

Abstract
Introduction:
Increased galectin-3 is associated with ischemic cardiomyopathy, although its role in early remodeling post-myocardial infarction (MI) has not been fully elucidated. There are no data demonstrating that blocking galectin-3 expression would have an impact on the heart and that its relationship to remodeling is not simply an epiphenomenon. The direct association between galectin-3 and myocardial inflammation, dysfunction, and adverse cardiovascular outcomes post-MI was examined using clinical and translational studies.
Methods:
We performed expression analysis of 9753 genes in murine model of acute MI. For galectin-3 loss of function studies, homozygous galectin-3 knock-out (KO) mice were subjected to coronary artery ligation procedure to induce acute MI (MI, N = 6; Sham, N = 6). For clinical validation, serum galectin-3 levels were measured in 96 patients with ST-elevation MI. Echocardiographic and angiographic parameters of myocardial dysfunction and 3-month composite outcome including mortality, recurrent MI, stroke, and heart failure hospitalization were measured.
Results:
In the infarct regions of murine models, galectin-3 was a robustly expressed gene. Elevated galectin-3 expression strongly correlated with macrophage-mediated genes. Galectin-3 KO mice showed reduced myocardial macrophage infiltration after acute MI. Galectin-3 levels were higher in patients with early systolic dysfunction, and predicted 3-month major adverse cardiovascular events (area under the curve [AUC]: 0.917 ± 0.063; P = .001).
Conclusions:
Galectin-3 is directly associated with early myocardial inflammation post-MI and may represent a potential target for therapeutic inhibition.
Keywords: Galectin-3, myocardial infarction, inflammation, fibrosis
Introduction
With advances in the treatment of acute myocardial infarction (MI), including more timely and complete revascularization, survival rates are improving. However, a subset of these patients develop ischemic cardiomyopathy as a consequence of progressive cardiac remodeling.1,2 Sudden myocardial tissue injury leads to loss of cardiomyocytes and the surrounding microvasculature. This triggers a tissue reparative response leading to steady deposition of myocardial proteins and fibroblast proliferation in place of the injured cardiomyocytes.3–5 Once acute tissue injury is complete, adaptive remodeling occurs in response to the insult.3,6 The acclimating ventricles frequently dilate, and the dead cardiomyocytes are progressively substituted by collagen fibers and fibroblasts.7 Ultimately, the pathophysiologic process of myocardial injury and repair leads to the development of ischemic cardiomyopathy and is associated with long-term adverse outcomes. Early initiation of therapy targeted to the remodeling process can therefore help prevent adverse adaptations and loss of myocardial function that leads to the development of heart failure (HF). Identifying potential mediators of this process will play a key role in developing therapeutic targets.
A recent consensus statement released by the American Heart Association (AHA) recognizes a few HF-associated biomarkers, and their serum level measurements can be a non-invasive method to gain knowledge about the severity of the disease and assist in the diagnosis, prognostication, and management of HF.8 These biomarkers comprise natriuretic peptides, highly sensitive troponin, soluble suppressor of tumorigenicity 2, mid-regional pro-adrenomedullin, galectin-3, cystatin-C, procalcitonin, and interleukin-6. The AHA highlights the necessity to further assess these markers for their therapeutic significance. Our study focuses on galectin-3, as the AHA consensus statement note that patients with chronic ambulatory HF and higher levels of galectin-3 are associated with mortality, although this association is weaker than other biomarkers in the chronic ambulatory HF setting. Prior studies have reported galectin-3 as a circulating biomarker of left ventricular (LV) dysfunction and poor outcomes after acute MI.9–14 However, a study by O’Donoghue15 showed that although there were associations between increasing levels of galectin-3 and the risk of cardiovascular death or HF, it did not remain a significant predictor after adjusting for traditional risk factors in contrast to other studied biomarkers.
Galectin-3 is a member of the β-galactoside-binding animal lectin family, with a specific amino acid sequence able to recognize β-galactose.16,17 Galectin-3 is secreted by macrophages and other cells, and stimulates the release of various growth factors as well as pro-inflammatory cytokines.18 After macrophage activation, cytosolic galectin-3 shifts to the plasma membrane where it is released.19 Once released, galectin-3 can function as an extracellular cytokine to activate cells by binding to its receptors.17 Compared with other pro-fibrotic agents, galectin-3 specifically increases the expression of stiff collagens.9 Stiff variants of collagen are non-contractile and contribute to cardiac dysfunction. Although galectin-3 has developed rapidly as a biomarker of chronic HF, there are limited data evaluating the expression of galectin-3 after acute MI.
The functional implications of galectin-3 release after ischemic myocardial injury and its relationship with other inflammatory and pro-fibrotic mediators of cardiac remodeling remain unclear. Due to the complex cellular and neurohumoral mechanisms involved in the remodeling process post-MI, it is difficult to determine whether galectin-3 is a biomarker of this process or a crucial mediator. It is possible that galectin-3 may play a key role in the remodeling process post-MI which is maladaptive and involves myocardial fibrosis and collagen deposition and, if true, could be a potential target for therapeutic inhibition. However, no studies to date have directly demonstrated its direct role in the remodeling process through process of antagonism or over-expression of galectin-3. Studies to better define the time course of galectin-3 expression post-MI and its relationship with development of myocardial dysfunction have been lacking. Correlation of galectin-3 expression with histologic changes in myocardial tissue may be best done with a translational approach because this would otherwise not be practical in patients. In this study, we use 2 pre-clinical models and examine serum samples in patients to demonstrate that galectin-3 expression is increased after acute MI and is associated with inflammation and development of myocardial dysfunction.
Methods
All pre-clinical procedures and protocols conformed to institutional guidelines for the care and use of animals in research and were reviewed and approved by the University at Buffalo Institutional Animal Care and Use Committee (IACUC). The human studies conformed to the local good clinical research practice and institutional review board (IRB) guidelines for the protection of human subjects. For human studies, written consent was obtained for the utilization of serum samples for biomarker studies, chart review for clinical data extraction, and follow-up phone calls. Patient identifiers were securely processed using our existing Health Insurance Portability and Accountability Act (HIPAA) guidelines.
Murine studies
Animal model
The microarray experiments were performed in mice (age 10-12 weeks, N = 24) using our study protocol described previously.20 For the real-time polymerase chain reaction (PCR) analysis, a separate subgroup of age-matched C57Bl/6 (MI, N = 7; Sham, N = 6) was used. For galectin-3 loss of function studies, homozygous galectin-3 knock-out (KO) mice and wild-type (WT) controls were purchased from Jackson Laboratory and subjected to acute MI induction (C57Bl/6 background, MI, N = 6; Sham, N = 6). Mice underwent permanent ligation of the left anterior descending (LAD) coronary artery producing an infarct in the anterior/anteroseptal walls of the LV. Concisely, mice were anesthetized with ketamine (1 mg/kg intramuscular) and xylazine (5 mg/kg subcutaneous), and were intubated to undergo a ligation procedure (6-0 prolene) of the left anterior coronary artery. The chest wall was closed with 5-0 silk sutures, and the mice were left to recover at 30°C. Sham surgeries were executed in the same manner, excluding the coronary artery ligation. To minimize discomfort, distress, and keep the pain to an absolute minimum, all studies were performed on anesthetized animals. Mice were continuously observed during procedure and for at least 1 hour into recovery. Euthanasia procedure conformed to the guidelines from the Panel on Euthanasia of the American Veterinary Medical Association.
Gene expression profile after acute MI
We induced acute MI in Swiss mice (aged 10-12 weeks, N = 24) as previously described.20 We have previously reported cDNA microarray results (Incyte mouse GEM-2 cDNA libraries with 9753 reporter genes) that showed an increased expression of galectin-3 and other genes related to macrophage activity.21
Microarray analyses were performed for 8 time-points to identify a comprehensive gene expression profile in both remote and infarct myocardial sections. To increase accuracy, we included only the reporter spots in which more than 40% of pixels displayed fluorescence greater than 2.5 times the local background. The data mining protocol and validation were adopted as outlined earlier.22 The Spearman rank-order correlation analysis was performed to measure the strength and direction of association between-galectin-3 and other overexpressed genes.
RNA isolation and real-time quantitative PCR
We performed additional studies on C57Bl6 mice for the quantitative analysis of galectin-3 expression 1 week after acute MI. Total RNA was isolated from mouse heart using Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA) following the manufacturer’s protocol. RNA purity (A260/A280 ratio ≥ 1.8) and concentration were measured using the NanoDrop. Total RNA was stored at −80°C. The RNA was reverse transcribed to cDNA using a reverse transcriptase kit (Quick-DNA Miniprep Kit; Zymo Research, Irvine, CA). In each quantitative polymerase chain reaction (qPCR) reaction, the cDNA equivalent of 5 ng RNA was used. The qPCR reactions contained FastStart Universal SYBR Green Master, Rox (Roche Life Science, Indianapolis, IN) and the forward and reverse primer each at a final concentration of 1 μM). The amplification protocol consisted of 3 segments: 10 minutes 95°C followed by 40 cycles of 30 seconds 95°C, 1 minute 58°C, and 1 minute 72°C; in segment 3, 1 minute 95°C, 30 seconds 55°C, and 30 seconds 95°C. The sequences of the primers for real-time PCR are shown in supplement (Table S1). The housekeeping gene GAPDH was used as the internal control. Relative expression of mRNA species was calculated using the comparative threshold (CT) cycle number method. Briefly, for each sample, a difference in CT values (ΔCT) is calculated for each mRNA by taking the mean CT of the duplicate tubes and subtracting the mean CT of the duplicate tubes for the reference RNA (GAPDH) measured on an aliquot from the same reverse transcription reaction. The ΔCT for the treated sample is then subtracted.
Immunohistochemistry and tissue morphometry
Macrophages were stained with mouse-specific anti-F4/80 antibody (Abcam, #AB6640, Cambridge, MA) that is known to react with murine macrophage glycoprotein). Galectin-3 was stained with mouse-specific anti-galectin-3 antibody (R&D AF1154, Minneapolis, MN). Serial sections were subjected to immunohistochemical staining with antibodies that detect macrophages (F4/80) and galectin-3, using species-matched positive and negative controls. The immunohistochemical studies were performed at Roswell Park Cancer Institute (RPCI) pathology resource core lab according to the standard manufacturer’s protocols. Investigators blinded to the study groups performed data analysis. For tissue morphometry, formalin-fixed paraffin-embedded mice hearts were sectioned into 5 µm thickness. These sections were stained using the Masson trichrome stain for the detection of myocardial fibrosis according to the manufacturer’s protocol. To estimate the degree of fibrosis, photomicrographs were obtained (Olympus BX45, Center Valley, PA), and the total area of fibrosis was quantified using NIH software, ImageJ (National Institute of Health; Bethesda, MD, USA).
Human studies
Patient selection
Written informed consent was granted by 96 patients to partake in an observational prospective study. The recruitment was performed from March to November 2016, at Buffalo General Medical Center and Gates Vascular Institute, Buffalo, NY, USA. Patients who presented with ST-elevation myocardial infarction (STEMI) and subsequently underwent primary percutaneous intervention (PCI), with the procedure performed within 90 minutes after initial medical contact with the patient, were eligible for inclusion in the study. Patients with malignancies at the time of recruitment or documented history of MI or stroke within the preceding 3 months were excluded. Ten healthy volunteers were also recruited as controls after providing informed consent. Institutional review board approval for the study protocol was obtained from University at Buffalo.
Serum galectin-3 measurements
For measurement of serum galectin-3 levels, serum samples were collected within 12 to 48 hours from initial presentation. Samples were centrifuged and then kept in heparinized test tubes in refrigerators at −800°C prior to performing galectin-3 assays. To measure serum galectin-3, a pre-coated galectin-3 specific 96-well strip microplate Enzyme-linked Immunosorbent Assays (ELISA) kit (R&D Systems; Minneapolis, MN, USA) was used, and optical densities were analyzed with standard galectin-3 concentrations (at ng/mL) based on the manufacturer’s recommendations.
Clinical outcomes
Baseline characteristics, available laboratory data, and outcomes were collected by a combination of chart review and 3-month follow-up phone interviews. Left ventricular ejection fraction (LVEF) measured by ventriculography at the time of PCI was recorded. Echocardiographic characteristics including LVEF, grade of diastolic dysfunction, E/E′ ratio, left atrial volume index (LAVI), left ventricular wall motion score index (LVSI) based on a 17-segment model, and percentage of normally functioning myocardium (%FM) were obtained from echocardiograms performed for routine clinical purposes as part of the patient’s hospitalization. The association between galectin-3 levels and baseline characteristics of study subjects was analyzed. Comparisons between galectin-3 levels in study subjects vs controls, and in patients with and without adverse outcomes were performed. The primary outcome measure was the composite endpoint of early major adverse cardiovascular events (MACEs), which is defined as all-cause mortality, stroke, recurrent MI, and/or hospitalization due to congestive heart failure (CHF) within 3 months. The secondary outcome measures were (a) cardiovascular death; (b) mildly (≤45%), moderately (≤40%), and severely (≤35%) reduced LVEF on ventriculography obtained during PCI; (c) echocardiographic variables obtained within 72-hour post-PCI, including the presence of diastolic dysfunction greater than stage 1, elevated E/E′ ratio of ≥15, moderately abnormal LAVI >35, LVSI >1.5, and %FM <50; and (d) length of hospitalization >48 hours.
Statistical analysis
Regarding microarray studies, we expressed data in densitometric units as fold-change over control. The microarray study design used a pooled tissue sample (N = 3) for 8 time-points, including infarct and remote zones as well as sham controls for which mRNA was isolated. A weighted gene pool was assumed to be present during hybridization. Group differences were analyzed using the Student t-test for normally distributed data. For clinical studies, data are expressed as mean ± standard error of mean (SEM) and percentage of patients for continuous and discrete variables, respectively. A chi-square (χ2) test or Fisher exact test was used to compare categorical variables and Mann-Whitney U or Kruskal-Wallis test was used to compare continuous variables with skewed distribution. The Spearman correlation was used to evaluate the association between Galectin-3 and other variables as applicable. Receiver-operating characteristic (ROC) curve analysis was conducted for galectin-3 to predict MACE. All the analyses were performed using SPSS software for Windows, version 23 (SPSS, Inc., IL, USA). Statistical significance was set at a 2-sided P value of <.05.
Results
Myocardial galectin-3 expression after acute MI was robust and dynamic
We identified 9753 genes that were expressed post-MI. Expression of galectin-3 was significantly increased along with other genes responsible for myocardial inflammation and fibrosis in the infarcted region compared with remote (Figure 1). Only subtle changes in galectin-3 expression were present at earlier time-points (2-4 days) in the remote myocardial segments, whereas infarcted regions showed more robust changes, with a peak increase of 7.8-fold at 2 weeks. Due to low specificity of the genomic data, we confirmed post-MI galectin-3 using a real-time quantitative PCR. The mice with acute MI had 22.5-fold (P < .05) increase of myocardial galectin-3 gene expression levels compared with Sham controls (Figure S1).
Figure 1.

Myocardial gene expression of galectin-3 after acute MI in mice. Panel A: Representative Trichrome-labeled section from infarct region. Panel B: Similarly stained section from remote region. The data columns represent fold-change of mRNA expression at different time-points of genomic profiling after acute MI induction. Panel C: Maximal galectin-3 expression was noted at 7-21 days with a chronic elevation present till 90 days post-MI. N = 3, each group; total, 8 time-points. *P < .05, 1-4 days vs 7-21 days; **P < .05, 7-21 days vs 45-90 days. Panel D: The remote region showed minimal and statistically non-significant galectin-3 mRNA expression over time. MI indicates myocardial infarction.
Time-specific Galectin-3 gene expression strongly correlated with other mediators of inflammation after acute MI
As our prior study demonstrated potential pro-inflammatory and fibrotic effects of galectin-3 in vitro, we performed time-dependent regression analyses to examine whether increased galectin-3 gene expression is associated with other macrophage-mediated genes (Table 1). Galectin-3 expression strongly correlated with other macrophage-specific genes, including macrosialin (CD68) and macrophage expressed gene-1 (Mpeg1). CD68 encodes a 110-kDa transmembrane glycoprotein that is highly expressed by human monocytes and tissue macrophages.23 CD68 expression in macrophages is useful for identifying macrophage-specific pathology as opposed to lymphocytes or neutrophils. In addition, Mpeg1 is expressed in high amounts by murine macrophages. Computer-assisted similarity searches indicate that this gene is a novel gene that shares ancestry to perforin, a cell lytic protein24 (Table 1).
Table 1.
Early (1-14 days) galectin-3 gene expression after acute MI in relation to other mediators of cardiac inflammation.
| Correlation with Galectin-3 mRNA (fold-increase of expression) | |
|---|---|
| Galectin-3 | rs = 1 (P = NA) |
| Neurohumoral | |
| Natriuretic peptide precursor | rs = –0.35 (P = .55) |
| Macrophage-mediated | |
| Macrophage expressed gene 1 | rs = 0.90 (P = .03*) |
| CD68 antigen (macrosialin) | rs = 0.98 (P = .001*) |
| Cathepsin S | rs = 0.97 (P = .004*) |
Abbreviations: MI, myocardial infarction; rs, the Spearman correlation coefficient.
P < .05.
Homozygous galectin-3 KO mice showed reduced myocardial macrophage infiltration after 1 week
To examine whether lack of galectin-3 activity is associated with alteration of post-MI myocardial inflammation, we induced acute MI in galectin-3 KO mice. First, we confirmed the phenotypic characteristics comparing the myocardial galectin-3 positivity. Wild-type mice showed abundant galectin-3 positive cells; KO mice showed no positivity (Figure 2, Panels A1-A4). Compared with WT mice with acute MI, there was morphologic evidence of reduced myocardial tissue damage on histology in galectin-3 KO mice. Furthermore, galectin-3 KO mice had significant reduction of myocardial macrophage infiltration as evidenced by reduced F4/80 positivity (Figure 2, Panels B1-B4).
Figure 2.

Post-MI myocardial galectin-3 expression and macrophage infiltration 1 week after LAD ligation. Wild-type mice showed scattered areas of galectin-3 positivity (Panel A1), which was absent in the myocardial sections obtained from galectin-3 KO mice (Panel A2). Panel A3 represents isotype control for galectin-3 antibody. Panel A4 represents quantitative analysis of Panels A1 and A2 (7 myocardial fields, each group). The macrophage-specific F4/80 staining in WT mice showed abundant positively stained cells in the infarct (red arrow) and peri-infarct (blue arrow) regions, especially in the areas of excess cardiomyocyte loss (Panel B1). The galectin-3 KO mice showed reduced F4/80 positive cellular infiltration compared with WT mice (Panel B2). Panel B3 represents isotype control for mouse F4/80 antibody. Panel B4 represents quantitative analysis of Panels B1 and B2 (12 myocardial fields, each group). KO indicates knock-out; LAD, left anterior descending; MI, myocardial infarction; WT, wild-type.
*P < .05.
Serum galectin-3 levels after acute MI were associated with early LV dysfunction
Of 96 patients, 92 underwent ventriculography at the time of PCI and 87 received an echocardiogram during their initial hospitalization. Serum levels of galectin-3 were progressively higher in patients with mildly (≤45%: 14.22 ± 1.04 vs >45%: 10.97 ± 0.96, P = .003), moderately (≤40%: 15.43 ± 1.54 vs >40%: 11.42 ± 0.79, P = .008), and severely (≤35%: 17.30 ± 1.52 vs >35%: 11.42 ± 0.77, P < .001) reduced LVEF on ventriculogram at the time of PCI (Figure 3). There was a negative correlation between LVEF and serum galectin-3 level (rs: –0.28, P = .006). Conversely, galectin-3 levels were not associated with elevated left ventricular end-diastolic pressure (LVEDP) during cardiac catheterization. Among echocardiographic measures, galectin-3 levels were found to be significantly higher in patients with stage II-IV diastolic dysfunction (P = .043), LVSI >1.5 (P = .006), and %FM <50 (P = .047). Comparisons between echocardiographic and angiographic measures of myocardial dysfunction are summarized in Table 2.
Figure 3.
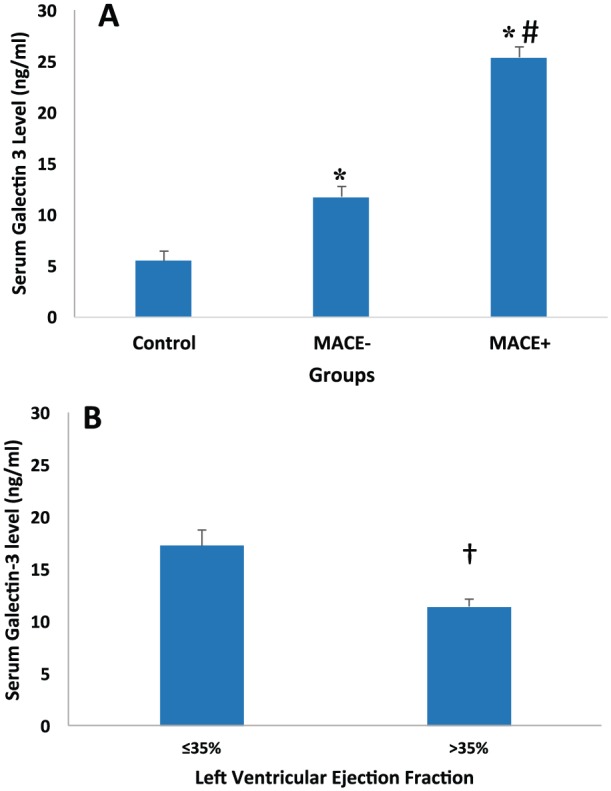
Comparison of serum galectin-3 level in healthy controls vs STEMI patients with either MACE-positive or MACE-negative outcome (Panel A) and in STEMI patients with reduced ejection fraction on ventriculogram (Panel B). Data are expressed as mean ± SEM. *P < .05 compared with control (Panel A); #P < .05 compared with MACE (Panel A); †P < .05 (Panel B). HF indicates heart failure; MACE, major adverse cardiovascular events defined as composite outcome of mortality, recurrent MI, stroke, and congestive HF hospitalization. MI, myocardial infarction; SEM, standard error of mean; STEMI, ST-elevation myocardial infarction.
Table 2.
Comparison of early outcomes including length of hospitalization and early echocardiographic (<72 hours) and angiographic (at time of PCI) parameters of myocardial dysfunction in STEMI patients in relation to galectin-3 level.
| Early outcomes | n | Serum galectin-3 levels | P value | |
|---|---|---|---|---|
| LVEF, % | ≤35% | 15 | 17.30 ± 1.52 | <.001 |
| >35% | 77 | 11.42 ± 0.77 | ||
| LVEDP, mm Hg | ≤20 mm Hg | 39 | 12.04 ± 0.99 | .962 |
| >20 mm Hg | 40 | 12.76 ± 1.32 | ||
| Diastolic dysfunction | ≤Stage I | 78 | 12.27 ± 0.78 | .043 |
| >Stage I | 6 | 17.31 ± 2.49 | ||
| LAVI, mL/m2 | ≤35 | 76 | 13.41 ± 1.11 | .446 |
| >35 | 8 | 14.19 ± 2.27 | ||
| E/E′ ratio | <15 | 70 | 12.53 ± 0.88 | .195 |
| ≥15 | 10 | 14.65 ± 1.95 | ||
| LVSI | ≤1.5 | 67 | 12.74 ± 1.21 | .006 |
| >1.5 | 18 | 16.40 ± 1.47 | ||
| %FM | <50 | 8 | 17.91 ± 2.15 | .018 |
| ≥50 | 77 | 13.06 ± 1.09 | ||
| Length of hospitalization (hours) | ≤48 | 38 | 11.15 ± 1.26 | .013 |
| >48 | 58 | 14.45 ± 1.25 | ||
Abbreviations: %FM, percentage of normally functioning myocardium; LAVI, left atrial volume index; LVEDP, left ventricular end-diastolic pressure (on ventriculogram); LVEF, left ventricular ejection fraction (on ventriculogram); LVSI, left ventricular score index; PCI, percutaneous intervention; STEMI, ST-elevation myocardial infarction.
Results are expressed as percentage, or mean ± SEM.
Serum galectin-3 levels at the time of MI were associated with adverse cardiovascular outcomes
First, we compared the baseline characteristics of all study patients to ascertain no major variability between MACE-positive and MACE-negative patients (Table 3). Compared with healthy controls, STEMI patients had significantly higher levels of circulating galectin-3 (ng/mL; healthy: 5.49 ± 0.46 vs MI: 13.14 ± 0.92, P < .001). Compared with MACE-negative patients, MACE-positive patients had significantly higher levels of serum galectin-3 (ng/mL, MACE negative: 11.71 ± 0.69 vs MACE positive: 25.42 ± 5.22, P < .001) (Figure 3). Comparison of circulating brain natriuretic peptide (BNP) (pg/mL, MACE negative: 178.39 ± 62.05 vs MACE positive: 470.17 ± 198.01, P = .013) also showed similar results. Compared with serum troponin and BNP levels, ROC curve analysis showed that galectin-3 had the greatest area under the curve (AUC) for predicting the development of MACE (0.917 ± 0.063, 95% CI: 0.794-1.0; P = .001) (Figure 4). Ten of the 96 STEMI patients were MACE positive; 5 of them were from cardiovascular death, 1 stroke, 3 MIs, and 1 CHF hospitalization. Patients who had cardiovascular mortality had the highest average level of galectin-3 among MACE-positive patients, with a significant difference compared with patients without cardiovascular mortality (ng/mL, non-cardiovascular: 12.17 ± 0.69 vs cardiovascular: 31.49 ± 9.96, P = .004).
Table 3.
Demographic characteristics of STEMI patients and comparison between patients with and without MACE.
| All patients (n = 96) |
MACE negative (n = 86) | MACE positive (n = 10) | |
|---|---|---|---|
| Baseline characteristics | |||
| Age, years | 61.79 ± 1.33 | 61.31 ± 1.41 | 65.90 ± 4.14 |
| Male sex, % | 65.6 | 67.4 | 50 |
| White, % | 93.8 | 93 | 100 |
| Medications used at baseline | |||
| ASA, % | 27.1 | 27.9 | 20 |
| Statin, % | 31.3 | 32.6 | 20 |
| Beta blocker, % | 26 | 27.9 | 10 |
| ACEi, % | 27.1 | 26.7 | 30 |
| Diuretic, % | 6.3 | 5.8 | 10 |
| Risk factors | |||
| HTN, % | 49 | 46.5 | 70 |
| DM, % | 26 | 23.3 | 50 |
| Smoking, % | 67.7 | 67.4 | 70 |
| Prior CHF, % | 5.2 | 4.7 | 10 |
| Prior MI, % | 15.6 | 16.3 | 10 |
| Labs | |||
| HbA1c, % | 6.49 ± 0.23 | 6.32 ± 0.21 | 8.0 ± 1.18 |
| LDL, mg/dL | 114.84 ± 5.03 | 114.58 ± 5.18 | 117.29 ± 20.56 |
| BNP, pg/mL | 218.18 ± 60.96 | 178.39 ± 62.05 | 470.17 ± 198.01* |
| CK, U/L | 891.01 ± 74.68 | 863.48 ± 74.41 | 1100.20 ± 311.28 |
| CK-MB, ng/mL | 54.35 ± 5.44 | 52.81 ± 5.34 | 67.63 ± 25.97 |
| Peak troponin, ng/mL | 92.65 ± 10.77 | 86.45 ± 11.01 | 145.99 ± 39.45 |
| Serum galectin-3, ng/mL | 13.144 ± 0.920 | 11.716 ± 0.692 | 25.427 ± 5.224 * |
| Left ventricular functional parameters | |||
| LVEF post-MI, % | 49.01 ± 1.35 | 49.50 ± 1.37 | 43.88 ± 5.98 |
| LVEDP, mm Hg | 22.77 ± 0.949 | 22.37 ± 1.02 | 23.44 ± 3.45 |
| E/E′ | 9.63 ± 0.44 | 9.19 ± 0.41 | 13.07 ± 1.92* |
| LAVI, mL/m2 | 22.88 ± 0.95 | 22.85 ± 0.97 | 23.11 ± 3.65 |
| LVSI | 1.34 ± 0.04 | 1.29 ± 0.31 | 1.67 ± 0.18 |
| %FM | 73.09 ± 2.58 | 75.59 ± 2.34 | 54.50 ± 12.08 |
Abbreviations: ACEi, angiotensin converting enzyme inhibitor; ASA, aspirin; BNP, brain natriuretic peptide; CHF, congestive heart failure, CK, creatine kinase; DM, diabetes mellitus; %FM, percentage of normally functioning myocardium; HbA1c, hemoglobin A1c; HF, heart failure; HTN, hypertension; LDL, low density lipoprotein; MACE: major adverse cardiovascular event, defined as composite outcome of mortality, recurrent MI, stroke, and congestive HF hospitalization; MI, myocardial infarction; LAVI, left atrial volume index; LVEDP, left ventricular end-diastolic pressure (on ventriculogram); LVEF, left ventricular ejection fraction (on ventriculogram); LVSI, left ventricular score index; STEMI, ST-elevation myocardial infarction.
Results are expressed as percentage, or mean ± SEM.
P < .05 vs MACE negative.
Figure 4.
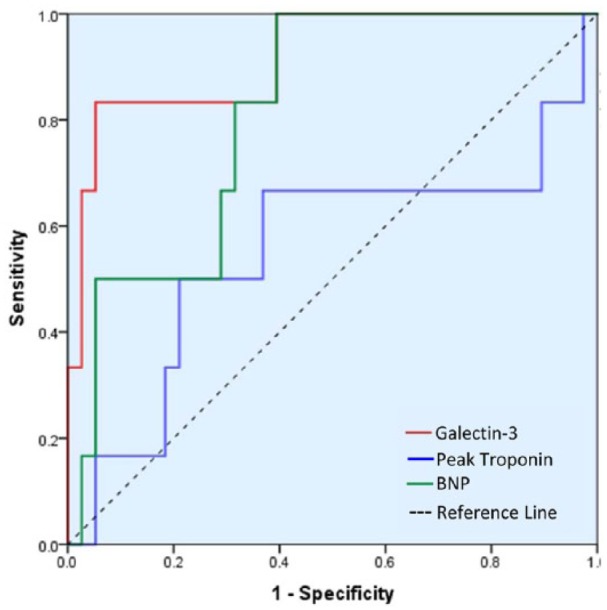
Receiver-operating characteristic curve showing prediction of positive MACE outcome with galectin-3 compared with peak troponin and BNP. Area under the curve for galectin-3 is 0.914 (CI: 0.789-1.0; P < .05), BNP is 0.847 (CI: 0.709-0.985; P < .05), and peak troponin is 0.554 (CI: 0.258-0.851; P = .674). BNP indicates brain natriuretic peptide; CI, confidence interval; MACE, major adverse cardiovascular event.
Major adverse cardiovascular event–positive patients also had significantly higher length of hospital stay compared with MACE-negative patients (days, MACE negative: 89.78 ± 14.24 vs MACE positive: 145.30 ± 45.41, P = .029). Furthermore, galectin-3 levels were higher in patients who were hospitalized for >48 hours (ng/mL, ≤48 hours: 11.15 ± 1.26 vs >48 hours: 14.45 ± 1.25, P = .013). Total length of hospital stay showed significant correlations with other variables, including galectin-3 (rs: 0.314, P = .002), BNP (rs 0.454, P = .002), peak troponin (rs: 0.341, P = .01), LVEF (rs: –0.297, P = .004), E-e’ ratio (rs: 0.229, P = .041), LVSI (rs: 0.011, P = .273), and age (rs: 0.352, P < .001). Galectin-3 levels were also higher in patients with a history of hypertension (HTN; P = .017), and in female patients (P = .027) (Table S2).
Given that the sampling time was not consistent, ranging between 12 and 48 hours from initial presentation, we performed a correlation analysis of galectin-3 level and sampling time. Galectin-3 level was weakly correlated with sampling time (rs: 0.25, P = .014). However, this is unlikely to be a confounding variable given that patients in MACE– versus MACE+ groups had no significant difference between their average sampling time (28.67 ± 0.89 hours vs 26.70 ± 2.56 hours, P = .532). This is also true for patients with LVEF ≤40% versus >40% (29.14 ± 1.96 hours vs 28.10 ± 0.97 hours, P = .797) and LVEF ≤35% versus >35% (29.23 ± 2.79 hours vs 28.07 ± 0.94 hours, P = .938).
Discussion
The mechanisms of progressive myocardial remodeling and loss of cardiac function following acute MI have not been fully elucidated. Acute MI per se is the main trigger; however, subsequent inflammation and fibrosis also contribute to the development of myocardial dysfunction.3,7,25,26 Our prior studies have identified the effects of macrophage activation, galectin-3 expression, and collagen deposition during the transition from the compensated state to HF.9 Whether galectin-3 is a silent bystander or a crucial mediator of HF is unclear. Although some clinical studies have demonstrated a strong association of elevated circulating galectin-3 levels with adverse outcomes in HF, coronary artery disease, and acute coronary syndrome,10–14 studies evaluating time and tissue-specific galectin-3 expression immediately following acute ischemic myocardial injury have been lacking. Perea and associates investigated the utility of galectin-3 for predicting post-infarct remodeling based on extracellular volume fraction mapping obtained by cardiac magnetic resonance imaging (MRI).27 Although this study demonstrated the relationship between galectin-3 and remote myocardial remodeling at 6 months, it did not provide conclusive evidence on whether galectin-3 was expressed after MI and its potential involvement in stimulating cardiac remodeling.
Justification of a translation approach
There are several potential challenges in translating pre-clinical findings to clinical research. First, rodents tolerate much larger MIs than what a human can survive. At the same time, remodeling in humans is altered by pharmacologic therapy and early reperfusion when compared with untreated animal models. This translational study uses both pre-clinical animal models and patient data to validate the relationship of galectin-3 with subsequent loss of myocardial function and adverse cardiovascular outcomes after MI.
Galectin-3 expression is expressed very early after acute MI and associated with regional and global LV dysfunction
Patients with reduced LVEF on ventriculography had higher levels of serum galectin-3 obtained within 48 hours of presentation. This is the first study to date to measure and correlate galectin-3 levels with LVEF this early after MI. This may indicate a very early role of galectin-3 in the myocardial response to ischemic injury. However, although a negative correlation between galectin-3 levels and LVEF was found, a direct causation for myocardial dysfunction cannot be implied at this point, as other factors contributing to a temporarily stunned myocardium may be playing a role. Di Tano and associates investigated the utility of galectin-3 in predicting LV remodeling (defined as a ≥15% increase in LV end-systolic volume) 48 hours after admission for MI, at 1 and 6 months.28 Higher baseline galectin-3 was associated to an increased risk of LV remodeling. Furthermore, patients with persistently elevated galectin-3 had worse outcomes, supporting a potential causal effect between galectin-3 and reduced LVEF. Although LVEF is a central indicator of cardiac function in ischemic heart disease, evaluation of regional function using alternate quantitative methods such as LVSI is also relevant. LVSI is of high prognostic value as it has been shown to predict mortality.29,30 In our study, patients with elevated LVSI, which is related to the extent and/or severity of wall motion abnormality, had higher levels of circulating galectin-3. Similarly, %FM, which measures the extent of myocardial dysfunction, was also associated with higher galectin-3 levels. Other studied early parameters of myocardial dysfunction included post-MI LVEDP,31 LAVI, diastolic dysfunction stage, and E/E′ ratio.32 Only diastolic dysfunction >stage I was shown to be associated with higher serum galectin-3 levels. However, this was not supported with other parameters of diastolic dysfunction such as E/E′ ratio or LVEDP.
Inflammation after MI: virtuous intentions, malign consequences?
Our results show that galectin-3 is associated with inflammation at a very early stage after acute MI. A similar expression profile was also noted for other inflammatory mediators, including macrophage-associated genes. Although some degree of inflammation is expected in the healing phase after acute MI, our study shows sustained activity of such mediators over a prolonged period of time. Such effects, although initially beneficial, may contribute to the development of myocardial dysfunction.25,33–35 Persistent inflammation through macrophage migration is also associated with vascular lesion formation via production of inflammatory cytokines and growth factors.36,37 Our studies show morphologic evidence of reduced myocardial tissue damage on histology in galectin-3 KO mice and the significant reduction of myocardial macrophage infiltration as evidenced by reduced CD68 positivity in KO mice. Although 1-week post-MI model may not fully examine the chronic myocardial remodeling, such model will provide crucial information on early myocardial damage, inflammation, and macrophage activity.
Galectin-3 increase in relation to major cardiovascular outcomes
Serum galectin-3 levels were also found to be significantly higher in patients with adverse cardiovascular outcomes at 3 months. Compared with known biomarkers (eg, troponin and BNP), galectin-3 was found to have the strongest predictive value for MACE based on ROC analysis. Among early clinical outcomes, our study is the first to show that patients who were hospitalized >48 hours also had significantly higher serum galectin-3 levels than those with shorter lengths of stay. Although other baseline variables and co-morbidities might have contributed to prolonged hospitalization, galectin-3 is likely to identify a sicker patient population at higher risk of poor outcome following MI.
Therapeutic implications of galectin-3 expression
A time-dependent serial genomic evaluation of more than 9000 unique genes identified galectin-3 as a robustly overexpressed protein after acute MI. In line with our prior study performed in a hypertensive rodent model of HF,9 gene expression profiling post-MI showed a strong association of expression of galectin-3 with other genes implicated in inflammation and loss of function. Galectin-3 expression may modulate ongoing inflammation through macrophage-derived cytokine mediators that may be the harbingers of adverse myocardial remodeling. One can hypothesize that blocking the expression or activity of galectin-3 could have therapeutic implications in preventing myocardial inflammation, adverse LV remodeling, and progressive LV dysfunction post-MI. However, further studies are needed to directly prove this hypothesis.
Clinical implications and translational outlook
The data presented in this study support the ability of galectin-3 expression to identify patients at higher risk of adverse cardiovascular outcomes and those who are likely to go on to have progressive LV dysfunction. The current study also explores a potential pathologic role of galectin-3 in stimulating myocardial inflammation that is involved with LV dysfunction. This work serves to underscore the need for therapeutic approaches that may halt the development of ischemic cardiomyopathy early in the process, and identifies galectin-3 as a potential target for such therapy. A better understanding of the molecular mechanisms underlying the dynamic expression of galectin-3 and the therapeutic potential by blocking galectin-3 is needed.
Supplementary Material
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR001412 to the University at Buffalo. Dr. Sharma is supported by Mentored Career Development Award from the NIH/NHLBI 1K08HL131987.
Declaration of Conflicting Interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: WM: Research design, patient enrollment and manuscript writing; MRC: Statistical analyses; SS and SM: Validation of genomics results with real-time PCR; CK, KF, TS, RuK, RoK: Patient enrollment, follow-up and clinical data extraction; SD: Serum retrieval and Enzyme-linked assays; WMB: Microarrays; BP: manuscript writing and conceptual critiques; SP: Immuno(histological) analyses; MK: Induction of acute MI in a murine model and UCS: Overall conceptual design, data analysis and mentorship.
Ethical approval: All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Informed consent: The study was approved by the University at Buffalo Institutional Review Board (IRB), and informed consent was obtained from all individual participants included in the study.
References
- 1. Guidry UC, Evans JC, Larson MG, Wilson PW, Murabito JM, Levy D. Temporal trends in event rates after Q-wave myocardial infarction: the Framingham Heart Study. Circulation. 1999;100:2054–2059. [DOI] [PubMed] [Google Scholar]
- 2. Steg PG, Dabbous OH, Feldman LJ, et al. ; Global Registry of Acute Coronary Events Investigators. Determinants and prognostic impact of heart failure complicating acute coronary syndromes: observations from the Global Registry of Acute Coronary Events (GRACE). Circulation. 2004;109:494–499. [DOI] [PubMed] [Google Scholar]
- 3. Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 4. Cleutjens JP, Blankesteijn WM, Daemen MJ, Smits JF. The infarcted myocardium: simply dead tissue, or a lively target for therapeutic interventions. Cardiovasc Res. 1999;44:232–241. [DOI] [PubMed] [Google Scholar]
- 5. Daskalopoulos EP, Janssen BJ, Blankesteijn WM. Myofibroblasts in the infarct area: concepts and challenges. Microsc Microanal. 2012;18:35–49. [DOI] [PubMed] [Google Scholar]
- 6. Yang F, Liu YH, Yang XP, Xu J, Kapke A, Carretero OA. Myocardial infarction and cardiac remodelling in mice. Exp Physiol. 2002;87:547–555. [DOI] [PubMed] [Google Scholar]
- 7. Weber KT, Sun Y, Diez J. Fibrosis: a living tissue and the infarcted heart. J Am Coll Cardiol. 2008;52:2029–2031. [DOI] [PubMed] [Google Scholar]
- 8. Chow SL, Maisel AS, Anand I, et al. Role of biomarkers for the prevention, assessment, and management of heart failure: a scientific statement from the American Heart Association. Circulation. 2017;135:e1054–e1091. [DOI] [PubMed] [Google Scholar]
- 9. Sharma UC, Pokharel S, van Brakel TJ, et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation. 2004;110:3121–3128. [DOI] [PubMed] [Google Scholar]
- 10. Ho JE, Liu C, Lyass A, et al. Galectin-3, a marker of cardiac fibrosis, predicts incident heart failure in the community. J Am Coll Cardiol. 2012;60:1249–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Daniels LB, Clopton P, Laughlin GA, Maisel AS, Barrett-Connor E. Galectin-3 is independently associated with cardiovascular mortality in community-dwelling older adults without known cardiovascular disease: the Rancho Bernardo study. Am Heart J. 2014;167:674.e1-682.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maiolino G, Rossitto G, Pedon L, et al. Galectin-3 predicts long-term cardiovascular death in high-risk patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2015;35:725–732. [DOI] [PubMed] [Google Scholar]
- 13. Tsai TH, Sung PH, Chang LT, et al. Value and level of galectin-3 in acute myocardial infarction patients undergoing primary percutaneous coronary intervention. J Atheroscler Thromb. 2012;19:1073–1082. [DOI] [PubMed] [Google Scholar]
- 14. Grandin EW, Jarolim P, Murphy SA, et al. Galectin-3 and the development of heart failure after acute coronary syndrome: pilot experience from PROVE IT-TIMI 22. Clin Chem. 2012;58:267–273. [DOI] [PubMed] [Google Scholar]
- 15. O’Donoghue ML, Morrow DA, Cannon CP, et al. Multimarker risk stratification in patients with acute myocardial infarction. J Am Heart Assoc. 2016;5:e002586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krzeminski M, Singh T, Andre S, et al. Human galectin-3 (Mac-2 antigen): defining molecular switches of affinity to natural glycoproteins, structural and dynamic aspects of glycan binding by flexible ligand docking and putative regulatory sequences in the proximal promoter region. Biochim Biophys Acta. 2011;1810:150–161. [DOI] [PubMed] [Google Scholar]
- 17. Ochieng J, Furtak V, Lukyanov P. Extracellular functions of galectin-3. Glycoconj J. 2004;19:527–535. [DOI] [PubMed] [Google Scholar]
- 18. Almkvist J, Karlsson A. Galectins as inflammatory mediators. Glycoconj J. 2004;19:575–581. [DOI] [PubMed] [Google Scholar]
- 19. Lin HM, Pestell RG, Raz A, Kim HR. Galectin-3 enhances cyclin D(1) promoter activity through SP1 and a cAMP-responsive element in human breast epithelial cells. Oncogene. 2002;21:8001–8010. [DOI] [PubMed] [Google Scholar]
- 20. Aartsen WM, Schuijt MP, Danser AH, Daemen MJ, Smits JF. The role of locally expressed angiotensin converting enzyme in cardiac remodeling after myocardial infarction in mice. Cardiovasc Res. 2002;56:205–213. [DOI] [PubMed] [Google Scholar]
- 21. Sharma UC, Mosleh W, Chaudhari MR, et al. Myocardial and serum galectin-3 expression dynamics marks post-myocardial infarction cardiac remodelling. Heart Lung Circ. 2016;26:736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schroen B, Heymans S, Sharma U, et al. Thrombospondin-2 is essential for myocardial matrix integrity: increased expression identifies failure-prone cardiac hypertrophy. Circ Res. 2004;95:515–522. [DOI] [PubMed] [Google Scholar]
- 23. Kurushima H, Ramprasad M, Kondratenko N, Foster DM, Quehenberger O, Steinberg D. Surface expression and rapid internalization of macrosialin (mouse CD68) on elicited mouse peritoneal macrophages. J Leukoc Biol. 2000;67:104–108. [DOI] [PubMed] [Google Scholar]
- 24. McCormack R, de Armas LR, Shiratsuchi M, Ramos JE, Podack ER. Inhibition of intracellular bacterial replication in fibroblasts is dependent on the perforin-like protein (perforin-2) encoded by macrophage-expressed gene 1. J Innate Immun. 2013;5:185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weber KT. From inflammation to fibrosis: a stiff stretch of highway. Hypertension. 2004;43:716–719. [DOI] [PubMed] [Google Scholar]
- 26. Pick R, Jalil JE, Janicki JS, Weber KT. The fibrillar nature and structure of isoproterenol-induced myocardial fibrosis in the rat. Am J Pathol. 1989;134:365–371. [PMC free article] [PubMed] [Google Scholar]
- 27. Perea RJ, Morales-Ruiz M, Ortiz-Perez JT, et al. Utility of galectin-3 in predicting post-infarct remodeling after acute myocardial infarction based on extracellular volume fraction mapping. Int J Cardiol. 2016;223:458–464. [DOI] [PubMed] [Google Scholar]
- 28. Di Tano G, Caretta G, De Maria R, et al. Galectin-3 predicts left ventricular remodelling after anterior-wall myocardial infarction treated by primary percutaneous coronary intervention. Heart. 2017;103:71–77. [DOI] [PubMed] [Google Scholar]
- 29. Klein P, Holman ER, Versteegh MI, et al. Wall motion score index predicts mortality and functional result after surgical ventricular restoration for advanced ischemic heart failure. Eur J Cardiothorac Surg. 2009;35:847–852; discussion 852-853. [DOI] [PubMed] [Google Scholar]
- 30. Eek C, Grenne B, Brunvand H, et al. Strain echocardiography and wall motion score index predicts final infarct size in patients with non-ST-segment-elevation myocardial infarction. Circ Cardiovasc Imag. 2010;3:187–194. [DOI] [PubMed] [Google Scholar]
- 31. White HD, Norris RM, Brown MA, Brandt PW, Whitlock RM, Wild CJ. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation. 1987;76:44–51. [DOI] [PubMed] [Google Scholar]
- 32. López Haldón J, Fernández Quero M, Mancha F, et al. Value of NT-ProBNP level and echocardiographic parameters in ST-segment elevation myocardial infarction treated by primary angioplasty: relationships between these variables and their usefulness as predictors of ventricular remodeling. Rev Esp Cardiol. 2010;63:1019–1027. [DOI] [PubMed] [Google Scholar]
- 33. Andersen MJ, Ersboll M, Bro-Jeppesen J, et al. Relationships between biomarkers and left ventricular filling pressures at rest and during exercise in patients after myocardial infarction. J Card Fail. 2014;20:959–967. [DOI] [PubMed] [Google Scholar]
- 34. Kuwahara F, Kai H, Tokuda K, et al. Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension. 2004;43:739–745. [DOI] [PubMed] [Google Scholar]
- 35. Matsubara LS, Matsubara BB, Okoshi MP, Franco M, Cicogna AC. Myocardial fibrosis rather than hypertrophy induces diastolic dysfunction in renovascular hypertensive rats. Can J Physiol Pharmacol. 1997;75:1328–1334. [PubMed] [Google Scholar]
- 36. Nicoletti A, Heudes D, Mandet C, Hinglais N, Bariety J, Michel JB. Inflammatory cells and myocardial fibrosis: spatial and temporal distribution in renovascular hypertensive rats. Cardiovasc Res. 1996;32:1096–1107. [DOI] [PubMed] [Google Scholar]
- 37. Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.