

Abstract
Background
We describe a female infant with Fragile-X syndrome, with a fully expanded FMR1 allele and preferential inactivation of the homologous X-chromosome carrying a de novo deletion. This unusual and rare case demonstrates the importance of a detailed genomic approach, the absence of which could be misguiding, and calls for reflection on the current clinical and diagnostic workup for developmental disabilities.
Case presentation
We present a female infant, referred for genetic testing due to psychomotor developmental delay without specific dysmorphic features or relevant family history. FMR1 mutation screening revealed a methylated full mutation and a normal but inactive FMR1 allele, which led to further investigation. Complete skewing of X-chromosome inactivation towards the paternally-inherited normal-sized FMR1 allele was found. No pathogenic variants were identified in the XIST promoter. Microarray analysis revealed a 439 kb deletion at Xq28, in a region known to be associated with extreme skewing of X-chromosome inactivation.
Conclusions
Overall results enable us to conclude that the developmental delay is the cumulative result of a methylated FMR1 full mutation on the active X-chromosome and the inactivation of the other homologue carrying the de novo 439 kb deletion. Our findings should be taken into consideration in future guidelines for the diagnostic workup on the diagnosis of intellectual disabilities, particularly in female infant cases.
Keywords: Developmental disabilities in females, FMR1 methylated full mutation, Fragile-X syndrome, Skewing of X-chromosome inactivation, Xq28 deletion
Background
Fragile-X syndrome (FXS, MIM #300624) is the most common cause of hereditary intellectual disability (ID) with an X-linked inheritance pattern and incomplete penetrance in females. FXS has been shown to be caused by an unstable CGG repeat within the 5’untranslated region of the fragile mental retardation-1 (FMR1) gene [1, 2]. This repeat is highly polymorphic with normal alleles harbouring 8 to 54 CGGs, while full expansions have more than 200 repeats [3]. The expansion within the full mutation range usually accompanied by abnormal methylation of the FMR1 gene promoter and repetitive region, reducing Fragile X mental retardation protein (FMRP) expression [4]. The physical, neurocognitive and behavioural FXS features are therefore the result of a typical loss-of-function mutation with epigenetic changes (histone modifications and DNA methylation), by mechanisms still not entirely understood [5]. A recent FXS epidemiologic study estimates the frequency of affected males at 1.4:10,000 and that of affected females at 0.9:10,000 [6]. The typical FXS phenotypic characteristics have been described in males. Around 50% of full mutation female carriers present some degree of cognitive impairment (from mild learning disability to severe cognitive dysfunction), but usually less severe than in FXS males [7, 8]. FXS should be considered in the presence of particular physical characteristics such as long face, large and protruding ears and macroorchidism, combined with ID or autistic behaviour. Besides familial cases, both males and females with ID, even those without the other clinical signs, should be tested for fragile-X because the pathognomonic FXS features are not always obvious or present [9]. Herein, we present the case of a female infant, referred for genetic consultation due to developmental delay and hyperactivity, without specific dysmorphic features or relevant family history. FMR1 mutation screening revealed the presence of a methylated full mutation and a normal but inactive allele, which prompted further investigation.
Case presentation
The proband was first referred to our genetics clinic at 11 months of age. She showed developmental delay and hyperactivity without specific dysmorphic features and with irrelevant family history. At 33 months the developmental profile was similarly delayed with limited speech and language acquisition. The proband had early intervention for speech and language as well as occupational therapy. The last evaluation at 44 months revealed remarkable inattentiveness besides persistence of developmental delay, poor language skills and a global developmental profile equivalent to 30 months and handling skills equivalent to 24 months. Irrelevant dysmorphisms included redundant eyelids, bulbous nose and protruding ears. Analytic studies showed slightly elevated creatine phosphokinase levels and normal creatinine metabolism. The parents and other relatives gave informed consent for samples to be used in this research study, approved by the medical ethics committee of the Centro Hospitalar do Porto (CHP, E.P.E.). Following the proband’s referral for genetic testing, a normal karyotype was found together with an FMR1 full mutation and a normal but inactive allele (Fig. 1). Co-segregation studies identified two at-risk females with FMR1 premutations and excluded FMR1 expansion in a maternal aunt. HUMARA testing [10], carried out on the proband’s peripheral blood, showed complete skewing of the X-chromosome inactivation (XCI) pattern. Further FMR1 analysis, by AmplideX® FMR1 mPCR, showed absence of size and/or methylation mosaicism (above 1%) and confirmed that the normal-sized allele was inactivated, suggesting that another cause was implicated in the skewing (Fig. 1) [11]. A good candidate for skewed XCI is XIST (MIM 314670), a non-protein coding gene, as a C to G transversion present in the minimal promoter (position − 43) underlies skewing in some families [12]. Several authors recognized that a C to A transversion at the same position results in skewing of XCI towards the active X homologue of heterozygous females [12, 13], while others found no such association [14]. XCI skewing in the proband was further investigated by sequencing the XIST promoter. No pathogenic variants were identified.
Fig. 1.
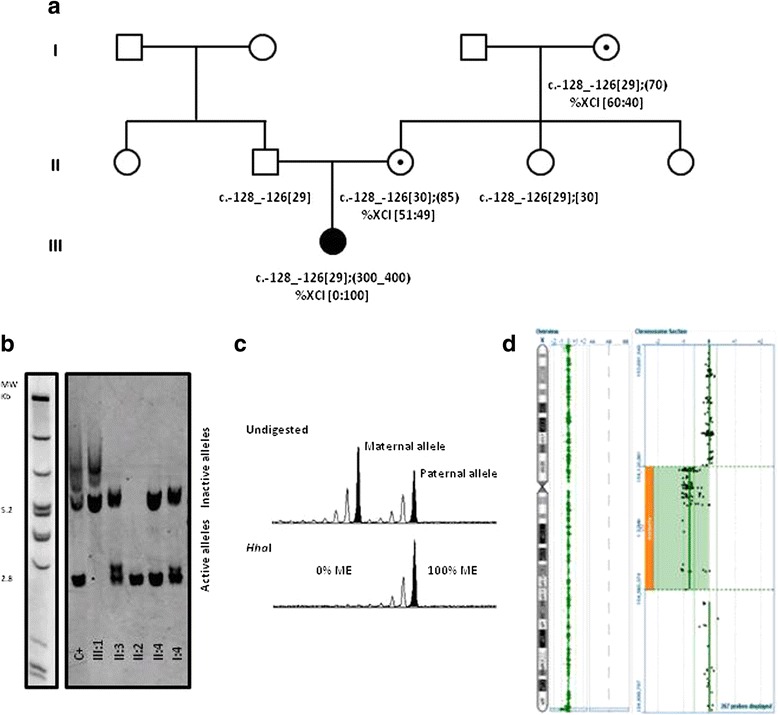
Pedigree and summary of overall laboratory findings. a – Proband’s pedigree (III:1) showing FMR1 genotyping and XCI ratio results. b – Southern blot analysis using GLFXDig1 probe (Gene LinkTM , Hawthorne, NY, US) and DNA Molecular Weight Markers II and III, DIG-labeled from Merck KGaA, Darmstadt, Germany; Female control with a full mutation (C+) c.-128_-126[30];[250_400]; Proband III:1 showing complete absence of normal, active FMR1 allele. Premutation carriers (II:3 and II:4) show four fragments corresponding to the normal active, expanded active (2.8 kb and above) and normal inactive and expanded inactive (5.2 kb and above) alleles [21]. c - HUMARA results obtained in proband’s leukocytes (III:1) [10]. HhaI completely digested the maternal allele indicating that the other allele is fully methylated (ME) and suggesting total skewing of the XCI pattern. The additional two females tested (II:3 and I:4) showed a normal XCI pattern (data not shown). d – Array Comparative Genomic Hybridisation (aCGH) performed using the Cytochip ISCA 8x60K (Cambridge Bluegnome, Illumina Inc., San Diego CA,USA) showed a deletion of 439Kb in chromosome X within band Xq28, classified as pathogenic, involving the genes: F8; CTD-2183H9.7; EEF1A1P31; CTD- 2183H9.3; FUNDC2; CMC4; MTCP1; BRCC3; RP11-143H17.1; VBP1; RP13-228 J13.9; RAB39B; CLIC2; RP13-228 J13.6; RP13-228 J13.10. Array design included a median resolution of 120Kb throughout the genome (Backbone) with increased density oligonucleotide probes in selected regions associated with clinically relevant phenotypes, in line with the International Standard Cytogenetic Array (ISCA) consortium. Analysis was performed using the Bluefuse software (Cambridge Bluegnome, Illumina Inc., San Diego CA, US) coupled with publicly available databases. Mutation nomenclature guidelines suggested by the Human Genome Variation Society (HGVS) (http://varnomen.hgvs.org/) were used. FMR1 reference sequence NM_002024.5
aCGH analysis was performed, revealing a 439 kb deletion in Xq28 (chrX:154,120,961–154,560,374 (hg19)) encompassing 16 genes. Similar deletions have been shown to be associated with extreme deviation of XCI, compatible with the skewing observed in our case [15, 16].
Discussion and conclusions
Diagnostic yield for chromosomal microarray analysis (CMA) in unexplained ID is between 15%- 20%, half of these carrying a de novo copy number variant [17]. CMA to assess DNA copy number is currently recommended as a first-tier test for postnatal evaluation of patients with developmental delay, intellectual disability, autism spectrum disorders and/or multiple congenital anomalies [17]. In this case, however, application of CMA technology in the first instance, revealing a de novo 439 kb deletion, could have misguided the diagnostic workup; for example, searching for hemizygous point mutations in the RAB39B and CLIC2 genes – included in this recurrently duplicated/deleted region – both of which have been implicated in ID [18, 19]. Although the deletion breakpoints were not sequenced in our case, according to previous publications, one can assume that they are within the directly orientated low-copy repeat (LCR) regions int22h-1 and int22h-2, located in the F8 gene (MIM #300841) [15]. There is no family history of haemophilia A, although the observed preferential XCI could explain the absence of haemophilic clinical signs in the proband. Another deleted gene in this region is VBP1 (MIM #300133), heterozygous deletions of which associate with high miscarriage rates in females without cognitive function involvement [16]. A methylated FMR1 full mutation was identified and according to Godler et al., the presence of an expansion is closely associated with an X-inactivation pattern skewed towards the mutated chromosome [20]. Here, we have showed that the developmental delay is the cumulative result of a methylated FMR1 full mutation on the active X-chromosome and the inactivation of the other homologue carrying the de novo Xq28 deletion, although we were unable to exclude FMR1 tissue mosaicism or the presence of other X-linked recessive pathogenic variants in genes involved in the Xq28 deletion. Overall, this report describes an atypical fragile-X female infant whose phenotype one may speculate should develop in a similar manner as that described for typical FXS males. This case poses additional challenges for genetic counseling and also calls for reflection on the clinical and diagnostic workup for developmental disabilities, particularly in infant females: a positive aCGH result should not hinder FMR1 sizing and methylation analysis and vice-versa. In the present case, first-tier aCGH could have misguided the clinical geneticist towards sequencing several genes involved in ID (e.g. RAB39B and CLIC2) resulting in a completely distinct diagnostic workflow.
Acknowledgements
The authors would like to thank the probands’ family for their motivation to take part in this study. Dr. Paula Jorge received a research grant “Bolsa de Investigação DEFI 2015”, CHP, E.P.E.
UMIB is supported by a multi-annual program form FCT Strategic Project UID/Multi/00215/2013.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- aCGH
Array Comparative Genomic Hybridisation
- CMA
Chromosomal microarray analysis
- FMR1
Fragile mental retardation-1 gene
- FMRP
Fragile mental retardation protein
- FXS
Fragile-X syndrome
- ID
Intellectual disability
- ISCA
International Standard Cytogenetic Array
- LCR
Low-copy repeat
- XCI
X-chromosome inactivation
Authors’ contributions
PJ: made substantial contributions to conception and design, been involved in drafting and critical revision, writing and final revision and agreed to be held responsible for all aspects, including integrity and accuracy of this work. EG: made substantial contributions to acquisition and data interpretation, been involved in drafting the manuscript, and agreed with the final version to be published and to be responsible for all aspects, including integrity and accuracy of this work. AG: made substantial contributions to acquisition and data analysis, been involved in designing the manuscript, and agreed with the final version to be published and to be accountable for all aspects, including integrity and accuracy of this work. IM: made substantial contributions to acquisition and data analysis, been involved in critical reviewing of the manuscript, and agreed with the final version to be published and to be accountable for all aspects, including integrity and accuracy of this work. NM: made substantial contributions to acquisition and data analysis, been involved in critical reviewing of the manuscript, and agreed with the final version to be published and to be accountable for all aspects, including integrity and accuracy of this work. BR: made substantial contributions to acquisition of data, been involved in drafting of the manuscript, and agreed with the final version to be published and to be accountable for all aspects, including integrity and accuracy of this work. HS: made substantial contributions to clinical assessment and medical data interpretation, been involved in drafting of the manuscript, and agreed with the final version to be published and to be accountable for all aspects, including integrity and accuracy of this work. JF: made substantial contributions to clinical assessment and medical data interpretation, been involved in drafting of the manuscript, and agreed with the final version to be published and to be accountable for all aspects, including integrity and accuracy of this work. GS: made substantial contributions to clinical assessment and medical data interpretation, been involved in the manuscript critical revision, and agreed with the final version to be published and to be accountable for all aspects, including integrity and accuracy of this work. CC: made substantial contributions to acquisition and data interpretation, been involved in drafting the manuscript, and agreed with the final version to be published and to be responsible for all aspects, including integrity and accuracy of this work. MRL: made substantial contributions to clinical assessment and medical data interpretation, been involved in the manuscript critical revision, and agreed with the final version to be published and to be accountable for all aspects, including integrity and accuracy of this work. VC: made substantial contributions to acquisition and data interpretation and analysis, been involved in the manuscript critical revision, and agreed with the final version to be published and to be responsible for all aspects, including integrity and accuracy of this work. RS: made substantial contributions to conception and design, been involved in manuscript critical revision, given final approval of the version to be published, and agreed to be held responsible for all aspects, including integrity and accuracy of this work.
Ethics approval and consent to participate
Proband’s parents, signed informed consent for the use of DNA samples in intellectual disability research. This study has been approved by the medical ethical committee of the Centro Hospitalar do Porto (CHP, E.P.E.) - REF 2014.203 (144-DEFI/173-CES).
Consent for publication
Proband’s parents, signed informed consent for publication that includes medical information and photographs.
Competing interests
Dr. Paula Jorge is an Associate Editor for BMC Medical Genetics. The remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Paula Jorge, Email: paula.jorge@chporto.min-saude.pt.
Elsa Garcia, Email: elsa.garcia@synlab.pt.
Ana Gonçalves, Email: ana.goncalves@chporto.min-saude.pt.
Isabel Marques, Email: isabel.medeiros.marques@gmail.com.
Nuno Maia, Email: n.maia84@gmail.com.
Bárbara Rodrigues, Email: be_rodrigues_@hotmail.com.
Helena Santos, Email: santoshelena@iol.pt.
Jacinta Fonseca, Email: jacintafonseca@hotmail.com.
Gabriela Soares, Email: gabriela.soares@chporto.min-saude.pt.
Cecília Correia, Email: cecilia.correia@ipo.min-saude.pt.
Margarida Reis-Lima, Email: margarida.reislima@gmail.com.
Vincenzo Cirigliano, Email: vincenzo.cirigliano@synlab.es.
Rosário Santos, Email: mrn.santos@gmail.com.
References
- 1.AJMH V, Pieretti M, Sutcliffe JS, Fu Y-H, DPA K, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang F, Eussen BE, G-JB v O, LAJ B, Riggins GJ, Chastain JL, Kunst CB, Galjaard H, Thomas Caskey C, Nelson DL, Oostra BA, Warren ST. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65(5):905–914. doi: 10.1016/0092-8674(91)90397-H. [DOI] [PubMed] [Google Scholar]
- 2.de Vries BB, Halley DJ, Oostra BA, Niermeijer MF. The fragile X syndrome. J Med Genet. 1998;35(7):579–589. doi: 10.1136/jmg.35.7.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biancalana V, Glaeser D, McQuaid S, Steinbach P. EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders. Eur J Hum Genet. 2015;23(4):417–425. doi: 10.1038/ejhg.2014.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin P, Warren ST. Understanding the molecular basis of fragile X syndrome. Hum Mol Genet. 2000;9(6):901–908. doi: 10.1093/hmg/9.6.901. [DOI] [PubMed] [Google Scholar]
- 5.Usdin K, Hayward BE, Kumari D, Lokanga RA, Sciascia N, Zhao XN. Repeat-mediated genetic and epigenetic changes at the FMR1 locus in the fragile X-related disorders. Front Genet. 2014;5:226. doi: 10.3389/fgene.2014.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunter J, Rivero-Arias O, Angelov A, Kim E, Fotheringham I, Leal J. Epidemiology of fragile X syndrome: a systematic review and meta-analysis. Am J Med Genet A. 2014;164a(7):1648–1658. doi: 10.1002/ajmg.a.36511. [DOI] [PubMed] [Google Scholar]
- 7.Macpherson J, Murray A, Webb J, Jacobs P. Fragile X syndrome: of POF and premutations. J Med Genet. 1999;36(2):171–172. [PMC free article] [PubMed] [Google Scholar]
- 8.Stembalska A, Laczmanska I, Gil J, Pesz KA. Fragile X syndrome in females - a familial case report and review of the literature. Dev Period Med. 2016;20(2):99–104. [PubMed] [Google Scholar]
- 9.Maia N, Loureiro JR, Oliveira B, Marques I, Santos R, Jorge P, Martins S. Contraction of fully expanded FMR1 alleles to the normal range: predisposing haplotype or rare events? J Hum Genet. 2017;62(2):269–275. doi: 10.1038/jhg.2016.122. [DOI] [PubMed] [Google Scholar]
- 10.Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51(6):1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 11.Grasso M, Boon EM, Filipovic-Sadic S, van Bunderen PA, Gennaro E, Cao R, Latham GJ, Hadd AG, Coviello DA. A novel methylation PCR that offers standardized determination of FMR1 methylation and CGG repeat length without southern blot analysis. J Mol Diagn. 2014;16(1):23–31. doi: 10.1016/j.jmoldx.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plenge RM, Hendrich BD, Schwartz C, Arena JF, Naumova A, Sapienza C, Winter RM, Willard HF. A promoter mutation in the XIST gene in two unrelated families with skewed X-chromosome inactivation. Nat Genet. 1997;17(3):353–356. doi: 10.1038/ng1197-353. [DOI] [PubMed] [Google Scholar]
- 13.Pugacheva EM, Tiwari VK, Abdullaev Z, Vostrov AA, Flanagan PT, Quitschke WW, Loukinov DI, Ohlsson R, Lobanenkov VV. Familial cases of point mutations in the XIST promoter reveal a correlation between CTCF binding and pre-emptive choices of X chromosome inactivation. Hum Mol Genet. 2005;14(7):953–965. doi: 10.1093/hmg/ddi089. [DOI] [PubMed] [Google Scholar]
- 14.Wang Z, Yan A, Lin Y, Xie H, Zhou C, Lan F. Familial skewed x chromosome inactivation in adrenoleukodystrophy manifesting heterozygotes from a Chinese pedigree. PLoS One. 2013;8(3):e57977. doi: 10.1371/journal.pone.0057977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Hattab AW, Fang P, Jin W, Hughes JR, Gibson JB, Patel GS, Grange DK, Manwaring LP, Patel A, Stankiewicz P, Cheung SW. Int22h-1/int22h-2-mediated Xq28 rearrangements: intellectual disability associated with duplications and in utero male lethality with deletions. J Med Genet. 2011;48(12):840–850. doi: 10.1136/jmedgenet-2011-100125. [DOI] [PubMed] [Google Scholar]
- 16.El-Hattab AW, Schaaf CP, Fang P, Roeder E, Kimonis VE, Church JA, Patel A, Cheung SW. Clinical characterization of int22h1/int22h2-mediated Xq28 duplication/deletion: new cases and literature review. BMC Med Genet. 2015;16:12. doi: 10.1186/s12881-015-0157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS, Martin CL, Ledbetter DH. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanmarsenille L, Giannandrea M, Fieremans N, Verbeeck J, Belet S, Raynaud M, Vogels A, Mannik K, Ounap K, Jacqueline V, Briault S, Van Esch H, D'Adamo P, Froyen G. Increased dosage of RAB39B affects neuronal development and could explain the cognitive impairment in male patients with distal Xq28 copy number gains. Hum Mutat. 2014;35(3):377–383. doi: 10.1002/humu.22497. [DOI] [PubMed] [Google Scholar]
- 19.Witham S, Takano K, Schwartz C, Alexov E. A missense mutation in CLIC2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins. 2011;79(8):2444–2454. doi: 10.1002/prot.23065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Godler DE, Inaba Y, Schwartz CE, Bui QM, Shi EZ, Li X, Herlihy AS, Skinner C, Hagerman RJ, Francis D, Amor DJ, Metcalfe SA, Hopper JL, Slater HR. Detection of skewed X-chromosome inactivation in fragile X syndrome and X chromosome aneuploidy using quantitative melt analysis. Expert Rev Mol Med. 2015;17:e13. doi: 10.1017/erm.2015.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jorge P, Oliveira B, Marques I, Santos R. Development and validation of a multiplex-PCR assay for X-linked intellectual disability. BMC Med Genet. 2013;14:80. doi: 10.1186/1471-2350-14-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.