

Abstract
Objective
To evaluate poly(GP), a dipeptide repeat protein, and neurofilament light chain (NfL) as biomarkers in presymptomatic C9orf72 repeat expansion carriers and patients with C9orf72‐associated frontotemporal dementia. Additionally, to investigate the relationship of poly(GP) with indicators of neurodegeneration as measured by NfL and grey matter volume.
Methods
We measured poly(GP) and NfL levels in cerebrospinal fluid (CSF) from 25 presymptomatic C9orf72 expansion carriers, 64 symptomatic expansion carriers with dementia, and 12 noncarriers. We explored associations with grey matter volumes using region of interest and voxel‐wise analyses.
Results
Poly(GP) was present in C9orf72 expansion carriers and absent in noncarriers (specificity 100%, sensitivity 97%). Presymptomatic carriers had lower poly(GP) levels than symptomatic carriers. NfL levels were higher in symptomatic carriers than in presymptomatic carriers and healthy noncarriers. NfL was highest in patients with concomitant motor neuron disease, and correlated with disease severity and survival. Associations between poly(GP) levels and small grey matter regions emerged but did not survive multiple comparison correction, while higher NfL levels were associated with atrophy in frontotemporoparietal cortices and the thalamus.
Interpretation
This study of C9orf72 expansion carriers reveals that: (1) poly(GP) levels discriminate presymptomatic and symptomatic expansion carriers from noncarriers, but are not associated with indicators of neurodegeneration; and (2) NfL levels are associated with grey matter atrophy, disease severity, and shorter survival. Together, poly(GP) and NfL show promise as complementary biomarkers for clinical trials for C9orf72‐associated frontotemporal dementia, with poly(GP) as a potential marker for target engagement and NfL as a marker of disease activity and progression.
Introduction
Frontotemporal dementia (FTD) is a neurodegenerative disorder characterized by early progressive behavioral and/or language deficits.1, 2 Up to 15% of patients with FTD concomitantly develop motor neuron disease (MND), and 10–20% of patients with MND develop FTD,3 suggesting that the two disorders lie on a clinical continuum. Pathogenic G4C2 repeat expansions in chromosome 9 open reading frame 72 (C9orf72) are the most common genetic cause of autosomal‐dominant FTD and amyotrophic lateral sclerosis (ALS).4, 5 Potential pathomechanisms include the loss of function of normal C9orf72 protein, and/or toxicity resulting from the accumulation of G4C2 transcripts that form RNA foci, interact with RNA‐binding proteins, and impair RNA processing.6 Expanded G4C2 transcripts also lead to the production of five dipeptide repeat (DPR) proteins through repeat‐associated non‐ATG (RAN) translation.7, 8 RAN translation of sense transcripts of the repeat produces poly(GA), poly(GR), and poly(GP), while RAN translation of antisense repeat transcripts produces poly(PA), poly(PR), and poly(GP).
Although promising drugs for C9orf72 expansions have emerged in preclinical studies, biomarkers for evaluating their efficacy have not been adequately assessed in humans. Disease‐modifying therapies advancing toward clinical trials include antisense oligonucleotides (ASOs) and small molecules that target G4C2 transcripts and consequently reduce G4C2 RNA foci and DPR proteins in C9orf72 patient‐derived cell models and animal models.9, 10, 11, 12, 13 In parallel with the rapid development of these potential therapeutics, biomarkers that measure target engagement, disease onset, and disease progression must be established for clinical trials to be successful. Previous studies suggest that poly(GP) is a promising marker of target engagement. This protein is detectable in the cerebrospinal fluid (CSF) of presymptomatic and symptomatic C9orf72 expansion carriers,10, 11, 14 and poly(GP) levels in CSF from (G4C2)66‐expressing mice correlate with ASO‐induced decreases in G4C2 RNA expression, RNA foci burden, and DPR protein levels within their brain.11 In addition, neurofilament light chain (NfL) is a potential marker of disease severity and progression for ALS, FTD, as well as other neurodegenerative diseases, including Alzheimer's disease.15, 16, 17 This marker for axonal injury is also increased in symptomatic but not presymptomatic C9orf72 expansion carriers, and correlates with prognosis and disease severity in genetic FTD.18, 19
Prior studies found that poly(GP) in CSF did not correlate with indicators of disease progression or neurodegeneration, yet these studies were largely conducted in patients with C9orf72‐associated ALS.11, 14, 20 In addition, no imaging data were available to assess potential relationships between CSF poly(GP) and grey matter atrophy. In this study, we investigated the clinical correlates of poly(GP) and NfL levels, and we explored associations between these biomarkers and grey matter volume in a large cohort of presymptomatic C9orf72 expansion carriers and patients with C9orf72‐associated dementia.
Materials and Methods
Subjects
We examined CSF from 101 subjects from C9orf72 families, which was collected among eight sites (five sites participating in the Genetic FTD Initiative (GENFI), the University of California, San Francisco, the VU University Medical Center, and IRCCS Fatebenefratelli; Table S1). These CSF samples were obtained from 64 patients with dementia caused by the C9orf72 repeat expansion (symptomatic carriers), and 37 healthy first‐degree family members of C9orf72 expansion carriers. The unaffected family members consisted of 25 presymptomatic C9orf72 expansion carriers and 12 noncarriers, and clinical investigators were blinded to mutation status. Family members were defined as unaffected if they had an absence of motor deficits, behavioral changes, and cognitive changes, as assessed by neurological examination, neuropsychological testing, and structured informant interviews (e.g., with a spouse or sibling). The presence of a pathogenic C9orf72 repeat expansion, defined as more than 30 repeats,4 was ascertained at the local sites. Symptomatic C9orf72 expansion carriers were diagnosed according to criteria for behavioral variant FTD (bvFTD, n = 47; 9 with concomitant MND)1 or primary progressive aphasia (PPA, n = 6; 2 with concomitant MND)2 at time of inclusion. Subjects with mild cognitive or behavioral symptoms who did not meet these diagnostic criteria for FTD were classified as having mild impairment (n = 9), and were included in the symptomatic carrier group. Among these nine subjects with mild symptomatology, seven had behavioral symptoms and two had memory symptoms. Lastly, two patients had dementia with a predominant memory presentation, without known behavioral or motor changes. ALS patients without cognitive or behavioral symptoms were not included in this study because the number of available subjects was too small to perform statistical analyses on this subgroup (n = 2). Of the 101 subjects, 33 were included in our previous study on CSF NfL.18
Age at disease onset was defined as the age when caregivers first noted a behavioral, motor, or cognitive change, and disease duration was defined as the interval between the age at disease onset and CSF collection. Mini‐Mental State Examination (MMSE) was used to measure global cognition,21 and the Clinical Dementia Rating scale (CDR) was used to assess symptom severity.22 All cognitive test scores were collected within 90 days of CSF collection.
Local ethics committees approved the study and all participants (or their legal representative) provided written informed consent.
CSF analyses
CSF was collected according to standardized local procedures and longitudinal samples were available from 10 C9orf72 expansion carriers who remained in the same clinical stage (presymptomatic or symptomatic) as when the baseline CSF was collected. Measurements of poly(GP) and NfL were performed blinded to clinical information. We performed each biomarker measurement in one laboratory to eliminate variability caused by testing at multiple sites. Poly(GP) was measured at the Mayo Clinic in Jacksonville, FL, and NfL was measured at the VU University medical center. CSF poly(GP) concentrations were measured in duplicate wells using a previously described immunoassay.11
CSF NfL was measured in duplicate using the enzyme‐linked immunosorbent assay of Uman Diagnostics (Umeå, Sweden), according to the manufacturer's instructions. Median intra‐ and interassay coefficient of variations were 1.5% (range: 0–11%) and 6.3% (range: 6.1–16.7%), respectively. Two samples had NfL levels that exceeded the upper limit of quantification of the assay (10,000 pg/mL). Since there was insufficient CSF available to measure NfL upon the dilution of these samples, they were excluded from the NfL analyses. Among the longitudinally collected samples, NfL measurements were not performed on two because of insufficient CSF volumes.
For a given clinical subgroup (noncarriers, presymptomatic carriers, and symptomatic carriers), CSF NfL and poly(GP) levels did not significantly differ among the different centers at which CSF was collected.
MRI acquisition and preprocessing
T1‐weighted MRI images (1,5 or 3 Tesla) captured within 3 months of CSF collection were included for imaging analyses (n = 72). After excluding poor quality scans (e.g., motion artifact, n = 6) and scans from subjects with structural abnormalities (including extensive white matter hyperintensities or lacunar infarcts, n = 3), scans were available for 63 subjects (11 noncarriers, 24 presymptomatic carriers, and 28 symptomatic carriers) from 11 different scanners across seven sites. The NfL level was unavailable for one symptomatic carrier. MRI images were analyzed using two methods: region of interest (ROI) analysis and voxel‐based morphometry (VBM). For ROI analysis, scans were parcellated into brain regions as previously described,23 using an atlas propagation and label fusion strategy,24 combining bilateral ROIs to calculate grey matter cortical (frontal, temporal, parietal, occipital, cingulate, insular), subcortical (hippocampus, amygdala, caudate, putamen, thalamus), and cerebellar volumes.25, 26 Whole brain volumes were calculated by combining all grey and white matter regions extracted from the automated brain segmentation method. All volumes were expressed as percentage of total intracranial volume (TIV), computed with SPM12 (Statistical Parametric Mapping, Wellcome Trust Centre for Neuroimaging, London, UK) running under Matlab R2014b (Math Works, Natick, MA).27 For the VBM preprocessing, T1 images were normalized using standard spatial normalization in SPM12 (http://www.fil.ion.ucl.ac.uk/spm/software/spm12/), modulated, corrected for nonlinear warping, then segmented into grey and white matter images. Grey matter images were smoothed using a half‐maximum isotropic Gaussian kernel with a size of 8 mm full‐width chosen due to the heterogeneity of scanners in the study.
Statistical analysis
Statistical analyses were performed in SPSS 21.0 for Windows (Armonk, NY) and graphs were drafted with GraphPad Prism 7 (La Jolla, CA). Test statistics were considered significant at P < 0.05 (two‐tailed). Since the poly(GP) concentrations were non‐normally distributed, a log‐transformation was applied after adding a constant of 0.1 to all values to avoid values of zero. CSF NfL was log‐transformed to normalize the data; three samples with levels >10,000 pg/mL [all had concomitant MND with either bvFTD (n = 2) or nonfluent variant PPA (n = 1)] continued to skew the data and were therefore set at 10,000 pg/mL prior to the transformation to allow further parametric analysis. Group comparisons of poly(GP) and NfL were first performed by Kruskal–Wallis (with Dunn's post hoc tests) or Mann–Whitney tests on raw data, followed by ANCOVAs on log‐transformed data with correction for age and gender. When longitudinal CSF samples were available, only the time‐point close to MRI or the first time point was used in group comparisons. Area under the curve (AUC) with a 95% confidence interval (CI), obtained by receiver operating characteristic analyses, was used to examine diagnostic performance, with optimal cut‐off levels at the highest (sensitivity + specificity‐1). Correlations with age at CSF collection, age at disease onset, disease duration, and cognitive scores were assessed with Spearman correlations on nontransformed data. Survival in patients was compared among tertiles of poly(GP) and NfL by Kaplan–Meier curves and Cox regressions adjusted for age, gender, presence of MND, and disease duration, with living patients included as censored data. The Cox regressions were also performed using poly(GP) or NfL as a continuous variable. Although we had a relatively small number of carriers with longitudinal samples (n = 10), exploratory analyses on poly(GP) and NfL change were undertaken by calculating the annual change [(second concentration–first concentration)/interval between time points] and testing these values against zero using a one‐sample Wilcoxon signed‐rank test.
All imaging analyses were controlled for age at CSF collection, gender, scanner, and TIV. Linear regressions were used to explore the associations between transformed poly(GP) or NfL and the ROIs. VBM analyses were conducted in SPM12 using subjects’ smoothed, modulated grey matter segments. Within SPM's general linear model framework, we used one‐sample t‐test designs in two separate analyses to correlate either log‐transformed poly(GP) levels or NfL with grey matter volume among all C9orf72 expansion carriers. We repeated these correlations between poly(GP) and NfL with grey matter in presymptomatic and symptomatic expansion carrier subgroups. Results were regarded as significant at P < 0.05 family‐wise error corrected for multiple comparisons (p fwe). When associations were not significant at this threshold, a less stringent threshold of P < 0.001 was used. Mean raw grey matter intensities were extracted from regions showing significant results at P < 0.001 using the MARSBAR toolbox for SPM8,28 and plotted against poly(GP) and NfL concentrations for visualization purposes.
Results
Demographic and clinical data
In total, 64 symptomatic C9orf72 repeat expansion carriers, 25 presymptomatic expansion carriers, and 12 healthy noncarriers were included in our study (Table 1). The clinical phenotypes of the symptomatic carriers were: bvFTD (n = 38), bvFTD‐MND (n = 9), subjects with mild impairment (n = 9), PPA [n = 6; four nonfluent variant PPA (two had concomitant MND), one with semantic variant PPA, and one with logopenic variant PPA], and dementia with a memory presentation (n = 2). For presymptomatic carriers, the clinical diagnoses of affected family members included: dementia only (n = 16), dementia and/or MND (n = 8), or MND only (n = 1). As expected, symptomatic carriers were older, performed worse on the MMSE, and had higher CDR sum of boxes (CDR‐SB) scores compared to both noncarriers and presymptomatic carriers (Table 1). The median age at disease onset in symptomatic carriers was 56 years but varied widely (17–76 years). The median disease duration at CSF collection was 2.8 years. Twenty‐four symptomatic carriers died during follow‐up with a median survival of 2.1 years after CSF collection (IQR: 1.0–4.2); the median follow‐up interval of patients living at follow‐up was 2.8 years (IQR: 1.2–4.5).
Table 1.
Demographic, clinical, and biochemical characteristics of noncarriers, presymptomatic, and symptomatic C9orf72 repeat expansion carriers
| Noncarriers, n = 12 | Presymptomatic carriers, n = 25 | Symptomatic carriers, n = 64 | P‐value | |
|---|---|---|---|---|
| Male : female, n | 7 : 5 | 8 : 17 | 35 : 29 | 0.13 |
| Age at CSF collection, years (IQR) | 44 (34–53) | 47 (41–57) | 60 (55–66)a | <0.001 |
| Age at onset, years (range) | n/a | n/a | 56 (17–76) | n/a |
| Disease duration, years (range) | n/a | n/a | 2.8 (0.5–28.9) | n/a |
| Concomitant MND, n | n/a | n/a | 11 | n/a |
| MMSE (IQR) | 30 (28–30) | 29 (29–30) | 25 (22–28)b | <0.001 |
| CDRc (IQR) | 0 (0–0) | 0 (0–0) | 1 (0.5–1)a | <0.001 |
| CDR–SBd (IQR) | 0 (0–0) | 0 (0–0) | 6 (3–8)a | <0.001 |
| CSF Poly(GP), ng/mL (IQR) | 0.00 (0.00–0.00) | 0.75 (0.33–1.50)e | 1.44 (0.49–2.51)e | <0.001 |
| CSF NfLf, pg/mL (IQR) | 333 (212–536) | 429 (336–830) | 1885 (848–2841)a | <0.001 |
| MRI available, n | 11 | 24 | 28 | n/a |
Medians are displayed for continuous variables, with according IQRs unless otherwise specified.
CSF, cerebrospinal fluid; IQR, interquartile range; MND, motor neuron disease; MRI, magnetic resonance imaging; n/a, not applicable; NfL, neurofilament light chain.
Higher than noncarriers and presymptomatic C9orf72 expansion carriers.
Lower than in noncarriers and presymptomatic C9orf72 expansion carriers.
Available in 9 noncarriers, 18 presymptomatic C9orf72 expansion carriers and 32 symptomatic carriers.
Available in 9 noncarriers, 18 presymptomatic and 28 symptomatic C9orf72 expansion carriers.
Higher than in noncarriers.
available in 12 noncarriers, 25 presymptomatic and 62 symptomatic C9orf72 repeat expansion carriers.
Poly(GP) levels
C9orf72 expansion carriers had higher poly(GP) levels than noncarriers (P < 0.001, Fig. 1A), which discriminated carriers from noncarriers with high accuracy (AUC 0.98, P < 0.001), with a specificity of 100%, and a sensitivity of 96.6% (cut‐off of >0.00 ng/mL, three expansion carriers fell below this cut‐off). Poly(GP) levels trended higher in symptomatic carriers compared to presymptomatic carriers (P = 0.10), and this observation became statistically significant upon correction for age and gender (post hoc Bonferroni corrected P = 0.04). Poly(GP) levels did not differ between males and females, among presymptomatic carriers grouped by their relatives’ diagnoses, among clinical diagnoses of the symptomatic carriers, nor between patients with or without concomitant MND (Fig. 1A and B). No significant associations were found between poly(GP) and age at CSF collection, age at disease onset, disease duration at time of CSF collection, MMSE, CDR, or CDR‐SB. No association between poly(GP) and survival was found.
Figure 1.
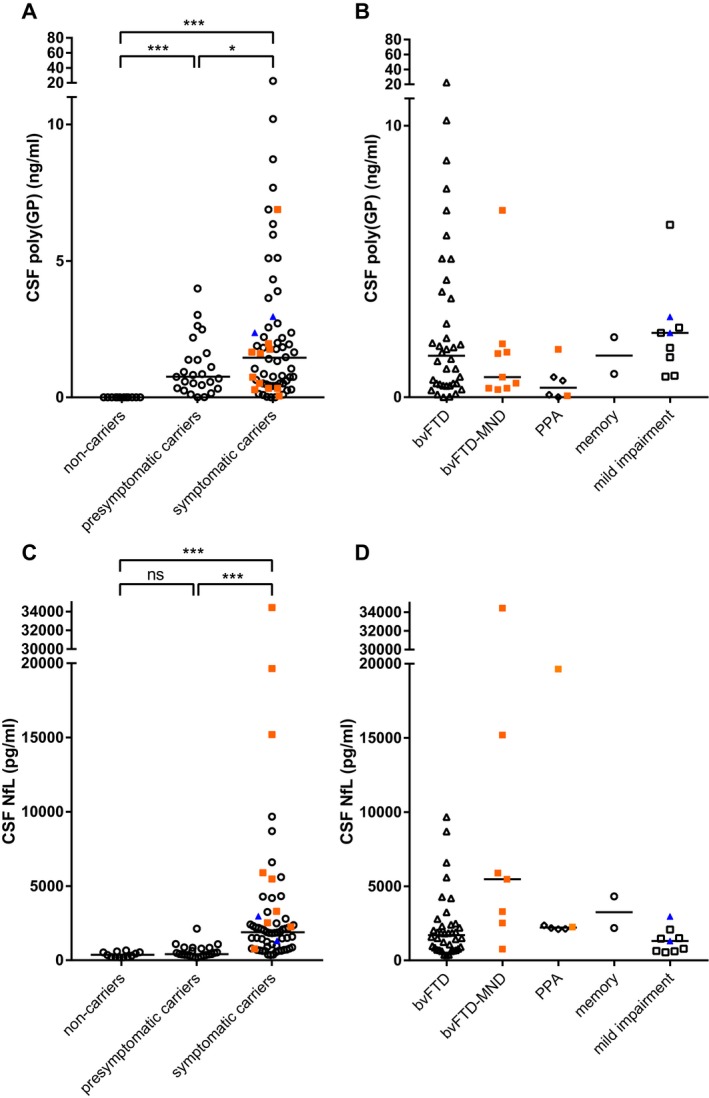
Poly(GP) and NfL levels by clinical stage and diagnosis. (A) Poly(GP) levels were higher in presymptomatic and symptomatic C9orf72 expansion carriers than in healthy noncarriers, and higher in symptomatic carriers than in presymptomatic carriers after correction for age and gender. (B) Poly(GP) levels did not differ between different diagnoses. (C) NfL levels were elevated in symptomatic C9orf72 repeat expansion carriers when compared to noncarriers and presymptomatic carriers. (D) NfL levels were highest in symptomatic carriers with concomitant MND. Patients with concomitant MND at CSF collection are displayed as orange filled squares, those who developed MND after collection are displayed as blue filled triangles. Horizontal lines represent group medians. P‐values from the ANCOVA analyses are displayed (corrected for age and gender) as follows: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ns: not significant.
In our exploratory longitudinal analysis of 10 C9orf72 expansion carriers, a modest but significant increase in poly(GP) was observed, and this was especially evident in the four presymptomatic carriers (P = 0.03, Fig. 2A). The median annual change in poly(GP) was 0.04 ng/mL, and the median time between the first and second samples was 2 years (range 1.0–5.4 years).
Figure 2.
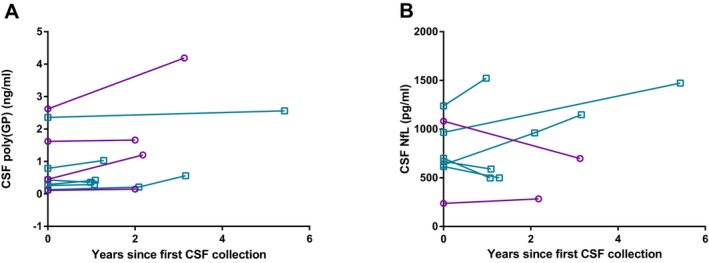
Longitudinal CSF poly(GP) and NfL levels. Longitudinal poly(GP) (A), but not NfL levels (B), increased significantly over time (P = 0.03 and P = 0.89, respectively) in presymptomatic (purple circles and connecting lines) and symptomatic (blue squares with connecting lines) C9orf72 repeat expansion carriers.
NfL levels
CSF NfL levels were significantly higher in symptomatic carriers than in presymptomatic carriers and noncarriers (both P < 0.001, Fig. 1C, Table 1), and did not differ between the latter two groups. High CSF NfL levels differentiated symptomatic from presymptomatic carriers with a specificity of 96.0% and a sensitivity of 65.4% (cut‐off at 1169 pg/mL, AUC 0.89, P < 0.001). Patients with concomitant MND at the time of CSF collection had higher NfL levels (median 5468 pg/mL) than those without concomitant MND (median 1819 pg/mL, P = 0.001, Fig. 1C and D). NfL did not differ between males and females, but did correlate with age in the total group and in each subgroup (entire cohort r s = 0.60, P < 0.001, carriers r s = 0.53, P < 0.001). NfL did not correlate with age at onset or disease duration at CSF collection, but negatively correlated with MMSE, and positively correlated with CDR and CDR‐SB in all carriers combined (MMSE r s = −0.57, P < 0.001, CDR r s = 0.73, P < 0.001, CDR‐SB r s = 0.72, P < 0.001), and in symptomatic carriers after stratification into presymptomatic versus symptomatic stage (MMSE r s = −0.42, P = 0.01, CDR r s = 0.39, P = 0.03, CDR‐SB r s = 0.43, P = 0.03). High NfL levels were associated with a poorer prognosis in terms of survival [Fig 3A, hazard ratio on NfL tertiles of 4.2 (95% CI: 2.0–8.6), P < 0.001].
Figure 3.
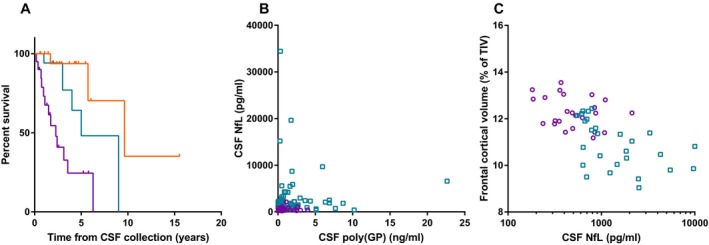
NfL predicts survival, does not correlate with poly(GP), and negatively correlates with frontal cortical volume. (A) Kaplan–Meier curve representing survival of symptomatic C9orf72 expansion carriers based on NfL levels; NfL levels were stratified into lowest (orange upper line), middle (blue middle line), and highest (purple lower line) tertiles; vertical ticks represent living patients censored at the date the patient was last known to be alive. Patients in the highest NfL tertile had the shortest survival. (B) NfL does not correlate with poly(GP) in presymptomatic (purple circles) or symptomatic (blue squares) C9orf72 expansion carriers, and (C) higher NfL levels significantly associated with lower frontal cortical volumes across presymptomatic (purple circles) and symptomatic (blue squares) C9orf72 expansion carriers combined (ROI analysis).
NfL did not correlate with poly(GP) in all carriers combined (P = 0.33, Fig. 3B), nor in presymptomatic or symptomatic carriers separately (P = 0.58 and P = 0.85, respectively). In an exploratory longitudinal analysis, NfL levels increased over time in some individuals but decreased in others, resulting in no significant longitudinal change at the group level (P = 0.89, n = 8, Fig. 2B). The different trajectories were not explained by evident differences in clinical characteristics.
Imaging associations
Associations of poly(GP) with grey matter volume
Although none of the correlations between grey matter volume and poly(GP) reached significance when corrected for multiple comparisons, trends of interest were noted. Across all C9orf72 expansion carriers combined, higher levels of poly(GP) trended with lower frontal and cingulate grey matter volumes from the ROI analysis (Table 2); this trend remained after excluding FTD patients with concomitant MND. The voxel‐wise grey matter analysis showed that poly(GP) tended to associate with regions of bilateral dorsomedial prefrontal and medial frontal cortices and lateral temporal cortex at P < 0.001 (Fig 4A, Table S2). Within these grey matter regions, bvFTD‐MND and bvFTD showed the lowest grey matter intensities (Fig 4B). For the presymptomatic carriers, higher poly(GP) levels tended to associate with lower temporal ROI volume (Table 2), and lower volume in medial prefrontal cortex and scattered regions within lateral temporal cortices in the VBM analysis (P < 0.001, Fig 4C, Table S2). For symptomatic carriers, higher poly(GP) levels trended with lower cingulate grey matter ROI volume (Table 2), and with lower volume in a small dorsomedial frontal cluster in the VBM analysis (P < 0.001, Fig 4D).
Table 2.
Associations between poly(GP) or NfL and grey matter regions of interest in C9orf72 expansion carriers
| ROI | poly(GP) | NfL | |||||
|---|---|---|---|---|---|---|---|
| All carriers, n = 52 | Presymptomatic carriers, n = 24 | Symptomatic carriers, n = 28 | All carriers, n = 51 | Presymptomatic carriers, n = 24 | Symptomatic carriers, n = 27 | ||
| Whole brain | β | −0.11 | −0.22 | −0.09 | 0.19 | −0.22 | 0.38 |
| P | 0.52 | 0.35 | 0.73 | 0.41 | 0.54 | 0.21 | |
| Frontal | β | −0.25 | −0.35 | −0.17 | −0.60 | −0.28 | −0.70 |
| P | 0.045 | 0.10 | 0.48 | <0.001 a | 0.38 | <0.001 a | |
| Temporal | β | −0.20 | −0.49 | −0.02 | −0.42 | −0.45 | −0.19 |
| P | 0.12 | 0.02 | 0.92 | 0.01 | 0.17 | 0.43 | |
| Parietal | β | −0.16 | −0.08 | −0.06 | −0.43 | −0.40 | −0.35 |
| P | 0.24 | 0.69 | 0.78 | 0.01 | 0.17 | 0.12 | |
| Occipital | β | −0.19 | 0.07 | −0.18 | −0.32 | 0.06 | −0.34 |
| P | 0.19 | 0.73 | 0.38 | 0.09 | 0.84 | 0.14 | |
| Cingulate | β | −0.31 | −0.05 | −0.51 | −0.07 | 0.04 | −0.07 |
| P | 0.02 | 0.81 | 0.02 | 0.69 | 0.88 | 0.79 | |
| Insula | β | 0.01 | −0.07 | 0.09 | −0.15 | −0.02 | −0.13 |
| P | 0.97 | 0.76 | 0.63 | 0.31 | 0.94 | 0.53 | |
| Cerebellum | β | −0.19 | −0.31 | −0.01 | 0.04 | −0.36 | 0.32 |
| P | 0.24 | 0.19 | 0.98 | 0.87 | 0.30 | 0.26 | |
| Hippocampus | β | −0.02 | −0.23 | −0.01 | 0.03 | 0.10 | −0.05 |
| P | 0.87 | 0.35 | 0.97 | 0.97 | 0.80 | 0.83 | |
| Amygdala | β | −0.23 | −0.24 | −0.88 | −0.35 | 0.10 | −0.32 |
| P | 0.15 | 0.29 | 0.39 | 0.10 | 0.77 | 0.28 | |
| Caudate nuclei | β | −0.25 | −0.35 | −0.23 | −0.10 | −0.21 | −0.27 |
| P | 0.07 | 0.05 | 0.30 | 0.62 | 0.45 | 0.30 | |
| Putamen | β | −0.24 | −0.12 | −0.31 | −0.33 | −0.27 | −0.41 |
| P | 0.06 | 0.62 | 0.11 | 0.05 | 0.53 | 0.06 | |
| Thalamus | β | −0.08 | −0.09 | −0.16 | −0.02 | 0.05 | 0.07 |
| P | 0.52 | 0.68 | 0.49 | 0.91 | 0.89 | 0.95 | |
Associations between poly(GP) (first three columns) and NfL (last three columns) concentrations and different brain and grey matter ROIs in C9orf72 repeat expansion carriers, by means of linear regression corrected for age, gender, and scanner. P‐values below 0.05 are bolded. NfL, neurofilament light chain; ROI, region of interest.
Significant after correction for multiple testing (Bonferroni corrected P‐value: P < 0.004).
Figure 4.
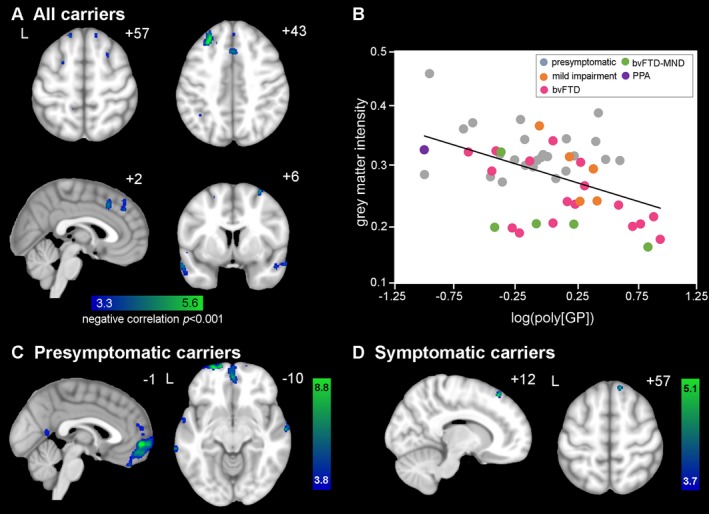
Voxel‐wise associations of grey matter deficits with higher poly(GP) levels. (A) Cross‐sectionally, regions in bilateral dorsolateral prefrontal and medial frontal cortices, and lateral temporal cortex showed lower grey matter volume associated with higher poly(GP) levels in the voxel‐wise analysis of C9orf72 repeat expansion carriers. Significant clusters were defined at a t‐threshold of P < 0.001 uncorrected, no significant clusters were found at p fwe < 0.05. Color bars represent t‐scores, and statistical maps are superimposed on the Montreal Neurological Institute template brain. The left side of the axial and coronal images corresponds to the left (L) side of the brain. (B) Mean grey matter intensity versus log‐transformed poly(GP) within the P < 0.001 map in (A), for 24 presymptomatic C9orf72 expansion carriers (grey dots), 4 mild impairment (orange dots), 17 bvFTD (pink dots), 5 bvFTD‐MND (green dots), and 1 PPA (purple dot) plotted for visualization purposes only. In general, FTD‐MND showed the lowest grey matter intensities compared to the other diagnostic groups. (C) For presymptomatic carriers, grey matter volume was negatively correlated with poly(GP) in medial prefrontal cortex and scattered regions within lateral temporal cortices. (D) For symptomatic carriers, a small dorsomedial frontal cluster showed lower grey matter volume associated with higher poly(GP) levels.
Associations of NfL levels with grey matter volume
Higher CSF NfL levels were associated with lower frontal (Fig. 3C), temporal, and parietal ROI grey matter volumes in all carriers combined, but the latter two ROIs did not survive multiple comparisons correction (Table 2). In the VBM analysis, higher NFL levels were associated with lower grey matter volumes in the ventral and dorsomedial prefrontal cortex, ventral and dorsal insula, anterior cingulate, caudate, medial thalamus, and several other frontotemporoparietal regions (P < 0.001, Fig 5A and B, Table S2). At p fwe < 0.05, higher NfL levels were associated with lower grey matter volume in small regions of dorsolateral prefrontal cortex, dorsal posterior insula, and the left caudate. In a subgroup analysis of presymptomatic carriers only, no significant correlations between grey matter ROIs and NfL emerged, and voxel‐wise, NfL levels correlated with grey matter deficits in the inferior and middle frontal gyrus, pre‐ and postcentral gyrus, operculum, superior temporal gyrus, lateral parietal regions, and the caudate only at P < 0.001 (Fig 5C, Table S2) with no significant regions at p fwe < 0.05. For symptomatic carriers, NfL significantly correlated with frontal cortex in the ROI analysis, and the VBM showed associations with bilateral dorsolateral prefrontal cortex, anterior and mid cingulate cortex, dorsal insula, pre‐ and postcentral gyrus, medial parietal regions and the caudate (P < 0.001, Fig 5D), with no significant regions at p fwe < 0.05.
Figure 5.
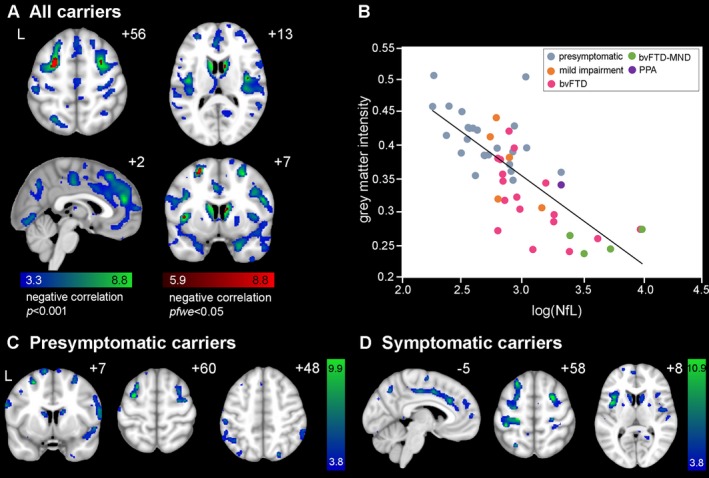
Voxel‐wise associations of grey matter deficits with higher NfL levels. (A) Widespread regions including ventral and dorsomedial prefrontal cortex, ventral and dorsal insula, anterior cingulate, caudate, and medial thalamus showed lower grey matter volume (voxel‐wise analysis) associated with higher NfL levels. Significant clusters were defined at a t‐threshold of P < 0.001 uncorrected (blue‐green colored) and p fwe < 0.05 (red colored). Color bars represent t‐scores, and statistical maps are superimposed on the Montreal Neurological Institute template brain. The left side of the axial and coronal images corresponds to the left (L) side of the brain. (B) Mean grey matter intensity versus log‐transformed NfL levels within the P < 0.001 map in (A), for 24 presymptomatic C9orf72 expansion carriers (grey dots), 4 mild impairment (orange dots), 17 bvFTD (pink dots), 4 bvFTD‐MND (green dots), and 1 PPA (purple dot) plotted for visualization purposes only. Both presymptomatic (C) and symptomatic carriers (D) show associations of NfL with grey matter atrophy in bilateral dorsolateral prefrontal cortex, parietal regions, and caudate. Symptomatic carriers (D) additionally show grey matter atrophy associated with NfL in key hubs targeted in bvFTD, including anterior and mid cingulate cortex and insula.
Discussion
In this study, CSF poly(GP), NfL, and grey matter volumes were determined in a cohort of 89 C9orf72 repeat expansion carriers to examine associations among these measures. Poly(GP) was detected in CSF of both presymptomatic and symptomatic C9orf72 expansion carriers, and not detected in noncarriers. In contrast, we found high NfL levels exclusively in symptomatic carriers, while levels in presymptomatic carriers remained similar to healthy noncarriers. Higher NfL levels correlated with greater disease severity as well as shorter survival. In addition, higher NfL levels associated with lower grey matter volumes in regions known to show smaller grey matter volume in presymptomatic and symptomatic carriers, but for poly(GP), only trends were observed. As such, CSF NfL and poly(GP) capture abnormalities during different phases of the disease, and may thus serve as complementary biomarkers for disease detection and future treatment monitoring.
Poly(GP) is a highly sensitive and specific biomarker for C9orf72 expansion carriers
We showed that CSF poly(GP) has high power to differentiate C9orf72 expansion carriers and noncarriers, consistent with previous reports.10, 11, 14 Interestingly, poly(GP) levels were undetectable in three C9orf72 expansion carriers, who had various clinical presentations, including a presymptomatic individual, a bvFTD patient, and a PPA patient. Future studies may inform whether alternative detection strategies with increased sensitivity would detect poly(GP) in these individuals, or whether their poly(GP) levels are truly negligible. Should postmortem tissue become available for these individuals and others, it would be of particular interest to evaluate how CSF poly(GP) levels compare to the frequency of DPR protein pathology and levels of repeat‐containing transcripts in the brain. As in previous studies,11, 14 we found that poly(GP) was detectable in CSF from presymptomatic C9orf72 expansion carriers, suggesting that DPR protein production emerges prior to neurodegeneration and that poly(GP) can be actively released from putatively healthy neurons. This notion is supported by in vitro experiments that show that DPR proteins are secreted from cultured cells.11, 29 Reports of autopsy studies in C9orf72‐patients have also described widespread DPR protein pathology prior to the formation of TDP‐43 inclusions and neuronal loss.30, 31, 32 These studies provide converging evidence that poly(GP) expression arises early in the lifespan of C9orf72 expansion carriers.
Poly(GP) levels are higher in symptomatic C9orf72 expansion carriers compared to presymptomatic carriers
Symptomatic carriers had higher poly(GP) levels compared to presymptomatic carriers after correcting for age and gender. In parallel, we found a modest increase in CSF poly(GP) over time in a small subset of 10 C9orf72 expansion carriers, most frequently in the presymptomatic subjects. In contrast, other studies had not identified a significant difference in poly(GP) levels between presymptomatic and symptomatic carriers,11, 14 nor an increase in poly(GP) levels over time.11 The time interval between repeated CSF collections was longer in the present exploratory analysis, suggesting that changes in poly(GP) might only emerge over longer periods. Measuring poly(GP) in larger cohorts over extended time periods as individuals convert from the presymptomatic to symptomatic phase will further help elucidate the temporal trajectory of poly(GP), and whether levels change relative to symptom onset.
We found that poly(GP) levels did not differ among clinical phenotypes, and did not correlate with age at disease onset or survival. Although there is a possibility of a type II error (i.e., a false‐negative association) given that several clinical subgroups had a small sample size, these data are in line with the lack of associations between clinical phenotypes and other C9orf72‐associated features, such as repeat size and RNA foci burden.33, 34 While we were unable to examine correlations between CSF poly(GP) and repeat length in blood because repeat length data were not available, such an analysis would likely be complicated given the substantial variation in repeat sizes among various tissues from the same individual.33 Furthermore, a previous study has shown no association between CSF poly(GP) and repeat length in blood,14 and we observed no association between poly(GP) levels in the cerebellum or frontal cortex and repeat length in these regions.35 Nonetheless, examining associations between antemortem CSF poly(GP) and repeat size, poly(GP) levels, and levels of other DPR proteins in various neuroanatomical regions will be of interest when postmortem tissue becomes available from a suitable number of cases. Also, as arginine‐containing DPR proteins, poly(GR) and poly(PR), are considered to be highly toxic,6 it is possible that elevated levels of these DPR proteins may correlate with clinical features and measures of neurodegeneration. The development of immunoassays quantifying these proteins remains technically challenging, but may lead to more insights into C9orf72 disease mechanisms.
NfL is normal in presymptomatic C9orf72 expansion carriers and is elevated in symptomatic carriers
Determining symptom onset in C9orf72 repeat expansion carriers is notoriously challenging. Presymptomatic carriers have a high incidence of psychiatric symptoms overlapping with bvFTD symptomatology,36 and some carriers have a mild, slowly progressive prodromal phase spanning several decades.37 Consequently, surrogate endpoints reflecting neurodegeneration are critical for assessing disease onset and the efficacy of therapeutic interventions. Our data show that NfL is elevated during the symptomatic phase of C9orf72‐associated dementia and correlate with indicators of disease severity (i.e., MMSE, CDR, and CDR‐SB), survival, and grey matter atrophy, consistent with other studies of NfL in sporadic and genetic FTD.18, 38, 39 In mouse models, NfL also correlates with disease severity, specifically with the burden of α‐synuclein, tau, or β‐amyloid inclusions, and NfL levels are attenuated with treatment.40 Thus, NfL could be utilized in clinical trials to stratify patients into more homogeneous groups with respect to disease severity and to assess the neuroprotective effect of therapeutic interventions. Furthermore, the strong association between CSF NfL and survival supports the use of NfL as a prognostic marker. NfL can now be reliably measured in serum and plasma, which is collected less invasively than CSF, making it promising for clinical use, especially when frequent measures are needed.17, 18, 41
Our small longitudinal study of CSF NfL levels did not show consistent changes over the time intervals assessed. Previous studies on ALS patients reported stable NfL levels, but showed an increase over time for a subset of patients with a rapid disease progression.17, 42, 43 Serum NfL levels have also been shown to progressively increase in sporadic PPA patients.44 The discrepancy between our and previous findings could be attributable to the small size of our longitudinal cohort, and the range of clinical phenotypes and disease durations among individuals at the time of sample collection. Given that the rate of neurodegeneration can differ not only throughout the course of disease but also among patients, additional longitudinal studies on larger cohorts of C9orf72 expansion carriers are needed to fully understand the temporal trajectory of NfL in relation to clinical changes.
Relationships between poly(GP) and indicators of neurodegeneration
This study did not show a significant relationship between poly(GP) levels and indicators of neurodegeneration, as reflected by NfL levels and grey matter volumes. Similarly, previous studies show no correlation between poly(GP) and NfL,14 nor neurofilament heavy chain (a different neurofilament subunit) in C9orf72‐associated ALS.20 Because previous studies have shown lower grey matter volumes in both presymptomatic and symptomatic carriers compared to controls,23, 36, 45, 46 we had hypothesized that higher levels of poly(GP) might be associated with lower grey matter volumes in C9orf72‐targeted regions. We found a trend toward higher poly(GP) with lower frontal and cingulate volume in our ROI analysis. In parallel with the ROI analysis, the voxel‐wise analysis showed that certain sparse regions of lower grey matter volumes tended to associate with higher poly(GP), which included regions in the bilateral dorsolateral prefrontal, medial frontal, and lateral temporal cortices. Although higher poly(GP) levels showed a relatively weak association with lower grey matter volumes in our analyses, the regions identified include those atrophied in C9orf72‐associated FTD patients,45, 47, 48, 49 and show reduced volume in presymptomatic C9orf72 expansion carriers.23, 36
Interestingly, our subgroup analysis of presymptomatic carriers showed a trend for association between higher poly(GP) and reduced bilateral medial orbitofrontal cortex, but symptomatic carriers showed only sparse regions that trended toward an association with high poly(GP). One potential explanation for this result is that DPR accumulation may arise focally during the presymptomatic phase and become widespread during the symptomatic phase, thus attenuating any potential relationship between grey matter atrophy and poly(GP) during the symptomatic phase. Overall, our results suggest that higher poly(GP) levels may be associated with some key foci in C9orf72‐associated regions, particularly for presymptomatic carriers, but poly(GP) levels are not a marker for neurodegeneration per se.
Higher NfL levels are associated with grey matter deficits
Elevated NfL levels were associated with lower grey matter volume based on both types of analysis. These data reveal vulnerable neuroanatomical structures during the presymptomatic and symptomatic stages, which include ventral and dorsomedial prefrontal cortex, ventral and dorsal insula, anterior cingulate, caudate, and the medial thalamus. These regions are highly anatomically congruent with atrophy patterns found in C9orf72‐associated frontotemporal dementia.45, 47, 48, 49, 50 Notably, several studies confirmed that the medial thalamus is a region affected across C9orf72 expansion carriers,50 even during the presymptomatic phase.23, 36, 46 Interestingly, our subgroup analysis of presymptomatic carriers showed that higher NfL levels were associated with smaller bilateral frontoparietal and caudate volumes, which may indicate that these regions are among the earliest regions of neurodegeneration, but longitudinal presymptomatic studies are needed to test this hypothesis.
Conclusion
The strength of this study is the use of a multimodal approach combining two CSF biomarkers with quantitative structural imaging metrics to investigate the relationship between poly(GP) and indicators of neurodegeneration in a large cohort of presymptomatic and symptomatic C9orf72 expansion carriers. To the best of our knowledge, this is the largest study thus far on poly(GP) in C9orf72‐associated FTD, and the first in examining associations between DPR protein levels and grey matter volume. We used two neuroimaging methods: ROI analyses, the metrics of which are reliable and can be readily analyzed by clinical groups, and voxel‐wise analyses, which can refine grey matter deficits in a more granular fashion. We show that poly(GP) and NfL are promising complementary biomarkers that capture the effects of the C9orf72 repeat expansion during different phases, and demonstrate different relationships with grey matter volume. While poly(GP) may display less utility as a prognostic or staging biomarker, it shows great promise as a pharmacodynamic biomarker for therapeutic approaches that target G4C2 RNA in preclinical models.11 Importantly, poly(GP) levels are detectable in presymptomatic carriers and would thus make the inclusion of this population in clinical trials more feasible. Because NfL reflects neurodegeneration and is associated with grey matter atrophy, it will be most useful for monitoring disease severity, and predicting disease progression and survival.
Author Contributions
L.H.M., T.F.G., and S.E.L. contributed to the study design, in the acquisition, analysis, and interpretation of data, drafted the manuscript and figures. A.C.S., L.C.J., S.R., contributed to data acquisition and analysis, and drafting of manuscript and figures. A.L.B., L.P., J.D.R., and J.C.v.S. contributed to the study design, in the acquisition and interpretation of data, and revised the manuscript. All other authors (L.D.K., J.M.P., J.L.P., E.L.v.E., E.G.D., S.F., C.G., H.J.R., R.S.V., D.G., Y.A.P., L.B., R.G., B.B., R.J.L., M.D.C., C.E.T., R.v.M., J.C.R., G.C., D.H.G., R.R., A.M.K., L.O., E.S., G.B., A.P., D.M.C., K.M.D., M.B., B.L.M.) contributed to the acquisition of data and/or in study coordination, and revised the manuscript.
Conflicts of Interest
T.F.G. and L.P. have a US patent on methods and materials for detecting C9orf72‐associated ALS and FTD using poly(GP) proteins (European patent filed). All other authors report no conflicts of interest relevant to this work.
Supporting information
Table S1. Number of subjects per site.
Table S2. Negative correlations between grey matter volume and poly(GP) or NfL for all, presymptomatic and symptomatic C9orf72 repeat expansion carriers – peak voxel regions from the voxel‐based morphometry analysis.
Acknowledgments
We thank all participants of this study, and the local research coordinators for the help in collecting the samples, clinical and imaging data. This study was supported by Deltaplan Dementie (The Netherlands Organisation for Health Research and Development, and Alzheimer Nederland, grant number 733050103), the European Joint Programme ‐ Neurodegenerative Disease Research and the Netherlands Organisation for Health Research and Development (PreFrontALS: 733051042, RiMod‐FTD 733051024), Alzheimer Nederland [L.H.M. (WE.09‐2014‐04) and C.T.], the Dioraphte Foundation, The Bluefield Project, the National Institutes of Health/National Institute of Neurological Disorders and Stroke [R21NS089979 (T.F.G.), R35NS097273 (L.P.), P01NS099114 (T.F.G, L.P.), U54NS092089 (A.L.B.), R35NS097261 (R.R.)], the Amyotrophic Lateral Sclerosis Association (T.F.G., L.P.), Robert Packard Center for ALS Research (L.P.), Target ALS Foundation (T.F.G., L.P.), Association for Frontotemporal Degeneration (L.P.), Muscular Dystrophy Association (T.F.G.), National Institute on Aging [P01AG019724 (B.L.M.); P50AG23501 (B.L.M.); AG032306 (H.J.R); K23AG039414 (S.E.L)], the Italian Ministry of Health [Ricerca Corrente (R.G., L.B., G.B.)], Spanish National Institute of Health Carlos III (ISCIII) under the aegis of the EU Joint Programme – Neurodegenerative Disease Research (JPND) [AC14/00013 (R.S.V.)] and Fundacio Marato de TV3 [20143810 (R.S.V.)], the Swedish Medical Research Council (C.G., L.O.), ALF Stockholm county council (C.G., L.O.), Swedish Brain Foundation (C.G., L.O.), Swedish Alzheimer Foundation (C.G., L.O.), Karolinska Institutet doctoral funding & Stratneuro (C.G., L.O.), Swedish Brain Power (C.G., L.O.), the association of Frontotemporal Dementia (C.T.) the Dutch Research Council (ZonMW, C.T.), Weston Brain Institute (C.T.) and Alzheimer's Drug Discovery Foundation (C.T.). The Dementia Research Centre at UCL is supported by Alzheimer's Research UK, Brain Research Trust, and The Wolfson Foundation. This work was supported by the NIHR Queen Square Dementia Biomedical Research Unit, the NIHR UCLH Biomedical Research Centre, and the MRC UK GENFI grant (MR/M023664/1). J.D.R. is supported by an MRC Clinician Scientist Fellowship (MR/M008525/1) and has received funding from the NIHR Rare Disease Translational Research Collaboration (BRC149/NS/MH). K.D. is supported by an Alzheimer's Society PhD Studentship (AS‐PhD‐2015‐005).
Funding Information
See Acknowledgment section for funding information.
This study was supported by Deltaplan Dementie (The Netherlands Organisation for Health Research and Development, and Alzheimer Nederland, grant number 733050103), the Netherlands Organisation for Health Research and Development (PreFrontALS: 733051042, RiMod‐FTD 733051024), Alzheimer Nederland [L.H.M. (WE.09‐2014‐04) and C.T.], the Dioraphte Foundation, The Bluefield Project, the National Institutes of Health National Institute of Neurological Disorders and Stroke [R21NS089979 (T.F.G.), R35NS097273 (L.P.), P01NS099114 (T.F.G, L.P.), U54NS092089 (A.L.B.), R35NS097261 (R.R.)], the Amyotrophic Lateral Sclerosis Association (T.F.G., L.P.), Robert Packard Center for ALS Research (L.P.), Target ALS Foundation (T.F.G., L.P.), Association for Frontotemporal Degeneration (L.P.), Muscular Dystrophy Association (T.F.G.), National Institute on Aging [P01AG019724 (B.L.M.); P50AG23501 (B.L.M.); AG032306 (H.J.R); K23AG039414 (S.E.L)], the Italian Ministry of Health [Ricerca Corrente (R.G., L.B., G.B.)], Spanish National Institute of Health Carlos III (ISCIII) under the aegis of the EU Joint Programme – Neurodegenerative Disease Research (JPND) [AC14/00013 (R.S.V.)] and Fundacio Marato de TV3 [20143810 (R.S.V.)], the Swedish Medical Research Council (C.G., L.O.), ALF Stockholm county council (C.G., L.O.), Swedish Brain Foundation (C.G., L.O.), Swedish Alzheimer Foundation (C.G., L.O.), Karolinska Institutet doctoral funding & Stratneuro (C.G., L.O.), Swedish Brain Power (C.G., L.O.), the association of Frontotemporal Dementia (C.T.) the Dutch Research Council (ZonMW, C.T.), Weston Brain Institute (C.T.) and Alzheimer's Drug Discovery Foundation (C.T.). The Dementia Research Centre at UCL is supported by Alzheimer's Research UK, Brain Research Trust, and The Wolfson Foundation. This work was supported by the NIHR Queen Square Dementia Biomedical Research Unit, the H Biomedical Research Centre, and the MRC UK GENFI grant (MR/M023664/1). J.D.R. is supported by an MRC Clinician Scientist Fellowship (MR/M008525/1) and has received funding from the NIHR Rare Disease Translational Research Collaboration (BRC149/NS/MH). K.D. is supported by an Alzheimer's Society PhD Studentship (AS‐PhD‐2015‐005).
Funding Statement
This work was funded by Deltaplan Dementie (The Netherlands Organisation for Health Research and Development, and Alzheimer Nederland grant 733050103; European Joint Programme ‐ Neurodegenerative Disease Research and the Netherlands Organisation for Health Research and Development grants PreFrontALS: 733051042 and RiMod‐FTD 733051024; Alzheimer Nederland grant WE.09‐2014‐04; Dioraphte Foundation grant ; National Institutes of Health/National Institute of Neurological Disorders and Stroke grants R21NS089979, R35NS097273, P01NS099114, U54NS092089, and R35NS097261; Amyotrophic Lateral Sclerosis Association grant ; Robert Packard Center for ALS Research grant ; Target ALS Foundation grant ; Association for Frontotemporal Degeneration grant ; Muscular Dystrophy Association grant ; National Institute on Aging grants P01AG019724, P50AG23501, AG032306, and K23AG039414; Italian Ministry of Health grant ; Spanish National Institute of Health Carlos III (ISCIII) under the aegis of the EU Joint Programme – Neurodegenerative Disease Research grant AC14/00013; Fundacio Marato de TV3 grant 20143810; Swedish Medical Research Council grant ; ALF Stockholm county council grant ; Swedish Brain Foundation grant ; Swedish Alzheimer Foundation grant ; Karolinska Institutet doctoral funding & Stratneuro grant ; Swedish Brain Power grant ; association of Frontotemporal Dementia grant ; Dutch Research Council grant ; Weston Brain Institute grant ; Alzheimer's Drug Discovery Foundation grant ; Alzheimer's Research UK grant ; Brain Research Trust grant ; The Wolfson Foundation grant ; NIHR Queen Square Dementia Biomedical Research Unit grant ; NIHR UCLH Biomedical Research Centre grant ; MRC UK GENFI grant MR/M023664/1; MRC Clinician Scientist Fellowship grant MR/M008525/1; NIHR Rare Disease Translational Research Collaboration grant BRC149/NS/MH; Alzheimer's Society PhD Studentship grant AS‐PhD‐2015‐005.
References
- 1. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134(Pt 9):2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gorno‐Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Woollacott IOC, Rohrer JD. The clinical spectrum of sporadic and familial forms of frontotemporal dementia. J Neurochem 2016;138:6–31. [DOI] [PubMed] [Google Scholar]
- 4. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Todd TW, Petrucelli L. Insights into the pathogenic mechanisms of Chromosome 9 open reading frame 72 (C9orf72) repeat expansions. J Neurochem 2016;138(Suppl):145–162. [DOI] [PubMed] [Google Scholar]
- 7. Ash PEA, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013;77:639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mori K, Arzberger T, Grässer FA, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 2013;126:881–893. [DOI] [PubMed] [Google Scholar]
- 9. Jiang J, Zhu Q, Gendron TF, et al. Gain of toxicity from ALS/FTD‐linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC‐containing RNAs. Neuron 2016;90:535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Su Z, Zhang Y, Gendron TF, et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)‐associated defects in c9FTD/ALS. Neuron 2014;83:1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gendron TF, Chew J, Stankowski JN, et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72‐associated amyotrophic lateral sclerosis. Sci Transl Med 2017;9:eaai7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013;80:415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sareen D, O'Rourke JG, Meera P, et al. Targeting RNA foci in iPSC‐derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 2013;5:208ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lehmer C, Oeckl P, Weishaupt JH, et al. Poly‐GP in cerebrospinal fluid links C9orf72‐associated dipeptide repeat expression to the asymptomatic phase of ALS/FTD. EMBO Mol Med 2017;9:859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zetterberg H, Skillbäck T, Mattsson N, et al. Association of cerebrospinal fluid neurofilament light concentration with Alzheimer disease progression. JAMA Neurol 2016;73:60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scherling CS, Hall T, Berisha F, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol 2014;75:116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lu C‐H, Macdonald‐Wallis C, Gray E, et al. Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology 2015;84:2247–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meeter LH, Dopper EG, Jiskoot LC, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol 2016;3:623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weydt P, Oeckl P, Huss A, et al. Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann Neurol 2016;79:152–158. [DOI] [PubMed] [Google Scholar]
- 20. Gendron TF, C9ORF72 Neurofilament Study Group ; Daughrity LM, et al. Phosphorylated neurofilament heavy chain: a biomarker of survival for C9ORF72‐associated amyotrophic lateral sclerosis. Ann Neurol 2017;82:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 22. Morris JC. The clinical dementia rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 23. Rohrer JD, Nicholas JM, Cash DM, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross‐sectional analysis. Lancet Neurol 2015;14:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cardoso MJ, Modat M, Wolz R, et al. Geodesic information flows: spatially‐variant graphs and their application to segmentation and fusion. IEEE Trans Med Imaging 2015;34:1976–1988. [DOI] [PubMed] [Google Scholar]
- 25. Diedrichsen J, Balsters JH, Flavell J, et al. A probabilistic MR atlas of the human cerebellum. NeuroImage 2009;46:39–46. [DOI] [PubMed] [Google Scholar]
- 26. Diedrichsen J, Maderwald S, Küper M, et al. Imaging the deep cerebellar nuclei: a probabilistic atlas and normalization procedure. NeuroImage 2011;54:1786–1794. [DOI] [PubMed] [Google Scholar]
- 27. Malone IB, Leung KK, Clegg S, et al. Accurate automatic estimation of total intracranial volume: a nuisance variable with less nuisance. NeuroImage 2015;104:366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brett M, Anton J‐L, Valabregue R, Poline J‐B. Region of interest analysis using an SPM toolbox [abstract]. Presented at the 8th International Conference on Functional Mapping of the Human Brain, June 2‐6, 2002, Sendai, Japan. Neuroimage 2002;16:xxxvi–lxvi.
- 29. Westergard T, Jensen BK, Wen X, et al. Cell‐to‐cell transmission of dipeptide repeat proteins linked to C9orf72‐ALS/FTD. Cell Rep 2016;17:645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Proudfoot M, Gutowski NJ, Edbauer D, et al. Early dipeptide repeat pathology in a frontotemporal dementia kindred with C9ORF72 mutation and intellectual disability. Acta Neuropathol 2014;127:451–458. [DOI] [PubMed] [Google Scholar]
- 31. Baborie A, Griffiths TD, Jaros E, et al. Accumulation of dipeptide repeat proteins predates that of TDP‐43 in frontotemporal lobar degeneration associated with hexanucleotide repeat expansions in C9ORF72 gene. Neuropathol Appl Neurobiol 2015;41:601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vatsavayai SC, Yoon SJ, Gardner RC, et al. Timing and significance of pathological features in C9orf72 expansion‐associated frontotemporal dementia. Brain 2016;139(Pt 12):3202–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Blitterswijk M, DeJesus‐Hernandez M, Niemantsverdriet E, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize‐72): a cross‐sectional cohort study. Lancet Neurol 2013;12:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. DeJesus‐Hernandez M, Finch NCA, Wang X, et al. In‐depth clinico‐pathological examination of RNA foci in a large cohort of C9ORF72 expansion carriers. Acta Neuropathol 2017;134:255–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gendron TF, van Blitterswijk M, Bieniek KF, et al. Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta Neuropathol 2015;130:559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee SE, Sias AC, Mandelli ML, et al. Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. Neuroimage Clin 2017;14:286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khan BK, Yokoyama JS, Takada LT, et al. Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry 2012;83:358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rohrer JD, Woollacott IOC, Dick KM, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology 2016;87:1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pijnenburg YAL, Verwey NA, van der Flier WM, et al. Discriminative and prognostic potential of cerebrospinal fluid phosphoTau/tau ratio and neurofilaments for frontotemporal dementia subtypes. Alzheimer's Dement (Amst) 2015;1:505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bacioglu M, Maia LF, Preische O, et al. Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron 2016;91:56–66. [DOI] [PubMed] [Google Scholar]
- 41. Gisslén M, Price RW, Andreasson U, et al. Plasma concentration of the neurofilament light protein (NFL) is a biomarker of CNS injury in HIV infection: a cross‐sectional study. EBioMedicine 2016;3:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steinacker P, Huss A, Mayer B, et al. Diagnostic and prognostic significance of neurofilament light chain NF‐L, but not progranulin and S100B, in the course of amyotrophic lateral sclerosis: data from the German MND‐net. Amyotroph. Lateral Scler. Front. Degener. 2017;18(1–2):112–119. [DOI] [PubMed] [Google Scholar]
- 43. Poesen K, De Schaepdryver M, Stubendorff B, et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology 2017;88:2302–2309. [DOI] [PubMed] [Google Scholar]
- 44. Steinacker P, Semler E, Anderl‐Straub S, et al. Neurofilament as a blood marker for diagnosis and monitoring of primary progressive aphasias. Neurology 2017;88:961–969. [DOI] [PubMed] [Google Scholar]
- 45. Boeve BF, Boylan KB, Graff‐Radford NR, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012;135:765–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Papma JM, Jiskoot LC, Panman JL, et al. Cognition and gray and white matter characteristics of presymptomatic C9orf72 repeat expansion. Neurology 2017;89:1256–1264. [DOI] [PubMed] [Google Scholar]
- 47. Lee SE, Khazenzon AM, Trujillo AJ, et al. Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain 2014;137:3047–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Whitwell JL, Weigand SD, Boeve BF, et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 2012;135(Pt 3):794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mahoney CJ, Beck J, Rohrer JD, et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain 2012;135:736–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sha SJ, Takada LT, Rankin KP, et al. Frontotemporal dementia due to C9ORF72 mutations: clinical and imaging features. Neurology 2012;79:1002–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Number of subjects per site.
Table S2. Negative correlations between grey matter volume and poly(GP) or NfL for all, presymptomatic and symptomatic C9orf72 repeat expansion carriers – peak voxel regions from the voxel‐based morphometry analysis.