

Abstract
The authors report a child with spinal muscular atrophy with respiratory distress type 1 (SMARD1). She presented atypically with hypothyroidism and heart failure due to septal defects that required early heart surgery and microcephaly in association with cerebral atrophy and thin corpus collosum. The subsequent asymmetrical onset of diaphragmatic paralysis, persistent hypotonia, and generalized muscle weakness led to the suspicion of spinal muscular atrophy with respiratory distress type 1. Sanger sequencing confirmed a compound heterozygous mutation in the Immunoglobulin Mu Binding Protein 2 (IGHMBP2) gene, with a known mutation c.2362C > T (p.Arg788*) and a novel frameshift mutation c.2048delG (p.Gly683A1afs*50). Serial nerve conduction study and electromyography confirmed progressive sensorimotor polyneuropathy and neuronopathy. In summary, this case report describes a child with spinal muscular atrophy with respiratory distress type 1 also with congenital cardiac disease and endocrine dysfunction, expanding the phenotypic spectrum of this condition. A high index of suspicion is needed in diagnosing this rare condition to guide the management and genetic counseling.
Keywords: SMARD1, IGHMBP2 gene, congenital heart disease, neuropathy, children
Spinal muscular atrophy with respiratory distress type 1 (SMARD1) is characterized by intrauterine growth restriction, muscle weakness, and progressive respiratory distress with diaphragmatic paralysis.1-4 It has been mapped to the Immunoglobulin Mu Binding Protein 2 (IGHMBP2) gene on chromosome 11q13.3, which encodes the immunoglobulin μ-binding protein 2.4
The authors report a child with microcephaly, hypothyroidism, and early progressive respiratory distress associated with heart failure on initial presentation. Subsequent bilateral diaphragmatic paralysis and progressive muscle weakness with predominant distal involvement led to the genetic confirmation of spinal muscular atrophy with respiratory distress type 1.
Case Report
The proposita is the first child of a nonconsanguineous Chinese couple. Antenatal history was unremarkable except for intrauterine growth restriction. She was born at full term via normal spontaneous delivery with a low birth weight of 1.9 kg. Initial examination showed microcephaly (head circumference 31 cm, <3rd percentile for age) and hypotonia. During her first month of life, slow oral feeding with poor weight gain was observed. Investigation at that time included echocardiogram which showed a moderate size secundum atrial septal defect (5 mm × 6 mm) and large ventricular septal defect (9 mm × 5 mm), and abnormal thyroid function with markedly elevated thyroid-stimulating hormone (932.8 MIU/L; normal reference: 1.12-4.47 MIU/L), and low free thyroxine level (<5.4 pmol/L; normal reference: 11.0-20.6 pmol/L). Thyroid scan study, antithyroglobulin and antithyroid peroxidase antibodies, were normal. After thyroxine replacement, her thyroid function normalized. As the heart failure progressed despite medical treatment, surgical closure for both the ventricular and atrial septal defects was performed at 3 months old. There was no residual lesion after the operation. However, she could not be weaned off from the ventilatory support despite the resolution of heart failure. Postoperative ultrasound confirmed left diaphragmatic paralysis, so diaphragmatic plication was performed 3 weeks after the cardiac surgery. The right diaphragm was unaffected at that time. Follow-up ultrasound, however, showed subsequent development of right diaphragmatic paralysis at 4 months of age. She was put on continuous mechanical ventilation via tracheostomy from the age of 5 months.
As she had persistent weakness, hypotonia, and decreased generalized movement with hyporeflexia, neuromuscular workup was initiated. Investigation at 5 months old showed normal creatine kinase level. The nerve conduction study confirmed sensorimotor mixed axonal and demyelinating polyneuropathy. The sural nerve biopsy showed axonal degeneration and secondary alteration of myelin (see Figure 1). Muscle biopsy from quadriceps showed nonspecific findings with a tiny focal group of small angulated atrophic fibers (see Figure 2). Initial genetic study of the Survival Of Motor Neuron 1, Telomeric (SMN1), Small Nuclear Ribonucleoprotein Polypeptide N (SNRPN), and Peripheral Myelin Protein 22 (PMP22) genes mutation and array-based comparative genomic hybridization, were all normal. Brain magnetic resonance imaging performed at 8 months old showed cerebral atrophy with delayed myelination pattern and diffusely thinned corpus callosum (see Figure 3). Metabolic workup including plasma amino acid, very-long-chain fatty acid, free carnitine and acylcarnitine profile, and lactate and pyruvate, were all unremarkable. Given persistent poor oral feeding, she required nasogastric tube feeding since first year of life and was put on gastrostomy feeding since 11 months old.
Figure 1.

Electron microscopy of sural nerve biopsy at 6 months of age showing (A) an axon with early degeneration, featuring flocculent quality of the axoplasm (TEM, ×23 000); (B) atrophic axon with thin myelin and associated debris underneath the myelin (left), adjacent to a relatively preserved axon with normal looking Schwann cell nucleus and myelin (right; TEM, ×11 000); (C) Low power magnification on electron microscopy shows isolated axonal degeneration and thin myelin that could be physiological at that time in life, and many unmyelinating fibers. The presence of collagen pockets could indicate loss of unmyelinated fibers.
Figure 2.
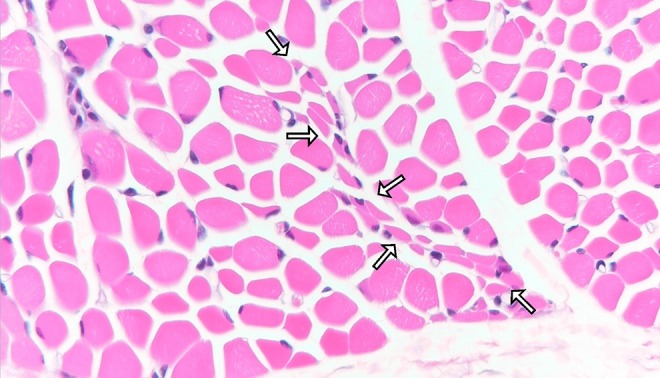
H&E staining of quadriceps muscle biopsy at 6 months of age showing a tiny group of small angulated atrophic fibers (enriched by arrows), which could suggest neuropathy (H&E, paraffin section, ×400).
Figure 3.
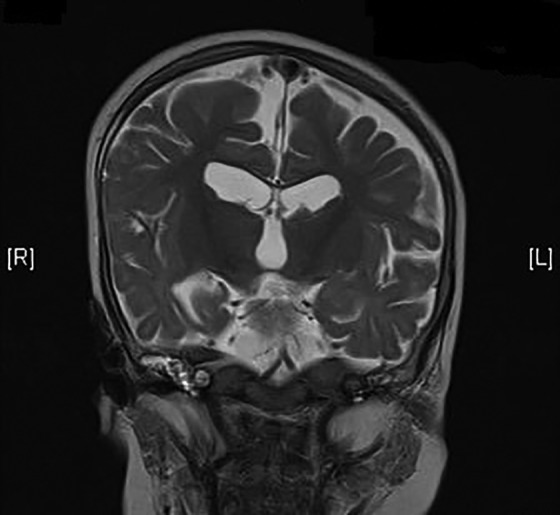
T2-weighted magnetic resonance imaging (MRI) of the brain at 8 months of age showing thin corpus callosum and cerebral atrophy.
At 22 months old, she was transferred to our hospital for further management. Initial examination showed her body weight and height at the 97th percentile and head circumference at the 3rd percentile for age. There was mild facial weakness but no ptosis or ophthalmoplegia. Her upper and lower extremities had generalized hypotonia, areflexia and weakness with minimal spontaneous movement except for mild active shoulder adduction and shrugging. There was also mild finger flexor tightness, but no obvious fingertip fat pad was seen. Spinal muscular atrophy with respiratory distress type 1 was suspected. Direct sequencing of the IGHMBP2 gene detected compound heterozygous mutations.
She further developed intermittent high blood pressure (>95th percentile) at 23 months of age. Investigations at that time included echocardiogram and blood tests on renal and thyroid function were all normal. There was no evidence of renal artery stenosis on renal mercaptoacetyltriglycine (MAG3) scan. The urine catecholamine profile, however, was found to be mildly elevated: norepinephrine: 210 to 370 nmol/mmolCr (normal < 120 nmol/mmolCr), normetanephrine: 143 nmol/mmolCr (normal < 40 nmol/mmolCr), metanephrine: 133 nmol/mmolCr (normal < 60 nmol/mmolCr), homovanillic acid: 11 to 17 µmol/mmolCr (normal < 8.1 µmol/mmolCr), vanillylmandelic acid: 10 to 20 µmol/mmolCr (normal < 10 µmol/mmolCr). Computed tomography scan of the abdomen and metaiodobenzylguanidine (MIBG) scan confirmed no evidence of an adrenal tumor. The blood pressure spontaneously normalized without the use of antihypertensive treatment. She started to have recurrent urinary tract infections since the age of 32 months old. Subsequent voiding cytogram confirmed small bladder volume and bilateral vesicoureteric reflux with dilated renal pelvis. An antivesicoureteric reflux surgery was planned but family declined. She also had the problem of excessive sweating and constipation. At the age of 4, she was on continuous mechanical ventilation via tracheostomy and gastrostomy feeding. She did not have any head control but had good social smile and could listen to conversation. She could not understand 1 stage command or indicate choice by word, eye pointing or facial expression. She died of urosepsis and heart failure at the age of 4 years and 7 months.
Electrophysiological Study
The proposita had repeated nerve conduction studies and electromyography (EMG) at 5, 11, and 22 months old. The initial study at 5 months old confirmed no sensory nerve action potential response from bilateral median, ulnar, and sural nerves. Motor nerve conduction study at that time yielded compound muscle action potential responses from the bilateral median and ulnar nerves, but no response from bilateral tibial and peroneal nerves. The median nerves compound muscle action potential amplitudes upon distal stimulation (left 1.4 mV, right 0.5 mV, age-appropriate norm5: 4.76 ± 1.13 mV) and the conduction velocities (left 21 m/s, right 18 m/s; age appropriate norm5: 37 ± 4.38 m/s) were decreased. The ulnar nerves compound muscle action potential amplitude upon distal stimulation (left 1.9 mV, right 0.1 mV, age-appropriate norm5: 6.61 ± 1.72 mV) and conduction velocities (left 18 m/s, right 16 m/s; age appropriate norm5: 40.5 ± 4.24 m/s) were also decreased. Upon the follow-up study at 11 months old, compound muscle action potential responses from the bilateral median and ulnar nerves could no longer be obtained. Needle EMG performed at 5 months old with activation at the right rectus femoris muscles reported normal motor unit action potential (MUAP) morphology with normal recruitment. When the test was repeated at 11 months old with activation at both left rectus femoris and right flexor digitorum superficialis muscles, there were new findings of abnormal spontaneous discharges with fibrillation and incomplete recruitment. Follow-up study at 22 months old at the right deltoid and right quadriceps muscles showed infrequent fibrillation at rest, but an increase in MUAP duration (9-10 milliseconds) with polyphasia on activation with decrease recruitment and incomplete interference pattern compatible to chronic denervation (see Figure 4).
Figure 4.

Electromyography (EMG) at right deltoid at 22 months: Upon activation, motor unit potentials has prolonged duration (10 milliseconds) and polyphasic morphology with amplitude up to 800 μV. Interference pattern was incomplete.
Genetic Study Findings
The genetic study was carried out by direct Sanger sequencing. Two heterozygous mutations were identified in the IGHMBP2 gene (see Figure 5). The first mutation c.2362C > T (p.Arg788*) is a known nonsense mutation causing spinal muscular atrophy with respiratory distress type 1.6 The second mutation c.2048delG (p.Gly683Alafs*50) was a novel mutation with deletion at coding nucleotide 2048, which resulted in reading frameshift. The predicted termination of the protein translation would stop at 50 amino acids downstream from the mutation site (codon 683) and the translated protein, if any, would be more than 200 amino acids shorter than the wild-type protein and is expected to be nonfunctional. This mutation, is therefore predicted to be disease-causing, with premature termination of protein translation resulted in truncated protein product. The function of this truncated protein is likely to be adversely affected. Father was confirmed to have the p.Arg788* mutation as a carrier. However, mother preferred no genetic testing so we could not verify her carrier status.
Figure 5.

Electropherograms of the IGHMBP2 gene of the proband: (upper panel) c.2048delG (p.Gly683Alafs*50); (lower panel) c.2362C>T(p.Arg788*). Both in same direction. The heterozygous sites are denoted by the letter N and arrows.
Discussion
Spinal muscular atrophy with respiratory distress type 1, also known as distal spinal muscular atrophy type 1, is a severe, rare form of autosomal recessive disease with severe infantile onset and high mortality. It is characterized by intrauterine growth restriction, profound hypotonia and weakness with absent reflexes, and abnormal finger fat pads. It is especially marked by the presence of progressive respiratory distress with diaphragmatic paralysis,1-3 and the respiratory failure usually manifested between 6 weeks and 6 months in combination with either diaphragmatic eventration or premature birth.7 Our patient exhibited some typical features described in children with spinal muscular atrophy with respiratory distress type 1. These include the presence of intrauterine growth restriction, profound hypotonia, oromotor dysfunction, progressive respiratory distress, bilateral diaphragmatic paralysis with initial unilateral presentation, significant generalized weakness, and later, autonomic dysfunction. On the other hand, she also had the very atypical associated presentation of congenital heart disease with septal defects, thyroid dysfunction, and microcephaly with thin corpus collosum, which have not been reported in previous SMARD1 literature. The early-onset hypothyroidism and heart failure symptoms were initially thought to be the main cause of her respiratory distress and poor feeding. This, together with the fact that her diaphragmatic paralysis occurred only after open heart surgery and was originally thought to be a postoperative complication (which can occur in 0.3%-2.8% of cases after cardiac surgery8), explained why spinal muscular atrophy with respiratory distress type 1 was not suspected in the first place.
Sanger sequencing confirmed the compound heterozygous mutations in the IGHMBP2 gene in our patient. The first mutation c.2362C>T (p.Arg788*) is a known mutation that had been earlier reported,6 whereas the second mutation c.2048delG (p.Gly683Alafs*50) is a novel variant. A defect in the IGHMBP2 (immunoglobulin-mu binding protein 2) gene, which is located on chromosome 11q13.2-13.4, causes a dysfunction of its encoded multidomain protein involved in immunoglobulin class switching and transcription as well as pre-mRNA processing and regulation of DNA replication.4,7,9 However, how the dysfunction of such a protein leads to nerve and neuronal degeneration is still not fully understood.
The serial nerve conduction studies in our patient confirmed severe combined axonal and demyelinating polyneuropathy affecting both motor and sensory nerves since very young age and had a progressive course. The EMG findings suggested a neurogenic progression with the initial grossly normal findings at 5 months old to the subsequent chronic denervation pattern when the study was repeated at 22 months old. The findings are compatible to an underlying neurogenic process with Wallerian degeneration of nerve fibers and axonal atrophy causing neurogenic muscle atrophy.10 While some reports highlighted the preservation of sensory nerve responses1 and predominant lower limb involvement11,12 in patients with spinal muscular atrophy with respiratory distress type 1, the findings in our patient showed combined sensorimotor peripheral nerve involvement in agreement with Majid et al. study10. In contrast to previous studies, our patient had significant upper as well as lower limb involvement since very young age.
Our patient also had autonomic dysfunction with intermittent hypertension, neurogenic bladder, excessive sweating, and constipation. Of interest was the finding of persistent elevated urinary catecholamine level during the workup for possible causes of hypertension. This has been reported in patients with spinal muscular atrophy during episodes of tachycardia13 and in patients with motor neuron disease,14,15 but not previously in patients with spinal muscular atrophy with respiratory distress type 1. While it was suggested that in patients with motor neuron disease the rise in noradrenaline level is secondary to central autonomic network abnormality,14 other studies in muscular dystrophy suggested that creatinine-based measurements have pitfalls for patients with neuromuscular diseases.16 The persistently elevated urine catecholamine/creatinine profile could be secondary to poor muscle bulk with our patient had a persistently low serum creatinine levels (12-18 mmol/L, reference range: 19-59 mmol/L), thereby causing a falsely positive result. The normal MIBG scan findings and the above possible explanation prevent further unnecessary investigation.
Conclusion
We present a child with spinal muscular atrophy with respiratory distress type 1 with compound heterozygous IGHMBP2 mutations compatible with an autosomal recessive inheritance. The association of multisystem abnormalities, including congenital heart disease, microcephaly, thin corpus collosum, and abnormal thyroid function, observed in our patient has not been reported previously. The findings expand the phenotypic presentation for spinal muscular atrophy with respiratory distress type 1 and affirm a high index of suspicion is required in the diagnosis of this rare neuromuscular condition.
Footnotes
Author Contributions: Dr. Annie TG Chiu wrote the first draft of the manuscript, coordinated with the co-authors in data collection, and contributed to the update of the manuscript, and gave final approval of the version to be published. Dr. Sophelia HS Chan wrote and revised the manuscript, oversaw the study, contributed to the data collection and interpretation, and gave final approval of the version to be published. Dr Wu SP contributed to data collection, revising the manuscript and gave final approval of the version to be published. Dr SH Ting contributed to the data collection and figure preparation for the muscle and nerve biopsy results and figures, and gave final approval of the version to be published. Dr. Brain HY Chung contributed to the revising the manuscript and gave final approval of the version to be published. Dr. Angel OK Chan contributed to the data collection and the preparation of the genetic study result and figure, and gave final approval of the version to be published. Dr. Virginia CN Wong contributed to revising the manuscript and gave final approval of the version to be published.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Annie Ting Gee Chiu, MBBS(HK), MRCPCH  http://orcid.org/0000-0003-0249-9725
http://orcid.org/0000-0003-0249-9725
Ethical Approval: Being a case report involving a single case, the IRB approval was not required as per the hospital policy.
References
- 1. van der Pol WL, Talim B, Pitt M, von Au K. 190th ENMC International Workshop: Spinal muscular atrophy with respiratory distress/distal spinal muscular atrophy type 1: 11-13 May 2012, Naarden, the Netherlands. Neuromuscul Disord. 2013;23(7):602–609. [DOI] [PubMed] [Google Scholar]
- 2. Kaindl AM, Guenther UP, Rudnik-Schoneborn S, et al. Spinal muscular atrophy with respiratory distress type 1 (SMARD1). J Child Neurol. 2008;23(2):199–204. [DOI] [PubMed] [Google Scholar]
- 3. Eckart M, Guenther UP, Idkowiak J, et al. The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Pediatrics. 2012;129(1): e148–e156. [DOI] [PubMed] [Google Scholar]
- 4. Grohmann K, Schuelke M, Diers A, et al. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat Genet. 2001;29(1):75–77. [DOI] [PubMed] [Google Scholar]
- 5. Cai F, Zhang J. Study of nerve conduction and late responses in normal Chinese infants, children, and adults. J Child Neurol. 1997;12(1):13–18. [DOI] [PubMed] [Google Scholar]
- 6. Fanos V, Cuccu A, Nemolato S, Marinelli V, Faa G. A new nonsense mutation of the IGHMBP2 gene responsible for the first case of SMARD1 in a Sardinian patient with giant cell hepatitis. Neuropediatrics. 2010;41(3):132–134. [DOI] [PubMed] [Google Scholar]
- 7. Guenther UP, Varon R, Schlicke M, et al. Clinical and mutational profile in spinal muscular atrophy with respiratory distress (SMARD): defining novel phenotypes through hierarchical cluster analysis. Hum Mutat. 2007;28(8):808–815. [DOI] [PubMed] [Google Scholar]
- 8. Joho-Arreola AL, Bauersfeld U, Stauffer UG, Baenziger O, Bernet V. Incidence and treatment of diaphragmatic paralysis after cardiac surgery in children. Eur J Cardiothorac Surg. 2005;27(1):53–57. [DOI] [PubMed] [Google Scholar]
- 9. Lim SC, Bowler MW, Lai TF, Song H. The Ighmbp2 helicase structure reveals the molecular basis for disease-causing mutations in DMSA1. Nucleic Acids Res. 2012;40(21):11009–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Majid A, Talat K, Colin L, Caroline R, Helen K, Christian de G. Heterogeneity in spinal muscular atrophy with respiratory distress type 1. J Pediatr Neurosci. 2012;7(3):197–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. San Millan B, Fernandez JM, Navarro C, Reparaz A, Teijeira S. Spinal muscular atrophy with respiratory distress type 1 (SMARD1) Report of a Spanish case with extended clinicopathological follow-up. Clin Neuropathol. 2016;35(2):58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pitt M, Houlden H, Jacobs J, et al. Severe infantile neuropathy with diaphragmatic weakness and its relationship to SMARD1. Brain. 2003;126(pt 12):2682–2692. [DOI] [PubMed] [Google Scholar]
- 13. Hachiya Y, Arai H, Hayashi M, et al. Autonomic dysfunction in cases of spinal muscular atrophy type 1 with long survival. Brain Dev. 2005;27(8):574–578. [DOI] [PubMed] [Google Scholar]
- 14. Ohno T, Shimizu T, Kato S, Hayashi H, Hirai S. Effect of tamsulosin hydrochloride on sympathetic hyperactivity in amyotrophic lateral sclerosis. Auton Neurosci. 2001;88(1-2):94–98. [DOI] [PubMed] [Google Scholar]
- 15. Shimizu T, Hayashi H, Kato S, Hayashi M, Tanabe H, Oda M. Circulatory collapse and sudden death in respirator-dependent amyotrophic lateral sclerosis. J Neurol Sci. 1994;124(1):45–55. [DOI] [PubMed] [Google Scholar]
- 16. Braat E, Hoste L, De Waele L, et al. Renal function in children and adolescents with Duchenne muscular dystrophy. Neuromuscul Disord. 2015;25(5):381–387. [DOI] [PubMed] [Google Scholar]