

Abstract
Objective
Cerebral palsy is a common, heterogeneous neurodevelopmental disorder that causes movement and postural disabilities. Recent studies have suggested genetic diseases can be misdiagnosed as cerebral palsy. We hypothesized that two simple criteria, that is, full‐term births and nonspecific brain MRI findings, are keys to extracting masqueraders among cerebral palsy cases due to the following: (1) preterm infants are susceptible to multiple environmental factors and therefore demonstrate an increased risk of cerebral palsy and (2) brain MRI assessment is essential for excluding environmental causes and other particular disorders.
Methods
A total of 107 patients—all full‐term births—without specific findings on brain MRI were identified among 897 patients diagnosed with cerebral palsy who were followed at our center. DNA samples were available for 17 of the 107 cases for trio whole‐exome sequencing and array comparative genomic hybridization. We prioritized variants in genes known to be relevant in neurodevelopmental diseases and evaluated their pathogenicity according to the American College of Medical Genetics guidelines.
Results
Pathogenic/likely pathogenic candidate variants were identified in 9 of 17 cases (52.9%) within eight genes: CTNNB1,CYP2U1,SPAST,GNAO1,CACNA1A,AMPD2,STXBP1, and SCN2A. Five identified variants had previously been reported. No pathogenic copy number variations were identified. The AMPD2 missense variant and the splice‐site variants in CTNNB1 and AMPD2 were validated by in vitro functional experiments.
Interpretation
The high rate of detecting causative genetic variants (52.9%) suggests that patients diagnosed with cerebral palsy in full‐term births without specific MRI findings may include genetic diseases masquerading as cerebral palsy.
Introduction
Cerebral palsy (CP) is the most common physical disability in childhood and is defined as “a group of permanent disorders of the development of movement and posture, causing activity limitation, that are attributed to nonprogressive disturbances that occurred in the developing fetal or infant brain”.1 Both genetic and environmental factors contribute to the etiology of CP. Recent studies have suggested that a larger number of rare pathogenic genetic variants2 or pathogenic copy number variations (CNVs)3 contribute to CP than previously expected.4 This suggests that “masquerading” genetic diseases can be misdiagnosed as CP.5 To distinguish these masqueraders from CP is essential for delivering appropriate therapy, prognosis, and genetic counseling. However, there is lack of consensus as to which types of unexplained CP should undergo genetic investigation. A recent meta‐analysis showed that preterm (<37 weeks) infants had a dramatically increased risk of CP,6 which suggests that full‐term CP may include more masqueraders than preterm CP. To our knowledge, no genetic studies have specifically focused on full‐term CP. In addition, brain MRI findings are essential for correctly assessing CP by excluding cases caused by apparent environmental factors or cases with other disorders5 (e.g., porencephaly with COL4A1 variants7 and cerebral infarction with CBS variants8) for which other diagnostic approaches could be used, such as metabolic screening and target sequencing.
In this study, we performed trio whole‐exome sequencing (WES) analysis and array comparative genomic hybridization (aCGH) analysis to identify genetic abnormality, focusing on a subgroup of CP patients isolated by two simple criteria, (1) gestational age of 37 weeks or more and (2) normal or nonspecific brain MRI findings.
Methods
Participants
Eight hundred and ninety‐seven cases of cerebral palsy, including spastic CP (hemiplegia/diplegia/triplegia/quadriplegia), ataxic CP, dyskinetic CP, and mixed type CP, were followed at the Takuto Rehabilitation Center for Children, Sendai, Japan between September 1, 1990 and March 31, 2016, as previously reported.9 Of these cases, 107 families were also recruited for inclusion with at least one family member of the family diagnosed with CP, according to the following criteria: (1) diagnosis of CP without a definitive acquired cause, (2) gestational age of 37 weeks or more, (3) no brain MRI findings that can aid in the diagnosis. Among these cases, 17 patients with both parents were available for trio WES, and written consent was obtained from all individuals (Fig. 1). This study was approved by the ethics committee of Tohoku University Hospital and Takuto Rehabilitation Center for Children.
Figure 1.
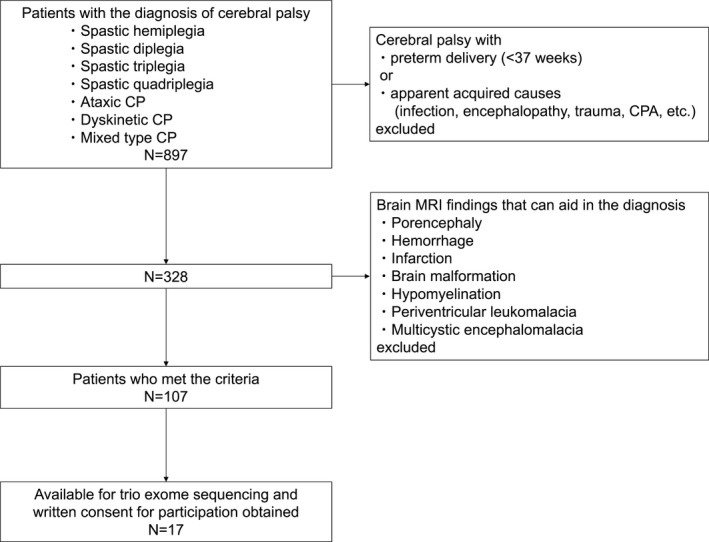
Patient flowchart. CP, cerebral palsy; CPA, cardiac pulmonary arrest.
Clinical features of case series
For each patient, demographic and clinical data were collected from the patient's chart, which included gender, age, age at diagnosis, details of pregnancy and birth, gestational weeks, birth weight, head circumference, neonatal events, type of CP, comorbidities, brain MRI findings, and functional classification scores for CP, such as the Gross Motor Function Classification Scale,10 the Manual Ability Classification System,11 and the Communication Function Classification System.12 The types of CP (spastic diplegia, ataxic type, diskinetic type, and mixed type) were classified based on previous reports,9, 13 and the brain MRI was performed as previously described.9
aCGH analysis
Microdeletions and microduplications were evaluated using SurePrint G3 CGH+SNP Microarray Kit 4 × 180K (ISCA) (Agilent Technologies, Santa Clara, CA, USA). CNVs were detected using CytoGenomics 2.5 (Agilent Technologies), and their pathogenicity was assessed using the International Standards for Cytogenomic Arrays (ISCA) database, the Database of Genomic Variants,14 and our in‐house database (n = 100).
WES analysis
We performed trio‐based WES for 17 families. Genomic DNA was captured using SureSelectXT Human All Exon V5 (50 Mb) or V6 (60 Mb) Kits (Agilent Technologies) and sequenced on a HiSeq2500 (Illumina) with 126‐base pair paired‐end reads. The reads were mapped to the hg19 human reference using Burrows‐Wheeler Aligner (BWA) 0.6.2‐r12615 and single‐nucleotide variants (SNVs), and insertions and/or deletions (indels) were called using the Genome Analysis Toolkit (GATK) v. 1.6–13.16 After quality filtering steps, variants were annotated using ANNOVAR.17 We prioritized the variants identified in enrolled CP patients as described below (Fig. 2). Nonsynonymous SNVs, splice‐site variants and indels were retained. We excluded variants present in the 1000 Genomes database, the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP6500), the Exome Aggregation Consortium (ExAC) database version 0.3 (all and East Asian populations), and the Human Genetic Variation Database (HGVD)18, 19 to discover rare de novo variants. For the autosomal recessive and X‐linked models, we excluded variants with an allele frequency equal to or greater than 1% in the 1000 Genomes database, the Exome Sequencing Project (ESP) 6500, the ExAC database version 0.3 (all and East Asian populations) and the HGVD. In the autosomal recessive and X‐linked models, autosomal variants with homozygotes found in the databases (or variants on the Chromosome X with hemizygotes in the databases) were filtered. Sorting intolerant from tolerant (SIFT),20 (polymorphism phenotypinb v2) PolyPhen2 HVAR,21 the Genomic Evolutionary Rate Profiling (GERP),22 and combined annotation‐dependent depletion (CADD)23 scores were used to predict nucleotide‐level conservation and the impact of amino acid substitutions. After extracting rare variants (listed in Table S1), only the rare variants that were present in Online Mendelian Inheritance in Man (OMIM) linked to neurodevelopmental diseases were prioritized as candidate variants. Each candidate variant was interpreted according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines.24 All the candidate variants were validated by Sanger sequencing, and the primers used for Sanger sequencing are listed in Table S2.
Figure 2.
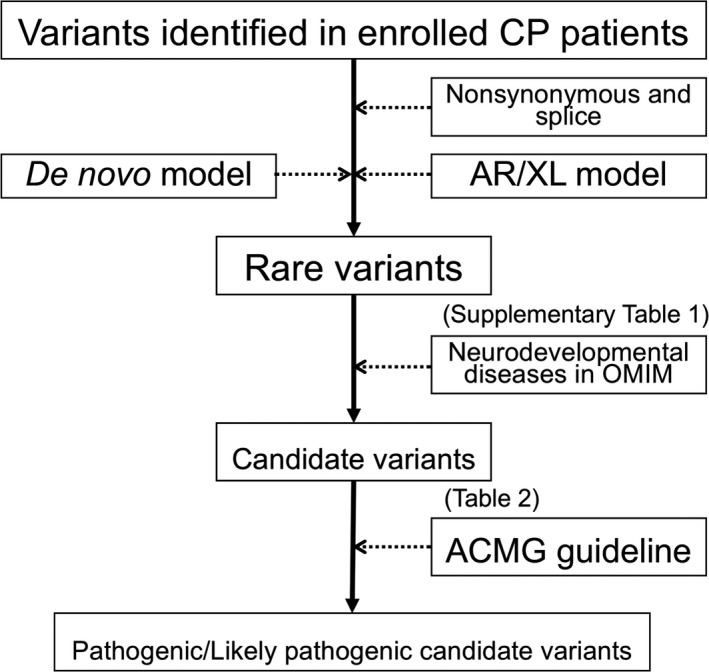
Schematic map of variants filtering. Rare variants and candidate variants in this figure are shown in Table S1 and Table 2, respectively. GQ, genotype quality; AAR, alternative allele ratio; AR, autosomal recessive; XL, X‐linked; ACMG, the American College of Medical Genetics and Genomics* “Databases” include the 1000 Genomes database (all and East Asian populations), the ESP6500, the ExAC database version 0.3 (all and East Asian populations) and the HGVD.
Extraction of RNA and reverse transcription‐polymerase chain reaction (RT‐PCR)
To demonstrate aberrant splicing of CTNNB1 (case 3) and AMPD2 (case 11) derived from each variant, whole blood was collected using a PAXgene RNA Blood RNA Kit (QIAGEN, Hilden, Germany) from each patient and two healthy controls. The RNA samples were extracted from fresh blood samples using a QIAamp RNA Blood Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer's instructions. The cDNA was synthesized using a PrimeScript RT (reverse transcription) reagent Kit with gDNA Eraser (TaKaRa, Shiga, Japan) according to the manufacturer's instructions. CTNNB1 and AMPD2 cDNA were amplified using TaKaRa ExTaq Hot Start Version (TaKaRa, Shiga, Japan), followed by agarose gel electrophoresis and analyzed by bidirectional Sanger sequencing. The primers are listed in Table S2.
AMP deaminase assay
White blood cells (WBCs) were extracted from the heparinized whole blood of two patients (case 3 and case 11) and two healthy volunteers to establish lymphoblastoid cell lines (LCLs) infected with Epstein–Barr virus (EBV) as previously described.25 AMPD2 enzyme activity in the WBCs was analyzed using these EBV‐infected LCLs (EBV‐LCLs). AMP deaminase activity in EBV‐LCL cells was assessed as previously described26 with minor modifications. Briefly, the cells were lysed in ice‐cold buffer A (250 mmol/L sucrose, 150 mmol/L KCl, 20 mmol/L imidazole HCl), and the protein concentration in the supernatant was measured with a Pierce BCA Protein Assay Kit (Thermo Scientific, Waltham, MA, USA). Extracts containing 2 μg of total protein were mixed with 50 mmol/L sodium citrate (pH 7), 50 mmol/L KCl, and 4 mmol/L AMP. For each protein extract a reaction mixture without AMP was prepared for background quantification. After 1 hour of incubation at 25°C, 50 μL of the previous reaction mixture and 100 μL of reagent A (10 mmol/L phenol, 20 mmol/L sodium nitroprusside) were transferred to the wells of a 96‐well plate containing the previous reaction samples and 100 μL of reagent B (12.5 mmol/L sodium hydroxide, 20 mmol/L dibasic sodium phosphate, 0.01% sodium hypochlorite) was then added. After 3 h of incubation at 25°C, the absorbance of the samples was measured at 625 nm using a SPECTRA Max 190 microplate reader (Molecular Devices, Sunnyvale, CA) against a reagent blank. An ammonia standard was prepared from ammonium sulfate to quantify the amount of ammonia formed by the cell extracts during incubation with AMP. To minimize variability in each experiment, the results were calculated from the ratio of absorbance in each sample to the average concentration in three control samples (case 3 and 2 healthy controls). Statistical analysis was performed using one‐way analysis of variance with a Dunnett's test for unequal variance.
Results
Characteristics of the participants
A total of 17 cases and their parents were enrolled and analyzed, as shown in Figure 1. Overall, 11 participants were male (64.7%) with a median age of 9.5 years (range, 3–30 years) and all participants were Japanese and nonconsanguineous. The median age at diagnosis of CP and follow‐up period was 40 months (range, 16–178 months) and 84 months (range, 26–358 months), respectively. This case series was classified into four CP types: spastic diplegia (11, 64.7%), dyskinetic type (3, 17.6%), ataxic type (2, 11.8%), and mixed type (1, 5.9%). All exhibited sporadic onset under 2 years of age, and their condition was nonprogressive, which is compatible with the definition of CP.1 None of the family histories of the cases were significant. Only case 2 underwent neonatal resuscitation with mask‐bag ventilation. Ten cases (58.8%) had normal brain MRI findings, and the others had nonspecific findings. The nonspecific brain MRI findings are shown (Fig. 3). All cases except case 4 (94.1%) exhibited intellectual disability at the last follow‐up. The clinical characteristics of these cases are described in Table 1 (see also Appendix S1).
Figure 3.
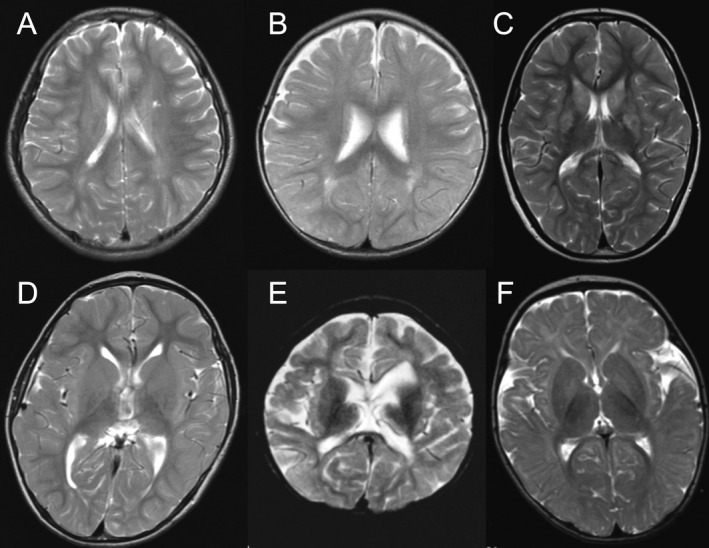
Nonspecific brain findings on T2‐weighted MRI. (A) Case 5; mild hyperintensity of the deep white matter around the body of the left lateral ventricle. (B) Case 7; slight hyperintensity of the left pars triangularis. (C) Case 11; hyperintensity in the striatum and a thin corpus callosum. (D) Case 13; a thin corpus callosum and mildly enlarged lateral ventricle. (E) Case 14; a thin corpus callosum and cerebral white matter atrophy. (F) Case 16; slight bilateral hyperintensities in the pallidum.
Table 1.
Clinical features of case series
| Case | Gender | Age, y | Age at CP diagnosis, m | Follow ‐up period, m | CP type | GW | BW at birth (SD), g | HC at birth (SD), cm | Perinatal complications | Brain MRI findings | Intellectual disability | Epilepsy | Autistic features | Other comorbidities | GMFCS | MACS | CFCS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 8 | 23 | 91 | D | 40 | 3066 (−0.5) | 34.0 (+0.5) | – | Normal | Yes | No | No | Optic nerve atrophy | 5 | 5 | 3 |
| 2 | M | 30 | 74 | 358 | S | 41 | 3310 (−0.3) | N/D |
Neonatal Resuscitation MAS/PDA |
Normal | Yes | Yes | No | Microcephaly | 2 | 4 | 4 |
| 3 | M | 7 | 58 | 87 | S | 40 | 3428 (+0.7) | 35.0 (+1.1) | – | Normal | Yes | No | No | Hyperekplexia | 4 | 3 | 3 |
| 4 | M | 9 | 51 | 63 | S | 38 | 3208 (−0.4) | 33.5 (−0.5) | – | Normal | No | No | No | – | 3 | 1 | 1 |
| 5 | M | 22 | 38 | 260 | S | 37 | 2890 (−0.4) | 32.5 (−0.5) | – | DWMH | Yes | No | No | – | 4 | 5 | 2 |
| 6 | M | 7 | 30 | 64 | S | 37 | 3182 (+1.0) | N/D | – | Normal | Yes | No | No | – | 2 | 1 | 1 |
| 7 | F | 7 | 16 | 81 | D | 38 | 2780 (−0.6) | 32.5 (−0.5) | – | DWMH | Yes | No | No | – | 5 | 5 | 4 |
| 8 | M | 15 | 164 | 26 | S | 40 | 3120 (−0.5) | 34.0 (−0.5) | – | Normal | Yes | No | Yes | Irritability | 1 | 2 | 2 |
| 9 | F | 13 | 116 | 46 | AT | 41 | 3548 (+1.3) | 32.5 (−0.5) | – | Normal | Yes | Yes | No | 4 | 4 | 4 | |
| 10 | M | 8 | 26 | 87 | S | 40 | 3086 (+0.0) | 33.0 (−0.3) | – | Normal | Yes | No | No | – | 4 | 3 | 2 |
| 11 | F | 5 | 22 | 51 | S | 38 | 2554 (−0.6) | 30.0 (−2.2) | – | BG hyperintensityWM atrophy | Yes | No | Yes | Irritability | 4 | 4 | 5 |
| 12 | F | 3 | 31 | 32 | AT | 39 | 3134 (+0.4) | 33.0 (−1.1) | − | Normal | Yes | No | No | Intention tremor | 2 | 4 | 4 |
| 13 | M | 10 | 40 | 84 | S | 40 | 3304 (+0.6) | 31.8 (−1.2) | − | WM atrophy | Yes | No | Yes | − | 1 | 2 | 2 |
| 14 | F | 22 | 27 | 263 | S | 40 | 2930 (−0.2) | 31.5 (−1.4) | − | Cortical atrophy | Yes | Yes | No | − | 4 | 4 | 5 |
| 15 | F | 6 | 56 | 82 | Mix (S, AT) | 37 | 1736 (−2.6) | 31.0 (−1.2) | SGA | Normal | Yes | No | No | VATER association | 2 | 3 | 3 |
| 16 | M | 11 | 25 | 130 | D | 37 | 3298 (+1.7) | 35.5 (+2.1) | – | BG hyperintensity | Yes | No | No | – | 4 | 3 | 3 |
| 17 | M | 19 | 178 | 76 | S | 37 | 3094 (+1.1) | 36.0 (+2.5) | – | Normal | Yes | Yes | Yes | Hemolysis | 2 | 4 | 5 |
CP, cerebral palsy; GW, gestational weeks; BW, birth weight; SD, standard deviation; HC, head circumference; MRI, magnetic resonance imaging; GMFCS, gross motor function classification; MACS, manual ability classification system; CFCS, communication function classification system; S, spastic diplegia; D, dyskinetic; AT, ataxic; N/D, no data; MAS, meconium aspiration syndrome; PDA, patent ductus arteriosus; SGA, small for gestational age; DWMH, Deep White matter hyperintensity; BG, basal ganglia; WM, white matter; ASD, autism spectrum disorder.
aCGH analysis
No CNVs were suspected to be pathogenically related to the phenotype of CP with aCGH. Only case 2 was identified as having X chromosome duplication and was confirmed to have XXY karyotypes in G‐banding, which is not closely related to CP.
WES analysis
The average mean depth was ×114. We obtained 17 clinically relevant candidate variants in 64.7% of the cases (11 of 17 cases) (Table 2). Of these cases, eight variants in seven cases within six genes: CTNNB1, CYP2U1, SPAST, GNAO1, CACNA1A, and AMPD2, were classified as pathogenic (41.2%) and the other two variants in two cases within two genes: STXBP1 and SCN2A were likely pathogenic (11.8%) according to the ACMG standards and guidelines (Table 2). Most of the pathogenic/likely pathogenic variants identified in the nine cases were de novo (77.8%, 7 of 9). Two of the cases had autosomal recessive inheritance including one case of compound heterozygosity. One identified gene, SPAST, was found in two cases (case 6 and 10). Among the causative variants identified in the nine patients, the variants in SPAST, GNAO1, CACNA1A, and STXBP1 have been previously reported,27, 28, 29, 30, 31, 32 whereas the variants in the other genes (CTNNB1, CYP2U1, AMPD2, and SCN2A) were novel. Of all the identified candidate genes, only SPAST, STXBP1, and SPTBN2 have been previously reported in CP patients.33, 34, 35 In five cases, we identified eight unknown significant candidate variants of five genes (UBA1, AMER1, FAT4, GPR98, and SPTBN2). None of the variants except p.(Arg524Trp) in SPTBN2 were associated with any evidence, such as functional analysis, previous reports, evolutionary conservation, or computational predictive programs, suggesting that they were a cause of disease. Thus, we did not pursue those candidate variants in this study.
Table 2.
Cases with pathogenic or potentially pathogenic variants identified by trio WES analysis and the result of aCGH analysis
| Case | CNVs | Gene | OMIM | OMIM disease | Previous report of CP | Inheritance | Position (hg19) | Effect on Protein | Protein change | Previously reported vaiant | SIFT score | Polyphen2 HVAR | CADD phred | GERP‐RS | ExAC | Pathogenicity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pathogenic/likely pathogenic candidate variants | ||||||||||||||||
| 3 | No | CTNNB1 | 114550 | Intellectual disability | No | De novo | Chr3:41275790 T > C | Splice site change | p.Glu562AlafsTer11 | No | NA | NA | 22.9 | 5.66 | 0 | Pathogenic |
| 5 | No | CYP2U1 | 610670 | Spastic paraplegia | No | AR‐homozygous | Chr4:108866283 del C | Frame shift | p.(Phe218fsTer42) | No | NA | NA | NA | NA | 0 | Pathogenic |
| 6 | No | SPAST | 604277 | Spastic paraplegia | Yes | De novo | Chr2:32361662 C > T | Missense | p.(Leu426Phe) | Yes | 0.002 | 0.999 | 32 | 5.62 | 0 | Pathogenic |
| 7 | No | GNAO1 | 139311 | EIEE | No | De novo | Chr16:56385308 G > A | Missense | p.(Glu246Lys) | Yes | 0 | 1 | 22.7 | 5.91 | 0 | Pathogenic |
| 9 | No | CACNA1A | 601011 | Spinocerebellar ataxia EIEE | No | De novo | Chr19:13476262 G > A | Missense | p.(Ser218Leu) | Yes | 0.002 | 0.998 | 33 | 5.55 | 0 | Pathogenic |
| 10 | No | SPAST | 604277 | Spastic paraplegia | Yes | De novo | Chr2:32366975 G > A | Missense | p.(Arg499His) | Yes | 0 | 1 | 34 | 4.98 | 0 | Pathogenic |
| 11 | No | AMPD2 | 102771 |
Spastic paraplegia Pontocerebellar hypoplasia |
No | AR‐Compound heterozygous | Chr1:110168415 G > A | Splice site change | p.Leu173Ter | No | NA | NA | 25.9 | 4.59 | 0 | Pathogenic |
| Chr1:110171419 C > T | Missense | p.(Pro456Leu) | No | 0 | 1 | 34 | 5.08 | 0 | Pathogenic | |||||||
| 12 | No | STXBP1 | 612164 | EIEE | Yes | De novo | Chr9:130428484 C > G | Missense | p.(Arg235Gly) | Yes | 0 | 1 | 34 | 5.72 | 0 | Likely Pathogenic |
| 17 | No | SCN2A | 607745 | EIEE | No | De novo | Chr2:166245610 T > C | Missense | p.(Leu1765Pro) | No | 0 | 1 | 25.6 | 5.72 | 0 | Likely Pathogenic |
| Unknown significance candidate variants | ||||||||||||||||
| 2 | dupX | UBA1 | 301830 | Spinal muscular atrophy | No | X‐linked | ChrX:47069086 C > T | Missense | p.(Thr668Ile) | No | 0.05 | 0.003 | 16.1 | 5.22 | 0 |
Unknown Significance |
| 6 | No | AMER1 | 3006471 | Osteopathia striata with cranial sclerosis | No | X‐linked | ChrX:63412347 G > A | Missense | p.(Pro274Ser) | No | 0.07 | 0.001 | 0.61 | 0.174 | 0.00001 | UnknownSignificance |
| 7 | No | FAT4 | 615546 | Van Maldergem syndrome | No | Compound heterozygous | Chr4:126238810 C > G | Missense | p.(Pro415Arg) | No | 0.18 | 1 | 18.22 | 4.55 | 0.0002 |
Unknown Significance |
| Chr4:126411082 A > G | Missense | p.(Met4370Val) | No | 1 | 0 | 0.027 | 0.09 | 0.0012 |
Unknown Significance |
|||||||
| 15 | No | GPR98 | 602851 | Usher syndrome type2C | No | Compound heterozygous | Chr5:89979443 A > G | Missense | p.(His1902Arg) | No | 0.32 | 0 | 9.526 | −0.074 | 0 |
Unknown Significance |
| Chr5:90086965 A > G | Missense | p.(Ile4773Met) | No | 0.095 | 0.859 | 21.5 | −10.8 | 0.0001 | UnknownSignificance | |||||||
| 17 | No | SPTBN2 | 604985 | Spinocerebellar ataxia | Yes | Compound heterozygous | Chr11:66455748 C > T | Missense | p.(Arg2089Lys) | No | 0.443 | 0.009 | 0.037 | −6.45 | 0.0002 |
Unknown Significance |
| Chr11:66475070 G > A | Missense | p.(Arg524Trp) | No | 0 | 1 | 34 | 4.47 | 0 |
Unknown Significance |
|||||||
CNV, copy number variation; OMIM, Online Mendelian Inheritance in Man; CP, cerebral palsy; dupX, duplicate X chromosome; AR, autosomal recessive; EIEE, early infantile epileptic encephalopathy; NA, not applicable.
cDNA analysis
We identified two splicing candidate variants (CTNNB1, Chr3: 41275790 T > C and AMPD2, Chr1: 110168415 G > A) in case 3 and case 11, respectively. We explored the effect of these variants on mRNA transcription using reverse transcription polymerase chain reaction (RT‐PCR). We obtained each wild‐type transcript and aberrant transcripts (Fig. 4). Sequence analysis of these cDNAs showed that translation of each aberrant transcript would have catenin beta‐1 and AMP deaminase 2 prematurely terminated. The variant in case 3 of CTNNB1 resulted in a frameshift followed by a premature termination at codon 572 of the mutant protein, which was preceded by an abnormal sequence of 10 amino acids (p.Glu562AlafsTer11) (Fig. 4A and B). Another variant in case 11 of AMPD2 also resulted in a premature termination at codon 216, which is the position immediately after the variant (Fig. 4C and D). These results support the pathogenicity of each variant.
Figure 4.
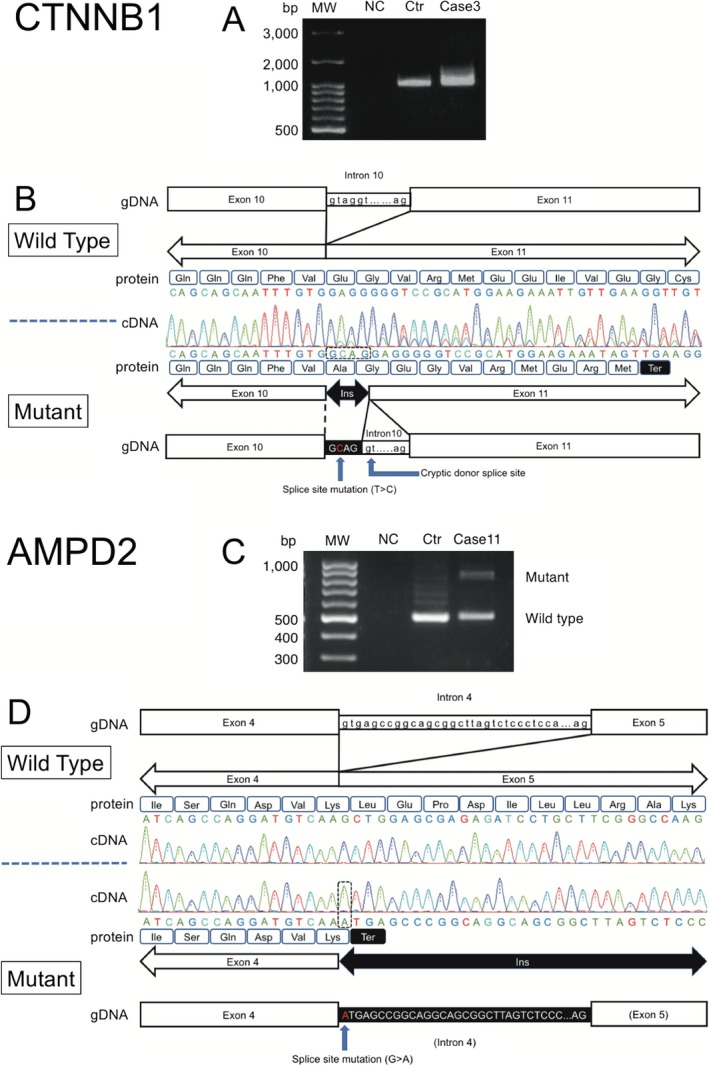
Analysis of the splice‐site variant of CTNNB1 in case 3 and AMPD2 in case 11. (A) RT‐PCR product of partial CTNNB1 run on 2% agarose gel electrophoresis. RT‐PCR products obtained from RNA of peripheral blood lymphocytes. (B) CTNNB1 sequence analysis of cDNA. The locations and corresponding cDNA sequencing results focusing on the exon 10–11 junction in the wild‐type (top) and CTNNB1 mutant cDNA (bottom) obtained from RT‐PCR. The corresponding translated amino acid sequences for the actual and hypothetical mutant products are also shown. The locations of the variants and stop codons are marked in the mutant diagram. (C) RT‐PCR product of partial AMPD2 from electrophoresis. (D) AMPD2 sequence analysis of cDNA. The locations and corresponding cDNA sequencing results focusing on the exon 4–5 junction in the wild‐type (top) and AMPD2 mutant cDNA (bottom) obtained from RT‐PCR. The corresponding translated amino acid sequences for the actual and hypothetical mutant products are also shown. The locations of the variant and stop codons are marked in the mutant diagram. bp, base pair; MW, molecular weight; NC, negative control; Ctr, control.
AMP deaminase assay
AMPD2 encodes one of three known AMP deaminase homologues, which convert AMP to IMP, and is necessary for guanine nucleotide biosynthesis and protein translation.26 To validate the pathogenicity of AMPD2 compound heterozygous variants in case 11, we performed an AMP deaminase activity assay. We found a significant reduction in AMP deaminase activity by approximately 60% in EBV‐LCL case 11 compared to two healthy controls and case 3 (Fig. 5A and B).
Figure 5.
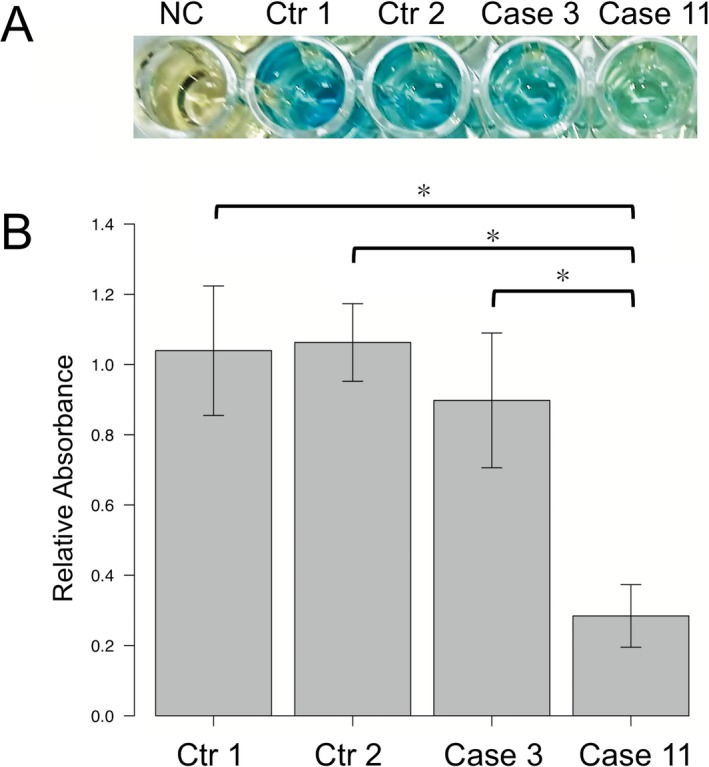
AMP deaminase assay. (A) Blue intensity represents relative activity. NC, Negative control; Ctr, Control. (B) Quantification of relative absorbance ± SD for six independent experiments measured at 625 nm. *P < 0.001. a Dunnett's test for unequal variance.
Discussion
A comprehensive genomic analysis by aCGH and trio WES was performed in 17 unexplained CP patients without a history of preterm delivery or specific MRI findings. We found that nine patients (52.9%) had pathogenic/likely pathogenic variants according to the ACMG guidelines and that these variants were de novo in seven patients and recessively inherited in two patients. The gene identified in eight CP masqueraders consisted of three hereditary spastic paraplegia (HSP) genes (CYP2U1, SPAST, and AMPD2), four genes associated with early infantile epileptic encephalopathy (EIEE) and neurodevelopmental diseases (SCN2A, CACNA1A, GNAO1, and STXBP1) and one other gene (CTNNB1). We confirmed the pathogenicity of two splice‐site variants in AMPD2 and CTNNB1 and one missense variant in AMPD2 using in vitro functional analysis. In a previous report, candidate variants were identified in 14 of 98 unselected CP patients (14.3%),2 indicating that our detection rate was significantly higher (P < 0.005, Fisher's exact test). Furthermore, if the variants of the previous study had been prioritized according to the ACMG guidelines and only genes known to be relevant to neurodevelopmental disorders were retained, the detection rate of candidate pathogenic/likely pathogenic genes for CP would have decreased to 4% (4 in 98 patients), contributing to an even larger difference compared to our study. Therefore, full‐term CP includes more masqueraders than the general population of CP patients, which suggests that efficient genetic testing can be developed for full‐term CP patients using trio WES analysis. Moreover, similar result have been reported by a recent study, according to which 11 of 78 patients with neurodevelopmental disorders diagnosed using WES exhibited CP‐like syndrome.35
No significant pathogenic CNVs were identified with aCGH in this study, unlike other previous reports,3, 36 in which pathogenic CNVs were identified in 9.6–31% of patients with aCGH. This wide range of proportions could be explained by variations in case selection or differences in the criteria used to evaluate pathogenicity. A high rate of pathologic CNVs (16%) was reported in children who were born small for their gestational age (SGA) with a persistent short stature.37 SGA patients enrolled in the previous CP cohorts may have contributed to the higher detection rate of CNVs compared to our CP cohorts, which contained only one SGA patient. Our findings indicate that the diagnostic yield of aCGH may be low in full‐term CP; however, further research with a larger sample is needed for establishing recommendations for the diagnostic testing strategy.
We identified candidate pathogenic variants of three genes known to cause HSP in four patients (CYP2U1 in case 5; SPAST in cases 6 and 10; and AMPD2 in case 11). HSP is a heterogeneous neurological disorder characterized by progressive spasticity and weakness of the lower limbs.38 To date, 55 HSP genes have been identified. HSP is clinically classified as either the pure form or complicated form, the latter of which is associated with additional neurological features such as ataxia, intellectual disability, neuropathy, and epilepsy.38 Present cases (case 5, 6, 10, and 11) all showed nonprogressive spastic diplegia during an extended follow‐up period (range, 51–260 months), which is consistent with criteria for CP but not HSP.1, 38 This suggests that these HSP genes can cause not only HSP, but also the spastic type of CP masqueraders. A homozygous nonsense variant was identified in case 5 in CYP2U1, which was reported to cause SPG56.38 Symptoms of case 5 included intellectual disability and spastic diplegia, which resembled the phenotype of SPG56.38 In cases 6 and 10, we found two known de novo variants in SPAST,27, 28 which is the most commonly mutated gene (SPG4) in HSP.37 Cases 6 and 10 developed a mild form of spastic diplegia, which was consistent with the severity of other patients who had variants in SPAST. Case 11 had compound heterozygous variants and low deaminase activity in her EBV‐LCL cells. A frameshift AMPD2 variant was recently identified in a patient with complicated HSP,39 whereas AMPD2 variants were mainly reported in pontocerebellar hypoplasia.26 Our results, together with a previous report, extend the phenotypic spectrum of AMPD2 variants (see Appendix S1).
Four candidate pathogenic/likely pathogenic variants of genes for EIEE and neurodevelopmental diseases were identified in four patients, including GNAO1 in case 7, CACNA1A in case 9, STXBP1 in case 12, and SCN2A in case 17. GNAO1 variants cause both EIEE and neurodevelopmental disorders,29 which manifested in case 7 as an intellectual disability, although no seizures occurred. We identified p.(Ser218Leu) variants in case 9, diagnosed as ataxic CP. The p.Phe1502del variant of CACNA1A has been reported to cause congenital ataxia, which has similar characteristics to case 9.40 CACNA1A variants cause various neuropsychiatric diseases. The p.(Ser218Leu) variant has also been associated with familial hemiplegic migraines and cerebellar ataxia,30 as well as EIEE.31 If the cerebellar ataxia develops congenitally and appears stable, it may be due to ataxic CP masqueraders, as in case 9. STXBP1 has been primarily reported to be a causative gene for EIEE. Additionally, various phenotypic features were recently described, including autistic features, spasticity, hypotonia, ataxia, and dyskinesia. Because approximately 5% of patients with variants of STXBP1 do not have seizures and a patient with the same variant as that in case 12 has been reported to show similar phenotypic symptoms, including ataxia, wide‐based gait, intention tremor, and intellectual disability without epilepsy,32 it is likely that candidate variant in STXBP1 caused ataxic CP with such phenotype in case 12. Some patients with SCN2A variants show intellectual disability, autistic behaviors, involuntary movement, and lower limb spasticity in addition to epilepsy,41 which explains the symptoms of case 17. Furthermore, case 17 was also a compound heterozygote of SPTBN2 variants. SPTBN2 has been identified in both autosomal dominant and autosomal recessive spinocerebellar ataxia,42 together with CP.34 Although both SPTBN2 variants in case 17 were classified as having uncertain significance and the patient's clinical presentation was consistent with SCN2A‐related disorder, we cannot eliminate the possibility that those variants in SPTNBN2 could contribute to the patient's phenotype. In addition to genes for HSP and EIEE/neurodevelopmental disorders, we identified a variant of CTNNB1 in case 3. Although the variants of CTNNB1 have been reported to cause autosomal dominant mental retardation type 19, most of the patients with this variant show signs of other clinical features, including progressive spastic diplegia, hypotonia, microcephaly, craniofacial abnormalities, and hyperekplexia.43 Case 3 had intellectual disability and spastic diplegia with hyperekplexia, which corresponded with the phenotype for CTNNB1‐related diseases.
Although the phenotypes are quite similar to CP, six of the eight genes identified as having candidate pathogenic/likely pathogenic variants (CTNNB1, CYP2U1, GNAO1, CACNA1A, AMPD2, and SCN2A) have not been reported to be the causative genes of CP potentially because they do not fit the definition of CP, such as the presence of regression or an older age onset. Therefore, these gene variants may be found in a wide clinical spectrum of neurodevelopmental diseases, such as HSP, complicated forms of intellectual disability, EIEE, and CP masqueraders.
The identification of the causative genes may affect patient management. For example, deep brain stimulation has been reported to have successfully improved movement disorder in two siblings with the GNAO1 variant.44 In addition to the case report, the patient of case 7 can be treated using deep brain stimulation. Moreover, spinocerebellar ataxia type 6, caused by the same causative gene (CACNA1A) as that in case 9, could be potentially treated using the miRNA‐mediated therapy in the future, as was reported in the mouse model.45
There are certain limitations of this study. First, the sample size was small in this highly selected case series. Although the yield in this study was significantly higher than that reported in unselected CP patients,2 our interpretation is limited because no comparison was made with the general CP population. As recent studies have not reported information on gestational ages or brain MRI of patients,2 we were not able to find CP patients who met our criteria to calculate a diagnostic yield for comparison with our results. Second, there are some gaps between the CP phenotype in this case series and that identified in their variants or genes. Functional validations were not performed in all known or pathogenic/likely pathogenic variants identified in this study. Moreover, functional experiments in the animal model and the identification of other patients with the same phenotype and gene are necessary to confirm our results. A lack of such validation leaves the possibility that those gene variants are not a monogenic cause, rather a contributor to the multifactorial disease, although these variants were prioritized following the ACMG guidelines. Third, the small sample size in our cohort also limited statistical comparisons between the genetic and clinical findings, including CP subtypes and intellectual disability. In terms of CP subtypes, four of the six patients with spasticity contained the variants in genes (CYP2U1, SPAST, and AMPD2) known to cause spastic paraplegia, while the other two contained the variants in genes (CTNNB1 and SCN2A) known to cause other neurodevelopmental disorders. Both genes have not been reported to cause spastic CP; however, the spasticity of patients has been reported previously.32, 41 The patients with other CP subtypes, including ataxic type and dyskinetic type, showed no tendency toward spastic diplegia. With respect to intellectual disability, the morbidity was higher (16/17, 94.1%) than previously reported (77.5%),46 suggesting a representative feature of our cohort, although it is challenging to accurately measure the intellectual disability,47 and this margin is insignificant owing to the small sample size. Larger CP cohort analyses including full‐term and preterm infants with diverse datasets (e.g., the Apgar score) classified using the brain MRI findings (normal to extensive abnormality, such as PVL) are needed to clarify the genetic epidemiology of CP and CP masqueraders.
In summary, our study showed the possibility that a subset of CP patients categorized by two simple criteria include CP masqueraders despite several limitations. Further genetic investigations of larger cohorts of CP patients are required to help support our results or identify other CP subsets that may likely include masqueraders. This could eventually aid the further characterization of the molecular mechanisms of CP and clinically similar disorders.
Author Contributions
Y.T., A.K., K.H., Y. N.‐U., Y.K., A.O., and S.K. conceived and designed the study. Y.T., A.K., K.H., Y.N.‐U., T.I., S.S.‐Y., T.M., M.A., S.S.‐M., Y.O., W.E., N.T., and Y.K. recruited patients and collected clinical information and samples. Y.T. and A.K. performed in vitro experiments and array comparative genome hybridization. Y.T., A.K., T.N., R.F., M.S., K.N., and Y.A. performed whole‐exome sequencing and analyzed the data. Y.T., A.K., K.H., and S.K. drafted the manuscript and figures. All authors critically evaluated the manuscript and approved the final version of the manuscript.
Conflict of Interest
Nothing to report.
Supporting information
Table S1. Rare variants.
Appendix S1. Case descriptions.
Table S2. The primers used for Sanger sequencing.
Acknowledgments
The authors thank the patients, families, and doctors who participated in this study. We thank Yoko Chiba, Kumi Ito, Miyuki Tsuda, Mami Kikuchi, Makiko Nakagawa, Yoko Tateda, and Kiyotaka Kuroda for technical assistance. We also acknowledge the support from the Biomedical Research Core of Tohoku University Graduate School of Medicine and Biomedical Research Unit of Tohoku University Hospital. This work was supported by JSPS KAKENHI Grant Number JP16K09983 (to AK and KH) and a research grant for Initiative on Rare and Undiagnosed Diseases in Pediatrics (IRUD‐P) from Japan Agency for Medical Research and Development.
Funding Statement
This work was funded by JSPS KAKENHI grant JP16K09983; Japan Agency for Medical Research and Development grant .
Contributor Information
Atsuo Kikuchi, Email: akikuchi-thk@umin.ac.jp.
Kazuhiro Haginoya, Email: khaginoya@kha.biglobe.ne.jp.
References
- 1. Rosenbaum P, Paneth N, Leviton A, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol Suppl 2007;109:8–14. [PubMed] [Google Scholar]
- 2. McMichael G, Bainbridge MN, Haan E, et al. Whole‐exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol Psych 2015;20:176–182. [DOI] [PubMed] [Google Scholar]
- 3. Oskoui M, Gazzellone MJ, Thiruvahindrapuram B, et al. Clinically relevant copy number variations detected in cerebral palsy. Nat Commun 2015;6:7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. MacLennan AH, Thompson SC, Gecz J. Cerebral palsy: causes, pathways, and the role of genetic variants. Am J Obstet Gynecol 2015;213:779–788. [DOI] [PubMed] [Google Scholar]
- 5. Lee RW, Poretti A, Cohen JS, et al. A diagnostic approach for cerebral palsy in the genomic era. Neuromolecular Med 2014;16:821–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oskoui M, Coutinho F, Dykeman J, et al. An update on the prevalence of cerebral palsy: a systematic review and meta‐analysis. Dev Med Child Neurol 2013;55:509–519. [DOI] [PubMed] [Google Scholar]
- 7. Gould DB, Phalan FC, Breedveld GJ, et al. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science 2005;308:1167–1171. [DOI] [PubMed] [Google Scholar]
- 8. Cardo E, Campistol J, Caritg J, et al. Fatal haemorrhagic infarct in an infant with homocystinuria. Dev Med Child Neurol 1999;41:132–135. [DOI] [PubMed] [Google Scholar]
- 9. Numata Y, Onuma A, Kobayashi Y, et al. Brain magnetic resonance imaging and motor and intellectual functioning in 86 patients born at term with spastic diplegia. Dev Med Child Neurol 2013;55:167–172. [DOI] [PubMed] [Google Scholar]
- 10. Palisano RJ, Rosenbaum P, Bartlett D, Livingston MH. Content validity of the expanded and revised gross motor function classification system. Dev Med Child Neurol 2008;50:744–750. [DOI] [PubMed] [Google Scholar]
- 11. Eliasson AC, Krumlinde‐Sundholm L, Rosblad B, et al. The Manual Ability Classification System (MACS) for children with cerebral palsy: scale development and evidence of validity and reliability. Dev Med Child Neurol 2006;48:549–554. [DOI] [PubMed] [Google Scholar]
- 12. Hidecker MJ, Paneth N, Rosenbaum PL, et al. Developing and validating the communication function classification system for individuals with cerebral palsy. Dev Med Child Neurol 2011;53:704–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Christine C, Dolk H, Platt MJ, et al. Recommendations from the SCPE collaborative group for defining and classifying cerebral palsy. Dev Med Child Neurol Suppl 2007;109:35–38. [DOI] [PubMed] [Google Scholar]
- 14. MacDonald JR, Ziman R, Yuen RK, et al. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 2014;42:D986–D992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Narahara M, Higasa K, Nakamura S, et al. Large‐scale East‐Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS One 2014;9:e100924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Higasa K, Miyake N, Yoshimura J, et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet 2016;61:547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cooper GM, Goode DL, Ng SB, et al. Single‐nucleotide evolutionary constraint scores highlight disease‐causing mutations. Nat Methods 2010;7:250–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hui‐Yuen J, McAllister S, Koganti S, et al. Establishment of Epstein‐Barr virus growth‐transformed lymphoblastoid cell lines. J Vis Exp 2011;57:e3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Akizu N, Cantagrel V, Schroth J, et al. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell 2013;154:505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Braschinsky M, Tamm R, Beetz C, et al. Unique spectrum of SPAST variants in Estonian HSP patients: presence of benign missense changes but lack of exonic rearrangements. BMC Neurol 2010;10:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crippa F, Panzeri C, Martinuzzi A, et al. Eight novel mutations in SPG4 in a large sample of patients with hereditary spastic paraplegia. Arch Neurol 2006;63:750–755. [DOI] [PubMed] [Google Scholar]
- 29. Saitsu H, Fukai R, Ben‐Zeev B, et al. Phenotypic spectrum of GNAO1 variants: epileptic encephalopathy to involuntary movements with severe developmental delay. Eur J Hum Genet 2016;24:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kors EE, Terwindt GM, Vermeulen FL, et al. Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann Neurol 2001;49:753–760. [DOI] [PubMed] [Google Scholar]
- 31. Epi KC; Epilepsy Phenome/Genome P , Genome P, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stamberger H, Nikanorova M, Willemsen MH, et al. STXBP1 encephalopathy: a neurodevelopmental disorder including epilepsy. Neurology 2016;86:954–962. [DOI] [PubMed] [Google Scholar]
- 33. Blair MA, Riddle ME, Wells JF, et al. Infantile onset of hereditary spastic paraplegia poorly predicts the genotype. Pediatr Neurol 2007;36:382–386. [DOI] [PubMed] [Google Scholar]
- 34. Parolin Schnekenberg R, Perkins EM, Miller JW, et al. De novo point mutations in patients diagnosed with ataxic cerebral palsy. Brain 2015;138:1817–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Srivastava S, Cohen JS, Vernon H, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol 2014;76:473–483. [DOI] [PubMed] [Google Scholar]
- 36. Segel R, Ben‐Pazi H, Zeligson S, et al. Copy number variations in cryptogenic cerebral palsy. Neurology 2015;84:1660–1668. [DOI] [PubMed] [Google Scholar]
- 37. Canton AP, Costa SS, Rodrigues TC, et al. Genome‐wide screening of copy number variants in children born small for gestational age reveals several candidate genes involved in growth pathways. Eur J Endocrinol 2014;171:253–262. [DOI] [PubMed] [Google Scholar]
- 38. Lo Giudice T, Lombardi F, Santorelli FM, et al. Hereditary spastic paraplegia: clinical‐genetic characteristics and evolving molecular mechanisms. Exp Neurol 2014;261:518–539. [DOI] [PubMed] [Google Scholar]
- 39. Novarino G, Fenstermaker AG, Zaki MS, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014;343:506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Garcia Segarra N, Gautschi I, Mittaz‐Crettol L, et al. Congenital ataxia and hemiplegic migraine with cerebral edema associated with a novel gain of function mutation in the calcium channel CACNA1A. J Neurol Sci 2014;342:69–78. [DOI] [PubMed] [Google Scholar]
- 41. Howell KB, McMahon JM, Carvill GL, et al. SCN2A encephalopathy: a major cause of epilepsy of infancy with migrating focal seizures. Neurology 2015;85:958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ikeda Y, Dick KA, Weatherspoon MR, et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet 2006;38:184–190. [DOI] [PubMed] [Google Scholar]
- 43. Kuechler A, Willemsen MH, Albrecht B, et al. De novo mutations in beta‐catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum. Hum Genet 2015;134:97–109. [DOI] [PubMed] [Google Scholar]
- 44. Kulkarni N, Tang S, Bhardwaj R, et al. Progressive movement disorder in brothers carrying a GNAO1 mutation responsive to deep brain stimulation. J Child Neurol 2016;31:211–214. [DOI] [PubMed] [Google Scholar]
- 45. Miyazaki Y, Du X, Muramatsu S, Gomez CM. An miRNA‐mediated therapy for SCA6 blocks IRES‐driven translation of the CACNA1A second cistron. Sci Transl Med 2016;8:347ra94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gabis LV, Tsubary NM, Leon O, et al. Assessment of abilities and comorbidities in children with cerebral palsy. J Child Neurol 2015;30:1640–1645. [DOI] [PubMed] [Google Scholar]
- 47. Sherwell S, Reid SM, Reddihough DS, et al. Measuring intellectual ability in children with cerebral palsy: can we do better? Res Dev Disabil 2014;35:2558–2567. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Rare variants.
Appendix S1. Case descriptions.
Table S2. The primers used for Sanger sequencing.