adverse experiences37,38. Furthermore, recent studies revealed that healthy subjects carrying the CACNA1C risk variant rs1006737 also show decreased prefrontal cortex activation and hippocampal dysfunction39,40. Even though it is not consistently observed41,42, the majority of evidence suggests that the non-coding risk SNP rs1006737 is associated with enhanced CACNA1C expression and a gain of CaV1.2 function10,12,43, suggesting deregulated Ca2+ signaling in the affected population and a significantly increased probability of psychiatric diseases13. These findings are further confirmed by several animal studies demonstrating that, besides an increase in anxiety-associated behavior44, Cacna1c heterozygous and forebrain-specific knockout mice exhibit an antidepressant-like phenotype, resilience to chronic stress, and a protective phenotype against mania-related behavior45–48. In addition to the data based on genetic modifications, the pharmacological inhibition of L-type calcium channels has repeatedly been linked to anti-stress and antidepressant-like effects, and reduced despair-like behavior in rodents49–51. Moreover, dihydropyridine compounds with good central nervous system (CNS) penetration, such as nimodipine showed promising mood stabilizing effects in humans with affective disorders, suggesting that LTCC antagonists represent an additional option in the pharmacotherapy of psychiatric diseases, and especially patients carrying the CACNA1C risk SNP could benefit from this alternative treatment strategy13,43,52. Elevated cytosolic calcium levels, which may be associated with the CACNA1C risk SNP rs 1006737, represent a major upstream pathway that impairs mitochondrial function in BD53. In this regard, it has been reviewed recently that systemic administration of the DHP isradipine suppresses cytosolic calcium transients, increases mitochondrial mass and lowers mitophagy in dopaminergic neurons54. These findings in vivo are in line with our own results in vitro, revealing an enhanced resistance against oxidative stress and beneficial effects on mitochondrial function in conditions of reduced Cacna1c expression and LTCC blockade with nimodipine.
As reported previously, impaired cellular adaptation to environmental stress constitutes another major trigger in the pathophysiology of psychiatric disorders2. In particular, it has been demonstrated in animal models that psychosocial stress results in depression-like behavior through the activation of oxidative and apoptotic mechanisms, including increased ROS levels, lipid peroxidation, reduced Bcl-2 expression, and increased caspase-322. Additionally, in human post-mortem studies a downregulation of several antioxidant genes and an increase in protein oxidation have been observed in the hippocampus and prefrontal cortex of patients with BD55,56. Hence, the applied model of glutamate-induced oxidative stress in hippocampal HT22 cells constitutes a suitable system reflecting a common cellular response to environmental stress in the development and course of psychiatric illnesses.
It has been repeatedly proposed that interactions between genetic and environmental risk factors significantly contribute to the complex and heterogeneous clinical manifestations of affective disorders2. Our study provides valuable insights into a gene-stress-interaction by showing that reduced Cacna1c expression, which represents a beneficial genetic factor, mediated neuroprotective effects against oxidative stress, particularly at the level of mitochondria. Mitochondrial function is essential for cellular resilience and adaptation to oxidative stress. Here, siRNA-mediated knockdown of Cacna1c preserved mitochondrial morphology, ATP values, respiration, membrane potential, and calcium homeostasis, and, furthermore, diminished excessive ROS formation in the employed model of oxidative glutamate toxicity. These findings are highly relevant as increasing evidence suggests a crucial role for mitochondrial dysfunction and related key determinants of cellular stress, such as intracellular Ca2+ deregulation and ROS formation in the development of mood disorders24.
Furthermore, our findings indicate a possible link between Cacna1c expression and mitochondrial function in oxytosis. In this paradigm of oxidative cell death, we found that both Cacna1c gene silencing and pharmacological inhibition of LTCC using nimodipine significantly diminished the glutamate-induced rise in mitochondrial calcium levels. We conclude that this effect was attributed to a reduced cellular calcium influx, thereby leading to a mild inhibition of complex IV and also affecting the downstream mitochondrial Ca2+ influx. We showed previously, however, that Ca2+ influx through the plasma membrane only partially contributed to mitochondrial damage and cell death in the applied model of glutamate-induced oxidative stress57. Based on the pronounced protective effects of Cacna1c knockdown and LTCC blockade, it has to be taken into consideration that, beyond its influence on calcium homeostasis at the plasma membrane, Cacna1c is possibly involved in further regulatory pathways at the level of mitochondria. For example, the calcium-dependent cytosolic phosphatase calcineurin dephosphorylates Drp1 thereby promoting its translocation to mitochondria and triggering detrimental mitochondrial fission; a process that is likely inhibited by Cacna1c gene silencing58. Moreover, both Cacna1c knockdown and LTCC blockade probably lead to altered CaV1.2-dependent gene transcription regulation and may suppress toxic mitochondrial Ca2+ uptake via transcriptional repression of enhanced MCU expression following glutamate treatment in HT22 cells35. Conversely, it has been described in arterial myocytes that mitochondria stimulate NF-κB-dependent CaV1.2 expression via elevated mitochondrial calcium concentration and subsequently induced mitochondrial ROS generation59. In this regard, it is suggested that silencing of Cacna1c possibly counteracts the respective feed-forward signaling cascade. In mast cells, however, Cacna1c knockdown augmented apoptosis after IgE stimulation by disrupting mitochondrial integrity, emphasizing that the maintenance of an appropriate mitochondrial calcium concentration is also essential for cell survival60.
We also observed that Cacna1c downregulation led to a reduced formation of lipid peroxides in HT22 cells upon glutamate exposure, indicating a partial action upstream of mitochondria. But since mitochondrial damage acts as an amplification factor of lipid peroxidation, mitochondria might still represent the main point of action of Cacna1c silencing in this model of oxidative glutamate toxicity26.
Overall, our findings suggest that the GWAS-confirmed psychiatric risk gene CACNA1C plays a major role in oxidative stress pathways with particular impact on mitochondrial integrity and function. However, the precise molecular mechanisms underlying the effects of CACNA1C regulation on mitochondrial performance in neurons and the emerging link to neuronal plasticity, maintenance and function in the context of affective disorders remain to be elucidated.
Materials and methods
Cell culture
HT22 cells were immortalized from primary hippocampal neurons using a temperature-sensitive SV-40 T antigen and selected from HT4 cells based on sensitivity to glutamate61,62. Since HT22 cells do not express functional ionotropic glutamate receptors, glutamate-induced cell death is mediated via an oxidative stress-dependent pathway called oxytosis. Oxidative glutamate toxicity in HT22 cells involves the inhibition of the glutamate/cystine antiporter, a subsequent depletion of glutathione, an increase in ROS production, a consecutive impairment of mitochondria, and an influx of Ca2+, which ultimately leads to neuronal cell death25–27,31.
HT22 cells were cultured at 37 °C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich, Munich, Germany) supplemented with 10% fetal bovine serum, 20 mM HEPES, 100 units/ml penicillin, 100 µg/ml streptomycin, and 2 mM glutamine. The cells were transfected with an optimized concentration of 40 nM siRNA using Lipofectamine RNAiMAX (Thermo Fisher Scientific, Darmstadt, Germany) in antibiotic-free growth medium. The siRNA sequences were as follows: scrambled Cacna1c siRNA (siScr, 5′-AUUGGCGCACUAUUAGACU-3′), Cacna1c siRNA (siCacna1c, 5′-GGAAAGCUCUAUACCUGUU-3′). The plated cells were treated with millimolar concentrations of glutamate for 15 to 18 h unless otherwise stated and assayed 30 to 48 h after transfection. The L-type calcium channel (LTCC) blocker nimodipine (100 mM stock solution in DMSO; Cayman Chemical, Ann Arbor, Michigan, USA) was used in a final concentration of 100 µM and applied together with 6 to 8 mM glutamate for 16 h.
RT-PCR
HT22 cells were seeded at a density of 180,000 cells/well in 6-well plates. Total RNA was isolated with the InviTrap Spin Universal RNA Mini Kit (Stratec Biomedical, Birkenfeld, Germany) 24 h after siRNA transfection. Reverse transcription PCR (RT-PCR) was performed using SuperScript III One-Step RT-PCR System (Thermo Fisher Scientific, Darmstadt, Germany) and the following primers: Cacna1c (502 bp) forward 5′-CAACGCAACCATCTCTACCG-3′ and reverse 5′-CAGTGCCTTCACGTC-GAATC-3′; Gapdh (399 bp) forward 5′-CGTCTTCACCACCATGGAGAAGGC-3′ and reverse 5′-AAGGCCATGCCAGTGAGCTTCCC-3′. The amplified products were visualized under UV light on a 1.5% agarose gel.
Protein analysis
Protein extraction and Western blot were performed according to established protocols31. Briefly, cells were lysed 48 h after transfection in 0.25 M D-mannitol, 0.05 M Tris base, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and 1% Triton X-100 supplemented with protease and phosphatase inhibitor cocktail tablets (Roche Diagnostics, Mannheim, Germany). The total protein amount was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Darmstadt, Germany) and 60 µg protein were loaded on a 7.5% polyacrylamide gel. After electrophoresis, the proteins were transferred from the gel onto a PVDF membrane (Roche Diagnostics, Mannheim, Germany). Then, the membranes were incubated with anti-CaV1.2 antibody (1:200; Alomone Labs, Jerusalem, Israel). Vinculin was detected as a loading control using an anti-Vinculin antibody (1:20,000; Sigma-Aldrich, Munich, Germany).
Determination of total glutathione
The amount of total glutathione in HT22 cells was quantified using the luminescence-based GSH/GSSG-Glo Assay (Promega, Mannheim, Germany) in accordance with the manufacturer’s instructions. HT22 cells were plated in a density of 6,000 cells/well in white-with-clear-bottom 96-well plates. Twenty-four hours after seeding, the cells were incubated for 2, 4, and 6 h with 10 mM glutamate. Absolute glutathione concentrations were calculated from a GSH standard curve.
Mitochondrial morphology
The evaluation of mitochondrial morphology was carried out as reported and validated previously with minor modifications25. In brief, 14,000 cells/well were seeded in 8-well ibiTreat plates (Ibidi, Martinsried, Germany). To visualize mitochondria, cells were incubated with 0.2 µM MitoTracker Deep Red (Thermo Fisher Scientific, Darmstadt, Germany) for 30 min before adding glutamate. After the fixation with 4% paraformaldehyde, images were acquired using a fluorescence microscope (DMI6000 B; Leica Microsystems, Wetzlar, Germany) at a 63× magnification. To generate reliable results according to previous validation, 500 cells per condition were counted and classified by their mitochondrial morphology into three different categories. Category 1 is defined by elongated and equally distributed mitochondria. Category 2 is characterized by partially fragmented mitochondria that are still distributed throughout the cytosol. Finally, Category 3 is represented by strongly fragmented mitochondria that are located around the nucleus. Three independent experiments were performed and the investigator was always blinded to the treatment history.
ATP measurement
The determination of ATP levels was performed using the ViaLight Plus Kit (Lonza, Verviers, Belgium) according to the manufacturer’s protocol. Briefly, cells were lysed, transferred to a white-walled 96-well plate and the luciferase containing ATP monitoring reagent was added. The resulting luminescence was detected with a FLUOstar OPTIMA reader (BMG Labtech, Ortenberg, Germany).
Measurement of OCR
The OCR as an indicator of mitochondrial respiration was measured with a Seahorse XFe96 Analyzer (Agilent Technologies, Waldbronn, Germany). The system allows for the consecutive injection of different modulators of the electron transport chain. The compounds and final concentrations used are as follows: oligomycin 3 µM, FCCP 0.5 µM, rotenone 0.1 µM, and antimycin A 1 µM. Three basal and three measurements after each injection were recorded. HT22 cells were assayed in DMEM containing 25 mM D-glucose, 2 mM glutamine, and 1 mM sodium pyruvate with an adjusted pH of 7.35.
Flow cytometry
Several parameters associated with glutamate-induced oxidative stress, such as lipid peroxidation, mitochondrial ROS formation, mitochondrial membrane potential, mitochondrial Ca2+ levels, and cell death, were analyzed with a Guava easyCyte 6–2 L flow cytometer (Merck Millipore, Darmstadt, Germany) using different fluorescent dyes. HT22 cells were plated at 30,000–55,000 cells/well in a 24-well format. Measurements were performed in triplicates, each with a total count of 10,000 cells, and are representative of a least three independent experiments.
Lipid peroxidation
BODIPY 581/591 C11 (4,4-difluoro-5-(4-phenyl-1,3-butadienyl)-4-bora-3a,4a-diaza-s-indacene-3-undecanoic acid; Thermo Fisher Scientific, Darmstadt, Germany) was utilized as a sensor for the oxidation of lipids and membranes. Here, HT22 cells were treated with 9 mM glutamate 24 h after siRNA transfection. Following 8 h of glutamate treatment, the cells were stained with 2 µM BODIPY dye for 1 h at 37 °C.
Mitochondrial superoxide formation
Superoxides are the predominant reactive oxygen species in mitochondria and were detected by incubation of the cells with 1.25 µM MitoSOX Red indicator (Thermo Fisher Scientific, Darmstadt, Germany) for 30 min at 37 °C27.
Mitochondrial membrane potential
Mitochondrial membrane depolarization was determined using the MitoPT TMRE Kit (ImmunoChemistry Technologies, Hamburg, Germany) following the recommendations of the manufacturer. HT22 cells were collected and stained with 0.2 µM TMRE (tetramethylrhodamine ethyl ester) for 30 min at 37 °C. The depolarizing reagent CCCP (carbonyl cyanide 3-chlorophenylhydrazone) was used in a final concentration of 50 µM and served as additional positive control.
Mitochondrial calcium concentration
To selectively study mitochondrial calcium levels Rhod-2 AM (rhodamine-2 acetoxymethyl ester; Thermo Fisher Scientific, Darmstadt, Germany) was reduced to Dihydrorhod-2 AM. Then, HT22 cells were harvested and incubated with 2 µM of the reduced dye in DMEM without serum for 30 min.
Cell death
Cell death was evaluated using the Annexin V-FITC Detection Kit (Promokine, Heidelberg, Germany) in accordance with the manufacturer’s instructions. The two containing dyes Annexin V and PI stain early apoptotic and dead cells respectively. Shortly, HT22 cells were trypsinized and the cell suspensions incubated with 5 µl/ml of each dye for 5 min at room temperature.
Cell viability
Cell viability was assessed by a colorimetric assay where MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide, 0.5 mg/ml; Sigma-Aldrich, Munich, Germany) is NADH-dependently reduced to a purple colored formazan product, which is then quantified by absorbance measurement at 570 nm with a reference filter at 630 nm.
As described previously, the xCELLigence Real-Time Cell Analysis (RTCA; Roche Diagnostics, Mannheim, Germany) system allows cell viability to be measured continuously in real time via electrical impedance readout63. Briefly, attached and proliferating cells cause a high and further increasing impedance signal, whereas the detachment of dying cells leads to a decrease in electrical impedance. Changes in the impedance are depicted as normalized cell index. For both assays HT22 cells were seeded in 96-well plates at 6,000-8,000 cells per well.
Statistical analysis
All data are presented as mean values with standard deviation (SD). Multiple comparisons were performed by one-way analysis of variance (one-way ANOVA) followed by Scheffé’s post hoc test. All experiments were performed in 3 to 8 replicates per treatment condition, as indicated, and were independently repeated at least twice for confirmation of the results. The parameters presented were calculated using the WinSTAT statistics Add-In for Microsoft Excel (R. Fitch Software, Bad Krozingen, Germany). A statistical significant difference was assumed at p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
Electronic supplementary material
Acknowledgements
This work is part of the DFG FOR 2107 and supported by a DFG funding to CC (CU 43/9-1). The authors thank Katharina Elsässer for her excellent technical support and Emma Esser for careful editing of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by R. Killick
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41420-018-0061-6.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.World Health Organization. Mental disorders. Fact sheet. http://www.who.int/mediacentre/factsheets/fs396/en/ (2017).
- 2.Keers R, Uher R. Gene-environment interaction in major depression and antidepressant treatment response. Curr. Psychiatry Rep. 2012;14:129–137. doi: 10.1007/s11920-011-0251-x. [DOI] [PubMed] [Google Scholar]
- 3.Kupfer DJ, Frank E, Phillips ML. Major depressive disorder: new clinical, neurobiological, and treatment perspectives. Lancet. 2012;379:1045–1055. doi: 10.1016/S0140-6736(11)60602-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Merikangas KR, Low NCP. The epidemiology of mood disorders. Curr. Psychiatry Rep. 2004;6:411–421. doi: 10.1007/s11920-004-0004-1. [DOI] [PubMed] [Google Scholar]
- 5.Lohoff FW. Overview of the genetics of major depressive disorder. Curr. Psychiatry Rep. 2010;12:539–546. doi: 10.1007/s11920-010-0150-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnett JH, Smoller JW. The genetics of bipolar disorder. Neuroscience. 2009;164:331–343. doi: 10.1016/j.neuroscience.2009.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferreira MAR, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green EK, et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol. Psychiatry. 2010;15:1016–1022. doi: 10.1038/mp.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bigos KL, et al. Genetic variation in CACNA1C affects brain circuitries related to mental illness. Arch. Gen. Psychiatry. 2010;67:939–945. doi: 10.1001/archgenpsychiatry.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heyes S, et al. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog. Neurobiol. 2015;134:36–54. doi: 10.1016/j.pneurobio.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshimizu T, et al. Functional implications of a psychiatric risk variant within CACNA1C in induced human neurons. Mol. Psychiatry. 2015;20:162–169. doi: 10.1038/mp.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhat S, et al. CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog. Neurobiol. 2012;99:1–14. doi: 10.1016/j.pneurobio.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berridge MJ. Calcium signalling and psychiatric disease: bipolar disorder and schizophrenia. Cell Tissue Res. 2014;357:477–492. doi: 10.1007/s00441-014-1806-z. [DOI] [PubMed] [Google Scholar]
- 15.Harrison PJ. Molecular neurobiological clues to the pathogenesis of bipolar disorder. Curr. Opin. Neurobiol. 2016;36:1–6. doi: 10.1016/j.conb.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kabir ZD, Lee AS, Rajadhyaksha AM. L-type Ca2+channels in mood, cognition and addiction: integrating human and rodent studies with a focus on behavioural endophenotypes. J. Physiol. 2016;594:5823–5837. doi: 10.1113/JP270673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hagberg H, et al. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 2015;11:192–208. doi: 10.1038/nrneurol.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nanni V, Uher R, Danese A. Childhood maltreatment predicts unfavorable course of illness and treatment outcome in depression: a meta-analysis. Am. J. Psychiatry. 2012;169:141–151. doi: 10.1176/appi.ajp.2011.11020335. [DOI] [PubMed] [Google Scholar]
- 19.Pignon B, et al. Prevalence and clinical severity of mood disorders among first-, second- and third-generation migrants. J. Affect. Disord. 2017;210:174–180. doi: 10.1016/j.jad.2016.12.039. [DOI] [PubMed] [Google Scholar]
- 20.Salim S. Oxidative stress and the central nervous system. J. Pharmacol. Exp. Ther. 2017;360:201–205. doi: 10.1124/jpet.116.237503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim HW, Rapoport SI, Rao JS. Altered expression of apoptotic factors and synaptic markers in postmortem brain from bipolar disorder patients. Neurobiol. Dis. 2010;37:596–603. doi: 10.1016/j.nbd.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kubera M, Obuchowicz E, Goehler L, Brzeszcz J, Maes M. In animal models, psychosocial stress-induced (neuro)inflammation, apoptosis and reduced neurogenesis are associated to the onset of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2011;35:744–759. doi: 10.1016/j.pnpbp.2010.08.026. [DOI] [PubMed] [Google Scholar]
- 23.Manji H, et al. Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci. 2012;13:293–307. doi: 10.1038/nrn3229. [DOI] [PubMed] [Google Scholar]
- 24.Scaini G, et al. Mitochondrial dysfunction in bipolar disorder: evidence, pathophysiology and translational implications. Neurosci. Biobehav. Rev. 2016;68:694–713. doi: 10.1016/j.neubiorev.2016.06.040. [DOI] [PubMed] [Google Scholar]
- 25.Grohm J, Plesnila N, Culmsee C. Bid mediates fission, membrane permeabilization and peri-nuclear accumulation of mitochondria as a prerequisite for oxidative neuronal cell death. Brain. Behav. Immun. 2010;24:831–838. doi: 10.1016/j.bbi.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 26.Tobaben S, et al. Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons. Cell Death. Differ. 2011;18:282–292. doi: 10.1038/cdd.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neitemeier S, et al. BID links ferroptosis to mitochondrial cell death pathways. Redox. Biol. 2017;12:558–570. doi: 10.1016/j.redox.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Culmsee C, Landshamer S. Molecular insights into mechanisms of the cell death program: role in the progression of neurodegenerative disorders. Curr. Alzheimer Res. 2006;3:269–283. doi: 10.2174/156720506778249461. [DOI] [PubMed] [Google Scholar]
- 29.Williams A, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huber SM, et al. Ionizing radiation, ion transports, and radioresistance of cancer cells. Front. Physiol. 2013;4:212. doi: 10.3389/fphys.2013.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landshamer S, et al. Bid-induced release of AIF from mitochondria causes immediate neuronal cell death. Cell Death Differ. 2008;15:1553–1563. doi: 10.1038/cdd.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Braun N, et al. UCP-3 uncoupling protein confers hypoxia resistance to renal epithelial cells and is upregulated in renal cell carcinoma. Sci. Rep. 2015;5:13450. doi: 10.1038/srep13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Maher P, Schubert D. Requirement for cGMP in nerve cell death caused by glutathione depletion. J. Cell Biol. 1997;139:1317–1324. doi: 10.1083/jcb.139.5.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu J, et al. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013;4:2034. doi: 10.1038/ncomms3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birsoy K, et al. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 2015;162:540–551. doi: 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmaal L, et al. Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the ENIGMA Major Depressive Disorder Working Group. Mol. Psychiatry. 2017;22:900–909. doi: 10.1038/mp.2016.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frodl T, Reinhold E, Koutsouleris N, Reiser M, Meisenzahl EM. Interaction of childhood stress with hippocampus and prefrontal cortex volume reduction in major depression. J. Psychiatr. Res. 2010;44:799–807. doi: 10.1016/j.jpsychires.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 39.Erk S, et al. Replication of brain function effects of a genome-wide supported psychiatric risk variant in the CACNA1C gene and new multi-locus effects. Neuroimage. 2014;94:147–154. doi: 10.1016/j.neuroimage.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 40.Paulus FM, et al. Association of rs1006737 in CACNA1C with alterations in prefrontal activation and fronto-hippocampal connectivity. Hum. Brain Mapp. 2014;35:1190–1200. doi: 10.1002/hbm.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gershon ES, et al. A rare mutation of CACNA1C in a patient with bipolar disorder, and decreased gene expression associated with a bipolar-associated common SNP of CACNA1C in brain. Mol. Psychiatry. 2014;19:890–894. doi: 10.1038/mp.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roussos P, et al. A role for noncoding variation in schizophrenia. Cell Rep. 2014;9:1417–1429. doi: 10.1016/j.celrep.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cipriani A, et al. A systematic review of calcium channel antagonists in bipolar disorder and some considerations for their future development. Mol. Psychiatry. 2016;21:1324–1332. doi: 10.1038/mp.2016.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee AS, et al. Forebrain elimination of cacna1c mediates anxiety-like behavior in mice. Mol. Psychiatry. 2012;17:1054–1055. doi: 10.1038/mp.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dao DT, et al. Mood disorder susceptibility gene CACNA1C modifies mood-related behaviors in mice and interacts with sex to influence behavior in mice and diagnosis in humans. Biol. Psychiatry. 2010;68:801–810. doi: 10.1016/j.biopsych.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kabir ZD, et al. Cacna1c in the prefrontal cortex regulates depression-related behaviors via REDD1. Neuropsychopharmacology. 2017;42:2032–2042. doi: 10.1038/npp.2016.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Terrillion CE, et al. Reduced levels of Cacna1c attenuate mesolimbic dopamine system function. Genes. Brain Behav. 2017;16:495–505. doi: 10.1111/gbb.12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bavley CC, Fischer DK, Rizzo BK, Rajadhyaksha AM. Cav1.2 channels mediate persistent chronic stress-induced behavioral deficits that are associated with prefrontal cortex activation of the p25/Cdk5-glucocorticoid receptor pathway. Neurobiol. Stress. 2017;7:27–37. doi: 10.1016/j.ynstr.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar N, Singh N, Jaggi AS. Anti-stress effects of cilnidipine and nimodipine in immobilization subjected mice. Physiol. Behav. 2012;105:1148–1155. doi: 10.1016/j.physbeh.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 50.Cohen C, Perrault G, Sanger DJ. Assessment of the antidepressant-like effects of L-type voltage-dependent channel modulators. Behav. Pharmacol. 1997;8:629–638. doi: 10.1097/00008877-199711000-00019. [DOI] [PubMed] [Google Scholar]
- 51.Mogilnicka E, Czyrak A, Maj J. Dihydropyridine calcium channel antagonists reduce immobility in the mouse behavioral despair test; antidepressants facilitate nifedipine action. Eur. J. Pharmacol. 1987;138:413–416. doi: 10.1016/0014-2999(87)90480-8. [DOI] [PubMed] [Google Scholar]
- 52.Levy NA, Janicak PG. Calcium channel antagonists for the treatment of bipolar disorder. Bipolar Disord. 2000;2:108–119. doi: 10.1034/j.1399-5618.2000.020204.x. [DOI] [PubMed] [Google Scholar]
- 53.Machado AK, Pan AY, da Silva TM, Duong A, Andreazza AC. Upstream pathways controlling mitochondrial function in major psychosis. A focus on bipolar disorder. Can. J. Psychiatry. 2016;61:446–456. doi: 10.1177/0706743716648297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Surmeier DJ, Halliday GM, Simuni T. Calcium, mitochondrial dysfunction and slowing the progression of Parkinson’s disease. Exp. Neurol. 2017;298:202–209. doi: 10.1016/j.expneurol.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benes FM, Matzilevich D, Burke RE, Walsh J. The expression of proapoptosis genes is increased in bipolar disorder, but not in schizophrenia. Mol. Psychiatry. 2006;11:241–251. doi: 10.1038/sj.mp.4001758. [DOI] [PubMed] [Google Scholar]
- 56.Andreazza AC, Wang JF, Salmasi F, Shao L, Young LT. Specific subcellular changes in oxidative stress in prefrontal cortex from patients with bipolar disorder. J. Neurochem. 2013;127:552–561. doi: 10.1111/jnc.12316. [DOI] [PubMed] [Google Scholar]
- 57.Dolga AM, et al. Mitochondrial small conductance SK2 channels prevent glutamate-induced oxytosis and mitochondrial dysfunction. J. Biol. Chem. 2013;288:10792–10804. doi: 10.1074/jbc.M113.453522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cereghetti GM, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl Acad. Sci. USA. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Narayanan D, Xi Q, Pfeffer LM, Jaggar JH. Mitochondria control functional CaV1.2 expression in smooth muscle cells of cerebral arteries. Circ. Res. 2010;107:631–641. doi: 10.1161/CIRCRESAHA.110.224345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suzuki Y, Yoshimaru T, Inoue T, Ra C. Ca v 1.2 L-type Ca2+channel protects mast cells against activation-induced cell death by preventing mitochondrial integrity disruption. Mol. Immunol. 2009;46:2370–2380. doi: 10.1016/j.molimm.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 61.Morimoto BH, Koshland DE. Excitatory amino acid uptake and N-methyl-D-aspartate-mediated secretion in a neural cell line. Proc. Natl Acad. Sci. USA. 1990;87:3518–3521. doi: 10.1073/pnas.87.9.3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davis JB, Maher P. Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res. 1994;652:169–173. doi: 10.1016/0006-8993(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 63.Diemert S, et al. Impedance measurement for real time detection of neuronal cell death. J. Neurosci. Methods. 2012;203:69–77. doi: 10.1016/j.jneumeth.2011.09.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.