

Summary
Mutations in the human LMNA gene cause a collection of diseases known as laminopathies. These include myocardial diseases that exhibit age‐dependent penetrance of dysrhythmias and heart failure. The LMNA gene encodes A‐type lamins, intermediate filaments that support nuclear structure and organize the genome. Mechanisms by which mutant lamins cause age‐dependent heart defects are not well understood. To address this issue, we modeled human disease‐causing mutations in the Drosophila melanogaster Lamin C gene and expressed mutant Lamin C exclusively in the heart. This resulted in progressive cardiac dysfunction, loss of adipose tissue homeostasis, and a shortened adult lifespan. Within cardiac cells, mutant Lamin C aggregated in the cytoplasm, the CncC(Nrf2)/Keap1 redox sensing pathway was activated, mitochondria exhibited abnormal morphology, and the autophagy cargo receptor Ref2(P)/p62 was upregulated. Genetic analyses demonstrated that simultaneous over‐expression of the autophagy kinase Atg1 gene and an RNAi against CncC eliminated the cytoplasmic protein aggregates, restored cardiac function, and lengthened lifespan. These data suggest that simultaneously increasing rates of autophagy and blocking the Nrf2/Keap1 pathway are a potential therapeutic strategy for cardiac laminopathies.
Keywords: autophagy, cardiac aging, Drosophila aging model, lamins, Nrf2/Keap1 pathway, protein aggregation
1. INTRODUCTION
Many characteristics of normal aging appear to be accelerated in individuals with dominant mutations in the LMNA gene encoding A‐type lamins (Ahmed, Ikram, Bibi & Mir, 2017; Apte, Stick & Radmacher, 2017; Cenni et al., 2017; Ikeda et al., 2016; Scaffidi & Misteli, 2006). These include cardiac diseases with a broad range of arrhythmic disturbances, left ventricle dysfunction, and heart failure that show increasing penetrance with age (Captur et al., 2017; Liang, Grogan & Ackerman, 2016). To gain insights into how to delay the onset and/or prevent these cardiac defects during aging, a greater understanding of the molecular basis of the pathology is needed.
The human LMNA gene encodes the developmentally regulated and nearly ubiquitously expressed A‐type lamins, Lamin A and C, which are produced by alternate splicing (Burke & Stewart, 2013). Lamins are intermediate filaments that line the inner nuclear membrane, providing structural support for the nucleus and organizing the genomic DNA (Ahmed et al., 2017; Azibani, Muchir, Vignier, Bonne & Bertrand, 2014; Guenantin et al., 2014; Wang, Zabell, Koh & Tang, 2017; Worman, 2012). Over 400 mutations have been identified in the LMNA gene (Dittmer & Misteli, 2011; Gruenbaum & Foisner, 2015). In addition to cardiac diseases, these mutations cause skeletal muscular dystrophy with age‐dependent penetrance and early‐onset aging syndromes; collectively, these diseases are referred to as laminopathies (Politano et al., 2013).
Genetically tractable model organisms have been used to understand the functions of lamins. Mice lacking A‐type lamins have severe cardiac and skeletal muscle defects, in addition to a shortened lifespan (Ramos et al., 2012; Zhang, Kieckhaefer & Cao, 2013). Furthermore, studies on the mouse models demonstrated that the cardiac defects and shortened lifespan can be partially reversed by treatment with rapamycin and temsirolimus (a derivative of rapamycin) (Choi et al., 2012; Ramos et al., 2012). Studies in Drosophila demonstrated that mutant lamins, modeled after those that cause human disease, lead to cytoplasmic aggregation of nuclear envelope proteins and loss of redox homeostasis in larval body wall muscles (Dialynas et al., 2012, 2015). Consistent with these findings, human muscle biopsy tissues showed both cytoplasmic aggregation of nuclear envelope (NE) proteins and activation of the Nrf2/Keap‐1 signaling pathway. Thus, these models have phenotypes similar to the human disease condition.
Here, we developed a Drosophila model of cardiac laminopathies. Mutations in the human LMNA that cause dilated cardiomyopathy with conduction defects are often point mutations resulting in amino acid substitutions in residues conserved among species. We modeled these mutations in the Drosophila Lamin C gene (hereafter referred as LamC) and assayed for effects on the fruit fly heart. Mechanisms of cardiac development and function are shared between Drosophila and humans (Diop & Bodmer, 2015; Melkani et al., 2013; Zhu et al., 2017). Furthermore, Drosophila has successfully been used to identify the genetic basis of cardiac deterioration that arises due to aging and metabolic dysregulation (Diop & Bodmer, 2015; Gill, Le, Melkani & Panda, 2015; Melkani et al., 2013).
The cardiolaminopathy Drosophila models exhibited age‐dependent decline in cardiac function that resulted in a shortened adult lifespan. Defects were observed in the nucleus, cytoplasm, and mitochondria of cardiomyocytes. In addition, adults showed an age‐dependent increase in triglycerides. Many of these abnormal features are common in human cardiac laminopathies (Captur et al., 2017). The Drosophila models allowed for genetic tests of suppression and identified new potential therapeutic targets for individuals with cardiolaminopathy and other types of laminopathies.
2. RESULTS
2.1. Mutant LamC caused age‐dependent cardiac defects and a shortened adult lifespan
To determine the mechanistic basis of cardiolaminopathies, we expressed wild‐type and mutant Drosophila LamC transgenes (referred to as R205W and G489V hereafter) in the heart (Figure 1a). These amino acid substitutions are analogous to human Lamin A/C R190W and G449V, respectively. Mutations in LMNA that give rise to Lamin A/C R190W are associated with progressive cardiac defect (including conduction defects) and reduced cardiac performance (Arbustini et al., 2002; Heller et al., 2017; Hermida‐Prieto et al., 2004). Mutations in LMNA that give rise to Lamin A/C G449V cause congenital muscular dystrophy, which is characterized by skeletal muscle defects in childhood and age‐dependent dilated cardiomyopathy (Dialynas et al., 2015). Cardiac‐specific expression of Drosophila LamC was obtained using the Gal4/UAS system with a Hand‐Gal4 driver (Han & Olson, 2005; Melkani et al., 2013). Expression of mutant LamC, but not wild‐type, caused adult cardiac defects in flies possessing an otherwise wild‐type genetic background (Figure 1b, c). Thus, mutant lamins caused dominant defects in these models, similar to the human disease condition (Dittmer & Misteli, 2011; Gruenbaum & Foisner, 2015).
Figure 1.
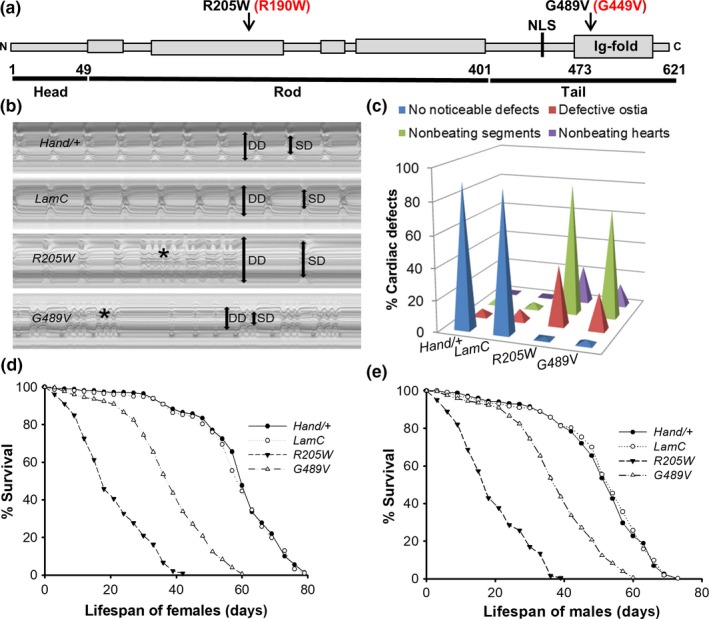
Drosophila Lamin C domain structure and effects of mutant LamC on cardiac function and lifespan. (a) Lamins contain a conserved structure with an N‐terminal head domain, coiled‐coil rod domain, and tail domain possessing an Ig‐fold. Amino acid substitutions in the rod and Ig‐fold domains used in this study are indicated. Numbering for Drosophila Lamin C is in black; the corresponding human disease‐causing amino acid substitution is in red. (b) M‐mode recordings (5‐s time periods) of dissected hearts from 3‐week‐old female Hand‐Gal4/+, wild‐type and mutant Lamin C (LamC) (R205W and G489V). M‐mode analysis revealed that R205W‐expressing hearts showed significant dilation and arrhythmias, whereas G489V‐expressing hearts showed restricted morphology and arrhythmias. Double‐headed arrows in the M‐mode traces indicate diastolic diameter (DD) and systolic diameter (SD) between the walls of the heart. (c) Summary of the qualitative cardiac defects from 3‐week‐old male and female adult controls (Hand/+ and wild‐type LamC) and mutants (R205W and G489V) showing the percent of flies exhibiting defective ostia, one or more noncontractile regions (conductive defect), and nonbeating hearts. (d and e) Cardiac‐specific expression of mutant LamC (R205W and G489V) resulted in a reduction in lifespan compared to controls expressing Hand/+ and wild‐type LamC controls (p < .001). Graphs indicating the percent survival for female adults (n = 150 for each group) versus age days posteclosion
Defective cardiac function was measured and quantitated according to established protocols (Gill et al., 2015; Melkani et al., 2013). M‐mode analysis was performed in which a specified region of one‐pixel width along the image of the heart was selected from each movie frame and aligned horizontally, generating a montage that portrays the movement of the heart walls over time (Figure 1b). Hearts from 3‐week‐old female flies expressing R205W and G489V showed significant dilation and restriction, respectively. Consistent with our finding, mutations in human LMNA result in dominant dilated, hypertrophic, and idiopathic cardiomyopathy (Arbustini et al., 2002; Heller et al., 2017; Marian, 2017). Furthermore, expression of mutant LamC resulted in dysrhythmic beating patterns when compared to hearts from age‐matched controls expressing wild‐type LamC and the Hand‐Gal4 driver alone (Figure 1b).
Expression of the mutant LamC resulted in defective ostia, noncontractile region(s) of the heart, and loss of heartbeat, an indicator of conduction defects (Figure 1c), which are observed in human LMNA patients (Arbustini et al., 2002; Brayson & Shanahan, 2017; Malhotra & Mason, 2009; Wolf et al., 2008). In addition to severe cardiac defects, heart‐specific expression of R205W and G489V had a drastic impact on the lifespan for both female and male adults compared to controls (Figure 1d, e). The half‐life of females expressing R205W and G489V was 21 and 39 days, respectively, compared to 58 days for females expressing wild‐type LamC (Figure 1d). Similarly, the half‐life of males expressing R205 and G489V was 18 and 38 days, respectively, compared to 56 days for males expressing wild‐type LamC (Figure 1e). Western analysis showed that similar levels of wild‐type and mutant LamC were expressed by the Hand‐Gal4 driver (Fig. S1a). Transgenic flies expressing wild‐type LamC had less than twofold higher level of LamC compared with nontransgenic controls. Importantly, the slightly elevated level of wild‐type LamC did not produce cardiac defects (Figure 1b, c). In contrast, the mutant versions of LamC expressed at levels similar to the exogenous wild‐type LamC produced obvious cardiac defects (Figure 1b, c). Thus, the cardiac defects were caused by mutant LamC and not increased total amounts of LamC. Furthermore, cardiac‐specific expression of mutant LamC did not result in noncardiac muscle defects as measured by adult flight (Fig. S1b). Therefore, the phenotypes were restricted to the muscle tissue in which the mutant LamC was expressed.
2.2. Mutant LamC caused progressive cardiac physiological dysfunction
Quantitative analysis of cardiac physiological parameters revealed progressive and severe cardiac defects upon expression of R205W and G489V (Figure 2). For example, the heart period (Figure 2a), arrhythmicity index (Figure 2b), diastolic interval (Figure 2c), and systolic interval (Figure 2d) of one‐ and 3‐week‐old female adults expressing R205W and G489V were significantly elevated compared to age‐matched controls. Furthermore, these parameters showed more severe deterioration in 3‐week‐old flies compared to one‐week‐old flies suggesting that the phenotypes were progressive (Figure 2a–d). Of note, only the systolic interval of hearts expressing the Hand‐Gal4 driver alone and the arrhythmicity index of hearts expressing wild‐type LamC and the Hand‐Gal4 driver alone showed subtle alterations in 3‐week‐old flies compared to their 1‐week‐old counterparts (Figure 2a, d). Due to the cardiac dilation caused by expression of R205W and the cardiac restriction caused by expression of G489V, both diastolic (Figure 2e) and systolic (Figure 2f) heart diameters were significantly enlarged and reduced at one and three weeks of age compared to age‐matched controls, respectively. The cardiac dilation and restriction reduced heart contractility (Figure 2e, f), which was further reflected by decreased fractional shortening (Figure 2g). Similar defects were observed in adult males (data not shown). Thus, these data demonstrate that heart‐specific expression of mutant LamC caused severe and progressive contractility‐related cardiac phenotypes.
Figure 2.
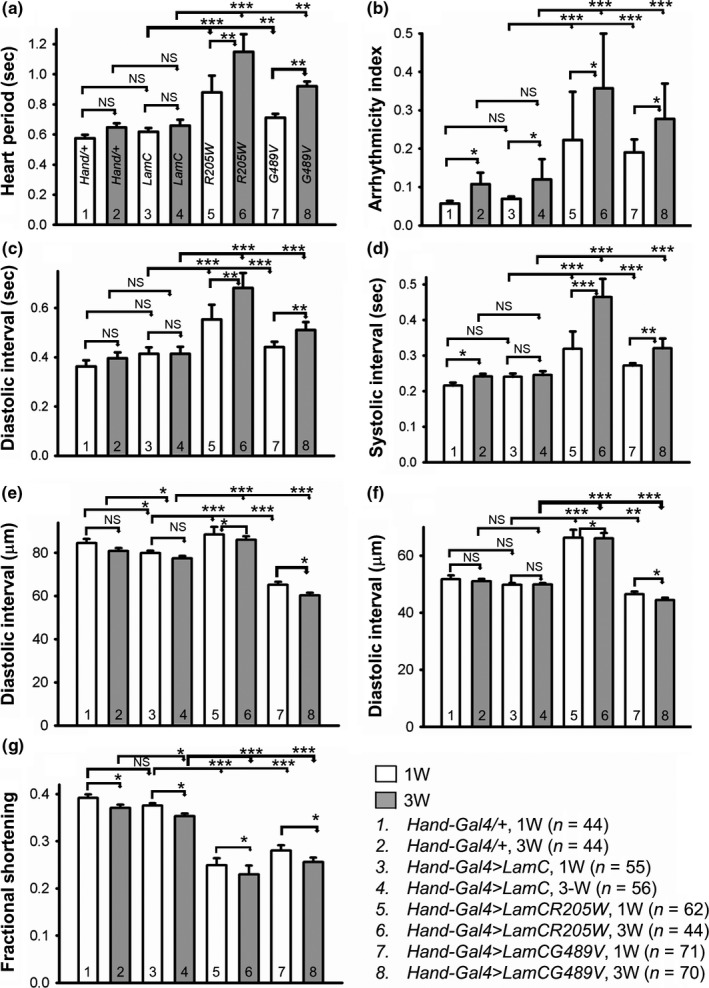
Mutant LamC caused progressive cardiac physiological dysfunction. One (1W) and three‐week (3W)‐old female adults (n = 44–71 per genotype) expressing Hand‐Gal4/+, wild‐type LamC, R205W, and G489V were assayed for the heart period (a), arrhythmia index (b), diastolic and systolic intervals (c and d), diastolic and systolic parameters (e and f), and fractional shortening (g). Minimal differences were observed between the Hand/+ and wild‐type LamC for the cardiac parameters measured. In contrast, significant differences were observed for all parameters (a–g) between the mutant LamC (R205W and G489V) and age‐matched controls. Data are shown as average ± s.e.m.s; statistical significance was determined using one‐way ANOVA and Tukey's post hoc test. For all parameters, statistical significance is denoted as follows: *p < .05; **p < .01; ***p < .001; NS = not significant
2.3. Mutant LamC caused nuclear blebbing, cytoplasmic aggregation of LamC and Otefin (Emerin), and disruption of the myofibrils
To examine the effects of mutant LamC expression at the cytological level, dissected hearts from three‐week‐old flies were fixed under relaxed conditions, incubated with antibodies specific for nuclear envelope proteins and/or fluorescent dyes to cytoskeletal proteins, and analyzed by confocal microscopy. Expression of wild‐type LamC showed normal nuclear morphology; LamC localized to the nuclear periphery and actin‐containing myofibrils were organized into a network (Figures 3a, b and S2a, b). In contrast, expression of the mutant LamC caused nuclear envelope blebbing, cytoplasmic aggregation of LamC, and disorganization of actin‐containing myofibrils (Figure 3a, b). The myofibrillar disorganization and LamC aggregation increased with age (Fig. S2a–c). Quantitation of LamC aggregates showed that the relative area occupied by the aggregates per total area surveyed was greater in hearts expressing the mutant LamC versus those expressing wild‐type LamC (Fig. S2c). Furthermore, the relative area occupied by aggregates increased with age (Fig. S2c). Immunostaining of hearts expressing wild‐type LamC with antibodies to Otefin (Ote) [a Drosophila orthologue of the human inner nuclear membrane LEM domain protein emerin (Barton, Lovander, Pinto & Geyer, 2016)] showed localization to the nuclear envelope as anticipated (Figure 3c). In contrast, hearts expressing mutant LamC showed greater amounts of cytoplasmic aggregation of Ote (Figures 3c and S2c). Thus, mutant LamC caused myofibril disorganization, nuclear blebbing, and cytoplasmic aggregation of nuclear envelope proteins.
Figure 3.
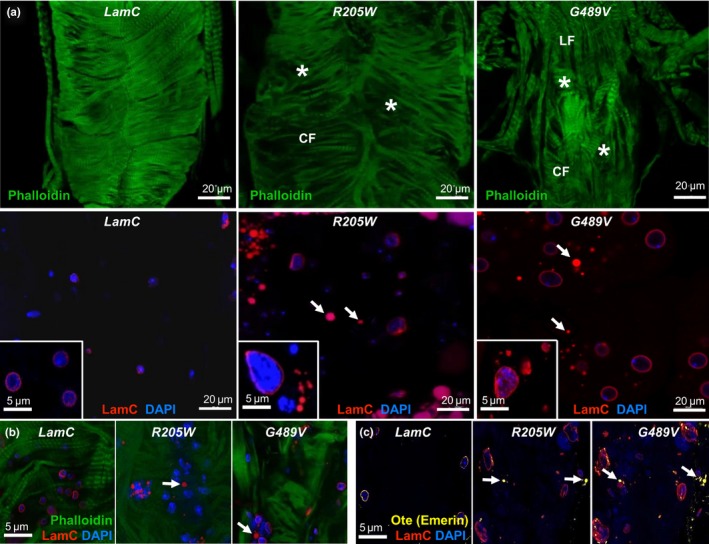
Mutant LamC caused myofibrillar disorganization, nuclear blebbing, and cytoplasmic aggregation of LamC and Otefin (orthologue of human Emerin). (a) Immunofluorescence of 3‐week‐old hearts stained with antibodies to LamC (red), phalloidin (F‐actin, green), and DAPI (DNA, blue). R205W and G489V caused disorganization of actin‐containing myofibrils that was not seen upon expression of wild‐type LamC (upper panels). CF and LF represent circumferential and longitudinal fibers, respectively. Asterisks represent myofibrillar disorganization (upper panels). Hearts expressing R205W and G489V that were stained with DAPI and anti‐LamC antibodies showed enlarged nuclei and cytoplasmic aggregation of LamC (arrows, lower panels). High magnification images are shown as insets. (b) Merged images of hearts expressing wild‐type LamC, R205W, and G489V stained with anti‐LamC antibodies (red) and DAPI (blue) showed cytoplasmic LamC aggregates (arrows) (c) Merged images of hearts expressing wild‐type LamC, R205W, and G489V stained with DAPI (DNA), anti‐LamC antibodies (red), and antibodies that recognize Otefin (yellow) showed cytoplasmic aggregates (arrows)
To determine the effects of LamC aggregation in cardiac tissue at the ultrastructural level, we employed transmission electron microscopy (TEM) (Figure 4a). TEM micrographs of a transverse section through the dorsal vessel of 3‐week‐old adults were prepared according to published procedures (Melkani et al., 2013). Adults expressing wild‐type LamC showed cardiomyocytes with characteristic discontinuous Z‐disks (“Z”). In contrast, similar aged adults expressing mutant LamC possessed hearts with severe myofibrillar degeneration (black asterisks) and poorly organized Z‐disks (Figure 4a, upper panels). Control hearts contained intact membrane‐bound nuclei (Figure 4a, white arrowhead, lower left panel), while hearts expressing R205W contained nuclear material (white asterisks) outside of membrane‐bound nuclei (Figure 4a, white arrowhead, lower middle panel). No intact nuclei were observed in hearts of similarly aged adults expressing G489V; however, membrane‐bound (black arrowhead) electron‐sparse structures were detected, which might represent degenerated nuclei and/or vacuoles (Figure 4a, lower right panel). Thus, the ultrastructural data were consistent with the abnormal phenotypes observed by confocal microscopy showing severe myofibrillar degeneration, nuclear morphological defects and revealed additional subcellular defects.
Figure 4.
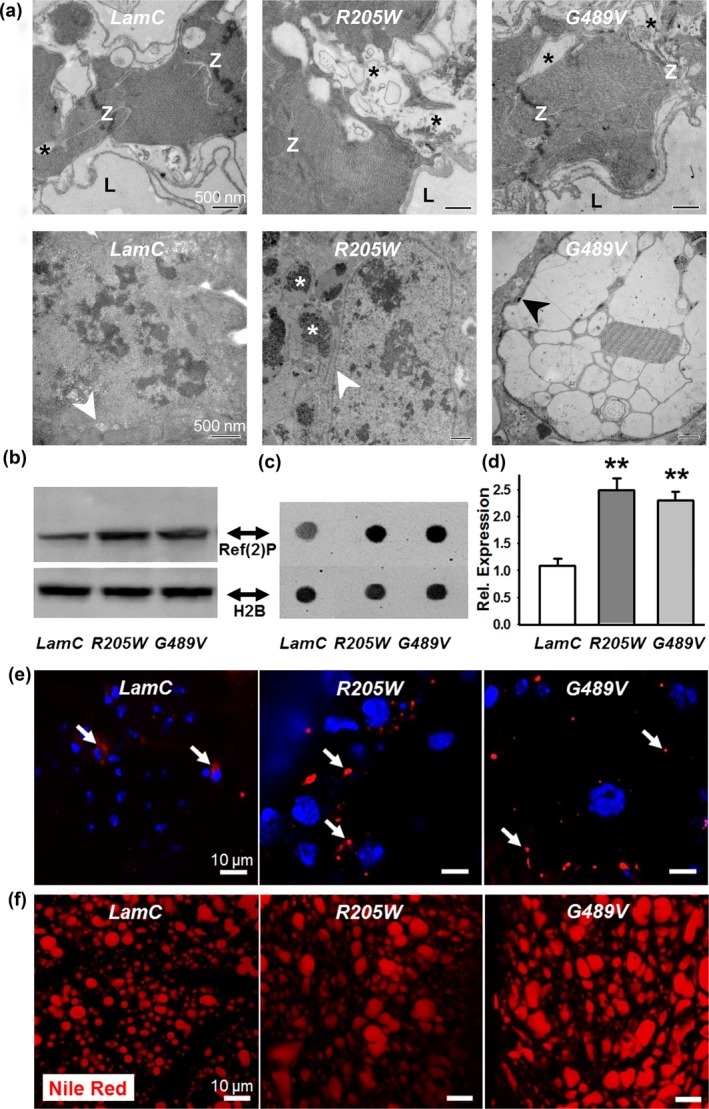
Cardiac‐specific expression of mutant LamC caused ultrastructural defects, upregulation of Ref(2)P, and altered adipose tissue homeostasis. (a) TEM micrographs of transverse sections through the heart of 3‐week‐old adults expressing wild‐type LamC showed contractile cardiomyocytes with characteristic Z‐disks (Z). Note that only minor myofibrillar degeneration was observed (black asterisk). In contrast, hearts expressing R205W and G489V showed substantial myofibrillar degeneration (black asterisks) and poorly organized Z‐disks (Z). The lumen (L) is a hemolymph‐containing compartment surrounded by contractile cardiomyocytes. In 3‐week‐old control hearts, intact membrane‐bound nuclei were detected. In contrast, hearts expressing R205W showed nuclear material outside of membrane‐bound nuclei (white asterisks). White arrows indicate the nuclear envelope. No intact nuclei were observed in 3‐week‐old hearts expressing G489V; however, membrane‐bound (black arrow) electron‐sparse structures were detected, which may constitute degenerated nuclei and/or vacuoles. Scale bars are 500 nm. (b) Western analysis of protein extracts from dissected hearts of 3‐week‐old female females expressing wild‐type and mutant LamC stained with antibodies that recognize Ref(2)P and histone H2B (loading control). (c) Representative antibody “dot blot” of protein extract from dissected hearts and stained with antibodies that recognize Ref(2)P and histone H2B. (d) Quantification of Ref(2)P staining, normalized to levels of histone H2B, in 3‐week‐old female flies hearts expressing R205W and G489V compared to same age flies expressing wild‐type LamC (**p < .01, n = 3 independent samples). (e) Cardiomyocytes of 3‐week‐old adults were stained with DAPI (blue) and antibodies to Ref(2)P (red). Hearts expressing mutant LamC showed Ref(2)P foci (arrows) that increased in number and size compared to those in flies expressing wild‐type LamC. (f) Adult fat bodies stained with Nile Red showed increased numbers and size of lipid droplets in expressing mutant LamC in cardiac tissue, relative to those expressing wild‐type LamC (nonautonomous effect)
2.4. Mutant LamC caused mislocalization of CncC (Nrf2) and increased levels of Ref(2)P (p62)
Abnormal cytoplasmic protein aggregation causes nuclear enrichment of the mammalian nuclear factor erythroid‐related factor 2 (Nrf2) in a mouse model of mutant αB‐crystallin‐induced cardiomyopathy (Kannan et al., 2013; Rajasekaran et al., 2011). Nrf2 and its cytoplasmic binding partner Keap1 function in cellular detoxification and are conserved in Drosophila (Deng & Kerppola, 2013, 2014). In a Drosophila model of noncardiac muscle laminopathies, Cap‐and‐collar C (CncC) [the orthologue of mammalian nuclear erythroid 2‐related factor 2 (Nrf2)] redox transcriptional regulator (Deng & Kerppola, 2013, 2014) accumulated in myonuclei and activated cellular detoxification genes (Dialynas et al., 2015). To determine whether CncC accumulated in this cardiac model of laminopathies, hearts expressing wild‐type and mutant LamC were stained with antibody to CncC (Deng & Kerppola, 2013, 2014). Hearts expressing wild‐type LamC showed CncC localization within the cytoplasm (Fig. S3). In contrast, hearts expressing mutant LamC showed CncC localization within the nucleus and cytoplasm (Fig. S3). Thus, mutant LamC caused nuclear enrichment of CncC.
Translocation of Nrf2 (CncC) into the nucleus in the Drosophila model of noncardiac muscle laminopathies was driven by cytoplasmic protein aggregation (Dialynas et al., 2015). Abundant protein aggregates increased levels of the mammalian autophagy cargo receptor p62 and the Drosophila orthologue Ref(2)P (Nezis et al., 2008). The p62 [Ref(2)P] protein binds Keap1, sequestering it from Nrf2 (CncC) in the cytoplasm, thereby allowing Nrf2 (CncC) to translocate into the nucleus (Dialynas et al., 2015; Jain et al., 2010). Antibodies showed elevated levels of Ref(2)P in protein extracts from hearts expressing mutant LamC via western and antibody dot blots (Figure 4b–d) as well as in immunostained tissues relative to hearts expressing wild‐type LamC (Figure 4e, red foci) Thus, cardiac‐specific expression of mutant LamC elevated levels of Ref(2)P, promoting CncC nuclear translocation.
Nuclear CncC (Nrf2) is a characteristic of redox imbalance (Deng & Kerppola, 2013, 2014). To determine the redox status of the hearts, reduced (GSH) and oxidized (GSSH) glutathione were measured (Anderson, 1985; Dialynas et al., 2015). The absolute values of both did not show differences between flies expressing wild‐type LamC and G489V (Fig. S4a); however, the ratio of GSH:GSSG was lower in 3‐week‐old adults expressing G489V compared to 3‐week‐old adults expressing wild‐type LamC, suggesting an oxidative redox imbalance at this age (Fig. S4a).
2.5. Mutant LamC disrupted adipose tissue homeostasis
Mutations in the human LMNA gene cause dysregulation of adipose tissue referred to as lipodystrophy (Wiltshire, Hegele, Innes & Brownell, 2013). These individuals experience age‐related loss of subcutaneous fat from their lower body and increased visceral fat in the upper portion of their body. While performing Drosophila heart dissections, we noted an age‐dependent increase in size of the fat bodies of adults expressing mutant LamC, relative to controls. Fat body tissue is thought to function similar to vertebrate adipose and liver tissues. The increased adipose tissue was apparent upon staining adults with the lipophilic dye Nile Red (Fowler & Greenspan, 1985; Lee, Bassel‐Duby & Olson, 2014). Nile Red staining showed increased lipid droplet size in 3‐week‐old adults expressing R205W and G489V, relative to controls (Figures 4f and S4b, upper panels). These cytological findings were supported by quantitative analysis of total triglycerides in adults. One‐day‐old adults with cardiac‐specific expression of wild‐type LamC and G489V showed no difference in triglyceride levels (Fig. S4b, lower panel). In contrast, 2‐ to 5‐week‐old adults showed significant increases in triglyceride levels. These data demonstrate an age‐dependent increase in total triglycerides.
2.6. Suppression of mutant LamC‐induced heart defects by cardiac‐specific upregulation of autophagy and downregulation of CncC (Nrf2)
The pathological features exhibited by the Drosophila cardiolaminopathy model included cytoplasmic aggregation of LamC, upregulation of p62, and nuclear enrichment of CncC (Figures 3 and 4). Genetic tools available in Drosophila allow for tissue‐specific over‐expression and RNAi knockdown (KD) of candidate genes that might suppress the cardiac abnormalities (Table S1). Logical candidates include those that increase autophagy (to eliminate cytoplasmic protein aggregates) and block the CncC/Keap1 signaling pathway. Over‐expression (OE) of an Atg1 transgene encoding a kinase that upregulates autophagy (Neufeld, 2007), suppressed the cardiac defects associated with mutant LamC (Figures 5, S5, S6). OE of Atg1 lowered the heart period, reduced cardiac arrhythmia, and enhanced cardiac performance (shown as fractional shortening) (Figure 5a–c). Importantly, Atg1 OE in a wild‐type LamC genetic background did not alter cardiac dysfunction (Figure 5a–c). Inhibition of Atg1 was achieved by expressing a transgene encoding a dominant negative (DN) Atg1 mutant (Neufeld, 2007), which enhanced the defective cardiac phenotypes caused by expression of G489V (Figures 5, S5, S6). It should be noted that expression of Atg1 DN in a wild‐type LamC genetic background produced mild cardiac phenotypes (Figure 5a–c). Thus, Atg1 OE suppressed the cardiac phenotypes and Atg1 DN enhanced cardiac defects caused by G489V.
Figure 5.
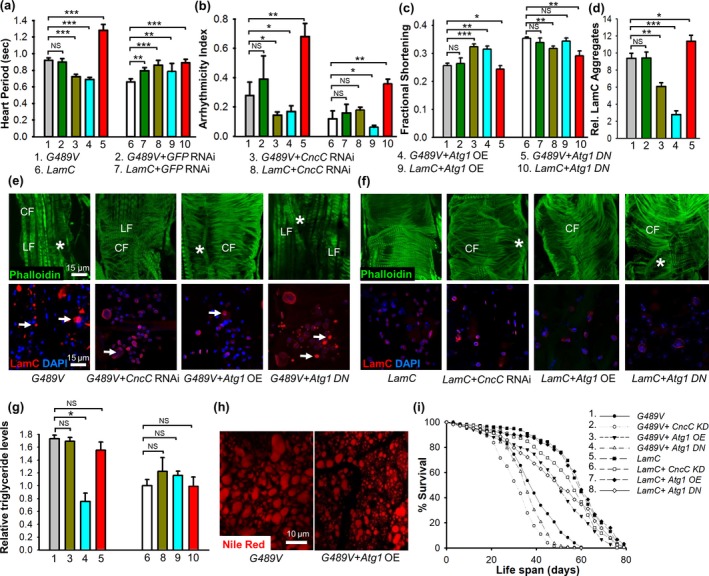
Cardiac‐specific expression of Atg1 and knockdown of CncC suppressed the abnormal cardiac, fat, and aging phenotypes caused by mutant LamC. Atg1 OE in hearts of 3‐week‐old adults (n = 30–70 per genotype) expressing G489V suppressed the heart period defects (a), cardiac dysrhythmia (b), and enhanced cardiac performance as represented by fractional shortening (c). In contrast, expression of Atg1 DN in hearts of 3‐week‐old adults expressing G489V enhanced the cardiac period and cardiac dysrhythmia and caused further deterioration of the heart (a–c). Cardiac‐specific expression of a CncC RNAi under the same conditions enhanced cardiac performance and suppressed heart period and cardiac dysrhythmia. An RNAi against GFP showed no effect on these cardiac parameters (a–c). These genetic modifiers had little to no effect on the physiology of hearts expressing wild‐type LamC (a–c). (d) Suppression of cardiac defects correlated with a reduction in cytoplasmic LamC aggregates. Cardiac‐specific expression of Atg1 and CncC RNAi suppressed the cytoplasmic aggregates, whereas Atg1 DN and GFP RNAi did not. The relative area of LamC aggregates/total area surveyed in confocal images was plotted. (e) Representative confocal images of the hearts stained with phalloidin (green), antibody against LamC (red), and DAPI (blue) showed a reduction in LamC protein aggregates. Cardiac‐specific of expression of CncC RNAi and Atg1 OE suppressed cytoplasmic LamC aggregates (arrows) and myofibrillar disorganization (*) in 3‐week‐old adults. Cardiac‐specific expression of Atg1 DN resulted in increased LamC aggregates and further deterioration of the organization of the actin‐containing myofibrils organization. Contractile circumferential fiber (CF) was missing; however, noncontractile longitudinal fibers (LF) were retained. (f) These genetic manipulations had little to no effect in hearts expressing wild‐type LamC (g) Cardiac‐specific Atg1 OE lowered the levels of total triglycerides. In contrast, Atg1 DN and RNAi against CncC showed no effect on triglyceride levels. Numbers correspond to the numbered genotypes in panels (a‐c). (h) Representative confocal images of adipose tissue from adults with cardiac‐specific expression of G489V and Atg1 OE stained with Nile Red showed a reduction in lipid droplets compared to the control expressing G489V alone. (i) Atg1 OE lengthened lifespan of adults with cardiac‐specific expression of G489V. The lifespan of adults (150–250 female per genotype) was determined for adults of the indicated genotypes. Atg1 OE suppressed the G489V‐induced shortened lifespan, whereas Atg1 DN and a CncC RNAi did not alter lifespan. The effect of these genetic modulators on lifespan of flies with cardiac‐specific expression of wild‐type LamC was used as a control. Statistical significance in A‐D, G, and F is denoted as follows: *p < .05; **p < .01; ***p < .001; NS = not significant
We next determined whether nuclear enrichment of CncC contributed to the cardiac phenotypes. Cardiac‐specific expression of an RNAi against CncC (referred as KD) suppressed the G489V‐induced cardiac physiological defects (Figure 5a–c, S5, S6a). It should be noted that cardiac‐specific RNAi against CncC, as well as a negative control RNAi against GFP, produced subtle cardiac defects in a wild‐type LamC background (Figure 5a–c and Fig. S5), suggesting that activation of the RNAi pathway in this context has a minor negative effect on cardiac function. Taken together, these data demonstrate that knockdown of CncC suppressed the cardiac defects caused by G489V, suggesting that nuclear enrichment of CncC contributes to the cardiac pathology.
To determine the cytological changes that account for the genetic suppression of the heart defects, confocal microscopy was used to image cardiac cells. Cardiac‐specific KD of CncC in hearts expressing G489V reduced cytoplasmic LamC aggregates, restored nuclear morphology, and promoted organization of actin‐containing myofibrils (Figure 5d–f). Likewise, Atg1 OE reduced cytoplasmic LamC aggregates, restored nuclear morphology, and promoted actin organization (Figure 5d–f). In contrast, expression of Atg1 DN (Neufeld, 2007) increased the number of LamC aggregates, compared to age‐matched adults (Figure 5d, e). Moreover, expression of the Atg1 DN in combination with G489V resulted in nearly complete loss of contractile circumferential fiber (CF); however, the noncontractile longitudinal fibers remained largely intact (Figure 5d–f). Cardiac‐specific KD of CncC, Atg1 OE, and the Atg1 DN resulted in subtle cytological defects in the wild‐type LamC background compared to age‐matched controls. Overall, these results suggest that increasing autophagy and reducing CncC/Keap1 redox signaling suppress cytological defects caused by G489V.
To test the impact of increasing autophagy and blocking the CncC/Keap1 pathway on adipose tissue homeostasis, both biochemical and cytological approaches were used. The impaired adipose tissue homeostasis caused by G489V was suppressed upon cardiac‐specific Atg1 OE, resulting in decreased triglycerides and smaller lipid droplets (Figure 5g, h). Cardiac‐specific expression of Atg1 DN had a minor negative impact on the elevated triglyceride levels (Figure 5g, h). In contrast to the results obtained by modulation of autophagy, cardiac‐specific KD of CncC did not alter G489V‐induced adipose tissue homeostasis defects (Figure 5g). Moreover, cardiac‐specific Atg1 OE and CncC KD did not alter adipose tissue homeostasis in a wild‐type LamC background (Figure 5g). Thus, increasing autophagy suppressed both the cardiac phenotypes and adipose tissue phenotypes induced by G489V. These results suggest that adipose accumulation might be due to the lack of fat utilization by the heart. However, CncC RNAi suppressed the cardiac defects, but not the adipose tissue accumulation, suggesting that the elevated triglycerides are not due to loss of cardiac function per se and might be due to altered systemic lipid homeostasis.
We next determined whether restoration of cardiac function and maintenance of adipose tissue homeostasis impacted the lifespan of adults with cardiac‐specific expression of G489V. Atg1 OE extended the lifespan of adults expressing G489V, whereas cardiac‐specific expression of Atg1 DN resulted in further shortening of the lifespan (Figure 5i). Cardiac‐specific expression of CncC RNAi did not alter the lifespan of adults expressing G489V, whereas it shortened the lifespan in adults expressing wild‐type LamC (Figure 5i). Taken together, these results indicated that cardiac‐specific expression of CncC RNAi suppressed LamC cytoplasmic aggregates, nuclear shape, and cytoskeletal defects, but did not suppress fat accumulation and the shortened lifespan caused by G489V. In contrast, Atg1 OE showed a nearly complete suppression of the abnormal phenotypes and restoration of lifespan.
To determine the generalizability of these findings, we performed similar genetic tests using flies that express the R205W transgene. Atg1 OE suppressed the cardiac dysfunction (Fig. S7a–c), cytoplasmic aggregates (Fig. S7d), and shortened lifespan (Fig. S7e). Collectively, these results are similar to the suppression observed for flies expressing the G489V transgene. In contrast, cardiac‐specific KD of CncC did not suppress mutant phenotypes caused by R205W (Fig. S7a–e). These data suggest that different mutant versions of LamC might have different effects on redox homeostasis, a topic for further investigation.
2.7. Interplay between autophagy and Nrf2/Keap1 signaling in suppression of cardiac defects
To determine whether an interplay exists between autophagy and CncC/Keap1 signaling, Atg1 (over‐expression and loss of function) was co‐expressed with an RNAi against CncC in cardiac tissue expressing wild‐type and mutant LamC. Atg1 OE in combination with CncC RNAi (referred to hereafter as the “double treatment”) suppressed the physiological and cytological defects in the heart, restored adipose tissue homeostasis, and sustained lifespan (Figures 6 and S6, S8, S9). Abnormalities in cardiac contractility (Figure 6c), cardiac arrhythmias (Figure 6b), and heart period were suppressed. Additionally, cardiac parameters such as cardiac systolic and diastolic diameters and intervals were more similar to those of hearts expressing wild‐type LamC than those without the double treatment (Fig. S8). The cardiac defects of adults expressing G489V were enhanced upon cardiac expression of Atg1 DN and an RNAi against CncC (Figure 6a–c and Fig. S8). Moreover, expression of Atg1 DN and an RNAi against CncC resulted in altered cardiac physiology (Figure 6a–c and Fig. S8). Taken together, these results support an interplay between autophagy and the CncC/Keap1 pathway in which Atg1 OE enhances the effects of CncC RNAi and profoundly improves cardiac function and lifespan, suggesting modulation of both redox signaling and autophagy as an avenue for therapy.
Figure 6.
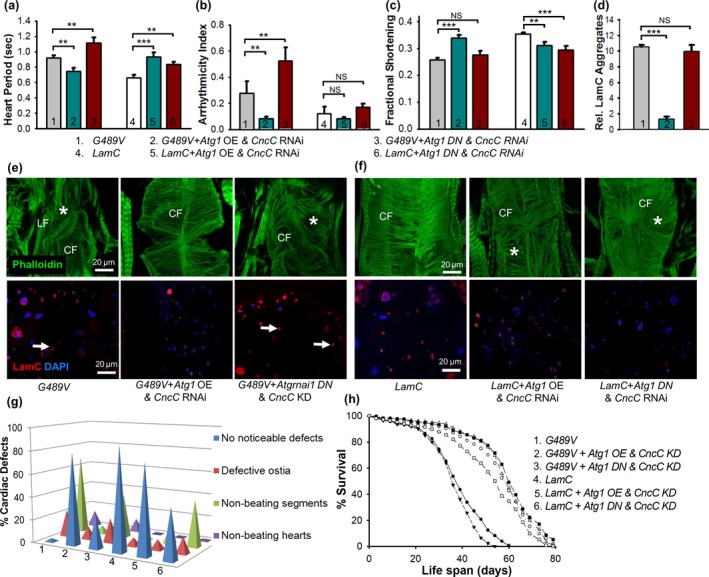
An interplay between autophagy and CncC/Keap1 suppressed cardiac dysfunction caused by mutant LamC. (a–c) The cardiac function of 3‐week‐old flies (n = 33–70 for each genotype) expressing wild‐type and mutant LamC was compared. Hearts expressing G489V, Atg1 OE, and RNAi against CncC showed a reduction in lower heart period (a), cardiac dysrhythmia (b), and enhanced cardiac performance as represented as fractional shortening (c) compared to hearts expressing G489V alone. Expression of G489V in combination with Atg1 DN and a CncC RNAi (KD) resulted in enhancement of cardiac period and dysrhythmia and increased deterioration compared with hearts expressing G489V alone. (d) Atg1 OE and CncC RNAi suppressed the LamC cytoplasmic aggregates. Quantification of the cytoplasmic aggregates was performed by taking the relative average area of the aggregates per the total area of the confocal images surveyed. (e–f) Representative confocal images of hearts from 3‐week‐old adults stained with phalloidin (green), antibodies against LamC (red), and DAPI (blue) showed that simultaneous OE of Atg1 and a CncC RNAi resulted in nearly complete suppression of cytoplasmic LamC aggregation (represented by arrows) and myofibrillar disorganization (represented by *) caused by G489V. In contrast, simultaneous expression of Atg1 DN and a CncC RNAi enhanced the disorganization of the actin‐containing myofibrils, with contractile circumferential fibers (CF) completely disorganized. The impact of this genetic combination resulted in subtle myofibrillar disorganization in the wild‐type LamC background, but did not alter nuclear morphology and LamC nuclear envelope localization. (g) Simultaneous cardiac‐specific expression of Atg1 and a CncC RNAi suppressed all of the cardiac parameters analyzed. (h) Simultaneous cardiac‐specific Atg1 OE with a CncC RNAi completely suppressed the G489V‐induced shortening of lifespan (150–250 adults were assayed per genotype). Expression of the Atg1 DN in combination with a CncC RNAi did not rescue the shortened lifespan caused by G489V. Statistical significance in A‐D is denoted as follows: *p < .05; **p < .01; ***p < 0.001; NS = not significant
To determine whether the suppression of cardiac dysfunction observed with the double treatment suppressed the cardiac cellular phenotypes, hearts were stained with antibodies to LamC. There was an absence of cytoplasmic LamC in the suppressed hearts. In addition, the nuclear morphological defects, myofibrillar disorganization, and nuclear enrichment of CncC were suppressed (Figure 6d and S9a, c). In contrast, simultaneous cardiac‐specific expression of Atg1 DN and a CncC RNAi did not suppress LamC cytoplasmic aggregates, nuclear defects, and myofibrillar disorganization (Figure 6d). In addition, enrichment of CncC was enhanced (S9D). In controls expressing wild‐type LamC, the double treatment caused minor myofibrillar disorganization, but had no apparent effect on LamC localization and nuclear morphology (Figure 6f). Collectively, these findings demonstrate that upregulation of autophagy and blocking the CncC/Keap1 pathway ameliorates the cardiac cellular defects caused by mutant LamC.
Simultaneous manipulation of autophagy and the CncC/Keap1 pathway was also tested for effects on adipose tissue homeostasis. The double treatment reduced total triglyceride levels in adults expressing G489V (Fig. S9e). In contrast, simultaneous expression of Atg1 DN and CncC RNAi did not alter the elevated triglyceride levels (Fig. S9e). Furthermore, these genetic combinations did not significantly change triglyceride levels in the LamC background (Fig. S9e). We reason that the suppressive effect of the double treatment on triglycerides is likely through increased autophagy as a similar effect was observed with Atg1 OE alone.
We next determined whether restoration of cardiac function and maintenance of adipose tissue homeostasis impacted the lifespan of G489V‐expressing adults (Figure 6h). The double treatment completely restored lifespan (Figure 6h). In contrast, simultaneous expression of Atg1 DN and an RNAi against CncC caused further reduction in the lifespan compared to flies expressing G489V alone (Figure 6h). As a control, the double treatment had little effect on the lifespan of adults with cardiac‐specific expression of wild‐type LamC. However, cardiac‐specific expression of Atg1 DN, CncC RNAi, and wild‐type LamC reduced lifespan compared to LamC alone. Taken together, these findings demonstrated that simultaneous over‐expression of Atg1 in combination with expression of CncC RNAi suppressed cytoplasmic aggregation of LamC, restored cardiac contractility, and improved lifespan.
3. DISCUSSION
Mutations in the human LMNA gene are associated with a collection of diseases called laminopathies in which the most common manifestation is progressive cardiac disease (Brayson & Shanahan, 2017; Heller et al., 2017; Marian, 2017; Naetar, Ferraioli & Foisner, 2017). We have generated Drosophila melanogaster models of age‐dependent cardiac dysfunction. In these models, mutations synonymous with those causing disease in humans were introduced into Drosophila LamC. Cardiac‐specific expression of mutant LamC resulted in (1) cardiac contractility, conduction, and physiological defects, (2) abnormal nuclear envelope morphology, (3) cytoplasmic LamC aggregation, (4) nuclear enrichment of the redox transcriptional regulator CncC (mammalian Nrf2), (5) and upregulation of autophagy cargo receptor Ref(2)P (mammalian p62) (Figures 1, 2, 3, 4, S3). These cardiac defects were enhanced with age (Figures 2 and S2) and accompanied by increased adipose tissue in the adult fat bodies (Figure 4f) and a shortened lifespan (Figure 1d, e).
To understand the mechanistic basis of cardiolaminopathy and identify genetic suppressors, we took advantage of powerful genetic tools available in Drosophila. The presence of cytoplasmic LamC aggregates prompted us to determine whether increasing autophagy would suppress the cardiac defects. Cardiac‐specific upregulation of autophagy (Atg1 OE) suppressed G489V‐induced cardiac defects (Figures 5 and S5–S6). Consistent with this, decreased autophagy due to expression of Atg1 DN resulted in enhanced deterioration of G489V‐induced cardiac dysfunction (Figures 5 and S5). Interestingly, cardiac‐specific Atg5 OE and Atg8a OE, two factors that also promote autophagy, showed little to no suppression of G489V‐induced heart dysfunction (Table S1), suggesting that Atg1 might be rate limiting in this context. Our findings are consistent with studies in mouse laminopathy models in which rapamycin and temsirolimus had beneficial effects on heart and skeletal muscle through inhibition of AKT/mTOR signaling (Choi et al., 2012; Ramos et al., 2012). Our findings are depicted in a model (Figure 7) in which cytoplasmic aggregation of mutant LamC results in upregulation of p62, which in turn inhibits autophagy via activation of TOR and inactivation of AMPK (Mihaylova & Shaw, 2011). AMPK inactivation leads to the activation of PI3K/Akt/mTOR pathway and inhibition of autophagy (Porta, Paglino & Mosca, 2014) Atg1 OE promoted clearance of the LamC aggregates and restored proteostasis in these Drosophila models. Thus, our data suggest that mutant LamC reduces autophagy, resulting in impairment of cellular proteostasis that leads to cardiac dysfunction.
Figure 7.
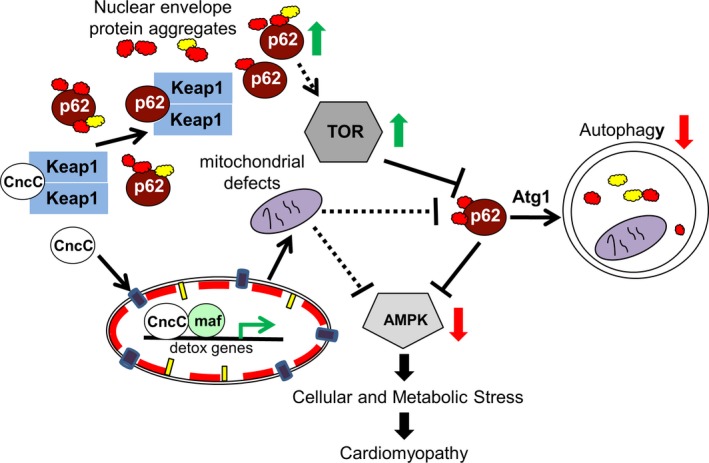
Model for the interactions between the autophagy and CncC/Keap1 signaling pathway in mutant lamin‐induced cardiac disease. Cellular and metabolic stress is triggered by the abnormal aggregation of nuclear envelope proteins in the cytoplasm (red and yellow circles with irregular margins), leading to increased levels of Ref(2)P (p62). Sequestration of Keap1 by Ref(2)P allows CncC (Nrf2) to enter the nucleus and activate antioxidant genes. Exhaustion of the antioxidant system is hypothesized to cause redox imbalance, potentially through mitochondrial dysfunction and dysregulation of energy homeostasis. Upregulation of Ref(2)P leads to activation of TOR, inactivation of autophagy, and inactivation of AMPK. Simultaneous over‐expression of Atg1 and inhibition of CncC suppressed cytoplasmic aggregation of LamC and restored cardiac contractility and lifespan
Cardiac‐specific expression of mutant LamC altered CncC subcellular localization (Fig. S3). Previously, Drosophila larval body wall muscles expressing G489V were shown to experience reductive stress, an atypical redox state characterized by high levels of reduced glutathione and NADPH, and upregulation CncC target genes (Dialynas et al., 2015). Cardiac‐specific CncC RNAi in the wild‐type LamC background did not produce major cardiac defects (Figures 5 and S5). Consistent with this, Nrf2 deficiency in mice does not compromise cardiac and skeletal muscle performance (Kannan et al., 2013; Rajasekaran et al., 2011). Cardiac‐specific CncC RNAi suppressed G489V‐induced cardiac dysfunction and reduced cytoplasmic LamC aggregation, but not R205W‐induced defects (Figures 5, S5‐S6, S7). However, cardiac‐specific RNAi against CncC did not affect G489V‐induced adipose tissue accumulation and lifespan shortening. Similar to the nuclear enrichment of CncC in hearts expressing G489V (Fig. S3), human muscle biopsy tissue from an individual with a point mutation in the LMNA gene that results in G449V (analogous to Drosophila G489V) showed nuclear enrichment of Nrf2 (Dialynas et al., 2015). Disruption of Nrf2/Keap1 signaling has also been reported for Hutchinson–Gilford progeria, an early‐onset aging disease caused by mutations in LMNA (Kubben et al., 2016). In this case, however, the thickened nuclear lamina traps Nrf2 at the nuclear envelope that results in a failure to activate Nrf2 target genes, leading to oxidative stress (Kubben et al., 2016). In our studies, we observed CncC nuclear enrichment; however, a redox imbalance was not readily observed at the three‐time points investigated (Fig. S4b). This might indicate that there is a window of time in disease progression in which redox imbalance occurs and that mechanisms are in place to re‐establish homeostasis.
It has been postulated that there is cross‐talk between autophagy and Nrf2/Keap1 signaling (Jain et al., 2010; Stepkowski & Kruszewski, 2011). We tested for this by manipulating autophagy and CncC (Nrf2) alone and in combination. CncC RNAi suppressed the cardiac defects caused by G489V, but not the lipid accumulation and lifespan shortening, suggesting the latter two phenotypes are not specifically due to loss of cardiac function (Figures 5 and S5–S6). In contrast, Atg1 OE suppressed the cardiac and adipose tissue defects and lengthened the lifespan. The double treatment (simultaneous Atg1 OE and RNAi knockdown of CncC) gave the most robust suppression of the mutant phenotypes (Figures 6 and S6–S9) and completely restored the lifespan (Figure 6h). Interestingly, Atg1 DN and RNAi knockdown of CncC simultaneously did not further deteriorate or improve the mutant phenotypes. Taken together, these data suggest that autophagy plays a key role in suppression of the G498V‐induced phenotypes and that knockdown on CncC enhances this suppression.
Our findings support a model whereby autophagy and Nrf2 signaling are central to cardiac health (Figure 7). We propose that cytoplasmic aggregation of LamC increases levels of Ref(2)P (p62), which competitively binds to Keap1 (Dialynas et al., 2015; Jain et al., 2010), resulting in CncC (Nrf2) translocation to the nucleus. Inside the nucleus, Nrf2 regulates genes involved in detoxification (Dialynas et al., 2015; Jain et al., 2010). Continued expression of antioxidant genes results in the disruption of redox homeostasis, defective mitochondria, and dysregulation of energy homeostasis/energy sensor such as AMPK and its downstream targets. Simultaneously, upregulation of Ref(2)P (p62) causes inhibition of autophagy via activation of TOR, which leads to the inactivation of AMPK (Mihaylova & Shaw, 2011). AMPK inactivation in combination with activation of the TOR pathway causes cellular and metabolic stress that leads to cardiomyopathy. In support of our model, transcriptomics data from muscle tissue of an individual with muscular dystrophy expressing Lamin A/C G449V (analogous to Drosophila G489V) showed (1) upregulation of transcripts from Nrf2 target genes, (2) upregulation of genes encoding subunits of the mTOR complex, and (3) downregulation of AMPK (unpublished data), further demonstrating relevance of the Drosophila model for providing insights on human pathology.
4. EXPERIMENTAL PROCEDURES
Due to space limitation, experimental procedures are described in the Supplementary section.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
GCM and LLW designed the project, conducted experiments, evaluated data, and prepared the manuscript; SB, AST, MTO, GHY, DEC, SC, and MN conducted experiments and evaluated data. All authors have reviewed the manuscript.
Supporting information
ACKNOWLEDGMENTS
We thank Drs. Rolf Bodmer and Karen Ocorr, Sanford Burnham Medical Discovery Institute (SBMDI) for use of equipment. We are grateful for technical training and thoughtful discussions with Drs. Rolf Bodmer and Sreehari Kalvakuri, SBMDI. This work was supported by a National Institutes of Health NIH grant AG049494 to GCM and NIH grant AR064894 and Muscular Dystrophy Association grant 44728 to LLW. AST is a Fellow of the Rees‐Stealy Research Foundation and the San Diego State University Heart Institute.
Bhide S, Trujillo AS, O'Connor MT, et al. Increasing autophagy and blocking Nrf2 suppress laminopathy‐induced age‐dependent cardiac dysfunction and shortened lifespan. Aging Cell. 2018;17:e12747 10.1111/acel.12747
Contributor Information
Lori L. Wallrath, Email: lori-wallrath@uiowa.edu
Girish C. Melkani, Email: gmelkani@mail.sdsu.edu
REFERENCES
- Ahmed, M. S. , Ikram, S. , Bibi, N. , & Mir, A. (2017). Hutchinson‐Gilford Progeria Syndrome: A premature aging disease. Molecular Neurobiology, [Epub ahead of print]. 10.1007/s12035-017-0610-7 [DOI] [PubMed] [Google Scholar]
- Anderson, M. E. (1985). Determination of glutathione and glutathione disulfide in biological samples. Methods in Enzymology, 113, 548–555. [DOI] [PubMed] [Google Scholar]
- Apte, K. , Stick, R. , & Radmacher, M. (2017). Mechanics in human fibroblasts and progeria: Lamin A mutation E145K results in stiffening of nuclei. Journal of Molecular Recognition, 30(2). [DOI] [PubMed] [Google Scholar]
- Arbustini, E. , Pilotto, A. , Repetto, A. , Grasso, M. , Negri, A. , Diegoli, M. , … Tavazzi, L. (2002). Autosomal dominant dilated cardiomyopathy with atrioventricular block: A lamin A/C defect‐related disease. Journal of the American College of Cardiology, 39(6), 981–990. [DOI] [PubMed] [Google Scholar]
- Azibani, F. , Muchir, A. , Vignier, N. , Bonne, G. , & Bertrand, A. T. (2014). Striated muscle laminopathies. Seminars in Cell & Developmental Biology, 29, 107–115. [DOI] [PubMed] [Google Scholar]
- Barton, L. J. , Lovander, K. E. , Pinto, B. S. , & Geyer, P. K. (2016). Drosophila male and female germline stem cell niches require the nuclear lamina protein Otefin. Developmental Biology, 415(1), 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayson, D. , & Shanahan, C. M. (2017). Current insights into LMNA cardiomyopathies: Existing models and missing LINCs. Nucleus, 8(1), 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke, B. , & Stewart, C. L. (2013). The nuclear lamins: Flexibility in function. Nature Reviews Molecular Cell Biology, 14(1), 13–24. [DOI] [PubMed] [Google Scholar]
- Captur, G. , Arbustini, E. , Bonne, G. , Syrris, P. , Mills, K. , Wahbi, K. , & Moon, J. C. (2017). Lamin and the heart. Heart, 104(6), 468–479. [DOI] [PubMed] [Google Scholar]
- Cenni, V. , D'Apice, M. R. , Garagnani, P. , Columbaro, M. , Novelli, G. , Franceschi, C. , & Lattanzi, G. (2017). Mandibuloacral dysplasia: A premature ageing disease with aspects of physiological ageing. Ageing Research Reviews, 42, 1–13. [DOI] [PubMed] [Google Scholar]
- Choi, J. C. , Muchir, A. , Wu, W. , Iwata, S. , Homma, S. , Morrow, J. P. , & Worman, H. J. (2012). Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Science Translational Medicine, 4(144), 144ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, H. , & Kerppola, T. K. (2013). Regulation of Drosophila metamorphosis by xenobiotic response regulators. PLoS Genetics, 9(2), e1003263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, H. , & Kerppola, T. K. (2014). Visualization of the Drosophila dKeap1‐CncC interaction on chromatin illumines cooperative, xenobiotic‐specific gene activation. Development, 141(16), 3277–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dialynas, G. , Flannery, K. M. , Zirbel, L. N. , Nagy, P. L. , Mathews, K. D. , Moore, S. A. , & Wallrath, L. L. (2012). LMNA variants cause cytoplasmic distribution of nuclear pore proteins in Drosophila and human muscle. Human Molecular Genetics, 21(7), 1544–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dialynas, G. , Shrestha, O. K. , Ponce, J. M. , Zwerger, M. , Thiemann, D. A. , Young, G. H. , … Wallrath, L. L. (2015). Myopathic lamin mutations cause reductive stress and activate the nrf2/keap‐1 pathway. PLoS Genetics, 11(5), e1005231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diop, S. B. , & Bodmer, R. (2015). Gaining insights into diabetic cardiomyopathy from Drosophila. Trends in Endocrinology and Metabolism, 26(11), 618–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer, T. A. , & Misteli, T. (2011). The lamin protein family. [Review]. Genome Biology, 12(5), 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler, S. D. , & Greenspan, P. (1985). Application of Nile red, a fluorescent hydrophobic probe, for the detection of neutral lipid deposits in tissue sections: Comparison with oil red O. Journal of Histochemistry and Cytochemistry, 33(8), 833–836. [DOI] [PubMed] [Google Scholar]
- Gill, S. , Le, H. D. , Melkani, G. C. , & Panda, S. (2015). Time‐restricted feeding attenuates age‐related cardiac decline in Drosophila. Science, 347(6227), 1265–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenbaum, Y. , & Foisner, R. (2015). Lamins: Nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annual Review of Biochemistry, 84, 131–164. [DOI] [PubMed] [Google Scholar]
- Guenantin, A. C. , Briand, N. , Bidault, G. , Afonso, P. , Bereziat, V. , Vatier, C. , … Vigouroux, C. (2014). Nuclear envelope‐related lipodystrophies. Seminars in Cell & Developmental Biology, 29, 148–157. [DOI] [PubMed] [Google Scholar]
- Han, Z. , & Olson, E. N. (2005). Hand is a direct target of Tinman and GATA factors during Drosophila cardiogenesis and hematopoiesis. Development, 132(15), 3525–3536. [DOI] [PubMed] [Google Scholar]
- Heller, F. , Dabaj, I. , Mah, J. K. , Bergounioux, J. , Essid, A. , Bonnemann, C. G. , … Wahbi, K. (2017). Cardiac manifestations of congenital LMNA‐related muscular dystrophy in children: Three case reports and recommendations for care. Cardiology in the Young, 27(6), 1076–1082. [DOI] [PubMed] [Google Scholar]
- Hermida‐Prieto, M. , Monserrat, L. , Castro‐Beiras, A. , Laredo, R. , Soler, R. , Peteiro, J. , … Crespo‐Leiro, M. (2004). Familial dilated cardiomyopathy and isolated left ventricular noncompaction associated with lamin A/C gene mutations. American Journal of Cardiology, 94(1), 50–54. [DOI] [PubMed] [Google Scholar]
- Ikeda, Y. , Kumagai, H. , Motozawa, Y. , Suzuki, J. , Akazawa, H. , & Komuro, I. (2016). Understanding vascular diseases: Lessons from premature aging syndromes. Canadian Journal of Cardiology, 32(5), 650–658. [DOI] [PubMed] [Google Scholar]
- Jain, A. , Lamark, T. , Sjottem, E. , Larsen, K. B. , Awuh, J. A. , Overvatn, A. , … Johansen, T. (2010). p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element‐driven gene transcription. Journal of Biological Chemistry, 285(29), 22576–22591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan, S. , Muthusamy, V. R. , Whitehead, K. J. , Wang, L. , Gomes, A. V. , Litwin, S. E. , … Rajasekaran, N. S. (2013). Nrf2 deficiency prevents reductive stress‐induced hypertrophic cardiomyopathy. Cardiovascular Research, 100(1), 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubben, N. , Zhang, W. , Wang, L. , Voss, T. C. , Yang, J. , Qu, J. , … Misteli, T. (2016). Repression of the antioxidant NRF2 pathway in premature aging. Cell, 165(6), 1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. H. , Bassel‐Duby, R. , & Olson, E. N. (2014). Heart‐ and muscle‐derived signaling system dependent on MED13 and Wingless controls obesity in Drosophila. Proceedings of the National Academy of Sciences of the United States of America, 111(26), 9491–9496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, J. J. , Grogan, M. , & Ackerman, M. J. (2016). LMNA‐mediated arrhythmogenic right ventricular cardiomyopathy and charcot‐marie‐tooth type 2B1: A patient‐discovered unifying diagnosis. Journal of Cardiovascular Electrophysiology, 27(7), 868–871. [DOI] [PubMed] [Google Scholar]
- Malhotra, R. , & Mason, P. K. (2009). Lamin A/C deficiency as a cause of familial dilated cardiomyopathy. Current Opinion in Cardiology, 24(3), 203–208. [DOI] [PubMed] [Google Scholar]
- Marian, A. J. (2017). Non‐syndromic cardiac progeria in a patient with the rare pathogenic p.Asp300Asn variant in the LMNA gene. BMC Medical Genetics, 18(1), 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkani, G. C. , Trujillo, A. S. , Ramos, R. , Bodmer, R. , Bernstein, S. I. , & Ocorr, K. (2013). Huntington's disease induced cardiac amyloidosis is reversed by modulating protein folding and oxidative stress pathways in the Drosophila heart. PLoS Genetics, 9(12), e1004024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova, M. M. , & Shaw, R. J. (2011). The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature Cell Biology, 13(9), 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naetar, N. , Ferraioli, S. , & Foisner, R. (2017). Lamins in the nuclear interior – life outside the lamina. Journal of Cell Science, 130(13), 2087–2096. [DOI] [PubMed] [Google Scholar]
- Neufeld, T. P. (2007). Contribution of Atg1‐dependent autophagy to TOR‐mediated cell growth and survival. Autophagy, 3(5), 477–479. [DOI] [PubMed] [Google Scholar]
- Nezis, I. P. , Simonsen, A. , Sagona, A. P. , Finley, K. , Gaumer, S. , Contamine, D. , … Brech, A. (2008). Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. Journal of Cell Biology, 180(6), 1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politano, L. , Carboni, N. , Madej‐Pilarczyk, A. , Marchel, M. , Nigro, G. , Fidziaoska, A. , … Hausmanowa‐Petrusewicz, I. (2013). Advances in basic and clinical research in laminopathies. Acta Myologica, 32(1), 18–22. [PMC free article] [PubMed] [Google Scholar]
- Porta, C. , Paglino, C. , & Mosca, A. (2014). Targeting PI3K/Akt/mTOR signaling in cancer. Frontiers Oncology, 4, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran, N. S. , Varadharaj, S. , Khanderao, G. D. , Davidson, C. J. , Kannan, S. , Firpo, M. A. , … Benjamin, I. J. (2011). Sustained activation of nuclear erythroid 2‐related factor 2/antioxidant response element signaling promotes reductive stress in the human mutant protein aggregation cardiomyopathy in mice. Antioxidants & Redox Signaling, 14(6), 957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos, F. J. , Chen, S. C. , Garelick, M. G. , Dai, D. F. , Liao, C. Y. , Schreiber, K. H. , … Kennedy, B. K. (2012). Rapamycin reverses elevated mTORC1 signaling in lamin A/C‐deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Science Translational Medicine, 4(144), 144ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi, P. , & Misteli, T. (2006). Lamin A‐dependent nuclear defects in human aging. Science, 312(5776), 1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepkowski, T. M. , & Kruszewski, M. K. (2011). Molecular cross‐talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radical Biology and Medicine, 50(9), 1186–1195. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Zabell, A. , Koh, W. , & Tang, W. H. (2017). Lamin A/C cardiomyopathies: Current understanding and novel treatment strategies. Current Treatment Options in Cardiovascular Medicine, 19(3), 21. [DOI] [PubMed] [Google Scholar]
- Wiltshire, K. M. , Hegele, R. A. , Innes, A. M. , & Brownell, A. K. (2013). Homozygous lamin A/C familial lipodystrophy R482Q mutation in autosomal recessive Emery Dreifuss muscular dystrophy. Neuromuscular Disorders, 23(3), 265–268. [DOI] [PubMed] [Google Scholar]
- Wolf, C. M. , Wang, L. , Alcalai, R. , Pizard, A. , Burgon, P. G. , Ahmad, F. , … Seidman, J. G. (2008). Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis‐triggered cardiac conduction system disease. Journal of Molecular and Cellular Cardiology, 44(2), 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worman, H. J. (2012). Nuclear lamins and laminopathies. Journal of Pathology, 226(2), 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Kieckhaefer, J. E. , & Cao, K. (2013). Mouse models of laminopathies. Aging Cell, 12(1), 2–10. [DOI] [PubMed] [Google Scholar]
- Zhu, S. , Han, Z. , Luo, Y. , Chen, Y. , Zeng, Q. , Wu, X. , & Yuan, W. (2017). Molecular mechanisms of heart failure: Insights from Drosophila. Heart Failure Reviews, 22(1), 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials