

Abstract
Tim17 and Tim23 are the main subunits of the TIM23 complex, one of the two major essential mitochondrial inner-membrane protein translocon machineries (TIMs). No chemical probes that specifically inhibit TIM23-dependent protein import were known to exist. Here we show that the natural product stendomycin, produced by Streptomyces hygroscopicus, is a potent and specific inhibitor of the TIM23 complex in yeast and mammalian cells. Furthermore, stendomycin-mediated blockage of the TIM23 complex does not alter normal processing of the major regulatory mitophagy kinase PINK1, but TIM23 is required to stabilize PINK1 on the outside of mitochondria to initiate mitophagy upon membrane depolarization.
Mitochondria fulfill many critical cellular functions, including the generation of energy, the maintenance of cellular homeostasis and the initiation of apoptosis1,2. The mammalian mitochondrial proteome comprises about 1,100 proteins, of which 13 are encoded by the mitochondrial DNA and the remainder are encoded by the nuclear genome. The latter enter the mitochondria via protein translocation complexes located on the outer (translocon outer membrane, TOM) and inner (translocon inner membrane, TIM) mitochondrial membrane3,4, making them essential for normal mitochondrial biogenesis. There are two major TIM complex-es—TIM22 and TIM23—named after their respective core subunits, the latter containing two core subunits, Tim17 and Tim23 in yeast and TIMM17A, TIMM17B1 or TIMM17B2 along with TIMM23 in mammalian cells. Altered synthesis, targeting, import, and processing of mitochondrial proteins, as well as mutations therein, lead to neurodegenerative and metabolic diseases5. Although TIM and TOM complexes are well characterized and genetic approaches have identified several mutants exerting specific phenotypes6, there are no selective chemicals available to rapidly interrogate and dissect individual aspects of mitochondrial protein import.
Results
Stendomycin targets the TIM23 translocon in yeast
Stendomycin, a lipopeptide produced by Streptomyces7 (Fig. 1a), was identified in a screen for low-molecular-weight compounds that inhibit Saccharomyces cerevisiae growth and was profiled in a genome-wide S. cerevisiae haploinsufficiency screen at a sublethal dose of 0.5 μM8. TIM17 and TIM23 heterozygotes, the core components of the TIM23 translocon, scored as highly sensitive to stendomycin (Fig. 1b). Conversely, the TIM22 heterozygote was unaffected in the profiling (Fig. 1b), suggesting that stendomycin specifically targets the TIM23, but not the TIM22, translocon.
Figure 1. Stendomycin targets the Tim17-Tim23 complex.
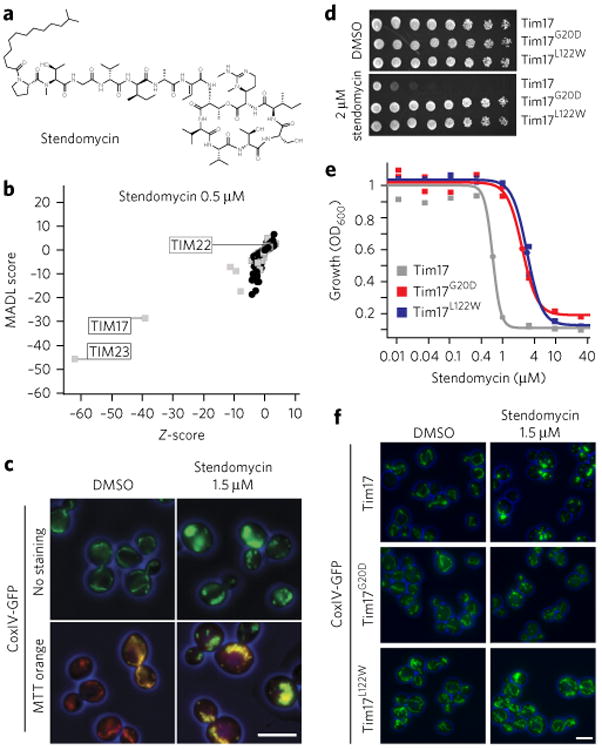
(a) Structure of stendomycin. (b) Haploinsufficiency profiling (HIP) of stendomycin at 0.5 μM. Each mutant strain in the pool is represented by a black circle (nonessential genes) or a gray square (essential genes). The y axis represents strain sensitivity (MADL, mean average distance from log of median), and x axis represents specificity (z-score). Plot shows data calculated from n = 2 technical replicates. (c) Yeast strains carrying a GFP-tagged mitochondrial marker, CoxIV, were treated with 1.5 μM of stendomycin or DMSO for 2 h at 30 °C, stained with mitotracker (MTT) orange, and assessed by fluorescence microscopy (scale bar, 5 μm). (d) Wild-type (Tim17), Tim17G20D and Tim17L122W strains were plated as serial dilutions on solid media (YPD) containing either stendomycin at minimum inhibitory concentration (MIC; 2 μM) or DMSO and incubated at 30 °C for 2 d. (e) Wild-type (Tim17; gray), Tim17G20D (red) and Tim17L122W (blue) strains were grown in duplicate for 18 h at 30 °C in YPD media with 2% DMSO containing a dilution series of stendomycin, and growth was assessed by absorbance at OD600. Dose–response curves were calculated by logistic regression. (f) Wild-type (Tim17), Tim17G20D and Tim17L122W strains were treated with 1.5 μM stendomycin or DMSO for 2 h at 30 °C and assessed for GFP using fluorescence microscopy. Experiments in c,d and f have been replicated twice with similar observations (scale bar, 5 μm).
As TIM17 and TIM23 are essential for mitochondrial function and morphology9, we monitored the mitochondrial markers CoxIV-GFP and mitotracker orange (MTT orange) following treatment with 1.5 μM stendomycin for 2 h. The yeast mitochondrial network collapsed upon stendomycin addition, although mitochondria maintained a membrane potential (ΔΨ), as the ΔΨ-dependent MTT orange was still colocalized primarily with CoxIV-GFP (Fig. 1c).
Selection of yeast mutants resistant to stendomycin identified two point mutations in Tim17—Tim17G20D and Tim17L122W (Fig. 1d)—conferring a three-fold resistance compared to wild type, with the effector concentrations for half-maximum response (EC50s) of stendomycin changing from 0.6 μM to 2.4 μM (Tim17G20D) and to 2.9 μM (Tim17L122W) (Fig. 1e). At stendomycin concentrations sufficient to substantially alter mitochondrial morphology in wild-type cells, the Tim17G20D and the Tim17L122W mutants displayed a normal mitochondrial morphology (Fig. 1f), suggesting that Tim17 is a likely target of stendomycin.
Stendomycin inhibits TIM23-dependent translocation in vitro
To add weight to this concept, we made use of in vitro protein translocation assays using purified yeast mitochondria from haploid wild-type, Tim17G20D and Tim17L122W cells incubated with radiolabeled TIM23 substrates destined for three final locations within mitochondria. Cytochrome (cyt) b2(1-167)-DHFR targets the intermembrane space (IMS); Coq2 and Mia40 target the inner mitochondrial membrane (IMM); and cyt b2(1-167)A63P-DHFR, Su9-DHFR and Hsp60 all target the matrix (ref 10). In addition, the import of the TIM22-dependent precursor AAC11 was analyzed. Although mitochondria from Tim17G20D cells behave similar to those from wild-type cells for import of all these proteins, Tim17L122W-derived mitochondria showed a slightly decreased protein import. Stendomycin (0.5 nM or 0.75 nM) strongly reduced the import of all assayed TIM23-dependent substrate precursors (Fig. 2a; Supplementary Results, Supplementary Fig. 1a), but not the TIM22-dependent AAC precursor (Fig. 2b; Supplementary Fig. 1b). The blocked TIM23-dependent precursor import was rescued in mitochondria from either Tim17G20D or Tim17L122W yeast, with the latter showing the greatest rescue (Fig. 2a).
Figure 2. Stendomycin blocks TIM23-mediated, but not TIM22-mediated, mitochondrial protein import.
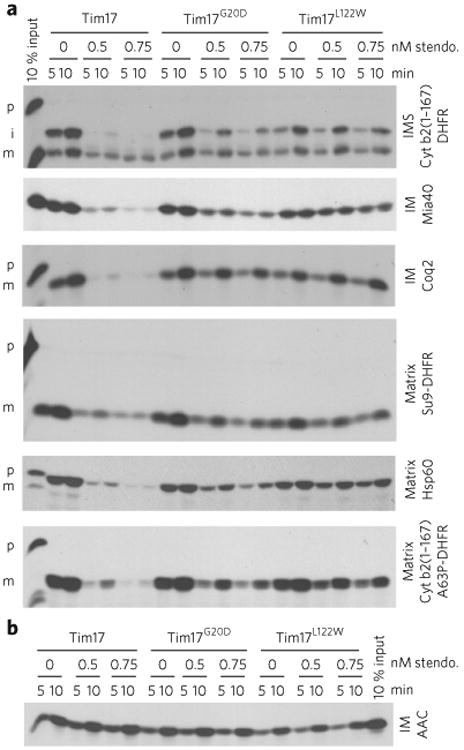
(a) Import assays were performed with radiolabeled TIM23 import pathway substrates into mitochondria isolated from wild-type (Tim17), Tim17G20D and Tim17L122W yeast strains. Time course assays were completed in the presence of 0.5 or 0.75 nM stendomycin (stendo.) or the vehicle control (1% DMSO), and nonimported precursor was removed by trypsin treatment and sodium carbonate extraction. p, precursor; i, intermediate; m, mature. IMS. intermembrane space; IM, inner membrane. (b) As in a, but assessing the TIM22 import pathway substrate AAC. Full gel scans are in Supplementary Information.
Stendomycin inhibits TIM23-dependent translocation in vivo
In agreement with the inhibition of protein translocation, stendomycin led to concentration-dependent accumulation of the precursors of two matrix proteins, Mge1 and Hsp60, in vivo (Fig. 3a). Additionally, crosslinking of the precursor protein cyt b2(1-167) A63P-DHFR during in vitro import enabled the identification of Tim17 and Tim23, as well as Hsp70 and Tim44, components of the protein-associated motor (PAM) complex, on the matrix side of the TIM23 complex12. Stendomycin treatment with a dose that reduces import disrupted these interactions, as did uncoupling of the membranes with carbonyl cyanide 3-chlorophenylhydrazone (CCCP; −ΔΨ; Fig. 3b). However, stendomycin did not affect the crosslink of AAC with components of the TIM22 complex import pathway such as Tim9 and Tim10 or Tom70 and Tom20 (Supplementary Fig. 2a). Consistent with the import data, in mitochondria from the two stendomycin-resistant Tim17 mutants the stendomycin-dependent disruption of the crosslinked complexes was partially rescued (Fig. 3b). However, these two stendomycin-resistant mutants were not resistant to the effects of the bona fide uncoupler CCCP. Thus, sten-domycin specifically targets TIM23 in vivo and in vitro to reduce the import of TIM23-dependent, but not TIM22-dependent, proteins into the IMM, IMS or matrix.
Figure 3. Stendomycin inhibits TIM23-dependent translocation in vivo.
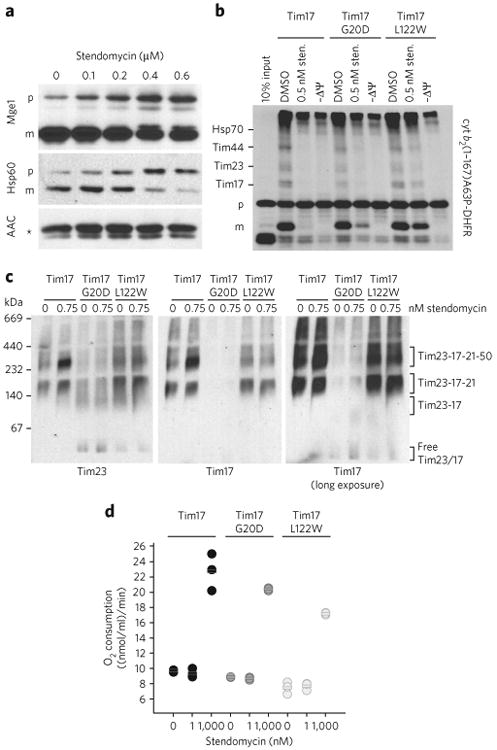
(a) In vivo accumulation of TIM23 complex substrates precursors. Wild-type yeast cells were grown in YPEG media and treated with the indicated concentration of stendomycin for 24 h. The accumulation of Hsp60 and Mge1 precursors was analyzed by immunoblot. The TIM22 complex substrate ACC was used as loading control. p, precursor; m, mature; * marks an unspecific reaction of the antibody. (b) cyt b2(1-167)A63P-DHFR was imported into mitochondria with the addition of 0.5 nM stendomycin or 1% DMSO. The membrane potential was disrupted by CCCP (−ΔΨ). Protein import was arrested by the addition of methotrexate, and cyt b2 (1-167)A63P-DHFR was crosslinked to components of the TIM23 import pathway by DSS. Crosslinked intermediates are indicated. p, precursor; m, mature. Assignment of crosslinked bands was performed as described21,22. (c) Mitochondria isolated from wild-type, Tim17G20D and Tim17L122W yeast strains were treated with 0.75 nM stendomycin for 15 min, solubilized with digitonin and separated by BN–PAGE. The TIM23 complex was analyzed by immunoblot using antibodies targeting Tim23 and Tim17 (see also Supplementary Fig. 2b). (d) Oxygen consumption of isolated mitochondria from indicated yeast strains was measured with an oxygen electrode. Respiration was initiated by the addition of NADH. 1 or 1,000 nM stendomycin or 1% DMSO was added once a stable respiration had been established. Respiration rates were quantitated based on the DMSO control (see Supplementary Fig. 3 for representative oxygraph plots). Bars represent mean values of n = 3 biological replicates (separate mitochondrial preparations), which are shown as circles (black, wild type; dark grey, Tim17G20D; light grey, Tim17L122W. Full gel scans are in Supplementary Information.
In addition, stendomycin causes accumulation of the TIM23 complex, containing Tim23–Tim17–Tim21–Tim50, in wild-type yeast (Fig. 3c; Supplementary Fig. 2b), without altering the steady state levels of its components (Supplementary Fig. 2c,d). Although TIM23 complex levels are not affected in Tim17L122W mitochondria, Tim17G20D mitochondria display reduced levels of the Tim23–Tim17–Tim21–Tim50 and Tim23–Tim17–Tim21 complexes and accumulation of the TIM23 core complex and free Tim17 and Tim23 (Fig. 3c). The mitochondria of Tim17G20D cells also show slightly decreased levels of Tim17 and Tim23 overall (Supplementary Fig. 2c,d), which is in agreement with recently published data reporting that mutations in the transmembrane segment TM1 (residues 18 to 35) of Tim17 lead to destabilized interactions with Tim 23 (ref. 13). Conversely, after stendomycin treatment of mitochondria from Tim17G20D and Tim17L122W strains, we could not observe any accumulation of the Tim23–Tim17–Tim21–Tim50 complex as we did for mitochondria isolated from wild-type yeast (Fig. 3c). These results show that increasing the amount of the TIM23 complex containing Tim50, but not that without Tim50, inhibits the import of all TIM23-dependent precursors tested14.
Stendomycin inhibits TIM23 import in vitro at doses that do not affect ΔΨ, because both TIM23- and TIM22-mediated import require ΔΨ, and at doses of stendomycin that inhibit the former, the latter retains its function (Figs. 2a,b and Fig. 3b). To confirm this observation, we assessed the ability of stendomycin to uncouple purified S. cerevisiae mitochondria, generating an increased oxygen consumption rate (OCR). At doses similar to those that specifically block TIM23 substrate import in vitro, no OCR increase was detectable in either wild-type or Tim17 mutant mitochondria (Fig. 3d; Supplementary Fig. 3). At higher doses, a similar OCR increase occurred in both wild-type and mutant Tim17 mitochondria, suggesting that stendomycin has additional effects on ΔΨ at higher doses that are independent of Tim17G20D and Tim17L122. Thus, stendomycin specifically targets the TIM23 translocon independently of its effects upon ΔΨ in S. cerevisiae.
Stendomycin targets TIM23 translocon in mammalian cells
As the amino acid sequence identity between yeast Tim17 and human TIMM17A, TIMM17B1, and TIMM17B2 is >45%, and both Gly20 and Leu122 are conserved residues (Supplementary Fig. 4), we assessed whether the mechanism of action of stendomycin was conserved in mammalian cells. The distribution of a TIM23-dependent reporter construct (mito-EGFP) together with the accumulation of the ΔΨ-dependent dye mitotracker red (MTT red) and the localization of a TIM23-independent mitochondrial marker, TOMM20, was evaluated. At 0.1 μM stendomycin, enhanced green fluorescent protein (EGFP)-mediated fluorescence accumulated primarily in the cytoplasm, a localization distinct from that of the mitochondrial EGFP fluorescence detected in DMSO-treated cells (Fig. 4a). The perturbed mito-EGFP localization at 0.1 μM was a result of neither a general alteration of mitochondria nor uncoupling, as ΔΨ-dependent TOMM20 and MTT red fluorescence were similar to that observed in DMSO-treated cells. However, at 1 μM stendomycin, both mito-EGFP and MTT red fluorescence were completely dispersed throughout the cell, and the mitochondrial network collapsed around the nucleus, as seen in the cells treated with 10 μM CCCP (Fig. 4a).
Figure 4. Mechanism of action of stendomycin is conserved.
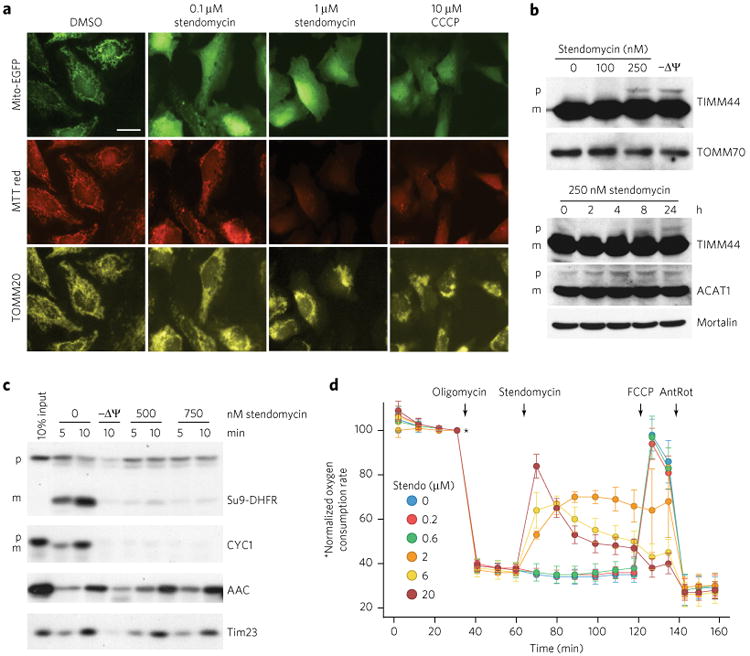
(a) HeLa cells were treated with stendomycin at doses noted or 0.1% DMSO before transfection with MTS-EGFP (mitochondrial targeting sequence of Cox8 fused to EGFP). 10 μM CCCP was used to disrupt the membrane potential (ΔΨ). 24 h after transfection, cells were stained with mitotracker (MTT) red, fixed and visualized for MTS-EGFP, MTT-red and TOMM20. Similar images were obtained in one independent experiment. Scale bar, 10 μM. (b) HeLa cells were treated with the indicated concentrations of stendomycin or with 10 μM CCCP (−ΔΨ) for 24 h (top panel) or with 250 nM stendomycin for the indicated time points (lower panel). Cells were lysed, and total extracts were separated by SDS-PAGE. The accumulation of the TIMM44 and ACAT1 precursors were analyzed by immunoblot. TOMM70 and Mortalin served as loading controls. p, precursor; m, mature. (c) In vitro import assays were performed using mitochondria isolated from HeLa cells and TIM23 and TIM22 import pathways substrates in the presence of 500 or 750 nM stendomycin or control (1% DMSO). Precursors include Su9-DHFR, CYC1, yACC (yeast ACC) and yTim23 (yeast TIM23). p, precursor; m, mature. 10 min time points were quantified and are displayed as percentages in Supplementary Figure 5a. (d) HeLa cells were assessed continuously for oxygen consumption while oligomycin (1 mM), stendomycin (at concentrations noted), FCCP (0.1 μM) and antimycin (1 μM)/rotenone (1 μM; AnRot) were sequentially administered. n = 8 technical replicates; Data represent mean and s.d. This data has been replicated with another passage of HeLa with similar observations. Full gel scans are in Supplementary Information.
Treatment of HeLa cells with stendomycin also caused accumulation of TIM23 substrate precursors TIMM44 and ACAT1 (Fig. 4b). Furthermore, translocation of labeled precursors of TIM23 substrates Su9-DHFR and CYC1 into mitochondria isolated from HeLa cells was reduced by stendomycin, whereas the TIM22 substrates AAC and Tim23 were unaffected (Fig. 4c; Supplementary Fig. 5a). Thus, stendomycin appears to specifically block TIM23-dependent, but not TIM22-dependent, mitochondrial protein import in an evolutionary conserved manner. Although stendomycin acts specifically on the TIM23 complex at low doses, at higher doses it appears to uncouple the IMM (which would also block import) in mammalian cells as well as in yeast.
To confirm the additional activity of stendomycin as an uncoupler, we assessed oxygen consumption in HeLa cells during ATP synthase inhibition (using oligomycin), where any increase in oxygen consumption is due exclusively to uncoupling of the proton gradient across the IMM (Fig. 4d). At a dose of 2 μM, stendomycin had an immediate uncoupling effect, which was more pronounced as the dosage increased. Uncoupling was not observed with stendomycin treatments of ≤0.6 μM over a 1-h time course. Mitochondria that had been uncoupled by stendomycin were refractory to further uncoupling by carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), as expected in the presence of oligomycin, which prevents the regeneration of the proton gradient across the inner mitochondrial membrane.
We then analyzed whether stendomycin uncouples mitochondria by acting on the adenine nucleotide translocator (ANT) or by permeabilizing mitochondrial membranes. Oxygen consumption of mitochondria isolated from wild-type yeast was initiated by NADH and ADP, and then inhibited by blocking ANT with carboxyatractyloside. Whereas the solvent control (DMSO) did not increase oxygen consumption, 1 μM stendomycin uncoupled the mitochondria as previously observed (Supplementary Fig. 5b). When we treated isolated yeast and HeLa cell mitochondria with stendomycin to analyze the release of proteins from mitochondria, no permeabilization of mitochondria could be observed (Supplementary Fig. 5c–e). The controls MB2—a small molecule previously identified to partially solubilize mitochondrial membranes—or detergent Triton X-100 lead to release of proteins into the supernatant. Therefore, the uncoupling of mitochondria by stendomycin was not a result of its effect on the ANT or the permeabilization of mitochondrial membranes. Finally, we determined the levels of ADP and ATP in HeLa cells following stendomycin treatment to assess whether blocking import could affect the ADP:ATP ratio. At high doses that cause uncoupling, there is a general loss of both ADP and ATP, indicating general inhibition. At doses that block only TIM23-dependent import, however, ATP levels are unaffected, whereas ADP levels are reduced (Supplementary Fig. 5f). This suggests that mitochondrial metabolism is compromised following TIM23 inhibition for 24 h.
PINK1 is normally processed in stendomycin treated cells
The steady state levels of the mitochondrial damage sensor PTEN-induced putative kinase 1 (PINK1) are very low because of its constitutive import into the inner mitochondrial membrane and subsequent cleavage by matrix processing peptidase (MPP) and the rhomboid protease presenilin-associated rhomboid like (PARL)15. This generates an N-terminal degron motif, leading to rapid degradation of PINK1 in the cytosol by the ubiquitin proteasome system. Damaged mitochondria that cannot maintain ΔΨ fail to import PINK1 to the IMM, leading to accumulation of uncleaved PINK1 in the outer mitochondrial membrane (OMM), where it recruits and activates Parkin and triggers mitophagy16. Although the mitochondrial targeting signal of PINK1 reaches the mitochondrial matrix, it is not known whether the TIM23 complex is essential for PINK1 trans-location. Having a potent TIM23-complex-specific chemical probe allowed us to evaluate whether PINK1 utilizes the TIM23 complex to reach the mitochondrial matrix to enable rapid turnover.
We assessed multiple markers of PINK1/Parkin-dependent mitophagy in GFP-Parkin HeLa cells transfected with a PINK1 or control siRNA and treated them with stendomycin or control compounds known to uncouple the respiratory chain and trigger mitophagy (CCCP or oligomycin and antimycin A). Stendomycin doses were selected to inhibit protein translocation only (0.2 μM) and to dissipate the ΔΨ (2 μM; Fig. 4a,d). Mitochondrial uncouplers known to induce mitophagy caused PINK1-dependent mitophagy when treated with 2 μM stendomycin; phospho S65-ubiquitin (p-Ub) and PINK1 levels increased with a concomitant decrease in CoxIV (inner membrane), TOMM20 (outer membrane), and NDP52 (nuclear dot protein 52; mitophagy adaptor protein) in cells expressing PINK1, but remained unaffected in cells in which PINK1 had been efficiently knocked down (Fig. 5a). However, treatment with 0.2 μM stendomycin, a concentration sufficient to block protein translocation into the inner membrane (Fig. 4a), did not cause the accumulation of uncleaved PINK1 at 4 or 20 h, nor did it alter any of the assessed mitophagy markers (Fig. 5a). To further dissect the potential processing of PINK1, PINK1 translocation into mitochondria purified from HeLa cells was evaluated by following the cleavage of radiolabeled PINK1 into its PINK1 f53 form17. As expected, CCCP prevented processing to PINK1 f53. Stendomycin, however, had little or no effect on the translocation and processing of PINK1 (Fig. 5b,c). Together, these results show that PINK1 sufficiently engages the inner membrane to be cleaved by PARL in the presence of stendomycin, suggesting that either TIM23-dependent PINK1 lateral diffusion is unaffected by stendomycin or that PINK1 can use alternative IMM machinery. If the former is true, this process is likely partially specific for PINK1, because stendomycin inhibits the lateral insertion of other IMM proteins such as CYC1 in HeLa cell mitochondria (Fig. 4c) or cyt b2-(1-167)-DHFR and Coq2 in yeast mitochondria (Fig. 2a).
Figure 5. Stendomycin inhibits mitochondrial inner-membrane translocation but does not stabilize PINK1 or trigger mitophagy.
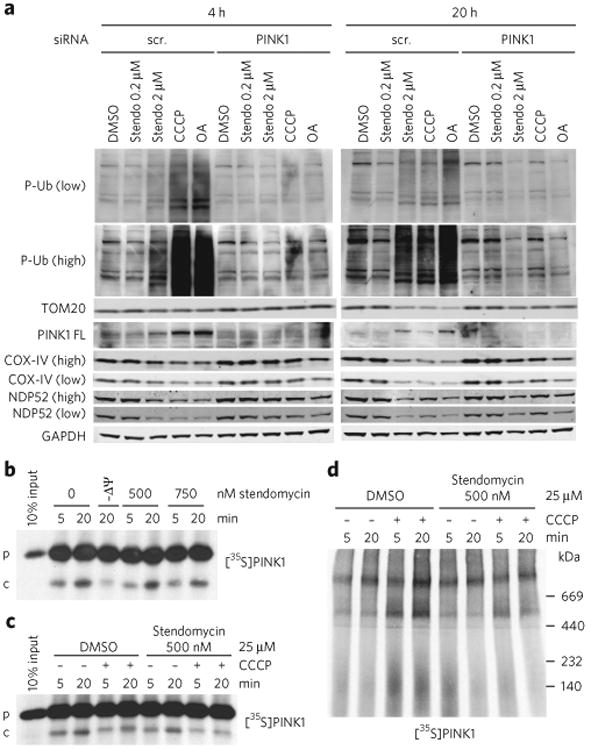
(a) HeLa cells expressing GFP-Parkin were transfected with control (scr, scrambled) or PINK1 siRNAs in the presence of DMSO (0.1%), stendomycin (stendo), CCCP (10 μM) or a combination of oligomycin (100 μM) and antimycin A (100 μM) (OA) for 4 or 20 h. Total cell lysates were analyzed by western blotting as indicated. Similar data was observed in one independent repeat for all markers shown. P-Ub, phospho Ser65-ubiquitin; PINK-FL, full-length PINK1. (b) In vitro import assays were performed as in Figure 4c using PINK1 as substrate. p, precursor; c, PARL-cleaved PINK1. (c) Radiolabeled human PINK1 was in vitro imported into isolated HeLa cell mitochondria in the presence or absence of 500 nM stendomycin for 5 and 20 min. The membrane potential was disrupted in indicated reaction by the addition of 25 μM CCCP. 1% DMSO served as the vehicle control. The membrane insertion of PINK1 was analyzed by alkali extraction for all reactions, which was followed by SDS-PAGE and autoradiography. (d) As in c but with mitochondria solubilized in digitonin and separated by BN-PAGE. PINK1 complexes were visualized by autoradiography. Full gel scans are in Supplementary Information.
It has been suggested that TOMM7, a subunit of the TOM complex, is not essential for regular PINK1 protein import in mammalian cells; however, upon mitochondrial depolarization, TOMM7 retains PINK1 in the TOM complex to activate mitophagy18,19. Here we show that the TIM23 that is complex inhibited by stendomycin does not seem to be involved in the regular import of PINK1 into mitochondria (Fig. 5a–c). To determine whether the TIM23 complex takes part in stabilizing PINK1 on mitochondria upon depolarization like TOMM7, we in vitro imported radiolabeled PINK1 into mitochondria from HeLa cells in the presence or absence of the mitochondrial ΔΨ and stendomycin. The disruption of the ΔΨ by CCCP leads to an accumulation of PINK1 at the TOM complex20, which was attenuated by the addition of stendomycin (Fig. 5d). Thus, although we cannot strictly rule out a role for TIM23 in PINK1 translocation, we conclude that the TIM23 complex inhibited by stendomycin is not needed for the import of Pink1, but is required to stabilize PINK1on the outside of mitochondria to initiate mitophagy upon depolarization. This is in agreement with the TOMM7 data and suggests that functional TOM and TIM are important for PINK1 stabilization following insults that reduce mitochondrial ΔΨ.
Discussion
Although stendomycin was first described in 1963 as an antifungal7, and its biosynthetic cluster has been delineated20, little is known about its mode of action. The genetic, cellular and biochemical data presented here propose that it is a specific and potent inhibitor of TIM23-dependent mitochondrial protein import. Stendomycin-dependent crosslinking of IMM components of the TIM23 machinery suggests that this large natural product can intimately engage the TIM23 complex, potentially by targeting Tim17. The stendomycin-resistant mutations identified here lie in two different transmembrane segments (TM) of Tim17: Tim17G20D in TM1 and Tim17L122W in TM4. Whereas Tim17L122W leads to a more pronounced rescue of translocation, as well as mitochondrial morphology, and shows normal TIM23 complex stability, Tim17G20D leads to a milder phenotype and a destabilized complex, confirming recently published data that mutations in the TM1 lead to destabilized interactions with Tim 23 (ref. 13). Importantly, the G20D mutation did not become rate limiting for the in vitro import of a set of substrates destined for various submitochondrial locations.
In addition, at higher doses, stendomycin exerts a further effect on mitochondria, causing a collapse of ΔΨ. Although we can clearly separate the two functions by dose, we do not yet understand how perturbation of the ΔΨ is achieved by stendomycin. Though higher doses may simply cause more drastic alterations in the TIM23 complex, it is plausible that at high concentrations the fatty acid moiety of stendomycin can interact with lipids, acting directly as a protonophore, a theory supported by the rapid effect on oxygen consumption seen within minutes of stendomycin addition to oligomycin pretreated cells; However, permeabilization assays with mitochondria isolated from yeast (Supplementary Fig. 5c,d) and HeLa (Supplementary Fig. 5e) could not detect any sign of membrane disruption.
Thus, stendomycin represents a potent and specific inhibitor of the TIM23 complex that, if used judiciously, will be of use in understanding the role of TIM23 regulated protein translocation in biology and diseases connected to altered mitochondrial homeostasis. As our data shows that the mechanism of action is conserved between yeast and humans, we applied stendomycin to propose that TIM23 is not essential for translocation and processing of the mitophagy marker PINK1, but is important to stabilize PINK1on the outside of mitochondria to initiate mitophagy upon depolarization.
Methods
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
Online Methods
Isolation of stendomycin
Stendomycin was isolated from the strain Streptomyces hygroscopicus NRRL 2751. The strain was cultivated in a stirred 100 L fermenter (medium: 20 g/L cerelose, 20 g/L glycerol, 10 g/L def. soybean meal, 10 g/L pharma media, 1 g/L (NH4)2SO4, 3 g/L CaCO3) for 6 d at 80 r.p.m. and 28 °C. The main culture was inoculated with 4% of a 3-d-old seed culture. After harvesting, the pH of the culture was adjusted to pH 5.5 and the entire culture broth extracted with 200 mL ethyl acetate. Stendomycin was purified by Sephadex LH-20 chromatography using methanol as a solvent, which was followed by RP18 chromatography using a formic acid and acetonitrile as mobile phase. Fractions containing stendomycin were evaporated, yielding in 160 mg stendomycin with a purity of >95%. The identity of stendomycin23 was confirmed by HR–MS and MS/MS spectroscopy and NMR measurements.
Antibodies
Antibodies against Phospho-Ser65Ub (ABS-1513; 1:500) were from Merck Millipore. Antibodies against PINK1 D8G3 (6946), COX-IV 3E11 (4850) and GAPDH (5174) were from Cell Signaling Technology (all 1:1,000). TOMM20 (56783 and FL-145) and NDP52 (ab68588; all 1:1,000) antibodies were from Abcam or Santa Cruz Biotechnology. Antibodies against aconitase (1:40,000), Tim17 (1:1,000), Tim23 (1:1,000), Tim44 (1:20,000) and Pam16 (1:1,000) were generated from recombinant protein by Pacific Immunology.
Haploinsufficiency profiling (HIP)
The HIP assay was performed in 24-well plates (Greiner; 662102), with 1,600 μl/well YPD. Experimental compounds were tested at n = 2 within the same plate, at or close to their IC30 concentration. Each plate contained two no-drug controls, one positive control (benomyl), ten experimental compounds in duplicates and one contamination control that received no cells. A standard experiment was 4 plates/40 experimental compounds processed robotically without human intervention. YPD and compound filled wells were inoculated with 250 yeast cells/strain (100 μl of a 1.5 OD600/ml culture) from an overnight log-phase preculture to start the experiment. The plates were pipetted with a standard 96 pipettor head by providing tip boxes preconfigured with a special tip pattern. Plates were incubated for 16 h in a robotic shaking incubator at 30 °C and 550 r.p.m., allowing for five doublings. 250 yeast cells/strain (120 μl of a 1.2 OD600/ml culture) were subsequently transferred into a preconfigured 24-well plate that was stored in a robotic plate reservoir at 4 °C until 30 min before its use, when it was pre-warmed to 30 °C. Once inoculated, the new plate was incubated at 30 °C at 550 r.p.m. to allow the next five yeast generations (generation 6–10), and the plate containing the first five doubling cultures was stored at 4 °C. This procedure was repeated two more times until the final plate containing the yeast with 20 generations was stored at 4 °C. By applying a fixed time/dilution scheme to the HIP assay and diluting entire plates at fixed times, we have simplified the procedure and managed to get optimal throughput. Careful IC30 determination and compound potency normalization resulted in very homogenous growth behaviors of the cultures in the 24-well plates. After each HIP experiment, growth curves recorded during the experiment were analyzed. Samples were excluded from further processing if curves indicated that the substance concentrations were too inhibitory for growth. An aliquot of 5 OD600 units of yeast/well from the HIP experiments were arrayed in 96-well plates and spun, and the supernatant was discarded. gDNA extraction was performed using the ChargeSwitch kit (Invitrogen; #18000) in a partially automated process. 150 μl/well of Zymolyase buffer (2 U Zymolyase, 50 ng RNase A, in 20 mM DTT and 20 mM Tris pH 7.5) was added, and the cells were incubated at 37 °C and 700 r.p.m. for 45 min. 300 μl/well lysis buffer (L18; Invitrogen) was added and the plate incubated at 56 °C and 700 r.p.m. for 30 min. 200 μl/well ice-cold precipitation buffer (N2; Invitrogen) was added, and the precipitate pelleted by centrifugation at 4 °C. The supernatant was saved to a new deep-well plate (AB-0932; ABgene) suitable to lock onto the Invitrogen MagnaRack magnet, and 40 μl/well of predispersed ChargeSwitch magnetic beads were added. The plate was incubated at room temperature for 5 min, which was followed by incubation on the MagnaRack for 5 min to pellet the beads. All supernatant was carefully aspirated. The deep-well plate was removed from the MagnaRack, and 500 μl/well wash buffer (W12; Invitrogen) was added and mixed to disperse the beads. The washing was repeated three times and the wash buffer completely removed. Finally, 70 μl of elution buffer (E6; Invitrogen) was added. The beads were resuspended by mixing and the plate incubated for 10 min at room temperature. The beads were pelleted for one last time by incubation on the magnet for 5 min, and the supernatant (containing the gDNA) was transferred to a new plate. The TAG PCR amplification and GenFlex Tag16K v2 hybridization protocol was used. The raw probe intensity values of the CEL are summarized and normalized to tag intensities. Tags with low intensity values in control samples are removed by computing an intensity value threshold based on the comparison of the correlation between the logarithmic intensity ratios for uptags and downtags across different intensity ranges. The tag intensities are then averaged to obtain a strain intensity value. To measure the relative abundance of each strain with respect to the averages of the control samples we compute logarithmic MAD (MADL) scores for each compound and concentration combination, and compute gene-wise z-scores (across all experiments), which are based on a robust parametric estimation of gene variability allowing for up to 15% outliers.
Yeast GFP microscopy
A CoxIV-GFP strain was grown in YPD to an OD600 of 0.5 and treated with 1.5 μM stendomycin for 90 min at 30 °C, then stained with Mitotracker Orange (Thermo Fisher Scientific) and subjected to fluorescence microscopy. To monitor phenotypes of the mutants, the plasmids isolated from the variomics library were transformed in the CoxIV-GFP strain and processed as described, omitting Mitotracker Orange staining.
Screen for stendomycin-resistant yeast mutants
Approximately 6.0 × 107 hap-loid MATa G418R Ura+ cells derived from the pooled variomic libraries24 were plated on a 150-mm plate of solid synthetic complete medium lacking uracil (SC-Ura) either with or without a test compound at MIC, with an average of 10,000 cells representing each variomic library. The plates were incubated at 30 °C for 3 or 4 d, until the resistant colonies appeared. Plasmids of resistant colonies were amplified and subjected to Sanger sequencing. Of the 96 analyzed clones, Tim17G20D was isolated 73 times and Tim17L122W was isolated 22 times. Genomic versions of haploid Tim17G20D and Tim17L122W were generated in BY4743 by transforming PCR-amplified mutant Tim17, selecting on stendomycin and sequence confirming the clones.
Yeast growth assays
Genomic alleles of TIM17 G20D and L122W were generated by homologous recombination and verified. Colony formation and liquid growth assays were performed as described25.
Cell culture and transfection
HeLa cells were cultured at 37 °C, 5% CO2, and maintained in Dulbecco's modified Eagle's medium (DMEM) with 4.5g/L glucose (Thermo Fisher Scientific) containing 10% FBS (heat-inactivated, Thermo Fisher Scientific) and 1% penicillin–streptomycin (Thermo Fisher Scientific). Cells were verified to be mycoplasma-free by routine testing.
Transfections were performed using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's instructions.
Full-length Parkin cDNA was cloned downstream of EGFP in pDEST CMV vectors (Thermo Fisher Scientific) and transfected into HeLa cells with Fugene HD (Roche) in complete medium following the manufacturers' specifications. A stable GFP-Parkin HeLa cell line originated from a pool selected with G418 (100 μg/ml; Thermo Fisher Scientific).
Immunostaining of cultured cells
HeLa cells were grown on cover slips and treated with 0.1% DMSO or stendomycin in indicated concentrations prior to transfection with MTS-EGFP. 24 h after transfection, the cells were stained with 50 μg/ml Mitotracker Red CMXROS (Thermo Fisher Scientific) for 30 min and then fixed in 4% paraformaldehyde (PFA) for 15 min at room temperature. After washing with PBS, the cells were permeabilized in 0.2% Triton X-100 for 10 min at room temperature and blocked for 30 min in 1% bovine serum albumin (BSA) in PBS. The primary and secondary antibody (Alexa Fluor 350; Life Technologies) was diluted in 1% BSA in PBS and incubated for 1 h at room temperature. The cells were washed between primary and secondary antibody applications in PBS. The cells were washed in PBS and mounted in Fluoromount-G (Southern Biotech). Pictures were taken on a Zeiss light microscope using a 40× objective. Pictures were processed with imageJ.
In vitro transcription and translation
All precursor proteins were in vitro transcribed and translated from a plasmid using the TnT SP6/T7 Quick Coupled Transcription/Translation System (Promega) and TRAN35S-LABEL, Metabolic Labeling Reagent (MP Biomedicals).
Isolation of mitochondria from yeast and in vitro protein import
Isolation of mitochondria from yeast was performed as previously described26. In vitro imports into yeast mitochondria were performed as previously described27. For the crosslinking of imported precursor to different components of the import machinery, methotrexate (2 μM) was added to energized mitochondria before the addition of radiolabeled precursor. After 5 min of import, 2 mM disuccinimidyl suberate (DSS) (Thermo Fisher Scientific) was added, and the import reaction was incubated for 30 min on ice. DSS was quenched with 100 mM Tris–HCl pH 8.0 and the mitochondria were re-isolated at 14,000 × g for 8 min. All samples were resuspended in 2× Laemmli buffer and resolved by SDS–PAGE. Imported proteins were detected by autoradiography. For the crosslinking of imported precursor to different components of the TIM22 complex import machinery, precursors were imported into mitochondria in the presence of 50 μM CCCP and 1 μM oligomycin. The pellets were washed in ice-cold breaking buffer 7.4 (0.6 M sorbitol, 20 mM K+ HEPES, pH 7.4) and recentrifuged. Pellets were resuspended in import buffer (-BSA, -Met) and subject to crosslinking with 1 mM ethylene glycol bis(succinimidyl succinate) (EGS) for 30 min at room temperature. The reactions were quenched with 100 mM Tris–HCl pH 8.0 for 10 min, centrifuged at 8,000 × g for 5 min, and analyzed by SDS–PAGE and autoradiography.
Blue native PAGE (BN–PAGE)
Mitochondria were lysed in BN lysis buffer (20 mM HEPES pH 7.4, 50 mM NaCl, 2.5 mM MgCl2, 10% glycerol, 0.1 mM EDTA, 0.5 mM PMSF, 1.5% digitonin ; AG scientific) at a concentration of 5 mg/ml for 30 min on ice. After a centrifugation at 21,000g for 10 min the mitochondrial extract was separated on a 5–15% BN–PAGE gel. Proteins were transferred to a PVDF membrane and detected by immunoblot using the indicated antibodies. Radioactive BN-gels were dried and imaged by autoradiography using a phosphoimager screen.
Isolation of mitochondria from cultured HeLa cells and protein import into mitochondria
HeLa cells were resuspended in homogenization buffer (20 mM HEPES pH 7.4, 220 mM mannitol, 70 mM sucrose, 0.2% bovine serum albumin and 0.5 mM PMSF), and the cells were disrupted with 30 strokes in a glass Teflon homogenizer. The cells were centrifuged at 800g. The supernatant was saved and the pellet re-extracted in homogenization buffer with another round of 30 strokes in a glass Teflon homogenizer followed by centrifugation at 800g. The supernatants were combined and centrifuged at 800g to pellet all remaining nuclei and unbroken cells. The supernatant was centrifuged at 8,000g for 10 min to obtain the mitochondria. The mitochondria were washed once in homogenization buffer without BSA and again pelleted at 8,000g. Ten micrograms of HeLa cell mitochondria were incubated in import buffer (20 mM HEPES pH 7.4, 220 mM mannitol, 70 mM sucrose, 1 mM ATP, 0.5 mM magnesium acetate, 20 mM sodium succinate, 5 mM NADH) for 5 min at 25 °C before the addition of the precursor protein. The membrane potential was disrupted by treatment with 50 μM CCCP. After adding the precursor, aliquots were taken at different time points and transferred to new tubes containing cold homogenization buffer and stored on ice until the last time point was taken. Trypsin was added to a final concentration of 10 μg/ml, and samples were incubated on ice for an additional 15 min. The trypsin digestion was terminated by adding soybean trypsin inhibitor to a final concentration of 250 μg/ml. Mitochondria were re-isolated by centrifugation at 10,000g for 5 min. Alkaline extraction was performed for membrane proteins in 100 mM Na2CO3 pH 11.5 for 30 min on ice. Membrane sheets containing the imported precursor protein were obtained by centrifugation at 21,000g for 30 min. Samples were resuspended in 2× SDS sample buffer and resolved by SDS–PAGE. For the analysis of imported proteins by BN–PAGE, mitochondria were not treated with trypsin; instead, they were washed once in 20 mM HEPES pH 7.4, 220 mM mannitol and 70 mM sucrose after the import and re-isolated by centrifugation at 10,000g for 10 min. The mitochondria were then lysed as described above. Imported proteins were detected by autoradiography.
Total protein extraction from yeast
Yeast was grown in YPEG media supplemented with 0.1% DMSO or different concentrations of stendomycin for 24 h at 30 °C. 1.3 OD600 (optical density at 600 nm) of cells were pelleted at 8,000 × g for 1 min, washed once with water and resuspended in 100 μl SUTE buffer (1% SDS, 8 M Urea, 10 mM Tris–HCl, pH 6.8, 10 mM EDTA, 1 mM PMSF and 10 μM MG132). After the adding approximately 100 μl of glass beads, the cells were ruptured by vortexing at full speed for 3 min at room temperature. The lysate was cleared by centrifugation at 21,000 × g for 10 min at room temperature and the supernatant was resolved by SDS–PAGE.
Membrane permeabilization assay
50 μg of yeast mitochondria were incubated in 20 mM HEPES/KOH pH 7.4, 0.6 M sorbitol, supplemented with either 1% DMSO, 100 μM stendomycin or 100 μM MitoBlock-2 for 30 min at 25 °C. Mitochondria were re-isolated by centrifugation at 10,000 × g for 10 min. The proteins in the supernatant were TCA–acetone precipitated. All pellets were resuspended in Thorner buffer (8 M urea, 5% SDS, 40 mM Tris pH 6.8, 0.1 mM EDTA, 5% mercaptoethanol) and resolved by SDS–PAGE. 50 μg HeLa mitochondria were resuspended in homogenizer buffer supplemented with 1% DMSO, stendomycin or 0.5% Triton X-100 and incubated at 25 °C for 30 min. Samples were then centrifuged at 10 000 × g and 4 °C for 5 min. The supernatants were separated from the pellets, and the proteins were TCA–acetone precipitated. Following precipitation, the supernatants were centrifuged at 20,800 × g and 4 °C for 10 min. Both the pellet and precipitated supernatants were resuspended in Thorner buffer and analyzed by SDS–PAGE and immunoblot.
Membrane potential and oxygen consumption
Purified mitochondria aliquots were thawed on ice. A Clark-type oxygen electrode in a stirred, thermostatically controlled chamber at 30 °C (Oxytherm; Hansatech) was used to measure oxygen consumption. Respiration was initiated on a suspension of 80 μg/mL mitochondria, 70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, and 1 mM EGTA, pH 7.2, after adding 2 mM NADH. Consumption rate was monitored for approximately 1.5–2 min. DMSO (1%) or stendomycin was then added, and respiration was measured for about another 1.5 min. Uncoupled respiration was achieved by adding 5 μM carbonyl cyanide 3-chlorophenylhydrazone (CCCP) to the chamber. To analyze stendomycin's effect on the adenine nucleotide translocator, ADP-dependent respiration was initiated with 2 mM NADH and 2 mM ADP. ADP-dependent respiration was stopped by the addition of 5 μg carboxyatractyloside (CAT) per milligram of purified mitochondria, and uncoupling was achieved by the addition of 1 μM stendomycin.
ADP:ATP assessment
ADP and ATP were measured in HeLa cell extracts as described in the instructions in kit #AB65313 from Abcam.
siRNA transfection and treatments
Cells were reverse transfected with 20 nM siRNA (Dharmacon On-Targetplus non-targeting and PINK1 SMART Pools; GE Healthcare) and 0.1% of Dharmafect1 transfection reagent (Dharmacon) for 4 d in complete transfection medium: DMEM with 4.5 g/L glucose (Thermo Fisher Scientific) containing 12.5% FBS (heat-inactivated; Thermo Fisher Scientific) according to the manufacturer's protocol.
At day three, DMSO (0.1%), CCCP, and antimycin A1 (all from Sigma) were added as indicated.
Western blotting
Cells were lysed in RIPA buffer (Thermo Fisher Scientific) supplemented with a protease–phosphatase inhibitor cocktail (CST). Protein concentration was determined with the Pierce BCA protein assay kit (Thermo Scientific). Samples were mixed with NuPAGE LDS sample buffer containing NuPAGE sample reducing agent, denatured for 10min at 70 °C and loaded onto NuPAGE Novex 4–12% Bis–Tris protein gels (Thermo Fisher Scientific). Proteins were transferred onto PVDF membranes. After blocking for 1h in Odyssey blocking buffer, membranes were incubated overnight at 4 °C with primary antibodies in Odyssey blocking buffer supplemented with 0.1% Tween-20 (Bio-Rad). Secondary antibodies (anti-rabbit IRDye 800CW from LI-COR; anti-mouse Alexa Fluor 680 from Thermo Fisher Scientific) were diluted in Odyssey blocking buffer supplemented with 0.1% Tween-20 and incubated with membranes for 1h at room temperature. Proteins were visualized using Odyssey infrared imaging system. Alternatively, membranes were blocked with PBS, 0.1% Tween-20 and 5% milk. Primary and secondary (anti-rabbit HRP or anti-mouse HRP from Thermo Fisher Scientific) antibodies were diluted in PBS, 0.1% Tween-20 and 5% milk and incubated overnight at 4 °C or for 1h at room temperature, respectively. Proteins were visualized on a FUJI ImageQuant LAS 4000 imager using the ECL Prime Western Blotting Detection Reagent (RPN2232; GE Healthcare Biosciences).
Isolated mitochondria from yeast were directly lysed in 1× Laemmli buffer and heated to 65 °C for 10 min. Different amounts of mitochondria were resolved by SDS–PAGE electrophoresis, transferred to PVDF membranes (EMD Millipore) and processed as above.
Data availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files) or are available from the corresponding author on reasonable request. A Life Sciences Reporting Summary for this paper is available.
Supplementary Material
Supplementary Fig. 2: Stendomycin alters the TIM23 complex (a) Radiolabeled AAC was imported into isolated yeast mitochondria of indicated strains in the absence of the membrane potential (-ΔΨ) and the presence of either 1% DMSO or 0.5 nM stendomycin. After the import, mitochondria were treated with 1 mM EGS to arrest import intermediates, followed by SDS-PAGE and autoradiography. Proteins crosslinked to AAC are indicated by an asterisk (22). (b) Mitochondria isolated from wild type yeast were treated with 0.75 or 1 nM of stendomycin for 15 min, solubilized with digitonin and separated by BN-PAGE. The TIM23 complex was analyzed by immunoblot using antibodies against Tim23 and Tim17. The locations of the different TIM23 complexes are indicated according to (13)(see also Fig. 3c) (c) Lysates as in (Fig. 3c) were separated by SDS-PAGE and analyzed by immunoblotting using antibodies against Tim17, Tim23, Tim44 and Aconitase as loading control. (d) Mitochondria isolated from wild type and Tim17 mutant yeast solubilized with digitonin and separated by SDS-PAGE. The TIM23 complex was analyzed by immunoblot using antibodies against Tim17, Tim23, Tim44 and Pam16, Aconitase was used as loading control
Supplementary Fig. 3: Stendomycin-resistant Tim17 mutations do not alter ΔΨat doses that block TIM23-dependent protein import Oxygen consumption of isolated mitochondria from indicated yeast strains was measured with an oxygen electrode. Respiration was initiated by the addition of NADH. 1 or 1000 nM stendomycin or 1% DMSO was added once a stable respiration had been established. Raw plots are shown, with quantification shown in Fig. 3d.
Supplementary Fig. 4: Tim17 is > 45% identical to TIMM17A and TIMM17B. Tim17 was aligned pairwise to TIMM17A, TIMM17B1 and TIMM17B2 using blast (https://blast.ncbi.nlm.nih.gov/BlastAlign.cgi) and the amino acid identity scores are given. The conserved residues, mutated in yeast Tim17 to give resistance to stendomycin, are highlighted.
Supplementary Fig. 5: Stendomycin does not uncouple via inhibition of ANT or membrane permeabilzation (a) In vitro import assays were performed as described in Fig. 4. 10 min time points were quantified and are displayed as percentages. (b) Oxygen consumption of isolated mitochondria from wild type yeast strains was measured with an oxygen electrode. Respiration was measured following sequential addition of NADH, ADP, carboxyatractyloside and then either DMSO or stendomycin. Slope rates are given in the table. (c) Yeast mitochondria were incubated with 100 μM stendomycin or 100 μM MB2 for 30 min in import buffer and released proteins (S) were separated from mitochondria (P) by centrifugation. Proteins were visualized by Coomassie staining. (d) As in (c), except immunoblot analysis was performed to determine the fractionation for aconitase, Osm1, AAC, and Tom70. As a control, treatment with the vehicle (1% DMSO) was included. (e) Isolated HeLa cell mitochondria were treated with 0.5% Triton X-100, 1% DMSO, or 1 μM stendomycin for 30 minutes followed by separation of the supernatant (S) and pellet (P) by centrifugation. Samples were subjected to SDS-PAGE and analyzed by immunoblotting for indicated proteins. (f) HeLa cells were incubated with stendomycin doses indicated for 24h and subjected to ADP:ATP level analysis using a luciferase reagent. n = 3 biological replicates; error bars are s.d.
Acknowledgments
The authors acknowledge the NIH GM61721 to C.M.K. and DFG STE 2045/1-1 to J.S. The authors also wish to thank the rest of the Novartis HIP HOP team.
Footnotes
Author contributions: I.F. and J.S. designed and performed experiments, analyzed the data and wrote the paper; M.G., L.G., M.A.C., C.P., R.C. and M.P., designed and performed experiments and analyzed the data; P.K. and D.H. supervised experiments and analyzed the data; J.B. designed experiments and analyzed the data; C.M.K. and S.B.H. designed experiments, analyzed the data and wrote the paper.
Competing financial interests: The authors declare competing financial interests: details accompany the online version of the paper.
Additional information: Any supplementary information, chemical compound information and source data are available in the online version of the paper.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Aoun M, Tiranti V. Mitochondria: a crossroads for lipid metabolism defect in neurodegeneration with brain iron accumulation diseases. Int J Biochem Cell Biol. 2015;63:25–31. doi: 10.1016/j.biocel.2015.01.018. [DOI] [PubMed] [Google Scholar]
- 2.Bhola PD, Letai A. Mitochondria-judges and executioners of cell death Sentences. Mol Cell. 2016;61:695–704. doi: 10.1016/j.molcel.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Endo T, Yamamoto H, Esaki M. Functional cooperation and separation of translocators in protein import into mitochondria, the double-membrane bounded organelles. J Cell Sci. 2003;116:3259–3267. doi: 10.1242/jcs.00667. [DOI] [PubMed] [Google Scholar]
- 4.Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacKenzie JA, Payne RM. Mitochondrial protein import and human health and disease. Biochim Biophys Acta. 2007;1772:509–523. doi: 10.1016/j.bbadis.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfanner N, Meijer M. Mitochondrial biogenesis: the Tom and Tim machine. Curr Biol. 1997;7:R100–R103. doi: 10.1016/s0960-9822(06)00048-0. [DOI] [PubMed] [Google Scholar]
- 7.Tompson RQ, Hughes MS. Stendomycin: A new antifungal antibiotic. J Antibiot (Tokyo) 1963;16:187–194. [PubMed] [Google Scholar]
- 8.Hoepfner D, et al. High-resolution chemical dissection of a model eukaryote reveals targets, pathways and gene functions. Microbiol Res. 2014;169:107–120. doi: 10.1016/j.micres.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Saleem A, Iqbal S, Zhang Y, Hood DA. Effect of p53 on mitochondrial morphology, import, and assembly in skeletal muscle. Am J Physiol Cell Physiol. 2015;308:C319–C329. doi: 10.1152/ajpcell.00253.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mokranjac D, Neupert W. Te many faces of the mitochondrial TIM23 complex. Biochim Biophys Acta. 2010;1797:1045–1054. doi: 10.1016/j.bbabio.2010.01.026. [DOI] [PubMed] [Google Scholar]
- 11.Sirrenberg C, Bauer MF, Guiard B, Neupert W, Brunner M. Import of carrier proteins into the mitochondrial inner membrane mediated by Tim22. Nature. 1996;384:582–585. doi: 10.1038/384582a0. [DOI] [PubMed] [Google Scholar]
- 12.Jensen RE, Johnson AE. Protein translocation: is Hsp70 pulling my chain? Curr Biol. 1999;9:R779–R782. doi: 10.1016/S0960-9822(00)80012-3. [DOI] [PubMed] [Google Scholar]
- 13.Demishtein-Zohary K, et al. Role of Tim17 in coupling the import motor to the translocation channel of the mitochondrial presequence translocase. eLife. 2017;6:e22696. doi: 10.7554/eLife.22696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chacinska A, et al. Mitochondrial presequence translocase: switching between TOM tethering and motor recruitment involves Tim21 and Tim17. Cell. 2005;120:817–829. doi: 10.1016/j.cell.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 15.Greene AW, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rüb C, Wilkening A, Voos W. Mitochondrial quality control by the Pink1/Parkin system. Cell Tissue Res. 2017;367:111–123. doi: 10.1007/s00441-016-2485-8. [DOI] [PubMed] [Google Scholar]
- 17.Becker D, Richter J, Tocilescu MA, Przedborski S, Voos W. Pink1 kinase and its membrane potential (ΔΨ)-dependent cleavage product both localize to outer mitochondrial membrane by unique targeting mode. J Biol Chem. 2012;287:22969–22987. doi: 10.1074/jbc.M112.365700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22:320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasson SA, et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature. 2013;504:291–295. doi: 10.1038/nature12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kersten RD, et al. A mass spectrometry-guided genome mining approach for natural product peptidogenomics. Nat Chem Biol. 2011;7:794–802. doi: 10.1038/nchembio.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ungermann C, Guiard B, Neupert W, Cyr DM. Te delta psi- and Hsp70/MIM44-dependent reaction cycle driving early steps of protein import into mitochondria. EMBO J. 1996;15:735–744. [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider HC, et al. Mitochondrial Hsp70/MIM44 complex facilitates protein import. Nature. 1994;371:768–774. doi: 10.1038/371768a0. [DOI] [PubMed] [Google Scholar]
- 23.Simorre JP, Genest D, Caille A, Ptak M. A 2D NMR study of the internal flexibility of the antifungal peptide stendomycin. Eur Biophys J. 1990;18:309–316. doi: 10.1007/BF00196921. [DOI] [PubMed] [Google Scholar]
- 24.Huang Z, et al. A functional variomics tool for discovering drug-resistance genes and drug targets. Cell Rep. 2013;3:577–585. doi: 10.1016/j.celrep.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pries V, et al. Advantages and challenges of phenotypic screens: the identification of two novel antifungal geranylgeranyltransferase I inhibitors. J Biomol Screen. 2016;21:306–315. doi: 10.1177/1087057115610488. [DOI] [PubMed] [Google Scholar]
- 26.Glick BS, Pon LA. Isolation of highly purified mitochondria from Saccharomyces cerevisiae. Methods Enzymol. 1995;260:213–223. doi: 10.1016/0076-6879(95)60139-2. [DOI] [PubMed] [Google Scholar]
- 27.Claypool SM, Whited K, Srijumnong S, Han X, Koehler CM. Barth syndrome mutations that cause tafazzin complex lability. J Cell Biol. 2011;192:447–462. doi: 10.1083/jcb.201008177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 2: Stendomycin alters the TIM23 complex (a) Radiolabeled AAC was imported into isolated yeast mitochondria of indicated strains in the absence of the membrane potential (-ΔΨ) and the presence of either 1% DMSO or 0.5 nM stendomycin. After the import, mitochondria were treated with 1 mM EGS to arrest import intermediates, followed by SDS-PAGE and autoradiography. Proteins crosslinked to AAC are indicated by an asterisk (22). (b) Mitochondria isolated from wild type yeast were treated with 0.75 or 1 nM of stendomycin for 15 min, solubilized with digitonin and separated by BN-PAGE. The TIM23 complex was analyzed by immunoblot using antibodies against Tim23 and Tim17. The locations of the different TIM23 complexes are indicated according to (13)(see also Fig. 3c) (c) Lysates as in (Fig. 3c) were separated by SDS-PAGE and analyzed by immunoblotting using antibodies against Tim17, Tim23, Tim44 and Aconitase as loading control. (d) Mitochondria isolated from wild type and Tim17 mutant yeast solubilized with digitonin and separated by SDS-PAGE. The TIM23 complex was analyzed by immunoblot using antibodies against Tim17, Tim23, Tim44 and Pam16, Aconitase was used as loading control
Supplementary Fig. 3: Stendomycin-resistant Tim17 mutations do not alter ΔΨat doses that block TIM23-dependent protein import Oxygen consumption of isolated mitochondria from indicated yeast strains was measured with an oxygen electrode. Respiration was initiated by the addition of NADH. 1 or 1000 nM stendomycin or 1% DMSO was added once a stable respiration had been established. Raw plots are shown, with quantification shown in Fig. 3d.
Supplementary Fig. 4: Tim17 is > 45% identical to TIMM17A and TIMM17B. Tim17 was aligned pairwise to TIMM17A, TIMM17B1 and TIMM17B2 using blast (https://blast.ncbi.nlm.nih.gov/BlastAlign.cgi) and the amino acid identity scores are given. The conserved residues, mutated in yeast Tim17 to give resistance to stendomycin, are highlighted.
Supplementary Fig. 5: Stendomycin does not uncouple via inhibition of ANT or membrane permeabilzation (a) In vitro import assays were performed as described in Fig. 4. 10 min time points were quantified and are displayed as percentages. (b) Oxygen consumption of isolated mitochondria from wild type yeast strains was measured with an oxygen electrode. Respiration was measured following sequential addition of NADH, ADP, carboxyatractyloside and then either DMSO or stendomycin. Slope rates are given in the table. (c) Yeast mitochondria were incubated with 100 μM stendomycin or 100 μM MB2 for 30 min in import buffer and released proteins (S) were separated from mitochondria (P) by centrifugation. Proteins were visualized by Coomassie staining. (d) As in (c), except immunoblot analysis was performed to determine the fractionation for aconitase, Osm1, AAC, and Tom70. As a control, treatment with the vehicle (1% DMSO) was included. (e) Isolated HeLa cell mitochondria were treated with 0.5% Triton X-100, 1% DMSO, or 1 μM stendomycin for 30 minutes followed by separation of the supernatant (S) and pellet (P) by centrifugation. Samples were subjected to SDS-PAGE and analyzed by immunoblotting for indicated proteins. (f) HeLa cells were incubated with stendomycin doses indicated for 24h and subjected to ADP:ATP level analysis using a luciferase reagent. n = 3 biological replicates; error bars are s.d.
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its supplementary information files) or are available from the corresponding author on reasonable request. A Life Sciences Reporting Summary for this paper is available.