

Abstract
One of the most paramount receptor-induced signal transduction mechanisms in hematopoietic cells is production of the lipid second messenger phosphatidylinositol(3,4,5)trisphosphate (PIP3) by class I phosphoinositide 3 kinases (PI3K). Defective PIP3 signaling impairs almost every aspect of hematopoiesis, including T cell development and function. Limiting PIP3 signaling is particularly important, because excessive PIP3 function in lymphocytes can transform them and cause blood cancers. Here, we review the key functions of PIP3 and related phosphoinositides in hematopoietic cells, with a special focus on those mechanisms dampening PIP3 production, turnover, or function. Recent studies have shown that beyond “canonical” turnover by the PIP3 phosphatases and tumor suppressors phosphatase and tensin homolog (PTEN) and SH2 domain-containing inositol-5-phosphatase-1 (SHIP-1/2), PIP3 function in hematopoietic cells can also be dampened through antagonism with the soluble PIP3 analogs inositol(1,3,4,5)tetrakisphosphate (IP4) and inositol-heptakisphosphate (IP7). Other evidence suggests that IP4 can promote PIP3 function in thymocytes. Moreover, IP4 or the kinases producing it limit store-operated Ca2+ entry through Orai channels in B cells, T cells, and neutrophils to control cell survival and function. We discuss current models for how soluble inositol phosphates can have such diverse functions and can govern as distinct processes as hematopoietic stem cell homeostasis, neutrophil macrophage and NK cell function, and development and function of B cells and T cells. Finally, we will review the pathological consequences of dysregulated IP4 activity in immune cells and highlight contributions of impaired inositol phosphate functions in disorders such as Kawasaki disease, common variable immunodeficiency, or blood cancer.
Keywords: phosphoinositide 3 kinase, AKT, SH2 domain-containing inositol-5-phosphatase, phosphatase and tensin homolog, ORAI, ITPKB/IP3-3KB/IP3KB, ITPKC/IP3-3KC/IP3KC, Kawasaki disease
Introduction
In one of the most paramount receptor-induced signal-transduction mechanisms, class I phosphoinositide 3 kinases (PI3K) phosphorylate the membrane-lipid phosphatidylinositol(4,5)bisphosphate [PI(4,5)P2, hereafter PIP2] into the lipid second messenger phosphatidylinositol(3,4,5)trisphosphate [PI(3,4,5)P3, hereafter PIP3, Figure 1]. By binding to their pleckstrin homology (PH) or certain other domains, PIP3 recruits key signaling effectors to cellular membranes, enabling their incorporation into signaling complexes and activation (1). Important examples in lymphocytes include the tyrosine kinase expressed in hepatocellular carcinoma (Tec)-family protein tyrosine kinases (TFK) IL-2-inducible T-cell kinase (Itk), Tec, and Bruton’s tyrosine kinase (Btk). TFK have essential functions in antigen–receptor signaling (2, 3). PIP3 also recruits the kinase Akt, a key promoter of cell survival, proliferation, differentiation, and activation. PI3K/Akt dysregulation contributes to immunodeficiencies, autoimmune diseases, allergies, and cancer (4–11). In this review, we discuss how immune cells use inositolphosphates (IPs) as soluble analogs of PIP3 and other phosphoinositides to control the functions of their lipid counterparts and other important cellular processes (Table 1).
Figure 1.
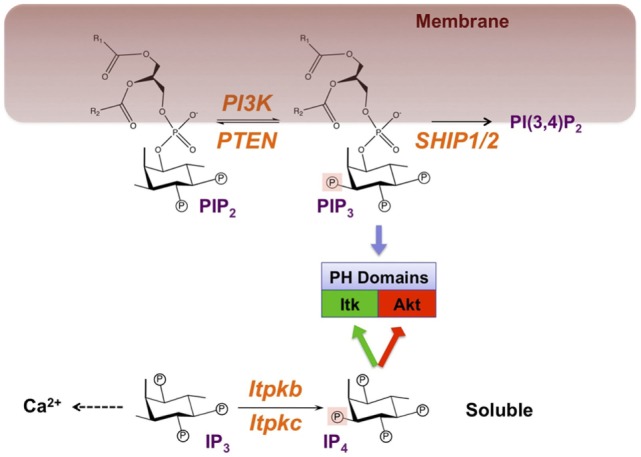
Symmetric signaling by phosphoinositide 3 kinase (PI3K) and Itpkb controls effector recruitment through the analogous but phase-separated pleckstrin homology (PH) domain ligands PIP3 and IP4. Engagement of antigen receptors activates PI3K, which phosphorylates the membrane-lipid phosphatidylinositol(4,5) bisphosphate (PIP2) on the 3-position of its cytoplasm-exposed inositol ring to generate phosphatidylinositol(3,4,5) trisphosphate (PIP3). Alternatively but not shown to emphasize the PI3K/Itpkb symmetry, phospholipase-Cγ1 (PLCγ1) can hydrolyze PIP2 into the second messengers diacylglycerol (DAG) and soluble inositol(1,4,5) trisphosphate (IP3). Canonically, PIP3 accumulation is limited through its removal by two families of phospholipid phosphatases: Phosphatase and tensin homolog (PTEN) which reverses the PI3K reaction, and SH2 domain-containing inositol polyphosphate-5-phosphatases (SHIP-1/2) which convert PIP3 into phospatidylinositol(3,4) bisphosphate [PI(3,4)P2]. Mainly through their IP headgroups, PIP2, PIP3, and PI(3,4)P2 can bind to PH and other domains in signaling proteins such as Itk and Akt, and recruit them to membranes. IP3 mobilizes Ca2+ but can also be phosphorylated at its 3-position into Inositol(1,3,4,5) tetrakisphosphate (IP4) by IP3 3-kinases (Itpka/b/c and inositol-phosphate-multikinase) (8, 19). Because it resembles the PIP3 headgroup, IP4 can also bind to certain PIP3-binding PH and other domains and promote (green) or inhibit (red) PIP3 binding. In CD4+CD8+ thymocytes, IP4 promotes PIP3 binding to the Itk/Tec PH domains to establish a feedback loop of PLCγ1 activation (20, 21). In neutrophils, NK cells, CD4−CD8− thymocytes undergoing β-selection and in hematopoietic stem cells (HSC), IP4 competition with PIP3 or PI(3,4)P2 for binding to its PH domain may limit Akt membrane recruitment and activation (22–27). IP4 can also inhibit RASA3/GAP1IP4BP-binding to PI(4,5)P2 or PIP3 (28, 29). Whether this occurs in immunocytes remains unknown. R1, R2, fatty acid side-chains. Circled P, phosphate moiety. Orange, enzymes with demonstrated physiological relevance in immunocytes.
Table 1.
Table of content.
| 1. Introduction |
| 2. Non-canonical antagonism by IP4 prevents excessive PI3K signaling in hematopoietic cells |
| 2.1. IP4 limits neutrophil function |
| 2.2. Itpkb limits myelopoiesis from GMP |
| 2.3. Itpkb dampens NK cell function |
| 2.4. Itpkb is required for HSC quiescence and longevity |
| 2.5. Itpkb is required for thymocyte β-selection by dampening Akt/mTORC1 function |
| 3. IP7 may antagonize PI3K in neutrophils |
| 4. IP4 may promote PI3K signaling to enable thymocyte positive selection |
| 5. IP4 dampens store-operated Ca2+ entry (SOCE) in immunocytes to promote survival and prevent inflammatory disease |
| 5.1. Itpkb is required for T cell viability and function |
| 5.2. Itpkc dampens Ca2+ mobilization in immune cells to prevent inflammatory disease |
| 5.3. Itpkb dampens SOCE in B cells |
| 5.4. Itpkb dampens SOCE in neutrophils |
| 6. Does Itpkb inhibition have therapeutic potential in human diseases? |
| 7. Conclusion and Perspectives |
Evidenced by the phenotypes of mice lacking the hematopoietically enriched PI3Kγ and PI3Kδ, reduced PIP3 signaling impairs most aspects of hematopoiesis, including hematopoietic stem cell (HSC) homeostasis and the development or function of T, B, and NK cells, myeloid mast cells, monocytes, granulocytes, and erythrocytes (4–9) (Figure 2). Limiting PIP3 signaling is particularly important, because excessive PIP3 function not only oppositely affects many of the same hematopoietic processes but can also transform lymphocytes and cause blood cancers. This is shown by the phenotypes of mice lacking the phosphoinositide-phosphatases phosphatase and tensin homolog (PTEN) or SH2 domain-containing inositol-5-phosphatase-1 (SHIP-1), which canonically limit PIP3 function by dephosphorylating it back into PIP2, or into PI(3,4)P2, respectively (8) (Figure 1). Moreover, PTEN is a pivotal tumor suppressor, and SHIP-1 and PTEN cooperatively suppress B cell lymphomagenesis (12). Besides SHIP-1, hematopoietic cells also express the closely related SHIP-2 (13–15). SHIP-2 dampens immunoglobulin-receptor signaling in macrophages and mast cells (16, 17). Its functions in lymphocytes remain to be elucidated. Highlighting the translational importance of preventing PIP3 hyperactivity in hematopoietic cells, the PI3Kδ inhibitor Idelalisib is approved for treating relapsed chronic lymphocytic leukemia (CLL), follicular B-cell non-Hodgkin lymphoma, and small lymphocytic lymphoma (18). Oncogenic PI3K mutations in 50% of human cancers, PTEN status as the second most-often mutated tumor suppressor gene in human cancers, and multiple efforts to therapeutically inhibit PI3K signaling for cancer, metabolic, and immune diseases further illustrate the broad therapeutic importance of preventing PIP3 hyperactivity (10, 11).
Figure 2.

Phosphoinositide 3-kinase (PI3K) loss-of-function or gain-of-function affects multiple stages of hematopoietic development, and mature hematopoietic cells. Hematopoiesis originates from quiescent, long-lived, and pluripotent hematopoietic stem cells (HSC) which reside in BM (BM) niches with low-metabolic and cell cycle activity (26, 30). After occasional division and activation, HSC daughter cells can differentiate through multiple hematopoietic progenitor cell stages including multipotent progenitors (MPP) into lymphoid or myeloid lineages. During lymphopoiesis, MPP-derived common lymphoid progenitors (CLP) give rise to the T cell, NK cell, and B cell lineages. CLP can also generate subsets of dendritic cells (DC), in particular plasmacytoid DC (pDC). CLPs initiate the B/T cell lineages through early thymic progenitors (ETP) and pro-B cells, respectively. ETPs develop through CD4−CD8− (DN) and CD4+CD8+ (DP) stages into mature T cells. In the bone marrow, pro-B cells develop via pre-B cells into immature B cells. These translocate into the spleen to mature through transitional stages into mature B cells. In myelopoiesis, MPP-derived common myeloid progenitors (CMP) give rise to granulocyte–monocyte progenitors (GMP) which in turn generate granulocytes, monocytes, and mast cells. Alternatively, CMP can give rise to megakaryocyte–erythrocyte progenitors (MEP), which in turn generate megakaryocytes and erythrocytes. CMP can also generate common DC precursors, which in turn generate most DC subsets (31). The map indicates major hematopoietic progenitors and mature cell types that are negatively (red font) or positively (green font) affected in mice deficient for the indicated PI3K isoforms, SHIP-1, or PTEN (4–9, 26, 30–34). Mixed red–green font indicates complex phenotypes with activation and inactivation components. Immune cells express multiple class I PI3K isoforms. Among those, mature T cell, B cell, NK cell, and mast cell functions or chemotaxis are particularly dependent on the protein tyrosine kinase-dependent receptor-activated PI3Kδ with contributions by the GPCR-activated PI3Kγ (32, 33). Monocyte/macrophage and granulocyte chemotaxis is critically dependent on PI3Kγ, with contributions by PI3Kδ and, in macrophages and neutrophilic granulocytes, PI3Kβ (33, 35). DC require PI3Kγ and δ for various aspects of their function (33). For detailed recent reviews of PI3K isoform functions in hematopoietic cells, see Ref. (32, 33).
Adding a non-canonical perspective to the mechanisms controlling PI3K function, we and others found that PIP3 activity in hematopoietic cells can also be dampened through antagonism with the soluble PIP3-analogs inositol(1,3,4,5)tetrakisphosphate (IP4, Figure 1) and inositol-heptakisphosphate, also called diphosphoinositol-pentakisphosphate (hereafter IP7) (22–27). Because IP4 is identical to the cytoplasm-exposed, PH domain-binding PIP3 headgroup, IP4 and PIP3 can compete for binding to the Akt PH domain. Similarly, IP7 can compete with PIP3 binding to PH domains (36, 37). Many PH domains bind PIP3 and IP4 with similar affinities, so IP4/PIP3 antagonism could be broadly relevant (1, 38). But how many PI3K functions are regulated by IP4 and IP7 remains a major open question (8, 38). We and others found that in HSC, T cell precursors, NK cells, and neutrophils, IP4 dampens PIP3 recruitment of Akt; IP7 dampens Akt recruitment in neutrophils (22–27). Other evidence suggests that IP4 may promote PIP3 function in thymocytes undergoing positive selection (20, 21). IP4 has additional functions in preventing anergy and death in developing B cells, apoptosis in peripheral T cells, and monocyte hyperactivity that may be unrelated to PI3K (29, 39–44). An emerging common mechanism controlling these different processes is the inhibition of store-operated Ca2+ entry (SOCE) through the plasma membrane by IP4, its metabolites, or the enzymes producing IP4.
IP4 is produced through phosphorylation of inositol(1,4,5)trisphosphate (IP3) by four IP3 3 kinases, three of which belong to the inositol trisphosphate kinase family (Itpka, Itpkb, and Itpkc, Figure 1) (8, 45). Hematopoietic functions of the fourth IP3 3-kinase, inositol phosphate multikinase (IPMK), remain unknown. IP3 is an important second messenger that mediates receptor-induced Ca2+ mobilization (46). Although many tissues can produce IP4, the hematopoietic system has proven particularly useful for elucidating its physiological functions. This may in part reflect a particularly high expression of the best studied IP3 3-kinase, Itpkb, in hematopoietic cells (8, 25). Itpkb is a major producer of IP4 in leukocytes, and several studies have used Itpkb−/− mice to show that IP4 deficiency profoundly affects hematopoietic cell development, homeostasis, survival, and function (Figure 3) (20–23, 25, 26, 28, 29, 39, 41–43, 47, 48). Itpkb is also abundant in the brain, which co-expresses Itpka. Itpka is not abundant in immune cells. Itpka deficiency caused neurological phenotypes in mice without reported immune defects (49, 50). No significant neurological phenotypes have been reported in Itpkb−/− mice (8, 45). Loss of the more broadly expressed Itpkc in mice hyperactivated macrophages and worsened coronary arteritis in a mouse model for Kawasaki disease (KD) (44), but did not affect other immunocytes as far as studied (44, 47, 51). But reduced ITPKC function in humans may hyperactivate T cells, B cells, and monocytes and promote KD (40, 44). Itpka/b mRNA expression is not affected by immunocyte activation, and Itpk expression profiles are overall comparable between mice and humans (15, 25, 52). Phorbol-12-myristate-13-acetate/ionomycin upregulated ITPKC mRNA in human PBMC and other cells (40).
Figure 3.

Itpks control multiple aspects of hematopoiesis. Shown are aspects of hematopoiesis affected positively (green symbols and fonts) or negatively (red symbols and fonts) by inactivation of Itpkb or Itpkc. Pink, human diseases associated with loss-of-function (lof) alleles of ITPKC (Kawasaki disease, KD) or ITPKB (common variable immunodeficiency, CVID). Abbreviations: cKO, conditional, gKO, germline knockout mice; AICD, activation-induced cell death; BMDM, bone marrow-derived macrophages. Hematopoietic cell stages and pathways are explained in the legend to Figure 2. For more details and references, see text. Mast cells express Itpkb and produce IP4 after stimulation (53). Small-molecule Itpk inhibition might augment their activation (54, 55), but the target selectivity of the low-affinity Itpk inhibitors used is unknown and genetic studies are needed. Adapted with permission from Ref. (8).
IP7 can be produced in several steps from IP4 or other precursors (8, 45). Among the required enzymes, deficiency in inositol hexakisphosphate kinase-1 (IP6K1) has unveiled important IP7 roles in dampening Akt function in neutrophils (24). IP4 and IP7 can both be metabolized into various other soluble IPs with unknown functions in lymphocytes, several of which were found in T cells (8, 45, 56).
Below, we review the impact of soluble IPs on hematopoietic cells in detail and discuss current models for how these interesting molecules can have such diverse functions (Table 1). Available data suggest that IP4 primarily engages two distinct mechanisms: non-canonical PIP3 antagonism to dampen PI3K signaling, and dampening of SOCE to restrict Ca2+ mobilization.
Non-Canonical Antagonism by IP4 Prevents Excessive PI3K Signaling in Hematopoietic Cells
Itpkb loss in mice causes either hyperactive or loss-of-function (lof) phenotypes in hematopoietic cells (Figure 3). Interestingly, most of the hyperactivation phenotypes appear to result at least in part from Akt hyperactivity due to reduced IP4 antagonism with PIP3.
IP4 Limits Neutrophil Function
The intriguing functions of Itpkb and IP4 as physiological antagonists of PI3K and PIP3 upstream of Akt were first described when the Luo and Schurmans labs characterized Akt gain-of-function phenotypes in Itpkb−/− neutrophilic granulocytes, an important component of the innate immune system (Figures 1 and 4) (57). Among Itpks, neutrophils mainly express Itpkb (8, 57). Stimulation with chemoattractants such as N-formyl-methionyl-leucyl-phenylalanine (fMLP) induced IP3 3-kinase activity and IP4 accumulation in neutrophils (23, 57, 58). Upon stimulation with fMLP or the complement factor C5a, bone marrow (BM)-derived neutrophils (BMN) from Itpkb−/− mice showed increased chemotaxis and superoxide production correlated with Akt hyperphosphorylation and actin hyperpolymerization (57). Akt PH domain–GFP fusion proteins co-precipitated IP4, IP5, and IP6. Treatment with cell-permeable IP4 had opposite effects to Itpkb knockout on neutrophils and inhibited fMLP-induced Akt PH domain membrane recruitment in HL60 promyelocytic leukemia cells. This suggested that Itpkb dampens chemoattractant-induced neutrophil activation, probably by producing IP4 which then competes with PIP3 or PI(3,4)P2 to inhibit Akt membrane recruitment and activation. Although elevated PI3K/Akt signaling promotes neutrophil viability (23), Itpkb−/− BMN had reduced viability in vitro (22). Thus, Itpkb loss probably caused additional defects in neutrophils. Indeed, despite initially reported normal fMLP-induced Ca2+ responses in Itpkb−/− neutrophils (22), follow-up work showed decreased Ca2+ store-release but enhanced SOCE (Figure 5) (22, 23, 57). It will be interesting to study if defective Ca2+ mobilization underlies the reduced viability.
Figure 4.
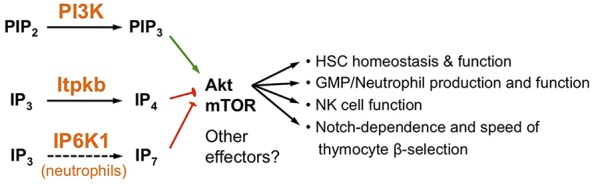
Non-canonical antagonism between phosphoinositide 3-kinase (PI3K) and Itpkb or inositol hexakisphosphate kinase-1 (IP6K1) controls multiple aspects of hematopoiesis. PI3K and Itpkb convert the analogous substrates PIP2 and IP3 into the analogous products PIP3 and IP4, respectively. By competing with PIP3 for Akt pleckstrin homology domain binding, IP4 then dampens PI3K-mediated Akt activation and signaling via mammalian or mechanistic target of rapamycin (mTOR). This ensures hematopoietic stem cell (HSC) homeostasis and function, warrants appropriate GMP/neutrophil and NK cell production and function, and establishes the Notch-dependence and kinetics of thymocyte β-selection (22, 23, 25–27). In neutrophils, IP6K1 can also antagonize PI3K activation of Akt by producing the additional PIP3 analog IP7 (24).
Figure 5.
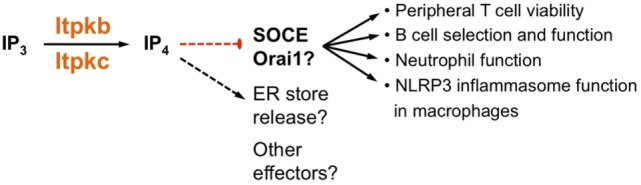
Itpkb controls immune cell biology by dampening store-operated Ca2+ entry (SOCE). Several studies suggest that in peripheral T cells, developing and mature B cells, neutrophils and macrophages, Itpkb or Itpkc dampen SOCE through Orai channels (23, 39–42, 44, 59). This may be required for T cell viability, for preventing B cell anergy, and for ensuring neutrophil function. The ability of an exogenously provided cell-permeable IP4-ester to reduce SOCE very quickly after administration would be consistent with direct SOCE inhibition through IP4 (39, 59). However, variably affected ER store release and previously published, complicated roles for Itpks, IP3, IP4, and IP4 metabolites in controlling Ca2+ mobilization in mammalian cells could point to alternate mechanisms and possible other effectors (8, 45, 60, 61). For more detailed discussions, see text.
The effects of Itpkb loss on in vivo neutrophil function were more complicated. In an acute peritonitis model, neutrophil recruitment into inflamed peritoneal cavities was augmented, but clearance of the injected bacteria was normal or slightly decreased even though in vitro, Itpkb−/− neutrophils killed serum-opsonized bacteria better than wild-type neutrophils (23, 57). The discrepancy likely reflects a reduced content of opsonizing IgG in the serum of Itpkb−/− mice due to defects in B cell function (29, 39, 41). Indeed, serum from Itpkb−/− mice facilitated killing of bacteria less efficiently than wild-type serum (23). Taken together, the data suggest that Itpkb limits neutrophil function, but the physiological consequences are complicated by contributions of defects in other immune cells in germline Itpkb−/− mice, and by diverse effects of Akt hyperactivation, Ca2+ dysregulation, and possibly other perturbed Itpkb/IP4 functions (8). Phenotypic similarities between Itpkb−/− and PTEN−/− mice include Akt hyperactivation, variably elevated migration, lung or peritoneal recruitment, superoxide production, and bacterial killing (62–65). They are consistent with a PI3K-counteracting Itpkb function. Phenotypic discrepancies such as the elevated viability of PTEN−/− neutrophils, or massive neutrophil organ-infiltration despite strongly impaired in vitro polarization and motility in SHIP−/− mice (62, 66, 67) might be explained by the aforementioned factors, or by distinct PTEN or SHIP functions that are unaffected by IP4 (25, 26). For example, SHIP-1 loss increases PIP3 levels but may also reduce production of its PH domain-binding product PI(3,4)P2, or perturb SHIP-1 scaffolding functions and protein interactions mediated by its adaptor domains (68). PTEN-loss causes PIP3 accumulation but may also reduce the levels of its product PI(4,5)P2, a PLC-substrate and protein ligand (69). IP4 can serve as a substrate for PTEN and SHIP-1 in vitro (8). Thus, the phenotypes of SHIP-1−/− and PTEN−/− mice might involve IP4 accumulation, which could limit the PI3K hyperactivation caused by loss of the PIP3 phosphatases. Moreover, PIP3 controls multiple effectors beyond Akt that can be differentially impacted by IP4, Itpks can have IP4-unrelated functions such as actin-bundling or removing IP3, and Itpkb can control different effectors depending on cell type and context. We discuss these possibilities in detail in Section “Conclusion and Perspectives.”
Itpkb Limits Myelopoiesis From GMP
Beyond neutrophil hyperactivation, Itpkb−/− mice also showed increased neutrophil production and peripheral blood numbers. This was associated with increased granulocyte–monocyte progenitor (GMP) proliferation and expansion and suggests that Itpkb restricts myeloid differentiation (22) (Figures 3 and 4). Hematopoietic progenitor cell-enriched BM cells from Itpkb−/− mice showed increased phosphorylation (activation) of Akt and its substrate, the cell-cycle inhibitor p21Cip1. Phosphorylation by Akt decreases cell cycle inhibition by p21Cip1, suggesting that Akt hyperactivation promotes GMP expansion by inhibiting p21Cip1. Consistent with this view, Akt is essential for myelopoiesis and can promote neutrophil and monocyte development (5, 26). While not formally proven, it is tempting to draw on the recently published HSC regulation by IP4 (26) and speculate that Itpkb limits GMP expansion and neutrophil production through IP4 antagonism with PIP3 for Akt PH domain binding and recruitment (Figure 4). To confirm this, conditional Itpkb disruption in GMP and phenotype-rescue studies with cell-permeable IP4 or Akt inhibitors will be important. Such studies can also rule out that the GMP phenotype results in part from the earlier HSC mobilization, or indirectly from the defective hematopoiesis and lymphopenia in Itpkb−/− mice (26).
Itpkb Dampens NK Cell Function
Other innate immunocytes highly impacted by intrinsic Itpkb loss are NK cells. These recognize and then kill virus-infected or cancer cells through imbalanced signaling by activating (aNKR) and inhibitory (iNKR) NK cell receptors [references in Ref. (25)]. iNKR engagement prevents inappropriate NK cell attack of normal body cells (70, 71). Virus infection or malignant transformation often downregulate iNKR ligands or upregulate aNKR ligands on target cells. The result is NK cell activation, the release of cytolytic granules, and secretion of pro-inflammatory cytokines and chemokines such as IFNγ. All aNKRs ultimately activate PI3K and/or phospholipase-Cγ (PLCγ). PI3K inactivation impairs NK cell maturation, IFNγ production, and cytotoxicity (1, 72–74).
aNKR ligation induced IP4 production in NK cells (75). Given the importance of PI3K in NK cells and the ability of IP4 to antagonize it, we assessed how loss of Itpkb and thus IP4 affects NK cells in Itpkb−/− mice. We found that Itpkb loss cell-autonomously elicited a more immature NKR repertoire and a reduced fraction of CD11b+CD27− most mature, long-lived NK cells compared with wild-type mice (25). Itpkb loss also increased the proportion of NK cells responding to NKR engagement and augmented effector functions, including IFNγ production, cytolytic granule release, and in vivo clearance of target cells lacking iNKR-engaging major histocompatibility complex I molecules. This was, at least in part, caused by defective dampening of PI3K-mediated Akt activation by IP4, because Itpkb−/− NK cells contained hyperactive Akt and treatment with cell-permeable IP4 or selective Akt- or PI3K inhibitors reversed both their Akt hyperactivation and hyperdegranulation (25). These data suggest that IP4 cell intrinsically promotes NK cell terminal maturation and acquisition of a mature NKR repertoire, but limits mature NK cell effector functions, in part by dampening Akt activity. Thus, non-canonical antagonism of PIP3 and IP4 is part of the important mechanisms preventing NK cell hyperactivity (Figure 4). Their limited understanding is a barrier to the development of safe and efficacious NK cell immunotherapies for cancer and virus infections (76, 77). In the future, it will be interesting to study possible IP4 roles in NK cell tolerance and to determine whether the Itpkb−/− NK cell phenotype arises exclusively from Akt hyperactivation or involves the deregulation of other NK cell-expressed PIP3 effectors, including Tec kinases or the guanine nucleotide exchange factor Vav (25).
Consistent with a PI3K gain-of-function phenotype in Itpkb−/− mice, loss of the NK cell-expressed PI3Kγ/δ caused an overall opposite phenotype with less CD11b+CD27+ NK cells, abnormal NKR repertoires, and reduced NKR-mediated IFNγ production and target cell lysis due to impaired NKR signaling and NK cell migration (1, 72–74). One important difference is that Itpkb promotes NK cell maturation but limits effector functions whereas PI3K promotes both processes (72–74). It will be important to elucidate the mechanistic underpinnings of this dichotomy. Among the PI3K-counteracting PIP3 phosphatases, SHIP-1 deficiency caused NKR repertoire changes distinct from those in Itpkb−/− mice and impaired effector functions including IFNγ secretion despite Akt hyperactivation (74, 78–81). However, the results were complicated by genetic background dependencies and NK cell dependence on both intrinsic and extrinsic SHIP-1 (82). PTEN knockdown in human NK cells mildly elevated cytolytic activity; PTEN overexpression reduced cytolysis by human and murine NK cells through mechanisms involving impaired immunological synapse formation without altering NK cell development and NKR repertoire in mice (83). However, overexpression artifacts may likely contribute to these differences from PI3K−/− mice. In another study, conditional PTEN deletion in murine NK cells did not strongly affect their maturation and NKR-induced IFNγ production, but caused NK cell hyperproliferation and hyperresponsiveness to the mobilizing chemoattractant S1P along with variable Akt/mammalian or mechanistic target of rapamycin (mTOR) hyperactivation. This resulted in premature BM egress and reduced lymphoid organ and liver, but elevated peripheral blood and lung NK cell numbers (84). Consistent with impaired tissue homing or -retention, PTEN−/− NK cells had an impaired ability to migrate to distal tumor sites, but cleared blood-borne tumor cells better than wild-type NK cells. The effects of Itpkb loss on NK cell migration remain to be elucidated. Based on the PTEN−/− phenotype and known PI3Kδ requirements for NK cell migration (1, 72–74), it will be interesting to study if reduced tissue homing or -retention contributes to the mildly reduced splenic NK cell numbers in Itpkb−/− mice (25). The NK cell phenotypic differences between SHIP−/− or PTEN−/− and Itpkb−/− mice could involve the factors discussed above in the neutrophil section, or NK cell-extrinsic contributions whose elimination requires conditional knockouts. Altogether, more detailed mechanistic and genetic studies to better discern the interplay between Itpkb, SHIP, and PTEN in controlling PI3K function in NK cells should prove exciting.
Itpkb Is Required for HSC Quiescence and Longevity
To warrant life-long hematopoiesis, HSC homeostasis must be tightly balanced between quiescence and activation (Figure 6). Persistent activation reduces HSC life span and pluripotency. This can cause immunodeficiencies, anemia, hematopoietic failure, blood cancer, and death (30).
Figure 6.
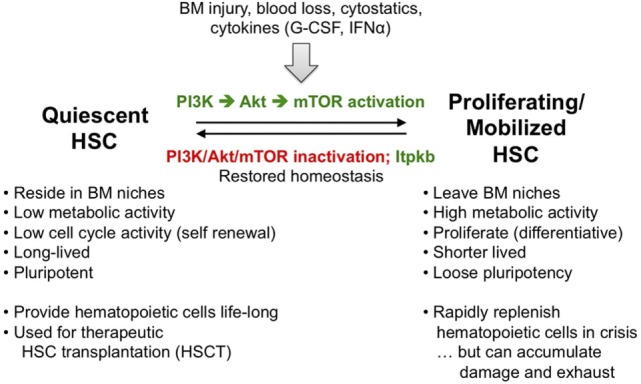
Hematopoietic stem cell (HSC) homeostasis is an exquisitely phosphoinositide 3-kinase (PI3K)-dependent process controlled by Itpkb. To ensure live-long hematopoiesis, HSC reside in BM niches and have low metabolic and cell cycle activity. As a consequence, HSC are long-lived and pluripotent. Stresses including BM injury, blood loss, exposure to cytostatic drugs or cytokines such as G-CSF or type 1 interferons activate and mobilize HSC to leave the BM niches, become metabolically active and proliferate. Some HSC daughter cells then differentiate into hematopoietic progenitors (Figure 2). As a consequence, activated HSC are short-lived and loose their pluripotency. This serves to rapidly replenish hematopoietic cells in a crisis or after HSC transplantation, but persistent HSC activation can lead to HSC damage and exhaustion, ultimately causing BM failure, anemia, immunodeficiencies, or blood cancer (85–87). To prevent this, resolution of HSC-activating stresses normally reverts them into quiescence once the activating stimuli subside. A key mediator of HSC activation that needs to be inactivated for re-entry into quiescence is PI3K signaling via Akt and downstream mammalian or mechanistic target of rapamycin (mTOR). In addition, we have identified Itpkb as a promoter of HSC quiescence and homeostasis that acts at least in part by inactivating Akt in HSC (26, 30).
Phosphoinositide 3-kinase is a key regulator of HSC homeostasis. PI3K, Akt, and downstream mTOR complex-1 (mTORC1) are required for HSC self-renewal and function, but also mediate HSC activation and mobilization out of their niches by stresses such as BM injury, blood loss, or treatment with cytostatics or cytokines. This serves to transiently increase hematopoiesis and augment immunocyte or erythrocyte production. Upon resolution of the stress, PI3K inactivation is required for HSC re-entry into quiescence. Excessive PI3K/Akt activity transiently expands HSC, followed by depletion and reduced long-term repopulating capability associated with variable myeloproliferative disease, T-cell acute lymphoblastic (T-ALL) or acute myeloblastic (AML) leukemia (30). Thus, PI3K/Akt activity in HSC needs to be tuned into an appropriate window. Although both PTEN and SHIP have been implicated, the relative importance of HSC-extrinsic vs. -intrinsic PTEN remains controversial, and SHIP-1 may primarily control HSC homeostasis extrinsically by acting in niche cells to prevent production of HSC mobilizing factors and ensure production of HSC-attracting CXCL12 (88).
Because HSC express Itpkb (22, 26), we hypothesized that Itpkb might dampen PI3K/Akt signaling in HSC through PIP3/IP4 antagonism to ensure their longevity. Supporting this view, young Itpkb−/− mice accumulated phenotypic HSC with a less quiescent, hyperproliferative phenotype (26). Itpkb−/− HSC underexpressed genes associated with stemness and quiescence, but overexpressed activation and differentiation-associated genes. They could home into the BM but had reduced persistence and colony-forming activity in vitro. In vivo, Itpkb−/− HSC had a massively reduced competitive long-term repopulating potential. Consistent with severely defective HSC longevity, aging Itpkb−/− mice lost HSC and other hematopoietic progenitors, and died prematurely with anemia (26).
Increased stem cell factor-mediated Akt/mTORC1 activation in Itpkb−/− HSC in vitro that could be prevented by treatment with cell-permeable IP4 or a small-molecule Akt inhibitor, and elevated mTORC1 activity in HSC in Itpkb−/− mice suggested that Itpkb dampens PI3K/Akt signaling in HSC via IP4. Moreover, Itpkb−/− HSC upregulated gene sets associated with Akt/mTORC1 hyperactivity, oxidative phosphorylation, and protein biosynthesis (26). HSC quiescence requires dampened protein biosynthesis and upstream PI3K/mTOR signaling (89). This suggests that the activation of Itpkb−/− HSC was at least in part caused by exaggerated metabolic activation and protein biosynthesis. Supporting this view, injection of the mTOR inhibitor rapamycin reversed the HSC hyperproliferation in Itpkb−/− mice (26). We proposed that Itpkb limits cytokine and PI3K/Akt/mTOR signaling in HSC to ensure longevity and prevent BM failure (Figures 4 and 6) (26, 30). Thus, Itpkb is a critical component of the mechanisms which tune PI3K activity in HSC appropriately to balance quiescence and activation.
The transient expansion but later depletion of HSC in Itpkb−/− mice is reminiscent of the phenotypes resulting from PTEN inactivation or expression of dominant-active Akt (90–92). However, T-ALL and AML have not been reported in Itpkb−/− mice (30). In addition, rapamycin reversed the HSC hyperproliferation in Itpkb−/− mice but did not rescue their colony-forming activity (26). The reasons remain to be determined, but could include differential effects of Itpkb inactivation, Akt activation, or PTEN loss on PI3K signaling in HSC, or, alternatively, a premature death of Itpkb−/− mice due to either anemia (26) or infections secondary to immunodeficiency (47) before blood cancer can develop. Itpkb loss might also impair signaling mechanisms required for colony-forming activity or cell transformation that are distinct from PI3K/mTORC1. But, rapamycin also reduced wild-type HSC colony-forming activity (26), and genetic studies suggest mTORC1 requirements for HSC regeneration and function (30). This might explain the difficulty of rescuing Itpkb−/− HSC function with mTORC1 inhibitors. More detailed biochemical and genetic studies will be needed to fully elucidate how Itpkb controls HSC biology. In particular, conditional Itpkb disruption in HSC and large mouse cohorts may help clarify whether Itpkb loss can transform blood cells, and whether HSC-extrinsic Itpkb inactivation contributes to the HSC defects in Itpkb−/− mice (30).
Itpkb Is Required for Thymocyte β-Selection by Dampening Akt/mTORC1 Function
Recently, we found that beyond innate immunocytes, the paradigm of Itpkb/PI3K antagonism upstream of Akt also applies to adaptive T lymphocytes (27). T cells develop in the thymus from HSC/CLP-derived early thymocyte progenitors (ETPs) through several CD4−CD8− “double negative” (DN) stages into CD4+CD8+ “double positive” (DP) thymocytes which then develop into CD4+ and CD8+ T cells (93, 94) (Figures 3 and 7A). To generate a diverse T cell repertoire reactive against many pathogens, the T cell receptor (TCR) α and β chain genes somatically rearrange in DN thymocytes. Productive rearrangement of one TCRβ-allele causes surface expression of a pre-TCR comprised of TCRβ, invariant pre-TCRα, and signal-transducing CD3 subunits on DN3a cells (95). If a pre-TCR is functional, its ligand-independent signaling triggers DN3 cell metabolic activation, proliferation and survival, allelic exclusion of the second TCRβ allele, initiation of TCRα gene rearrangements, and differentiation via CD8+ immature single-positive (ISP) into DP cells (93, 94). This “β-selection” ensures that only DN3 cells expressing a functional TCRβ chain develop further. It is the major cell-fate determining event for αβ T cells. Defective β-selection causes a DN3-block and severe immunodeficiency (4, 95).
Figure 7.

Non-canonical antagonism of phosphoinositide 3-kinase (PI3K) by Itpkb delays thymocyte β-selection and renders it Notch dependent. (A) T cells develop in the thymus from HSC and CLP-derived early thymocyte progenitors (ETPs) through several successive CD4−CD8− “double-negative” stages (DN2-DN4) and a CD8+ immature single-positive (ISP) stage into CD4+CD8+ double-positive (DP) thymocytes (93, 94). DP cells then undergo positive and negative selection to mature into CD4+ or CD8+ T cells. At the DN3a stage, expression of a pre-T cell receptor (TCR) composed of an invariant pre-TCRα (pTα) chain and a fully rearranged TCRβ chain triggers metabolic activation, proliferation, survival, β chain allelic exclusion, the initiation of TCRα chain somatic gene rearrangements, acquisition of the α/β T cell fate, and developmental progression to the DP stage. DP thymocytes express a mature TCR composed of fully rearranged α and β chains. The DN3-to-DP transition requires pre-TCR and costimulatory Notch signals. This process is termed β-selection, because it allows only DN3 cells expressing a functional TCRβ chain to survive and mature. (B) Based on studies in Itpkb−/− mice, we recently proposed a model in which pre-TCR and Notch signaling both activate PI3K to produce PIP3 in DN3 cells. PIP3 then recruits and activates Akt to increase glucose metabolism via the Akt/mammalian or mechanistic target of rapamycin (mTOR) pathway. This is required for β-selection. However, pre-TCR signaling also activates Itpkb to produce IP4, which competes with PIP3 for Akt pleckstrin homology domain binding and limits Akt recruitment and signaling in pre-TCR expressing DN3 cells. By limiting downstream glucose metabolism, this “IP4 brake” delays the kinetics of β-selection and renders this process dependent on Notch costimulation (27). (C) Without Itpkb, IP4 no more dampens Akt activation. In the presence of Notch signals, Akt is now hyperactivated and causes an accelerated DN3-to-DP cell differentiation (indicated by bold arrows). (D) In absence of Itpkb, pre-TCR signaling alone sufficiently activates Akt/mTOR to trigger DP cell development without Notch engagement (27).
β-Selection requires pre-TCR and co-stimulatory Notch signals, which promote DN3 cell metabolism, growth, survival, proliferation, and differentiation. Excessive Notch signaling, however, causes T-ALL. This is augmented by pre-TCR signals (6, 95–100). So, like cytokine signaling in HSC, pre-TCR/Notch signaling in DN3 cells needs to be tuned into an appropriate intensity window.
Both pre-TCR and Notch activate PI3K/Akt (4, 6, 97) (Figure 7B). PI3K/Akt are essential and rate limiting for β-selection by promoting glycolysis, proliferation, survival, and differentiation (6, 101–103). Pinpointing a need to limit PI3K/Akt signaling in DN3 cells for β-selection and its dependence on both pre-TCR and Notch, conditionally Pten−/− DN cells showed constitutively active Akt and accelerated development to DP cells. They could generate DP cells without pre-TCR or Notch signaling (104–108). But many details about how pre-TCR and Notch crosstalk via PI3K are controversial, and it remains unclear why pre-TCR signaling alone is insufficient for β-selection (4, 6, 108). The ability of IP4 to antagonize PIP3 binding to Akt and documented Itpkb expression and activation by TCR signaling in thymocytes (20, 28, 47, 48) prompted us to explore a role for Itpkb in this process.
We found that Itpkb−/− DN3 cells were pre-TCR hyperresponsive with Akt/mTOR-hyperactivation and metabolic hyperactivity (27). Mixed BM chimeras and in vitro studies showed a DN3 cell-intrinsic Itpkb requirement. In vitro and in vivo, Itpkb−/− DN3 cells showed an accelerated and Notch independent, but pre-TCR dependent differentiation into DP cells with wild-type like proliferation and viability. Pharmacological inhibition of Akt, mTOR, or glucose metabolism restored wild-type developmental kinetics and the Notch dependence of Itpkb−/− DN3 cells in fetal thymic organ cultures or OP9/OP9-DL1 cell co-cultures. Finally, Itpkb codisruption enabled the CD3-induced development of Rag2−/− DN3 cells into ISP and DP cells in mice injected with a γ-secretase inhibitor which blocks Notch signaling and impaired the maturation of Itpkb+/+Rag2−/− DN cells in vivo (27). So, Itpkb loss in DN3 cells reduced the Notch dependence of DN thymocyte development to DP cells in vitro and in vivo.
Itpkb−/− thymocytes had strongly reduced IP3 3-kinase activity and IP4 levels, but normal IP3 levels and Ca2+ mobilization (20, 28, 47). Based on the IP4/PIP3 antagonism in other immunocytes, we proposed that pre-TCR induced IP4/PIP3 antagonism governs β-selection by restricting PI3K/Akt/mTOR signaling and metabolic activation (27). In our model, Itpkb controls pre-TCR/Notch crosstalk through combined restriction of pre-TCR induced and Notch induced PI3K/Akt signaling (Figure 7B). This ensures that Akt is only activated to the extent needed for β-selection and only in an appropriate context: pre-TCR+ DN3 cells interacting with Notch-ligand expressing stromal cells in the thymus (93). This prevents premature differentiation. Without Itpkb, excessive Akt signaling accelerates DN3-to-DP development (Figure 7C). In the absence of Notch, Itpkb loss enables pre-TCR signaling alone to sufficiently activate Akt to rescue DN3-to-DP thymocyte development (Figure 7D). Altogether, non-canonical Itpkb antagonism with PI3K both delays thymocyte β-selection and renders it Notch-dependent.
Interestingly, Itpkb has distinct functions from SHIP and PTEN in β-selection. SHIP-1−/− early thymocytes develop normally (109). Conditionally Pten−/− DN cells have constitutively active Akt and generate DP cells without pre-TCR or Notch signaling (104–107). And constitutively active Akt allows DN3-to-DP cell development without pre-TCR or Notch-signaling, but not without both (97, 103, 105, 110). Notch may promote β-selection in part by inducing HES1 to repress PTEN, and c-Myc to promote proliferation (107). By contrast, Itpkb loss accelerates DN3 cell differentiation without significant effects on proliferation and viability, and overcomes the dependence of β-selection on Notch but not the pre-TCR (27). We hypothesize that the latter reflects the requirement for TCR signals to activate Itpkb and produce IP4 (28, 47, 48). Without pre-TCR signals, Itpkb is inactive and its loss has no further effect. Itpkb loss might also reduce less essential positive Itpkb roles in pre-TCR signaling, such as promoting Itk activation (20, 111). The PI3K-independent c-Myc induction by Notch (107) should be unaffected by IP4. This might explain why Itpkb loss overcomes Notch requirements and accelerates DN3 cell differentiation but not proliferation. The surprising lack of increased DN3/DN4 cell viability in Itpkb−/− mice might reflect differing degrees of Akt/mTOR hyperactivation in Pten−/−, dominant-active Akt1-expressing, and Itpkb−/− DN3/DN4 cells (27). Finally, the mechanistic differences between Itpkb, PTEN, and Notch regulation of β-selection, and the aforementioned death due to hematopoietic failure or infections (26, 47) might explain why Itpkb−/− mice do not present the leukemias/lymphomas caused by excessive signaling of Notch, PI3K, or Akt in DN3 cells (6, 95, 98). It will be interesting to study if combined deficiency in Itpkb and PTEN or SHIP causes earlier blood cancer development and increases its incidence.
Wrapping up this section, neutrophils, NK cells, HSC, DN3 thymocytes, and likely GMP provide examples where non-canonical antagonism of Itpkb and PI3K/Akt controls important physiological processes (Figure 4). Thus, IP4 antagonism with PIP3 is broadly important at least in hematopoietic cells. One major downstream process is metabolism, although other PIP3-regulated processes likely contribute depending on cell type and context. Additional roles for PIP3-independent functions of IP4 and Itpkb cannot be ruled out (8, 45). Consistent with these possibilities, the precise effects of Itpkb, SHIP, or PTEN inactivation in hematopoietic cells often differ. This underscores the distinct importance of Itpks and IP4 in controlling hematopoiesis.
IP7 may Antagonize PI3K in Neutrophils
Besides IP4, IP7 produced by IP6Ks can also compete with PIP3 for PH domain binding to dampen PI3K function. This was first shown in vitro and in Dictyostelium discoideum where IP6K1 deletion enhanced the membrane translocation of several PH domain-containing proteins and augmented downstream chemotactic signaling (36). A later study showed that through the same mechanism, IP6K1 and IP7 dampen Akt function in skeletal muscle, white adipose tissue and liver cells to limit insulin sensitivity (37). In IP6K1−/− mice, these organs showed elevated Akt/mTOR and reduced GSK3β signaling, resulting in insulin hypersensitivity and resistance to high-fat diet or aging-induced obesity. By contrast, IP7 treatment inhibited Akt phosphorylation and activation by PDK1 in a PH domain-dependent manner.
Expanding on these findings, the Luo lab demonstrated that IP7 can also dampen PIP3 signaling in neutrophils (24) (Figure 4). Neutrophils from IP6K1−/− mice or human neutrophils treated with a pharmacological IP6K1 inhibitor showed Akt hyperactivation after fMPL treatment, enhanced PIP3-mediated membrane recruitment of an ectopically expressed Akt PH domain, elevated phagocytic and bactericidal activity, and augmented Akt-dependent, NADPH-oxidase mediated superoxide production compared to wild-type or untreated neutrophils, respectively. By contrast, overexpression of wild-type but not catalytically inactive IP6K1 in neutrophil-like differentiated HL60 cells (dHL60 cells) caused IP7 overproduction and suppressed fMLP-induced Akt activation, membrane recruitment, and downstream superoxide production. And exogenous IP7 blocked PI3K-dependent superoxide production in neutrophils. Suggesting physiological relevance of these findings, IP6K1−/− mice had elevated peritoneal ROS but reduced intraperitoneal bacterial counts in two different acute peritonitis models at early timepoints post-bacterial infection when macrophages and lymphocytes are not yet recruited. This occurred despite attenuated peritoneal neutrophil accumulation, possibly secondary to accelerated bacterial clearance or chemoattractant deactivation by the elevated ROS. Surprisingly, IP6K1−/− neutrophils showed wild-type like cell adhesion, directionality, migration velocity, and recruitment to the peritoneal cavity upon adoptive transfer and had wild-type like viability in vitro, although these processes are PI3K dependent. This somewhat contrasts with the effects of PTEN loss in neutrophils and clould reflect different regulation of PI3K by PTEN and IP6K1 in these, non-redundant PI3K dampening by Itpkb in neutrophils, or the surprising but incomplete drop in neutrophil IP7 levels after fMLP stimulation (24). Comparing PIP3 vs. IP4 vs. IP7 amounts and resulting PI3K/Akt activity in neutrophils lacking PTEN vs. IP6K1 vs. Itpkb might further elucidate how differential PIP3 antagonism by these enzymes impacts nuances of PI3K signaling.
Despite similarly increased fMPL-induced Akt recruitment and superoxide production, Itpkb−/− and IP6K1−/− neutrophils showed several phenotypic differences. In particular, Itpkb−/− neutrophils had reduced in vitro viability, increased chemotaxis and peritoneal recruitment, and normal-to-reduced bacterial clearance in vivo (22, 57). By contrast, IP6K1−/− neutrophils showed unimpaired in vitro viability, migration, and peritoneal recruitment but improved bacterial clearance (24). The mechanistic underpinnings of these differences remain to be elucidated. It is tempting to speculate that they include the normal vs. elevated Ca2+ mobilization in IP6K1−/− vs. Itpkb−/− neutrophils (23, 24, 112), and potential differences in the serum content of opsonizing IgG due to defective B cell functions in Itpkb−/− mice (29, 39, 41). Whether IP6Ks and IP7 have functions in B cells is unknown. Moreover, without conditional knockout mice, differential contributions of possible phenotypes in other immune cells cannot be ruled out but might explain the improved bacterial clearance in IP6K1−/− mice despite attenuated neutrophil peritoneal accumulation (24). Partial redundancy between IP6K1 and the also neutrophil-expressed IP6K2 is another possibility (24). In addition, IP7 can bind multiple proteins including epigenetic regulators, and contrasting with IP4 can serve as a non-enzymatic protein phosphorylating agent (113–115). It remains to be elucidated whether these functions play roles in neutrophils. Finally, Ip6k1-mediated inorganic polyphosphate production in platelets promoted alveolar neutrophil accumulation during bacterial pneumonia (116). Distinct features of IP6K1 regulation in neutrophils, and of IP7 vs. inorganic polyphosphates, IP4 and PIP3 may also explain differences between the neutrophil phenotypes of IP6K1−/−, PTEN−/− (63–65), and SHIP−/− mice (62, 117), summarized above in the Itpkb section and in Ref. (112).
IP4 may Promote PI3K Signaling to Enable Thymocyte Positive Selection
The first hematopoietic defect in Itpkb−/− mice reported independently by the Schurmans/Erneux group and us was a severe T cell deficiency resulting from blocked thymocyte development at the DP stage (28, 47) (Figure 3). Studying the underlying molecular defect, we found evidence that IP4 may promote the PIP3-mediated membrane recruitment and activation of Itk downstream of the TCR by acting as a soluble PIP3 analog that binds the Itk PH domain and promotes PIP3 binding (20) (Figure 8). This was the first demonstration that IP4 has an important in vivo function and can act as a physiologically relevant PIP3 analog, and that Itpkb controls PI3K function in vivo.
Figure 8.

Feedback-activation of Itk/phospholipase-Cγ1 (PLCγ1) by IP4 may amplify mild T cell receptor (TCR) signals such that they can trigger thymocyte positive selection. Based on studies of Itpkb−/− mice and mathematical simulations of different signaling circuitries in DP cells (20, 21, 28, 56), we proposed a simplified model where TCR engagement on DP thymocytes activates proximal protein tyrosine kinases (SFK, ZAP), which then tyrosine-phosphorylate transmembrane adaptors (TMAPs, LAT). Among other events, this activates phosphoinositide 3-kinase (PI3K), which then phosphorylates membrane PIP2 into PIP3. By binding to their PH domains, PIP3 recruits PLCγ1 and its upstream activator, the Tec-family protein tyrosine kinase Itk into the TCR signalosome which also contains LAT and the adaptors SLP-76 and Gads. In this model, Itk is oligomeric with low PH domain affinity for PIP3 (dark gray). Therefore, initial Itk recruitment is limited and only triggers low-level PLCγ1 activation. PLCγ1 hydrolyzes PIP2 into low amounts of the second messenger diacylglycerol (DAG) and IP3. IP3 mobilizes Ca2+. Ca2+ binds calmodulin (CaM) which then binds to and activates Itpkb and calcineurin (CaN) (118). CaN dephosphorylates and activates the transcription factor NFAT (data not shown) (119). Itpkb phosphorylates IP3 into IP4. IP4 binding to one Itk subunit allosterically increases the PH domain affinity of all Itk subunits for PIP3 (light gray). This promotes Itk membrane recruitment, causing full PLCγ1 activation and sufficient DAG-production to activate Ras/Erk and trigger thymocyte positive selection. Itpkb loss perturbs this feedback activation through decreased IP4 production and Itk recruitment. As a result, insufficient DAG production impairs Ras/Erk activation and positive selection, causing a block of thymocyte development at the DP stage. Itpkb and IP4 may have additional functions in DP thymocytes, which also express additional IP3 3-kinases whose roles in positive/negative selection remain unclear (28, 47). For a detailed discussion, see text [adapted with permission from Ref. (8)].
In DP cells, TCR ligand-sensitivity is assessed through interactions with self-peptide/MHC complexes on thymic stromal cells. Insufficient TCR signals cause thymocyte death by neglect. Adequately mild signals cause DP cell survival and differentiation into CD4 and CD8 single-positive T cells. This “positive selection” ensures that only T cells with a functional TCR develop. Intermediate TCR signals “agonist-select” regulatory T cells. But excessive TCR signals in DP cells cause activation-induced cell death (AICD). This “negative selection” prevents the maturation of self-reactive T cells which could cause autoimmune diseases (120).
T cell receptor stimulation activates proximal protein tyrosine kinases, which then phosphorylate transmembrane adaptors including LAT. Their phosphotyrosine moieties subsequently bind and recruit downstream effectors including PI3K, Itk, and phospholipase-Cγ1 (PLCγ1) (8, 19). Itk recruitment also requires binding of its PH domain to membrane-PIP3 (19) (Figure 8). Itk/PLCγ1 co-recruitment to LAT allows Itk to phosphorylate and activate PLCγ1. PLCγ1 then hydrolyzes membrane PIP2 into diacylglycerol (DAG) and soluble IP3. DAG recruits PKCs, and RAS-GRP1 to activate Ras/Erk signaling. This is required for positive selection (8). IP3 binds to IP3 receptors in the ER to mobilize Ca2+. Alternatively, Itpkb can convert IP3 into IP4. In some cells, IP4 can also control Ca2+ mobilization (19, 45, 61, 121).
In Itpkb−/− mice, positive selection was severely blocked. Data about negative selection were negative or inconclusive (8, 20, 28, 47). As expected, Itpkb−/− DP cells showed reduced TCR-induced IP4 production. Although Itpkb loss was expected to cause IP3 accumulation and Itpkb−/− peripheral T cells showed elevated Ca2+ mobilization (59), Itpkb−/− DP cells produced normal amounts of IP3 and Ca2+ (28, 47). The inability of catalytically inactive Itpkb, but ability of exogenous IP4 to restore positive selection of Itpkb−/− DP cells suggested a specific IP4 requirement for this pivotal process (8, 20). Biochemical studies then showed that in Itpkb−/− DP cells, TCR-induced Erk activation was impaired because of defective Itk membrane recruitment and activation. This impaired PLCγ1 activation and DAG production (20, 28). Compensation of reduced IP3 turnover via Itpkb by reduced PLCγ1-mediated IP3-production might explain the normal IP3 levels in Itpkb−/− DP cells (20).
The dual ability of IP4 to bind to the Itk PH domain and impair PIP3-binding at high, but promote PIP3-binding at low, physiological concentrations then suggested that IP4 might be required for Itk membrane recruitment and activation by augmenting Itk PH domain binding to PIP3 (20). Although the precise mechanism remains to be fully elucidated, the ability of full-length Itk or its PH domain alone to oligomerize is consistent with a model where IP4 binding to one Itk-subunit induces allosteric changes in the other Itk-subunits that cooperatively increase the affinity of their PH domains for PIP3 (2, 3, 8, 20, 122) (Figure 8).
These data suggest that in DP cells, IP4 may establish a positive feedback loop of PLCγ1 activation by Itk that is required for the production of sufficient DAG to activate Ras/Erk and trigger positive selection (8, 20). Ca2+ controls signaling by binding to various proteins, including calmodulin (CaM). Ca2+/CaM can bind and activate Itpkb (45, 48, 123–126). TCR-induced IP4 production requires Itpkb, Ca2+, and CaM (8, 28, 47, 56, 127, 128). This and the inability of CaM-nonbinding mutant Itpkb to restore Itpkb−/− thymocyte maturation (20, 48) suggested that TCR-induced IP4 production in DP cells may involve an additional feed-forward loop of Itpkb activation by Ca2+/CaM downstream of PLCγ1 (8). We proposed that this combination of feedback- and feedforward-activation loops establishes an IP4- and Ca2+-dependent signal amplifier that allows mild TCR stimuli to trigger positive selection, but is dispensable for negative selection triggered by strong stimuli (8). This circuitry might also underlie a previously proposed signal splitter that directs selection outcome (129). Supporting this model, strong TCR stimuli rescued DAG production and Erk signaling in Itpkb−/− thymocytes (20, 28). However, detailed studies in sensitive models (130) will be required to conclusively determine how Itpkb loss affects negative and agonist selection.
Due to difficulties in quantifying Itk interactions with PIP3 and IP4, in monitoring Itk-oligomerization in vivo, and in generating non-oligomerizing Itk mutants, the physiological relevance of Itk oligomerization remains controversial (20, 122, 131–136) and many mechanistic details of how IP4 controls Itk remain to be elucidated. Providing conceptual support for the model in Figure 8, computational simulations of various circuitries involving mono- or oligomeric Itk indicated that those models which shared a cooperative-allosteric Itk regulation by IP4 involving oligomeric PH domains were most robust against variations of reactant amounts and kinetic rates at the single-cell level (21). Interestingly, some models predicted an additional benefit for Itk inhibition through PIP3 antagonism by high doses of IP4. Although high-dose IP4 can inhibit Itk PH domain binding to PIP3, it is unknown whether such high doses can be achieved in DP cells (20). Further exploration of bimodal Itk regulation by IP4 and of the physiological relevance of different Itk dimers remain important future research areas (21). Finally, recent evidence that in TCR-stimulated thymocytes, Itpkb is phosphorylated by Erk which is counteracted by the Ca2+-activated phosphatase calcineurin suggests complex additional circuitries whose physiological relevance remains to be elucidated (137).
Although Itpkb is pivotal for positive selection, residual Itpk activity and IP4 production in Itpkb−/− DP cells suggest relevance for other IP3 3-kinases and that complete IP4 loss could have more severe phenotypes (28, 47). This would be consistent with the broader roles of PI3K and Itk in thymocyte selection (2–5, 111, 138–142). Co-disruption of several IP3 3-kinases in DP cells will be required to address this question. Moreover, it will be important to generate conditionally Itpkb-deficient mice and exclude contributions of the defects in HSC (26) and β-selection (27) to the DP thymocyte defects in germline Itpkb−/− mice. Finally, differences in the selection phenotypes of Itpkb−/− and Itk−/− mice point toward possible roles for other IP4 targets, or for Itpkb interactions with actin (8, 45, 143, 144). Thus, deeper mechanistic studies should yield important additional insight.
IP4 Dampens Store-Operated Ca2+ Entry (SOCE) in Immunocytes to Promote Survival and Prevent Inflammatory Disease
Itpkb Is Required for T Cell Viability and Function
Peripheral T cells express all three Itpks. TCR stimulation induced IP3 3-kinase activity and IP4 production in Jurkat T cells (56, 127). To elucidate the functions of Itpkb and IP4 in peripheral T cells, two studies have used different approaches aimed to leave T cell development intact. The Cooke group combined studies of mice with tamoxifen-induced conditional Itpkb disruption (Itpkb cKO) with studies of the effects of a specific and selective, orally bioavailable pan-Itpk small-molecule inhibitor, GNF362 (59). Tamoxifen treatment of Itpkb cKO mice caused a mild defect in positive selection that contrasts with the severe block in germline Itpkb−/− (Itpkb gKO) mice (20, 28, 47). Indeed, Itpkb cKO mice had control-like numbers of splenic B and T cells. Compared to Cre+unfloxed controls, Itpkb cKO T cells had normal basal viability but underwent AICD after TCR stimulation (59). Intact cytokine production may suggest that this is their primary defect, consistent with rescued T cell viability and proliferation by FasL blockade. Supporting T cell malfunction, Itpkb cKO mice failed to generate antibody responses to T cell-dependent but not -independent antigens.
Following IP3-mediated Ca2+ release from ER stores, STIM1 proteins in the ER sense the resulting Ca2+ depletion, translocate close to the plasma membrane and activate Orai channels which mediate SOCE. This is essential for T cell activation (119). Interestingly, Itpkb cKO T cells showed enhanced SOCE, and treatment with high doses of cell-permeable IP4 rapidly inhibited SOCE in HEK293 cells overexpressing STIM1 and ORAI (59). The opposing effects of Itpkb loss and IP4 treatment on SOCE might suggest that Itpkb limits SOCE through IP4. Consistent with this view, GNF362-treatment blocked IP4 production in Jurkat T cells and enhanced TCR-induced SOCE in thymocytes and murine T cells. GNF362 also inhibited T cell proliferation and caused Itpkb-dependent AICD (59). In mice, GNF362 recapitulated the blocked T cell development seen in Itpkb−/− mice. Consistent with T cell inhibition, GNF362 inhibited joint swelling and secondary antibody responses in a rat antigen-induced arthritis model (59).
While the precise mechanism through which Itpkb and IP4 inhibit SOCE in T cells remains to be elucidated, elevated Ca2+ mobilization can induce pro-apoptotic genes to mediate AICD, and Orai1-deficient T cells are resistant to AICD (145). So, the elevated SOCE in Itpkb cKO T cells might explain their AICD (59). However, phenotype rescue through pharmacologic or genetic prevention of the SOCE elevation in Itpkb-inactivated T cells will be required to prove this. Otherwise, it remains possible that the AICD of Itpkb-inactivated T cells results at least in part from a hypersensitivity to TCR stimulation or generally increased TCR signals. Given the hyper-responsiveness of Itpkb−/− HSC, DN thymocytes, NK cells, and neutrophils to stimulation, this remains a possibility worth testing. Then again, based on the defective Itk/PLCγ1 activation in Itpkb−/− DP cells (20) and the Akt/mTOR hyperactivation in Itpkb−/− DN thymocytes (27), Itpkb-deficient peripheral T cells could have complex additional defects with loss-of-function and gain-of-function components that remain to be explored.
In an independent study, transient transgenic Itpkb expression partially rescued thymocyte development in another line of Itpkb gKO mice (43). These but not mice transiently expressing catalytically inactive Itpkb showed partially restored SP thymocytes. They also had low numbers of peripheral T cells with an activated/memory phenotype but decreased TCR-induced proliferation and survival, and increased cytokine secretion compared to wild-type mice. TCR-induced Ca2+ mobilization was not significantly altered.
The reduced proliferation and survival of Itpkb transgene-rescued Itpkb gKO T cells are consistent with the Itpkb cKO or GNF362-treated T cell phenotypes (59). However, the activated/memory phenotype and cytokine hypersecretion contrast with those. Possible reasons could be homeostatic expansion of the few transgene-rescued T cells, or confounding effects of infections. Moreover, transgenic Itpkb was expressed from the Lck proximal promoter which transiently expresses transgenes in DN and DP thymocytes but not in HSC (146). So, unrescued HSC defects in Itpkb gKO mice (26), the super-physiological amount of transgenic Itpkb in the rescued thymocytes (43), the incomplete rescue of thymocyte development, or low residual transgene expression in peripheral T cells could all possibly affect T cell phenotypes.
Wrapping up, both studies suggest that Itpkb and IP4 have critical functions in ensuring the survival and function of activated peripheral T cells (Figure 5). The underlying mechanism may involve IP4 dampening of SOCE, but the molecular details remain to be explored and other possibilities have not been ruled out. Clearly, further studies of how Itpkb controls T cell function should prove exciting.
Itpkc Dampens Ca2+ Mobilization in Immune Cells to Prevent Inflammatory Disease
Despite its broad expression, studies in Itpkc−/− mice have not yet unveiled lymphocyte phenotypes, and co-disruption of Itpkb and Itpkc did not worsen the thymocyte defects in Itpkb−/− mice. Itpkc−/− thymocytes showed unaltered IP3 3-kinase activity (47, 51). This argues against major Itpkc roles in adaptive immune responses in mice. By contrast, human population genetics suggest that ITPKC may limit Ca2+ mobilization in, and function of human T cells (Figure 5). In a seminal study (40), Onouchi et al. found an interesting association of a human ITPKC allele that reduced ITPKC mRNA splicing efficiency and abundance (ITPKClof) with increased susceptibility to KD, a multisystem inflammatory vasculitis that mainly affects coronary arteries (147). KD is the leading cause of childhood-acquired heart disease in developed countries (40). Several subsequent studies confirmed the ITPKClof genetic association, although others found no evidence for it, likely due to different subject cohorts with unknown confounding genetic and environmental influences (148).
Acute phase KD patients showed T cell infiltration into the coronary artery wall and IL-2 overproduction, suggesting T cell hyperactivation. PMA/ionomycin treatment upregulated ITPKC mRNA levels in human T cells, and ITPKC overexpression decreased, but ITPKC knockdown increased, phytohemagglutinin- and PMA-induced NFAT activation and IL-2 mRNA expression in Jurkat cells. This suggests that ITPKC inhibits human T cell activation upstream of the Ca2+-activated transcription factor NFAT (40, 149). Another important advance in our understanding of how ITPKC controls KD was provided by the recent finding that Itpkc limits Ca2+ mobilization in myeloid cells to restrict activation of the NLRP3 inflammasome (44). Compared to wild-type controls, bone marrow-derived macrophages from Itpkc−/− mice had elevated basal and ionomycin-induced Ca2+ levels and NLRP3 expression. They responded with NLRP3 hyper-induction and excessive release of pro-inflammatory IL-1β to in vitro activation by LPS/ATP or Lactobacillus casei cell wall extract (LCWE). In a LCWE-induced KD model, Itpkc−/− mice overproduced circulating IL-1β and developed a more severe disease compared to wild-type controls.
Ascribing human relevance to these findings, acute-phase KD patients had higher serum levels of IL-1β, IL-18, and their antagonists IL-1RA and IL-18BP than convalescent and age-matched febrile controls (44). Whole blood from acute-phase KD patients also hyperexpressed a gene signature suggesting NLRP3 activation. Interestingly, EBV-immortalized B cells from KD patients or healthy controls harboring homozygous ITPKClof had reduced Itpkc protein levels. They recapitulated the elevated basal and ionomycin-induced Ca2+ levels of murine Itpkc−/− macrophages, showed a more sustained Ca2+ mobilization, and overexpressed NLRP3 (44). They also overproduced mitochondrial superoxide, a Ca2+-dependent NLRP3-activator. So, Itpkc loss in human B cells associates with Ca2+ hypermobilization, which likely triggers superoxide-mediated NLRP3 activation. Acute phase KD patients carrying homozygous ITPKClof also showed elevated plasma concentrations and LPS/ATP-stimulated PBMC production of IL-1β and IL-1. This suggests that the NLRP3 hyperactivity caused overproduction of pro-inflammatory cytokines, similar to Itpkc loss in mice. Increased resistance to standard IVIG therapy in KD patients carrying ITPKClof supports pathological relevance of these effects (44). These observations suggest interesting similarities between KD and recurrent fever syndromes that may reflect causative NLRP3 hyperactivity. They may explain the efficacy of IL-1 blockade in recalcitrant KD and may identify IL-1β, IL-18, and their antagonists as much-needed biomarkers for early diagnosis (44).
Intriguingly, KD may not be the only disease affected by Itpkc. Recent studies found potential associations between ITPKC genetic variations and Hirschsprung disease, calcium nephrolithiasis, and cervical squamous cell carcinoma (150–152). Thus, further mechanistic studies of Itpkc biology are becoming exceedingly important.
Itpkb Dampens SOCE in B Cells
Chemically induced Itpkb gKO mice showed overall normal B cell development in the BM but had markedly reduced numbers of all splenic B cell subsets (39, 41). Further studies showed that Itpkb is essential for the selection of functional B cells. To avoid autoimmunity, B cells carrying a self-reactive B cell receptor (BCR) are tolerized through clonal deletion, functional inactivation (anergy), or BCR editing to a different antigen specificity (153). Mature B cells from Itpkb−/− mice shared many features with B cells from BCR and BCR-antigen transgenic anergy models (154). Examples are IgM downregulation, impaired BCR-driven proliferation, reduced upregulation of surface-CD69, CD86, and MHCII, and decreased antibody responses to T cell-independent antigens (29, 39, 41). Responses to LPS or CD40 stimulation were normal. In the HEL BCR transgenic model, Itpkb loss converted responses to mild BCR stimulation from activation to anergy, and responses to moderate stimuli from anergy to deletion (41). This resembles the effect of losing other inhibitors of BCR signaling, such as CD22, SH2 domain containing phosphatase-1, or the Src family protein tyrosine kinase Lyn (8, 153). In developing B cells, Itpkb thus prevents mild BCR stimuli from inducing tolerance and ensures that only B cells expressing self-reactive BCRs are tolerized.
The Schurmans group found overall similar changes in B cell development and impaired T cell-independent antibody responses in Itpkb gKO mice. This was associated with reduced in vitro survival of Itpkb−/− B cells, which upregulated pro-apoptotic Bim (29). Bim haploinsufficiency or transgenic expression of anti-apoptotic Bcl-2 increased B cell numbers in Itpkb−/− mice. Bcl-2 expressing Itpkb−/− B cells showed diminished BCR-induced Erk activation. The authors used data from non-lymphoid COS cells to suggest that IP4 increases B cell survival by sequestering the IP4-binding, Ras-inactivating protein RASA3/Gap1IP4BP (155) in the cytosol, resulting in sustained Ras/Erk activation, Bim-phosphorylation, and Bim-degradation (29). However, without confirmation in B cells, the physiological relevance of RASA3/Gap1IP4BP regulation by IP4 remains unclear. Later, the same group used 3-83μδ BCR transgenic mice to explore Itpkb roles in B cell tolerance (42). They found that in a context of mild BCR engagement, Itpkb loss impaired B cell maturation and viability, again associated with Bim upregulation. B cell deletion in a context of stronger BCR engagement was unimpaired. Overall, these findings support a shift from B cell functionality or anergy to deletion when Itpkb is lost. Although both BCR-transgenic models revealed surface IgM downregulation on Itpkb−/− B cells, some differences in the specific response patterns to increasing BCR engagement likely reflect different signaling capacities of the two transgenic BCRs.
In BCR-transgenic anergy models, constitutive expression of self-antigens causes BCR desensitization with defective activation of proximal Lyn/Syk kinases and downstream PLCγ2, IP3 production, and Ca2+ mobilization (8, 153). By contrast, chemically induced Itpkb−/− anergic B cells showed overall normal BCR activation of Lyn, Btk, PLCγ2, Erk1/2, and IKKα/β and control-like IP3-production, but increased SOCE (39). SOCE normalization by exogenous cell-permeable IP4 suggested that the elevated SOCE might result from impaired SOCE dampening by IP4 (Figure 5). The Schurman group initially reported reduced BCR- or ionomycin-induced Ca2+ influx in Itpkb−/− B cells (29). However, 3-83μδ BCR transgenic Itpkb−/− B cells showed an elevated BCR-induced Ca2+ influx compared to Itpkb+/+ controls, similar to chemically induced Itpkb−/−IgHEL transgenic and non-BCR transgenic mice (39, 41). The reason for the discrepant Ca2+ defects in the original Itpkb−/− mouse cohort remains unclear, but might include differentially augmented B cell deletion between the models, or effects of an altered Bim/Bcl-2 ratio on IP3-receptor function in those particular B cells (42, 156). Consistent with this view, Itpkb−/−IgHEL transgenic mice showed neither increased negative selection nor Bim accumulation (41). Alternate explanations might include differences in housing, health status, genetic background, or age of the mice used in the different studies.
Despite minor differences, all four studies support a pivotal Itpkb role in dampening BCR signaling to prevent aberrant B cell tolerization. By augmenting BCR signaling, Itpkb loss induces anergy of B cells expressing low-to-moderately self-reactive BCRs, but deletion of normally anergic B cells expressing more strongly self-reactive BCRs (41, 42). Thus, Itpkb feedback inhibits BCR signaling to broaden the repertoire of immature B cells that survive negative selection. This positions the BCR selection window appropriately to ensure a normal B cell repertoire that is further tuned through BCR editing. One prediction of this model would be an increased generation of self-reactive B cells which might eventually cause autoimmune disease. Reported diminished BCR light-chain editing in Itpkb−/− vs. wild-type B cells suggests that such autoreactive cells would probably not be “reprogrammed” through receptor editing (157). However, neither Itpkb−/− mice nor mixed radiation chimeras of Itpkb−/− BM with wild-type T, B, and myeloid cells have shown signs of autoimmunity (8). This could reflect perturbed positive Itpkb functions in peripheral B cells, or the premature death of Itpkb−/− mice due to HSC defects (26) or infections (47) before autoimmunity can develop. Conditional Itpkb disruption in developing vs. mature B cells might prevent some of these problems and help clarify this conundrum, in particular when combined with detailed analyses of the BCR repertoire.
Indeed, a recent study reported that after tamoxifen-induced Itpkb deletion in all cells, Itpkb cKO mice had near normal B cell numbers and T cell-independent immunization responses associated with reduced Ca2+ ER release but elevated SOCE in B cells (59). So, induced Itpkb loss recapitulated the SOCE increase in germline Itpkb−/− B cells but had no major effects on B cell development, homeostasis, viability, and function. Similarly, GNF362 pan-Itpk inhibitor treatment reduced BCR-induced Ca2+ ER release but augmented SOCE in wild-type B cells (59). These effects strikingly resemble those reported for Itpkb−/− neutrophils (23, 112). They also resemble the elevated SOCE in Itpkb cKO and GNF362-treated wild-type T cells, although ER release was not detected there (59). Interestingly, GNF362 still inhibited ER release in Itpkb−/− B cells, but without affecting SOCE (59). So, in murine B cells, SOCE is primarily dampened by Itpkb, but ER release requires additional IP3 3-kinases such as Itpkc, whose loss-of-function in human B cells elevated basal and ionomycin-induced Ca2+ levels (44).
The overall normal B cell homeostasis and function in Itpkb cKO mice suggest that the increased tolerance of Itpkb gKO B cells results from their altered development and selection. The precise functional consequences of Itpkb loss in mature B cells remain to be elucidated. Drawing on the phenotypes of EBV-transformed human B cells carrying the ITPKClof allele (44), one might expect NLRP3 hyperactivation. It will be interesting to assess if Itpkb cKO mice hyperproduce immunoglobulins or pro-inflammatory cytokines and develop inflammatory disease.
It is intriguing that the main molecular defect in Itpkb−/− and ITPKClof B cells is aberrant Ca2+ mobilization. While effects on basal Ca2+ levels and ER store-release are discrepant (possibly depending on model system and assay conditions), elevated SOCE emerges as a common effect (Figure 5). This suggests that the main function of Itpkb/c and IP4 in B cells is to inhibit BCR-induced Ca2+ signaling. The precise mechanism causing the elevated SOCE in Itpkb- or Itpkc-deficient B cells and other immune cells remains unknown. We discuss possibilities in Section “Conclusion and Perspectives.” Beyond elucidating this mechanism, establishing causality of the elevated SOCE for the B cell phenotypes remains important.
Itpkb’s pivotal role in controlling B cell development and function is further emphasized by the recent association of a microdeletion which causes ITPKB deficiency in humans with a common variable immunodeficiency (CVID) (158). A patient carrying this microdeletion expressed reduced ITPKB protein. He had reduced serum IgG and IgA, but normal IgM levels and suffered from recurrent skin infections and other symptoms. He did not respond to T cell-independent Streptococcus pneumoniae vaccinations and had decreased numbers of T, Treg, and NK cells, but normal B cell numbers with increased proportions of marginal zone, transitional, memory, and CD21low B cells. Antigen-induced lymphocyte proliferation and neutrophil oxidative burst were severely impaired. Although additional genes are likely affected by the microdeletion and incomplete ITPKB protein loss, two ITPKB missense mutations and a synonymous variant may all explain differences between this patient and the KO mice, mechanistic studies to confirm causality of the ITPKB mutation for the CVID should prove exciting.
Limiting hematopoietic cell-intrinsic PI3K signaling is critical for preventing blood cancers. In mice, SHIP-1 and PTEN deficiency in B cells caused B cell lymphoma associated with excessive PI3K/Akt signaling (12). Human diffuse large B-cell lymphoma (DLBCL) samples under-expressed PTEN and SHIP-1 (12, 159), and human mantle cell lymphoma samples under-expressed PTEN (160). Reduced PTEN expression or predicted oncogenic PI3Kα mutations associated with poor survival in DLBCL (159) and a third of Burkitt’s lymphomas have inactivating PTEN mutations (161). Although no significant changes in PI3K signaling in B cells have been reported in Itpkb−/− mice, it is attractive to speculate that Itpkb or redundant IP3 3-kinases could have tumor-suppressor functions by dampening PI3K signaling through IP4/PIP3 antagonism. Consistent with this view, a large-scale retroviral mutagenesis screen identified Itpkb as one of the 50 most important common insertion sites in murine lymphoma. Itpkb insertions were anti-correlated with insertions in Pik3cd encoding PI3Kδ (162, 163). But no blood cancer phenotypes have been reported in Itpkb−/− mice. As discussed before, this could reflect their premature death due to BM failure, anemia, or infections (26, 47), or partial Itpkb redundancy with Itpka, Itpkc, or IPMK. Conditional Itpkb disruption in the B cell lineage to avoid anemia and infections, or breeding Itpkb−/− mice into blood cancer models will be required to further explore possible Itpkb tumor-suppressor functions. Co-disruption of several IP33-kinases can address possible redundancy.
Consistent with an ITPKB tumor-suppressor function in human blood cancers, large-scale whole exome sequencing has identified three different ITPKB somatic mutations as candidate CLL drivers in 2% of human patients (164). Two frameshift mutations will remove the Itpkb catalytic domain and thus impair IP4 production; the effects of a T626S mutation remain to be explored. Several other studies have found ITPKB locus deletions, copy number reductions, or missense mutations in patients with DLBCL, Burkitt’s lymphoma, or transformed FL, which often progresses to DLBCL (165–170). Their pathological relevance and underlying mechanisms are unknown. Finally, another retroviral mutagenesis screen found insertions in Itpkb to synergize with a retrovirally expressed, AML-associated Runx1-mutant in promoting murine BM progenitor outgrowth (171). The same study found that ITPKB amplifications and mRNA upregulation associate with poor survival in human AML. However, retroviral insertion can activate or inactivate genes, Itpkb protein levels, function and causality were unassessed and in another study, Itpkb knockdown increased human AML cell expansion (172). Thus, the precise function of Itpkb in AML remains unclear.
Itpkb Dampens SOCE in Neutrophils
IP4 limitation of SOCE may not be limited to lymphocytes. This is suggested by the decreased Ca2+ store release but enhanced SOCE in Itpkb−/− neutrophils (Figure 5) (23), discussed above. Its functional consequences remain to be elucidated.
Does Itpkb Inhibition have Therapeutic Potential in Human Diseases?
The T and B cell defects in germline Itpkb−/− mice sparked efforts to develop specific and selective Itpkb small-molecule inhibitors as potential therapeutics for autoimmune disorders or transplant rejection, reviewed in detail in Ref. (8, 149). Consistent with the distinct structural features and biochemical properties of IP3 3-kinases, several different inhibitors have been developed. However, many lack the required potency, isoform selectivity, specificity, and oral bioavailability. Some show high Itpk selectivity over IPMK, but none is exclusively selective for Itpkb (54, 59, 149, 173–175). A possible utility of Itpk inhibitors for immunosuppression is also supported by the T cell impaired phenotypes of Itpkb cKO mice and mice treated with the oral pan-Itpk inhibitor GNF362, and by the GNF362 efficacy in a rat antigen-induced arthritis model (59). Interestingly, induced Itpkb deletion in adult mice, or GNF362 treatment of adults unveiled no major defects in B cell function. Thus, the efficacy of any treatment of adults with Itpk inhibitors might primarily rely on T cell inhibition, limiting the utility of this approach to T cell-mediated diseases. A therapeutic Itpk inhibitor would need to be exquisitely selective for Itpkb to avoid inhibition of Itpkc, whose lof hyperactivates T cells, B cells, and macrophages and has been implicated in human inflammatory KD, Hirschsprung disease, calcium nephrolithiasis, and cervical squamous cell carcinoma (150–152). GNF362 does inhibit Itpka and Itpkc (59), but the relevance of their co-inhibition for any phenotypes remains to be elucidated. Any therapeutic approach would also need to avoid the CVID, BM failure/anemia, and possibly neutrophil hyperactivity found in human patients or mice with persistent Itpkb lof (22, 23, 26, 57, 158), and the disruption of possible Itpkb tumor-suppressor functions discussed in the B cell section. Based on the common reversibility of drug-induced HSC mobilization (85–87) and the dependence of many neutrophil functions on B cell-produced immunoglobulins (22, 23, 57), transient or intermittent Itpkb inhibition might mitigate some of these liabilities and might possibly even be able to expand HSC for therapeutic engraftment (26, 30). Finally, further elucidation of potential Itpkb roles in Alzheimer’s disease (176, 177), multiple sclerosis (178), and malignant melanoma (179) might unveil additional therapeutic opportunities or liabilities for selective Itpkb inhibitors. It will be particularly interesting to study whether Itpkb-dependent immunological mechanisms contribute to these diseases.
Conclusion and Perspectives
The data reviewed above have identified Itpkb, Itpkc, and IP4 as critical regulators of the development and function of most hematopoietic and immune cell types (Figure 3). IP4 primarily acts through two mechanisms: non-canonical PIP3 antagonism to dampen PI3K signaling, and SOCE dampening to restrict Ca2+ mobilization. PIP3 antagonism has been relatively well established, but one remaining puzzle discussed above is why PI3K signaling appears normal in Itpkb−/− B cells. The precise molecular mechanism through which Itpks and IP4 inhibit SOCE, however, remains to be determined, and a formal proof that elevated SOCE causes the associated B cell, T cell, and neutrophil phenotypes is lacking. SOCE dampening might possibly include IP4-blockade of the polybasic region in STIM1 which mediates plasma membrane recruitment, IP3-turnover by Itpks, other controversial IP3 3-kinase or IP4-roles in Ca2+-mobilization, or other functions of IP4 or its metabolites (8, 45, 119).
SOCE-modulation, additional unknown mechanisms of Itpkb/c and IP4 action, or partial redundancy of Itpka-c and IPMK could explain some of the phenotypic discrepancies between mice or humans lacking Itpkb, Itpkc, SHIP, or PTEN, reviewed above for each affected cell type. Discussed in detail elsewhere (8, 19, 38, 45), additional relevant mechanisms might involve other lymphocyte-expressed IP4-binding proteins beyond Tec kinases and Akt, including PDK1 (180), RASA2/GAP1m, RASA3/GAP1IP4BP, centaurin-α1, cytohesins, or synaptotagmins. Indeed, impaired RASA3 sequestration from the plasma membrane by IP4 has been suggested to cause Ras/Erk hyperactivation in Itpkb−/−-deficient thymocytes and B cells, although whether this occurs in lymphocytes and is physiologically relevant remains to be shown (28, 29). Identifying the entire complement of IP4-binding proteins in hematopoietic cells, and delineating their functions, will be important for a more comprehensive elucidation of how this pivotal soluble messenger controls hematopoiesis. In particular, it will be interesting to explore why Itpkc may have distinct functions in murine vs. human T cells, and what determines which mechanisms Itpks or IP4 engage in a given cell type, and whether they promote (as in DP thymocytes) or dampen (as in DN thymocytes, peripheral T, B, and NK cells, HSC, GMP, monocytes/macrophages, and neutrophils) immunoreceptor signaling and immune cell function.
Possible explanations could include the Orai-mediated SOCE requirement in mature immunocytes but not thymocytes (119), or differing Itpk or IP4 functions in different cellular or signaling contexts, or after different intensities of the input signal (8). Itpkb controls SOCE in T cells but not DP thymocytes, and Tec kinases in DP thymocytes but not B cells (8, 25, 26, 45). Moreover, thymocyte positive selection is triggered by mild and/or transient TCR signals in DP thymocytes and requires IP4. Negative selection is mediated by strong and/or sustained TCR signals and might be less impaired in Itpkb−/− mice (20, 47, 181). Peripheral T cells also generate strong TCR signals that might be impacted differently by IP4 deficiency. Our mathematical modeling studies suggested that a combination of IP4 positive (at low concentrations) and negative (at high concentrations) feedback would make TCR signaling most robust (21). Thus, a re-evaluation of Itpk functions in immunoreceptor signaling circuitries from a systems-perspective might prove informative. Alternatively, the effects of Itpks and IP4 might depend on their cellular concentration, subcellular localization, posttranslational modification, or on the specific IP4 effectors or metabolites present in a cell. Distinct roles of different Itpks could also involve IP4-unrelated noncatalytic functions of Itpka/b but not Itpkc in actin bundling (8, 45, 144, 182).
Possible contributions of IP4 metabolites are illustrated by the role of IP7 in dampening PIP3 function in neutrophils (24). Moreover, a recent study unveiled its precursor inositol-hexakisphosphate (IP6) as a candidate regulator of the B cell expressed Tec-kinase Btk (183). In vitro, physiological IP6 concentrations activated Btk by binding to a specific site in its PH–Tec-homology (TH) domain unrelated to its PIP3-binding site. IP6 sandwiching between two PH–TH domains might enable transient Btk dimerization and activation. While the physiological relevance of this mechanism remains to be shown, it might provide a second example beyond IP4 regulation of Itk for how soluble IPs could promote PH domain function. Interestingly, both examples involve PH domain oligomerization (20, 183). Among the ~234 mammalian PH domains, only ~10% bind phosphoinositides, and only those of Itk and perhaps dynamin have been shown to oligomerize in cells (1). If PH domain oligomerization is required for their positive regulation by IPs, this mode of regulation might thus be rare. But then, soluble IP4 might promote PIP3 binding of Tec and RASA3, whose PH domain oligomerization status remains unknown (20). Thus, elucidating what determines whether an IP promotes or inhibits the function of a given PH domain, or does not affect it at all, remains an important future direction.
Beyond acting as protein ligands, inositol-pyrophosphates including IP7 can also act as non-enzymatic protein-phosphorylating agents (113–115). Whether this controls hematopoietic cell functions remains to be elucidated. Clearly, deciphering the functions of unstudied “inositol code” members in hematopoietic cells promises to open up exciting and unexpected novel biology (8, 56).
Given the paramount importance of PIP3 regulation through its turnover by SHIP and PTEN (10–12), one wonders whether IP4 and IP7 might also be controlled via turnover. In vitro, several phosphatases including SHIP-1/2 can dephosphorylate the 5-positions of IP3 and IP4, and PTEN can convert IP4 into IP3 (184–188). Whether this occurs in vivo is unknown, although Jurkat T cells contain an unknown IP4 5-phosphatase unrelated to SHIP-1 (60, 189). In vivo studies of IP4 turnover appear worthwhile.
Except for one recent study focused on peripheral T and B cells (59), most of the published data about in vivo IP3 3-kinase functions to date were obtained in germline knockout mice. The B cell tolerance in mice with constitutive but not acutely induced Itpkb inactivation (59) illustrates that some of the germline knockout phenotypes likely include secondary effects of earlier defects in hematopoiesis, or sustained extrinsic effects of Itpkb loss in other cell types. It will therefore be important to confirm developmental stage-specific cell-intrinsic Itpkb/c and IP4 functions in appropriate conditional knockout mice. Concluding, Itpks and IP4 clearly play exciting and important roles in hematopoietic cells, but much work remains to be done to fully elucidate the roles of the “inositol code.” We can expect fascinating results.
Author Contributions
ME and KS wrote this review, prepared figures and revised this review.
Conflict of Interest Statement
KS is an employee of Pfizer, Inc. The remaining coauthor declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Sabine Siegemund for help with Figure 6, Luise Westernberg for help with Figure 7, and Yina Huang for critical reading of this manuscript and valuable comments.
Footnotes
Funding. Parts of this work were supported by Pfizer, Inc., NIH grants AI070845, GM100785 and GM065230, and The Leukemia & Lymphoma Society Scholar Award 1440-11 to KS. ME received salary support through The Scripps Research Institute Graduate Program.
References
- 1.Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol (2008) 9(2):99–111. 10.1038/nrm2328 [DOI] [PubMed] [Google Scholar]
- 2.Prince AL, Yin CC, Enos ME, Felices M, Berg LJ. The Tec kinases Itk and Rlk regulate conventional versus innate T-cell development. Immunol Rev (2009) 228(1):115–31. 10.1111/j.1600-065X.2008.00746.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Readinger JA, Mueller KL, Venegas AM, Horai R, Schwartzberg PL. Tec kinases regulate T-lymphocyte development and function: new insights into the roles of Itk and Rlk/Txk. Immunol Rev (2009) 228(1):93–114. 10.1111/j.1600-065X.2008.00757.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Juntilla MM, Koretzky GA. Critical roles of the PI3K/Akt signaling pathway in T cell development. Immunol Lett (2008) 116(2):104–10. 10.1016/j.imlet.2007.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buitenhuis M, Coffer PJ. The role of the PI3K-PKB signaling module in regulation of hematopoiesis. Cell Cycle (2009) 8(4):560–6. 10.4161/cc.8.4.7654 [DOI] [PubMed] [Google Scholar]
- 6.Fayard E, Moncayo G, Hemmings BA, Hollander GA. Phosphatidylinositol 3-kinase signaling in thymocytes: the need for stringent control. Sci Signal (2010) 3(135):re5. 10.1126/scisignal.3135re5 [DOI] [PubMed] [Google Scholar]
- 7.Okkenhaug K, Fruman DA. PI3Ks in lymphocyte signaling and development. Curr Top Microbiol Immunol (2010) 346:57–85. 10.1007/82_2010_45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sauer K, Cooke MP. Regulation of immune cell development through soluble inositol-1,3,4,5-tetrakisphosphate. Nat Rev Immunol (2010) 10(4):257–71. 10.1038/nri2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patrussi L, Mariggiò S, Corda D, Baldari CT. The glycerophosphoinositols: from lipid metabolites to modulators of T-cell signaling. Front Immunol (2013) 4:213. 10.3389/fimmu.2013.00213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okkenhaug K, Graupera M, Vanhaesebroeck B. Targeting PI3K in cancer: impact on tumor cells, their protective stroma, angiogenesis, and immunotherapy. Cancer Discov (2016) 6(10):1090–105. 10.1158/2159-8290.CD-16-0716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT.The PI3K pathway in human disease. Cell (2017) 170(4):605–35. 10.1016/j.cell.2017.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miletic AV, Anzelon-Mills AN, Mills DM, Omori SA, Pedersen IM, Shin DM, et al. Coordinate suppression of B cell lymphoma by PTEN and SHIP phosphatases. J Exp Med (2010) 207(11):2407–20. 10.1084/jem.20091962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruyns C, Pesesse X, Moreau C, Blero D, Erneux C. The two SH2-domain-containing inositol 5-phosphatases SHIP1 and SHIP2 are coexpressed in human T lymphocytes. Biol Chem (1999) 380(7–8):969–74. 10.1515/BC.1999.120 [DOI] [PubMed] [Google Scholar]
- 14.Muraille E, Bruhns P, Pesesse X, Daeron M, Erneux C. The SH2 domain containing inositol 5-phosphatase SHIP2 associates to the immunoreceptor tyrosine-based inhibition motif of Fc gammaRIIB in B cells under negative signaling. Immunol Lett (2000) 72(1):7–15. 10.1016/S0165-2478(00)00162-0 [DOI] [PubMed] [Google Scholar]
- 15.Heng TS, Painter MW, Immunological Genome Project Consortium . The immunological genome project: networks of gene expression in immune cells. Nat Immunol (2008) 9(10):1091–4. 10.1038/ni1008-1091 [DOI] [PubMed] [Google Scholar]
- 16.Pengal RA, Ganesan LP, Fang H, Marsh CB, Anderson CL, Tridandapani S. SHIP-2 inositol phosphatase is inducibly expressed in human monocytes and serves to regulate Fcgamma receptor-mediated signaling. J Biol Chem (2003) 278(25):22657–63. 10.1074/jbc.M302907200 [DOI] [PubMed] [Google Scholar]
- 17.Leung WH, Bolland S. The inositol 5’-phosphatase SHIP-2 negatively regulates IgE-induced mast cell degranulation and cytokine production. J Immunol (2007) 179(1):95–102. 10.4049/jimmunol.179.1.95 [DOI] [PubMed] [Google Scholar]
- 18.Keating GM. Idelalisib: a review of its use in chronic lymphocytic leukaemia and indolent non-Hodgkin’s lymphoma. Target Oncol (2015) 10(1):141–51. 10.1007/s11523-015-0359-8 [DOI] [PubMed] [Google Scholar]
- 19.Huang YH, Sauer K. Lipid signaling in T-cell development and function. Cold Spring Harb Perspect Biol (2010) 2(11):a002428. 10.1101/cshperspect.a002428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang YH, Grasis JA, Miller AT, Xu R, Soonthornvacharin S, Andreotti AH, et al. Positive regulation of Itk PH domain function by soluble IP4. Science (2007) 316(5826):886–9. 10.1126/science.1138684 [DOI] [PubMed] [Google Scholar]
- 21.Mukherjee S, Rigaud S, Seok SC, Fu G, Prochenka A, Dworkin M, et al. In silico modeling of Itk activation kinetics in thymocytes suggests competing positive and negative IP4 mediated feedbacks increase robustness. PLoS One (2013) 8(9):e73937. 10.1371/journal.pone.0073937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jia Y, Loison F, Hattori H, Li Y, Erneux C, Park SY, et al. Inositol trisphosphate 3-kinase B (InsP3KB) as a physiological modulator of myelopoiesis. Proc Natl Acad Sci U S A (2008) 105(12):4739–44. 10.1073/pnas.0800218105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia Y, Schurmans S, Luo HR. Regulation of innate immunity by inositol 1,3,4,5-tetrakisphosphate. Cell Cycle (2008) 7(18):2803–8. 10.4161/cc.7.18.6688 [DOI] [PubMed] [Google Scholar]
- 24.Prasad A, Jia Y, Chakraborty A, Li Y, Jain SK, Zhong J, et al. Inositol hexakisphosphate kinase 1 regulates neutrophil function in innate immunity by inhibiting phosphatidylinositol-(3,4,5)-trisphosphate signaling. Nat Immunol (2011) 12(8):752–60. 10.1038/ni.2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sauer K, Park E, Siegemund S, French AR, Wahle JA, Sternberg L, et al. Inositol tetrakisphosphate limits NK cell effector functions by controlling PI3K signaling. Blood (2013) 121(2):286–97. 10.1182/blood-2012-05-429241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siegemund S, Rigaud S, Conche C, Broaten B, Schaffer L, Westernberg L, et al. IP3 3-kinase B controls hematopoietic stem cell homeostasis and prevents lethal hematopoietic failure in mice. Blood (2015) 125(18):2786–97. 10.1182/blood-2014-06-583187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westernberg L, Conche C, Huang YH, Rigaud S, Deng Y, Siegemund S, et al. Non-canonical antagonism of PI3K by the kinase Itpkb delays thymocyte beta-selection and renders it Notch-dependent. Elife (2016) 5:1–24. 10.7554/eLife.10786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wen BG, Pletcher MT, Warashina M, Choe SH, Ziaee N, Wiltshire T, et al. Inositol (1,4,5) trisphosphate 3 kinase B controls positive selection of T cells and modulates Erk activity. Proc Natl Acad Sci U S A (2004) 101(15):5604–9. 10.1073/pnas.0306907101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marechal Y, Pesesse X, Jia Y, Pouillon V, Perez-Morga D, Daniel J, et al. Inositol 1,3,4,5-tetrakisphosphate controls proapoptotic Bim gene expression and survival in B cells. Proc Natl Acad Sci U S A (2007) 104(35):13978–83. 10.1073/pnas.0704312104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rigaud S, Sauer K. IP 3 3-kinase B prevents bone marrow failure. Oncotarget (2015) 6(18):15706–7. 10.18632/oncotarget.4480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou H, Wu L. The development and function of dendritic cell populations and their regulation by miRNAs. Protein Cell (2017) 8(7):501–13. 10.1007/s13238-017-0398-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol (2013) 31:675–704. 10.1146/annurev-immunol-032712-095946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hawkins PT, Stephens LR. PI3K signalling in inflammation. Biochim Biophys Acta (2015) 1851(6):882–97. 10.1016/j.bbalip.2014.12.006 [DOI] [PubMed] [Google Scholar]
- 34.Sukhbaatar N, Hengstschlager M, Weichhart T. mTOR-mediated regulation of dendritic cell differentiation and function. Trends Immunol (2016) 37(11):778–89. 10.1016/j.it.2016.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kulkarni S, Sitaru C, Jakus Z, Anderson KE, Damoulakis G, Davidson K, et al. PI3Kbeta plays a critical role in neutrophil activation by immune complexes. Sci Signal (2011) 4(168):ra23. 10.1126/scisignal.2001617 [DOI] [PubMed] [Google Scholar]
- 36.Luo HR, Huang YE, Chen JC, Saiardi A, Iijima M, Ye K, et al. Inositol pyrophosphates mediate chemotaxis in dictyostelium via pleckstrin homology domain-PtdIns(3,4,5)P3 interactions. Cell (2003) 114(5):559–72. 10.1016/S0092-8674(03)00640-8 [DOI] [PubMed] [Google Scholar]
- 37.Chakraborty A, Koldobskiy MA, Bello NT, Maxwell M, Potter JJ, Juluri KR, et al. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell (2010) 143(6):897–910. 10.1016/j.cell.2010.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Hills LB, Huang YH. Lipid and protein co-regulation of PI3K effectors Akt and Itk in lymphocytes. Front Immunol (2015) 6:1–11. 10.3389/fimmu.2015.00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller AT, Sandberg M, Huang YH, Young M, Sutton S, Sauer K, et al. Production of Ins(1,3,4,5)P(4) mediated by the kinase Itpkb inhibits store-operated calcium channels and regulates B cell selection and activation. Nat Immunol (2007) 8(5):514–21. 10.1038/ni1458 [DOI] [PubMed] [Google Scholar]
- 40.Onouchi Y, Gunji T, Burns JC, Shimizu C, Newburger JW, Yashiro M, et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet (2008) 40(1):35–42. 10.1038/ng.2007.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller AT, Beisner DR, Liu D, Cooke MP. Inositol 1,4,5-trisphosphate 3-kinase B is a negative regulator of BCR signaling that controls B cell selection and tolerance induction. J Immunol (2009) 182(8):4696–704. 10.4049/jimmunol.0802850 [DOI] [PubMed] [Google Scholar]
- 42.Marechal Y, Queant S, Polizzi S, Pouillon V, Schurmans S. Inositol 1,4,5-trisphosphate 3-kinase B controls survival and prevents anergy in B cells. Immunobiology (2011) 216(1–2):103–9. 10.1016/j.imbio.2010.03.012 [DOI] [PubMed] [Google Scholar]
- 43.Pouillon V, Marechal Y, Frippiat C, Erneux C, Schurmans S. Inositol 1,4,5-trisphosphate 3-kinase B (Itpkb) controls survival, proliferation and cytokine production in mouse peripheral T cells. Adv Biol Regul (2013) 53(1):39–50. 10.1016/j.jbior.2012.08.001 [DOI] [PubMed] [Google Scholar]
- 44.Alphonse MP, Duong TT, Shumitzu C, Hoang TL, McCrindle BW, Franco A, et al. Inositol-triphosphate 3-kinase C mediates inflammasome activation and treatment response in Kawasaki disease. J Immunol (2016) 197(9):3481–9. 10.4049/jimmunol.1600388 [DOI] [PubMed] [Google Scholar]
- 45.Schell MJ. Inositol trisphosphate 3-kinases: focus on immune and neuronal signaling. Cell Mol Life Sci (2010) 67(11):1755–78. 10.1007/s00018-009-0238-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Streb H, Irvine RF, Berridge MJ, Schulz I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature (1983) 306(5938):67–9. 10.1038/306067a0 [DOI] [PubMed] [Google Scholar]
- 47.Pouillon V, Hascakova-Bartova R, Pajak B, Adam E, Bex F, Dewaste V, et al. Inositol 1,3,4,5-tetrakisphosphate is essential for T lymphocyte development. Nat Immunol (2003) 4(11):1136–43. 10.1038/ni980 [DOI] [PubMed] [Google Scholar]
- 48.Chamberlain PP, Sandberg ML, Sauer K, Cooke MP, Lesley SA, Spraggon G. Structural insights into enzyme regulation for inositol 1,4,5-trisphosphate 3-kinase B. Biochemistry (2005) 44(44):14486–93. 10.1021/bi051256q [DOI] [PubMed] [Google Scholar]
- 49.Jun K, Choi G, Yang SG, Choi KY, Kim H, Chan GC, et al. Enhanced hippocampal CA1 LTP but normal spatial learning in inositol 1,4,5-trisphosphate 3-kinase(A)-deficient mice. Learn Mem (1998) 5(4–5):317–30. [PMC free article] [PubMed] [Google Scholar]
- 50.Kim IH, Park SK, Hong ST, Jo YS, Kim EJ, Park EH, et al. Inositol 1,4,5-trisphosphate 3-kinase a functions as a scaffold for synaptic Rac signaling. J Neurosci (2009) 29(44):14039–49. 10.1523/JNEUROSCI.2483-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scoumanne A, Molina-Ortiz P, Monteyne D, Perez-Morga D, Erneux C, Schurmans S. Specific expression and function of inositol 1,4,5-trisphosphate 3-kinase C (ITPKC) in wild type and knock-out mice. Adv Biol Regul (2016) 62:1–10. 10.1016/j.jbior.2016.03.001 [DOI] [PubMed] [Google Scholar]
- 52.Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol (2009) 10(11):R130. 10.1186/gb-2009-10-11-r130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cunha-Melo JR, Dean NM, Moyer JD, Maeyama K, Beaven MA. The kinetics of phosphoinositide hydrolysis in rat basophilic leukemia (RBL-2H3) cells varies with the type of IgE receptor cross-linking agent used. J Biol Chem (1987) 262(24):11455–63. [PubMed] [Google Scholar]
- 54.Chang YT, Choi G, Bae YS, Burdett M, Moon HS, Lee JW, et al. Purine-based inhibitors of inositol-1,4,5-trisphosphate-3-kinase. Chembiochem (2002) 3(9):897–901. 10.1002/1439-7633(20020902)3:9<897::AID-CBIC897>3.0.CO;2-B [DOI] [PubMed] [Google Scholar]
- 55.Stokes AJ, Shimoda LM, Lee JW, Rillero C, Chang YT, Turner H. Fcepsilon RI control of Ras via inositol (1,4,5) trisphosphate 3-kinase and inositol tetrakisphosphate. Cell Signal (2006) 18(5):640–51. 10.1016/j.cellsig.2005.06.003 [DOI] [PubMed] [Google Scholar]
- 56.Sauer K, Huang YH, Ying H, Sandberg M, Mayr GW. Phosphoinositide analysis in lymphocyte activation. Curr Protoc Immunol (2009) Chapter 11:Unit 11.1. 10.1002/0471142735.im1101s87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jia Y, Subramanian KK, Erneux C, Pouillon V, Hattori H, Jo H, et al. Inositol 1,3,4,5-tetrakisphosphate negatively regulates phosphatidylinositol-3,4,5-trisphosphate signaling in neutrophils. Immunity (2007) 27(3):453–67. 10.1016/j.immuni.2007.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dillon SB, Murray JJ, Verghese MW, Snyderman R. Regulation of inositol phosphate metabolism in chemoattractant-stimulated human polymorphonuclear leukocytes. Definition of distinct dephosphorylation pathways for IP3 isomers. J Biol Chem (1987) 262(24):11546–52. [PubMed] [Google Scholar]
- 59.Miller AT, Dahlberg C, Sandberg ML, Wen BG, Beisner DR, Hoerter JA, et al. Inhibition of the inositol kinase Itpkb augments calcium signaling in lymphocytes and reveals a novel strategy to treat autoimmune disease. PLoS One (2015) 10(6):e0131071. 10.1371/journal.pone.0131071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hermosura MC, Takeuchi H, Fleig A, Riley AM, Potter BV, Hirata M, et al. InsP4 facilitates store-operated calcium influx by inhibition of InsP3 5-phosphatase. Nature (2000) 408(6813):735–40. 10.1038/35047115 [DOI] [PubMed] [Google Scholar]
- 61.Irvine R. Inositol phosphates: does IP(4) run a protection racket? Curr Biol (2001) 11(5):R172–4. 10.1016/S0960-9822(01)00086-0 [DOI] [PubMed] [Google Scholar]
- 62.Nishio M, Watanabe K, Sasaki J, Taya C, Takasuga S, Iizuka R, et al. Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat Cell Biol (2007) 9(1):36–44. 10.1038/ncb1515 [DOI] [PubMed] [Google Scholar]
- 63.Subramanian KK, Jia Y, Zhu D, Simms BT, Jo H, Hattori H, et al. Tumor suppressor PTEN is a physiologic suppressor of chemoattractant-mediated neutrophil functions. Blood (2007) 109(9):4028–37. 10.1182/blood-2006-10-055319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Y, Jia Y, Pichavant M, Loison F, Sarraj B, Kasorn A, et al. Targeted deletion of tumor suppressor PTEN augments neutrophil function and enhances host defense in neutropenia-associated pneumonia. Blood (2009) 113(20):4930–41. 10.1182/blood-2008-06-161414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sarraj B, Massberg S, Li Y, Kasorn A, Subramanian K, Loison F, et al. Myeloid-specific deletion of tumor suppressor PTEN augments neutrophil transendothelial migration during inflammation. J Immunol (2009) 182(11):7190–200. 10.4049/jimmunol.0802562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Helgason CD, Damen JE, Rosten P, Grewal R, Sorensen P, Chappel SM, et al. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev (1998) 12(11):1610–20. 10.1101/gad.12.11.1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu Q, Sasaki T, Kozieradzki I, Wakeham A, Itie A, Dumont DJ, et al. SHIP is a negative regulator of growth factor receptor-mediated PKB/Akt activation and myeloid cell survival. Genes Dev (1999) 13(7):786–91. 10.1101/gad.13.7.786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blunt MD, Ward SG. Pharmacological targeting of phosphoinositide lipid kinases and phosphatases in the immune system: success, disappointment, and new opportunities. Front Immunol (2012) 3:226. 10.3389/fimmu.2012.00226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hammond GR, Fischer MJ, Anderson KE, Holdich J, Koteci A, Balla T, et al. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science (2012) 337(6095):727–30. 10.1126/science.1222483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Joncker NT, Shifrin N, Delebecque F, Raulet DH. Mature natural killer cells reset their responsiveness when exposed to an altered MHC environment. J Exp Med (2010) 207(10):2065–72. 10.1084/jem.20100570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jonsson AH, Yang L, Kim S, Taffner SM, Yokoyama WM. Effects of MHC class I alleles on licensing of Ly49A+ NK cells. J Immunol (2010) 184(7):3424–32. 10.4049/jimmunol.0904057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jiang K, Zhong B, Gilvary DL, Corliss BC, Hong-Geller E, Wei S, et al. Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat Immunol (2000) 1(5):419–25. 10.1038/80859 [DOI] [PubMed] [Google Scholar]
- 73.Tassi I, Cella M, Gilfillan S, Turnbull I, Diacovo TG, Penninger JM, et al. p110gamma and p110delta phosphoinositide 3-kinase signaling pathways synergize to control development and functions of murine NK cells. Immunity (2007) 27(2):214–27. 10.1016/j.immuni.2007.07.014 [DOI] [PubMed] [Google Scholar]
- 74.Kerr WG, Colucci F. Inositol phospholipid signaling and the biology of natural killer cells. J Innate Immun (2011) 3(3):249–57. 10.1159/000323920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cassatella MA, Anegon I, Cuturi MC, Griskey P, Trinchieri G, Perussia B. Fc gamma R(CD16) interaction with ligand induces Ca2+ mobilization and phosphoinositide turnover in human natural killer cells. Role of Ca2+ in Fc gamma R(CD16)-induced transcription and expression of lymphokine genes. J Exp Med (1989) 169(2):549–67. 10.1084/jem.169.2.549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Terme M, Ullrich E, Delahaye NF, Chaput N, Zitvogel L. Natural killer cell-directed therapies: moving from unexpected results to successful strategies. Nat Immunol (2008) 9(5):486–94. 10.1038/ni1580 [DOI] [PubMed] [Google Scholar]
- 77.Romagne F, Vivier E. Natural killer cell-based therapies. F1000 Med Rep (2011) 3:9. 10.3410/M3-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang JW, Howson JM, Ghansah T, Desponts C, Ninos JM, May SL, et al. Influence of SHIP on the NK repertoire and allogeneic bone marrow transplantation. Science (2002) 295(5562):2094–7. 10.1126/science.1068438 [DOI] [PubMed] [Google Scholar]
- 79.Wahle JA, Paraiso KH, Costello AL, Goll EL, Sentman CL, Kerr WG. Cutting edge: dominance by an MHC-independent inhibitory receptor compromises NK killing of complex targets. J Immunol (2006) 176(12):7165–9. 10.4049/jimmunol.176.12.7165 [DOI] [PubMed] [Google Scholar]
- 80.Wahle JA, Paraiso KH, Kendig RD, Lawrence HR, Chen L, Wu J, et al. Inappropriate recruitment and activity by the Src homology region 2 domain-containing phosphatase 1 (SHP1) is responsible for receptor dominance in the SHIP-deficient NK cell. J Immunol (2007) 179(12):8009–15. 10.4049/jimmunol.179.12.8009 [DOI] [PubMed] [Google Scholar]
- 81.Fortenbery NR, Paraiso KH, Taniguchi M, Brooks C, Ibrahim L, Kerr WG. SHIP influences signals from CD48 and MHC class I ligands that regulate NK cell homeostasis, effector function, and repertoire formation. J Immunol (2010) 184(9):5065–74. 10.4049/jimmunol.0901862 [DOI] [PubMed] [Google Scholar]
- 82.Banh C, Miah SM, Kerr WG, Brossay L. Mouse natural killer cell development and maturation are differentially regulated by SHIP-1. Blood (2012) 120(23):4583–90. 10.1182/blood-2012-04-425009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Briercheck EL, Trotta R, Chen L, Hartlage AS, Cole JP, Cole TD, et al. PTEN is a negative regulator of NK cell cytolytic function. J Immunol (2015) 194(4):1832–40. 10.4049/jimmunol.1401224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leong JW, Schneider SE, Sullivan RP, Parikh BA, Anthony BA, Singh A, et al. PTEN regulates natural killer cell trafficking in vivo. Proc Natl Acad Sci U S A (2015) 112(7):E700–9. 10.1073/pnas.1413886112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell (2008) 135(6):1118–29. 10.1016/j.cell.2008.10.048 [DOI] [PubMed] [Google Scholar]
- 86.Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nat Rev Immunol (2010) 10(3):201–9. 10.1038/nri2726 [DOI] [PubMed] [Google Scholar]
- 87.Pietras EM, Warr MR, Passegue E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol (2011) 195(5):709–20. 10.1083/jcb.201102131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hazen AL, Smith MJ, Desponts C, Winter O, Moser K, Kerr WG. SHIP is required for a functional hematopoietic stem cell niche. Blood (2009) 113(13):2924–33. 10.1182/blood-2008-02-138008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Signer RA, Magee JA, Salic A, Morrison SJ. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature (2014) 509(7498):49–54. 10.1038/nature13035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature (2006) 441(7092):475–82. 10.1038/nature04703 [DOI] [PubMed] [Google Scholar]
- 91.Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature (2006) 441(7092):518–22. 10.1038/nature04747 [DOI] [PubMed] [Google Scholar]
- 92.Kharas MG, Okabe R, Ganis JJ, Gozo M, Khandan T, Paktinat M, et al. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood (2010) 115(7):1406–15. 10.1182/blood-2009-06-229443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Petrie HT, Zuniga-Pflucker JC. Zoned out: functional mapping of stromal signaling microenvironments in the thymus. Annu Rev Immunol (2007) 25:649–79. 10.1146/annurev.immunol.23.021704.115715 [DOI] [PubMed] [Google Scholar]
- 94.Xiong J, Armato MA, Yankee TM. Immature single-positive CD8+ thymocytes represent the transition from Notch-dependent to Notch-independent T-cell development. Int Immunol (2011) 23(1):55–64. 10.1093/intimm/dxq457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aifantis I, Mandal M, Sawai K, Ferrando A, Vilimas T. Regulation of T-cell progenitor survival and cell-cycle entry by the pre-T-cell receptor. Immunol Rev (2006) 209:159–69. 10.1111/j.0105-2896.2006.00343.x [DOI] [PubMed] [Google Scholar]
- 96.Ciofani M, Schmitt TM, Ciofani A, Michie AM, Cuburu N, Aublin A, et al. Obligatory role for cooperative signaling by pre-TCR and Notch during thymocyte differentiation. J Immunol (2004) 172(9):5230–9. 10.4049/jimmunol.172.9.5230 [DOI] [PubMed] [Google Scholar]
- 97.Ciofani M, Zuniga-Pflucker JC. Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat Immunol (2005) 6(9):881–8. 10.1038/ni1234 [DOI] [PubMed] [Google Scholar]
- 98.Campese AF, Garbe AI, Zhang F, Grassi F, Screpanti I, von Boehmer H. Notch1-dependent lymphomagenesis is assisted by but does not essentially require pre-TCR signaling. Blood (2006) 108(1):305–10. 10.1182/blood-2006-01-0143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Taghon T, Yui MA, Pant R, Diamond RA, Rothenberg EV. Developmental and molecular characterization of emerging beta- and gammadelta-selected pre-T cells in the adult mouse thymus. Immunity (2006) 24(1):53–64. 10.1016/j.immuni.2005.11.012 [DOI] [PubMed] [Google Scholar]
- 100.Tussiwand R, Engdahl C, Gehre N, Bosco N, Ceredig R, Rolink AG. The preTCR-dependent DN3 to DP transition requires Notch signaling, is improved by CXCL12 signaling and is inhibited by IL-7 signaling. Eur J Immunol (2011) 41(11):3371–80. 10.1002/eji.201141824 [DOI] [PubMed] [Google Scholar]
- 101.Fayard E, Gill J, Paolino M, Hynx D, Hollander GA, Hemmings BA. Deletion of PKBalpha/Akt1 affects thymic development. PLoS One (2007) 2(10):e992. 10.1371/journal.pone.0000992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Juntilla MM, Wofford JA, Birnbaum MJ, Rathmell JC, Koretzky GA. Akt1 and Akt2 are required for alphabeta thymocyte survival and differentiation. Proc Natl Acad Sci U S A (2007) 104(29):12105–10. 10.1073/pnas.0705285104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mao C, Tili EG, Dose M, Haks MC, Bear SE, Maroulakou I, et al. Unequal contribution of Akt isoforms in the double-negative to double-positive thymocyte transition. J Immunol (2007) 178(9):5443–53. 10.4049/jimmunol.178.9.5443 [DOI] [PubMed] [Google Scholar]
- 104.Hagenbeek TJ, Naspetti M, Malergue F, Garcon F, Nunes JA, Cleutjens KBJM, et al. The loss of PTEN allows TCR {alpha}{beta} lineage thymocytes to bypass IL-7 and pre-TCR-mediated signaling. J Exp Med (2004) 200(7):883–94. 10.1084/jem.20040495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kelly AP, Finlay DK, Hinton HJ, Clarke RG, Fiorini E, Radtke F, et al. Notch-induced T cell development requires phosphoinositide-dependent kinase 1. EMBO J (2007) 26(14):3441–50. 10.1038/sj.emboj.7601761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shiroki F, Matsuda S, Doi T, Fujiwara M, Mochizuki Y, Kadowaki T, et al. The p85alpha regulatory subunit of class IA phosphoinositide 3-kinase regulates beta-selection in thymocyte development. J Immunol (2007) 178(3):1349–56. 10.4049/jimmunol.178.3.1349 [DOI] [PubMed] [Google Scholar]
- 107.Wong GW, Knowles GC, Mak TW, Ferrando AA, Zuniga-Pflucker JC. HES1 opposes a PTEN-dependent check on survival, differentiation, and proliferation of TCRbeta-selected mouse thymocytes. Blood (2012) 120(7):1439–48. 10.1182/blood-2011-12-395319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hagenbeek TJ, Wu X, Choy L, Sanchez-Irizarry C, Seshagiri S, Stinson J, et al. Murine Pten(-/-) T-ALL requires non-redundant PI3K/mTOR and DLL4/Notch1 signals for maintenance and gammac/TCR signals for thymic exit. Cancer Lett (2014) 346(2):237–48. 10.1016/j.canlet.2013.12.027 [DOI] [PubMed] [Google Scholar]
- 109.Kashiwada M, Cattoretti G, McKeag L, Rouse T, Showalter BM, Al-Alem U, et al. Downstream of tyrosine kinases-1 and Src homology 2-containing inositol 5’-phosphatase are required for regulation of CD4+CD25+ T cell development. J Immunol (2006) 176(7):3958–65. 10.4049/jimmunol.176.7.3958 [DOI] [PubMed] [Google Scholar]
- 110.Lee K, Nam KT, Cho SH, Gudapati P, Hwang Y, Park DS, et al. Vital roles of mTOR complex 2 in Notch-driven thymocyte differentiation and leukemia. J Exp Med (2012) 209(4):713–28. 10.1084/jem.20111470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lucas JA, Felices M, Evans JW, Berg LJ. Subtle defects in pre-TCR signaling in the absence of the Tec kinase Itk. J Immunol (2007) 179(11):7561–7. 10.4049/jimmunol.179.11.7561 [DOI] [PubMed] [Google Scholar]
- 112.Luo HR, Mondal S. Molecular control of PtdIns(3,4,5)P3 signaling in neutrophils. EMBO Rep (2015) 16(2):149–63. 10.15252/embr.201439466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Saiardi A, Bhandari R, Resnick AC, Snowman AM, Snyder SH. Phosphorylation of proteins by inositol pyrophosphates. Science (2004) 306(5704):2101–5. 10.1126/science.1103344 [DOI] [PubMed] [Google Scholar]
- 114.Bhandari R, Saiardi A, Ahmadibeni Y, Snowman AM, Resnick AC, Kristiansen TZ, et al. Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proc Natl Acad Sci U S A (2007) 104(39):15305–10. 10.1073/pnas.0707338104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Burton A, Azevedo C, Andreassi C, Riccio A, Saiardi A. Inositol pyrophosphates regulate JMJD2C-dependent histone demethylation. Proc Natl Acad Sci U S A (2013) 110(47):18970–5. 10.1073/pnas.1309699110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hou Q, Liu F, Chakraborty A, Jia Y, Prasad A, Yu H, et al. Inhibition of IP6K1 suppresses neutrophil-mediated pulmonary damage in bacterial pneumonia. Sci Transl Med (2018) 10(435):eaal4045. 10.1126/scitranslmed.aal4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Strassheim D, Kim JY, Park JS, Mitra S, Abraham E. Involvement of SHIP in TLR2-induced neutrophil activation and acute lung injury. J Immunol (2005) 174(12):8064–71. 10.4049/jimmunol.174.12.8064 [DOI] [PubMed] [Google Scholar]
- 118.Medyouf H, Ghysdael J. The calcineurin/NFAT signaling pathway: a novel therapeutic target in leukemia and solid tumors. Cell Cycle (2008) 7(3):297–303. 10.4161/cc.7.3.5357 [DOI] [PubMed] [Google Scholar]
- 119.Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol (2015) 33:291–353. 10.1146/annurev-immunol-032414-112212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stritesky GL, Jameson SC, Hogquist KA. Selection of self-reactive T cells in the thymus. Annu Rev Immunol (2011) 30:95–114. 10.1146/annurev-immunol-020711-075035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Irvine RF, Schell MJ. Back in the water: the return of the inositol phosphates. Nat Rev Mol Cell Biol (2001) 2(5):327–38. 10.1038/35073015 [DOI] [PubMed] [Google Scholar]
- 122.Qi Q, Sahu N, August A. Tec kinase Itk forms membrane clusters specifically in the vicinity of recruiting receptors. J Biol Chem (2006) 281(50):38529–34. 10.1074/jbc.M609180200 [DOI] [PubMed] [Google Scholar]
- 123.Nalaskowski MM, Mayr GW. The families of kinases removing the Ca2+ releasing second messenger Ins(1,4,5)P3. Curr Mol Med (2004) 4(3):277–90. 10.2174/1566524043360726 [DOI] [PubMed] [Google Scholar]
- 124.Pattni K, Banting G. Ins(1,4,5)P3 metabolism and the family of IP3-3kinases. Cell Signal (2004) 16(6):643–54. 10.1016/j.cellsig.2003.10.009 [DOI] [PubMed] [Google Scholar]
- 125.Xia HJ, Yang G. Inositol 1,4,5-trisphosphate 3-kinases: functions and regulations. Cell Res (2005) 15(2):83–91. 10.1038/sj.cr.7290270 [DOI] [PubMed] [Google Scholar]
- 126.Irvine RF, Lloyd-Burton SM, Yu JC, Letcher AJ, Schell MJ. The regulation and function of inositol 1,4,5-trisphosphate 3-kinases. Adv Enzyme Regul (2006) 46:314–23. 10.1016/j.advenzreg.2006.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Imboden JB, Pattison G. Regulation of inositol 1,4,5-trisphosphate kinase activity after stimulation of human T cell antigen receptor. J Clin Invest (1987) 79(5):1538–41. 10.1172/JCI112986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zilberman Y, Howe LR, Moore JP, Hesketh TR, Metcalfe JC. Calcium regulates inositol 1,3,4,5-tetrakisphosphate production in lysed thymocytes and in intact cells stimulated with concanavalin A. EMBO J (1987) 6(4):957–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Neilson JR, Winslow MM, Hur EM, Crabtree GR. Calcineurin B1 is essential for positive but not negative selection during thymocyte development. Immunity (2004) 20(3):255–66. 10.1016/S1074-7613(04)00052-4 [DOI] [PubMed] [Google Scholar]
- 130.Fu G, Casas J, Rigaud S, Rybakin V, Lambolez F, Brzostek J, et al. Themis sets the signal threshold for positive and negative selection in T-cell development. Nature (2013) 504(7480):441–5. 10.1038/nature12718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Laederach A, Cradic KW, Fulton DB, Andreotti AH. Determinants of intra versus intermolecular self-association within the regulatory domains of Rlk and Itk. J Mol Biol (2003) 329(5):1011–20. 10.1016/S0022-2836(03)00531-X [DOI] [PubMed] [Google Scholar]
- 132.Colgan J, Asmal M, Neagu M, Yu B, Schneidkraut J, Lee Y, et al. Cyclophilin A regulates TCR signal strength in CD4+ T cells via a proline-directed conformational switch in Itk. Immunity (2004) 21(2):189–201. 10.1016/j.immuni.2004.07.005 [DOI] [PubMed] [Google Scholar]
- 133.Qi Q, August A. The Tec family kinase Itk exists as a folded monomer in vivo. J Biol Chem (2009) 284(43):29882–92. 10.1074/jbc.M109.003129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Severin A, Joseph RE, Boyken S, Fulton DB, Andreotti AH. Proline isomerization preorganizes the Itk SH2 domain for binding to the Itk SH3 domain. J Mol Biol (2009) 387(3):726–43. 10.1016/j.jmb.2009.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Andreotti AH, Schwartzberg PL, Joseph RE, Berg LJ. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb Perspect Biol (2010) 2(7):a002287. 10.1101/cshperspect.a002287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Min L, Wu W, Joseph RE, Fulton DB, Berg L, Andreotti AH. Disrupting the intermolecular self-association of Itk enhances T cell signaling. J Immunol (2010) 184(8):4228–35. 10.4049/jimmunol.0901908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hatano A, Matsumoto M, Nakayama KI. Phosphoproteomics analyses show subnetwork systems in T-cell receptor signaling. Genes Cells (2016) 21(10):1095–112. 10.1111/gtc.12406 [DOI] [PubMed] [Google Scholar]
- 138.Hawkins PT, Stephens LR. PI3Kgamma is a key regulator of inflammatory responses and cardiovascular homeostasis. Science (2007) 318(5847):64–6. 10.1126/science.1145420 [DOI] [PubMed] [Google Scholar]
- 139.Rommel C, Camps M, Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol (2007) 7(3):191–201. 10.1038/nri2036 [DOI] [PubMed] [Google Scholar]
- 140.Weichhart T, Saemann MD. The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann Rheum Dis (2008) 67(Suppl_3):iii70–4. 10.1136/ard.2008.098459 [DOI] [PubMed] [Google Scholar]
- 141.Felices M, Yin CC, Kosaka Y, Kang J, Berg LJ. Tec kinase Itk in gammadeltaT cells is pivotal for controlling IgE production in vivo. Proc Natl Acad Sci U S A (2009) 106(20):8308–13. 10.1073/pnas.0808459106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fruman DA, Bismuth G. Fine tuning the immune response with PI3K. Immunol Rev (2009) 228(1):253–72. 10.1111/j.1600-065X.2008.00750.x [DOI] [PubMed] [Google Scholar]
- 143.Nalaskowski MM, Fliegert R, Ernst O, Brehm MA, Fanick W, Windhorst S, et al. Human inositol 1,4,5-trisphosphate 3-kinase isoform B (IP3KB) is a nucleocytoplasmic shuttling protein specifically enriched at cortical actin filaments and at invaginations of the nuclear envelope. J Biol Chem (2011) 286(6):4500–10. 10.1074/jbc.M110.173062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Erneux C, Ghosh S, Koenig S. Inositol(1,4,5)P3 3-kinase isoenzymes: catalytic properties and importance of targeting to F-actin to understand function. Adv Biol Regul (2016) 60:135–43. 10.1016/j.jbior.2015.09.004 [DOI] [PubMed] [Google Scholar]
- 145.Kim KD, Srikanth S, Yee MK, Mock DC, Lawson GW, Gwack Y. ORAI1 deficiency impairs activated T cell death and enhances T cell survival. J Immunol (2011) 187(7):3620–30. 10.4049/jimmunol.1100847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shi J, Petrie HT. Activation kinetics and off-target effects of thymus-initiated cre transgenes. PLoS One (2012) 7(10):e46590. 10.1371/journal.pone.0046590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kawasaki T, Kosaki F, Okawa S, Shigematsu I, Yanagawa H. A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics (1974) 54(3):271–6. [PubMed] [Google Scholar]
- 148.Onouchi Y. The genetics of Kawasaki disease. Int J Rheum Dis (2018) 21(1):26–30. 10.1111/1756-185X.13218 [DOI] [PubMed] [Google Scholar]
- 149.Huang YH, Hoebe K, Sauer K. New therapeutic targets in immune disorders: ItpkB, Orai1 and UNC93B. Expert Opin Ther Targets (2008) 12(4):391–413. 10.1517/14728222.12.4.391 [DOI] [PubMed] [Google Scholar]
- 150.Yang YC, Chang TY, Chen TC, Chang SC, Chen WF, Chan HW, et al. Genetic polymorphisms in the ITPKC gene and cervical squamous cell carcinoma risk. Cancer Immunol Immunother (2012) 61(11):2153–9. 10.1007/s00262-012-1280-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kan WC, Chou YH, Chiu SJ, Hsu YW, Lu HF, Hsu W, et al. Study of the association between ITPKC genetic polymorphisms and calcium nephrolithiasis. Biomed Res Int (2014) 2014:397826. 10.1155/2014/397826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kim JH, Jung SM, Shin JG, Cheong HS, Seo JM, Kim DY, et al. Potential association between ITPKC genetic variations and Hirschsprung disease. Mol Biol Rep (2017) 44(3):307–13. 10.1007/s11033-017-4111-6 [DOI] [PubMed] [Google Scholar]
- 153.Hemon P, Renaudineau Y, Debant M, Le Goux N, Mukherjee S, Brooks W, et al. Calcium signaling: from normal B cell development to tolerance breakdown and autoimmunity. Clin Rev Allergy Immunol (2017) 53(2):141–65. 10.1007/s12016-017-8607-6 [DOI] [PubMed] [Google Scholar]
- 154.Cambier JC, Gauld SB, Merrell KT, Vilen BJ. B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat Rev Immunol (2007) 7(8):633–43. 10.1038/nri2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cullen PJ, Hsuan JJ, Truong O, Letcher AJ, Jackson TR, Dawson AP, et al. Identification of a specific Ins(1,3,4,5)P4-binding protein as a member of the GAP1 family. Nature (1995) 376(6540):527–30. 10.1038/376527a0 [DOI] [PubMed] [Google Scholar]
- 156.Schurmans S, Pouillon V, Marechal Y. Regulation of B cell survival, development and function by inositol 1,4,5-trisphosphate 3-kinase B (Itpkb). Adv Enzyme Regul (2011) 51(1):66–73. 10.1016/j.advenzreg.2010.08.001 [DOI] [PubMed] [Google Scholar]
- 157.Ramsey LB, Vegoe AL, Miller AT, Cooke MP, Farrar MA. Tonic BCR signaling represses receptor editing via Raf- and calcium-dependent signaling pathways. Immunol Lett (2011) 135(1–2):74–7. 10.1016/j.imlet.2010.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Louis AG, Yel L, Cao JN, Agrawal S, Gupta S. Common variable immunodeficiency associated with microdeletion of chromosome 1q42.1-q42.3 and inositol 1,4,5-trisphosphate kinase B (ITPKB) deficiency. Clin Transl Immunology (2016) 5(1):e59. 10.1038/cti.2015.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Abubaker J, Bavi PP, Al-Harbi S, Siraj AK, Al-Dayel F, Uddin S, et al. PIK3CA mutations are mutually exclusive with PTEN loss in diffuse large B-cell lymphoma. Leukemia (2007) 21(11):2368–70. 10.1038/sj.leu.2404873 [DOI] [PubMed] [Google Scholar]
- 160.Rudelius M, Pittaluga S, Nishizuka S, Pham TH, Fend F, Jaffe ES, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood (2006) 108(5):1668–76. 10.1182/blood-2006-04-015586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schmitz R, Ceribelli M, Pittaluga S, Wright G, Staudt LM. Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb Perspect Med (2014) 4(2):1–13. 10.1101/cshperspect.a014282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Uren AG, Kool J, Matentzoglu K, de Ridder J, Mattison J, van Uitert M, et al. Large-scale mutagenesis in p19(ARF)- and p53-deficient mice identifies cancer genes and their collaborative networks. Cell (2008) 133(4):727–41. 10.1016/j.cell.2008.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kool J, Berns A. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat Rev Cancer (2009) 9(6):389–99. 10.1038/nrc2647 [DOI] [PubMed] [Google Scholar]
- 164.Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell (2013) 152(4):714–26. 10.1016/j.cell.2013.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet (2011) 43(9):830–7. 10.1038/ng.892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A (2012) 109(10):3879–84. 10.1073/pnas.1121343109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature (2012) 490(7418):116–20. 10.1038/nature11378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of follicular lymphoma transformation. Cell Rep (2014) 6(1):130–40. 10.1016/j.celrep.2013.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Dubois S, Viailly PJ, Mareschal S, Bohers E, Bertrand P, Ruminy P, et al. Next-generation sequencing in diffuse large B-cell lymphoma highlights molecular divergence and therapeutic opportunities: a LYSA Study. Clin Cancer Res (2016) 22(12):2919–28. 10.1158/1078-0432.CCR-15-2305 [DOI] [PubMed] [Google Scholar]
- 170.Mareschal S, Dubois S, Viailly PJ, Bertrand P, Bohers E, Maingonnat C, et al. Whole exome sequencing of relapsed/refractory patients expands the repertoire of somatic mutations in diffuse large B-cell lymphoma. Genes Chromosomes Cancer (2016) 55(3):251–67. 10.1002/gcc.22328 [DOI] [PubMed] [Google Scholar]
- 171.Rae DT, Hocum JD, Bii V, Deeg HJ, Trobridge GD. A novel retroviral mutagenesis screen identifies prognostic genes in RUNX1 mediated myeloid leukemogenesis. Oncotarget (2015) 6(31):30664–74. 10.18632/oncotarget.5133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Jude JG, Spencer GJ, Huang X, Somerville TDD, Jones DR, Divecha N, et al. A targeted knockdown screen of genes coding for phosphoinositide modulators identifies PIP4K2A as required for acute myeloid leukemia cell proliferation and survival. Oncogene (2015) 34(10):1253–62. 10.1038/onc.2014.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Choi G, Chang Y-T, Chung S-K, Choi KY. Molecular interactions of all possible regioisomers of synthetic myo-inositol phosphates with inositol 1,4,5-trisphosphate 3-kinase. Bioorg Med Chem Lett (1997) 7(21):2709–14. 10.1016/S0960-894X(97)10060-9 [DOI] [Google Scholar]
- 174.Mayr GW, Windhorst S, Hillemeier K. Antiproliferative plant and synthetic polyphenolics are specific inhibitors of vertebrate inositol-1,4,5-trisphosphate 3-kinases and inositol polyphosphate multikinase. J Biol Chem (2005) 280(14):13229–40. 10.1074/jbc.M500545200 [DOI] [PubMed] [Google Scholar]
- 175.Poinas A, Backers K, Riley AM, Mills SJ, Moreau C, Potter BV, et al. Interaction of the catalytic domain of inositol 1,4,5-trisphosphate 3-kinase A with inositol phosphate analogues. Chembiochem (2005) 6(8):1449–57. 10.1002/cbic.200400443 [DOI] [PubMed] [Google Scholar]
- 176.Stygelbout V, Leroy K, Pouillon V, Ando K, D’Amico E, Jia Y, et al. Inositol trisphosphate 3-kinase B is increased in human Alzheimer brain and exacerbates mouse Alzheimer pathology. Brain (2014) 137(Pt 2):537–52. 10.1093/brain/awt344 [DOI] [PubMed] [Google Scholar]
- 177.Salta E, Sierksma A, Vanden Eynden E, De Strooper B. miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in Alzheimer’s brain. EMBO Mol Med (2016) 8(9):1005–18. 10.15252/emmm.201606520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tajouri L, Mellick AS, Tourtellotte A, Nagra RM, Griffiths LR. An examination of MS candidate genes identified as differentially regulated in multiple sclerosis plaque tissue, using absolute and comparative real-time Q-PCR analysis. Brain Res Brain Res Protoc (2005) 15(2):79–91. 10.1016/j.brainresprot.2005.04.003 [DOI] [PubMed] [Google Scholar]
- 179.Gyorffy B, Lage H. A web-based data warehouse on gene expression in human malignant melanoma. J Invest Dermatol (2007) 127(2):394–9. 10.1038/sj.jid.5700543 [DOI] [PubMed] [Google Scholar]
- 180.Komander D, Fairservice A, Deak M, Kular GS, Prescott AR, Peter Downes C, et al. Structural insights into the regulation of PDK1 by phosphoinositides and inositol phosphates. EMBO J (2004) 23(20):3918–28. 10.1038/sj.emboj.7600379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol (2003) 21:139–76. 10.1146/annurev.immunol.21.120601.141107 [DOI] [PubMed] [Google Scholar]
- 182.Windhorst S, Fliegert R, Blechner C, Mollmann K, Hosseini Z, Gunther T, et al. Inositol 1,4,5-trisphosphate 3-kinase-A is a new cell motility-promoting protein that increases the metastatic potential of tumor cells by two functional activities. J Biol Chem (2010) 285(8):5541–54. 10.1074/jbc.M109.047050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wang Q, Vogan EM, Nocka LM, Rosen CE, Zorn JA, Harrison SC, et al. Autoinhibition of Bruton’s tyrosine kinase (Btk) and activation by soluble inositol hexakisphosphate. Elife (2015) 4:1–31. 10.7554/eLife.06074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Erneux C, Govaerts C, Communi D, Pesesse X. The diversity and possible functions of the inositol polyphosphate 5-phosphatases. Biochim Biophys Acta (1998) 1436(1–2):185–99. 10.1016/S0005-2760(98)00132-5 [DOI] [PubMed] [Google Scholar]
- 185.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem (1998) 273(22):13375–8. 10.1074/jbc.273.22.13375 [DOI] [PubMed] [Google Scholar]
- 186.Pesesse X, Moreau C, Drayer AL, Woscholski R, Parker P, Erneux C. The SH2 domain containing inositol 5-phosphatase SHIP2 displays phosphatidylinositol 3,4,5-trisphosphate and inositol 1,3,4,5-tetrakisphosphate 5-phosphatase activity. FEBS Lett (1998) 437(3):301–3. 10.1016/S0014-5793(98)01255-1 [DOI] [PubMed] [Google Scholar]
- 187.Caffrey JJ, Darden T, Wenk MR, Shears SB. Expanding coincident signaling by PTEN through its inositol 1,3,4,5,6-pentakisphosphate 3-phosphatase activity. FEBS Lett (2001) 499(1–2):6–10. 10.1016/S0014-5793(01)02500-5 [DOI] [PubMed] [Google Scholar]
- 188.Ooms LM, Horan KA, Rahman P, Seaton G, Gurung R, Kethesparan DS, et al. The role of the inositol polyphosphate 5-phosphatases in cellular function and human disease. Biochem J (2009) 419(1):29–49. 10.1042/BJ20081673 [DOI] [PubMed] [Google Scholar]
- 189.Astoul E, Edmunds C, Cantrell DA, Ward SG. PI 3-K and T-cell activation: limitations of T-leukemic cell lines as signaling models. Trends Immunol (2001) 22(9):490–6. 10.1016/S1471-4906(01)01973-1 [DOI] [PubMed] [Google Scholar]