

Abstract
Purpose
The von Hippel-Lindau tumor suppressor is inactivated in the majority of clear cell renal cell carcinomas (ccRCCs), leading to inappropriate stabilization of hypoxia-inducible factor-2α (HIF-2α). PT2385 is a first-in-class HIF-2α antagonist. Objectives of this first-in-human study were to characterize the safety, pharmacokinetics, pharmacodynamics, and efficacy, and to identify the recommended phase II dose (RP2D) of PT2385.
Patients and Methods
Eligible patients had locally advanced or metastatic ccRCC that had progressed during one or more prior regimens that included a vascular endothelial growth factor inhibitor. PT2385 was administered orally at twice-per-day doses of 100 to 1,800 mg, according to a 3 + 3 dose-escalation design, followed by an expansion phase at the RP2D.
Results
The dose-escalation and expansion phases enrolled 26 and 25 patients, respectively. Patients were heavily pretreated, with a median of four (range, one to seven) prior therapies. No dose-limiting toxicity was observed at any dose. On the basis of safety, pharmacokinetic, and pharmacodynamic profiling, the RP2D was defined as 800 mg twice per day. PT2385 was well tolerated, with anemia (grade 1 to 2, 35%; grade 3, 10%), peripheral edema (grade 1 to 2, 37%; grade 3, 2%), and fatigue (grade 1 to 2, 37%; no grade 3 or 4) being the most common treatment-emergent adverse events. No patients discontinued treatment because of adverse events. Complete response, partial response, and stable disease as best response were achieved by 2%, 12%, and 52% of patients, respectively. At data cutoff, eight patients remained in the study, with 13 patients in the study for ≥ 1 year.
Conclusion
PT2385 has a favorable safety profile and is active in patients with heavily pretreated ccRCC, validating direct HIF-2α antagonism for the treatment of patients with ccRCC.
INTRODUCTION
Although targeted therapies have considerably improved the prognosis for patients with clear cell renal cell carcinoma (ccRCC),1 only 8%2 to 11.7% of patients3 with metastatic disease will survive 5 years. Thus, more effective therapies are needed.
Inactivation of the tumor suppressor gene von Hippel-Lindau (VHL) occurs in the majority of ccRCC patients.4-8 The VHL protein (pVHL) is a component of an E3 ubiquitin ligase that mediates protein degradation by the proteasome. Among the proteins targeted by pVHL is the transcription factor hypoxia-inducible factor-2α (HIF-2α), an oncogenic driver in ccRCC.4,8 Under normal oxygen tension, HIF-2α is hydroxylated on specific proline residues, a modification that is required for recognition by pVHL. As a result, HIF-2α is rapidly degraded in cells in normal oxygen conditions (Fig 1). In hypoxic conditions, the prolyl hydroxylases that perform the post-translational modification of HIF-2α are inactivated, and HIF-2α is stabilized. HIF-2α heterodimerizes with aryl hydrocarbon receptor nuclear translocator (ARNT; also known as HIF-1β) to form an active transcription factor. This allows increased expression of hypoxia-inducible genes, such as erythropoietin (EPO), which increase the production of red blood cells, resulting in increased oxygenation.9 Loss of pVHL function, through mutation, hypermethylation, deletion, or other genomic alterations of VHL, results in constitutive stabilization of HIF-2α even in normoxic conditions. In ccRCC tumor cells with pVHL deficiency, HIF-2α upregulates the expression of genes that are important to tumor growth and metastasis, including those that encode cyclin D1 (CCND1), vascular endothelial growth factor A (VEGFA), transforming growth factor α, and C-X-C chemokine receptor 4.8,10
Fig 1.

Hypoxia-inducible factor-2α (HIF-2α) inhibition and the von Hippel-Lindau (VHL)/HIF axis in renal cell carcinoma. Abbreviations: ARNT, aryl hydrocarbon receptor nuclear translocator; pVHL, VHL protein.
Currently approved therapies for ccRCC include inhibitors of vascular endothelial growth factor (VEGF) and VEGF receptor tyrosine kinases. These inhibitors provide therapeutic benefit because ccRCC is highly dependent on tumor angiogenesis through VEGF signaling. Because VEGFA is one of the target genes of HIF-2α, inhibition of HIF-2α transcriptional activity would be expected to inhibit tumor angiogenesis in ccRCC by inhibiting HIF-2α–induced upregulation of VEGFA expression. HIF-2α antagonism is also expected to impede tumor growth by inhibiting HIF-2α–induced upregulation of genes such as CCND1 and TGFA, which promote tumor cell proliferation but are independent of the VEGF-signaling pathway. Therefore, HIF-2α antagonism in ccRCC may provide greater efficacy than is achieved with VEGF receptor kinase inhibitors.
Structural analyses have identified a ligand-binding pocket in the Per-ARNT-Sim (PAS)-B domain of HIF-2α,11,12 an observation that was followed by the discovery of synthetic small molecules that occupy the pocket, including PT2385.13 PT2385 disrupts HIF-2α/ARNT heterodimerization and inhibits HIF-2α target gene expression. In a mouse xenograft model of ccRCC, PT2385 treatment resulted in decreased expression of HIF-2α target genes, decreased circulating human VEGF-A protein, and increased tumor cell apoptosis.13 PT2385 treatment also resulted in regression of mouse tumor xenografts derived from the VHL-deficient human renal cell cancer (RCC) cell line 786-O, whereas treatment with the standard-of-care VEGFR tyrosine kinase inhibitor sunitinib resulted in tumor stasis. In addition, PT2385 inhibited growth of xenografts derived from a human ccRCC tumor refractory to sunitinib and the mammalian target of rapamycin inhibitor everolimus.13 On the basis of these results, a first-in-human, phase I, dose-escalation trial of the first-in-class oral HIF-2α antagonist PT2385 was conducted in patients with advanced ccRCC previously treated with one or more VEGF inhibitors.
PATIENTS AND METHODS
Patients
This study enrolled patients at six centers between November 2014 and February 2016 and is still ongoing. The data cutoff date for this report is March 9, 2017. Patients had locally advanced or metastatic RCC with a clear cell component and had disease progression during treatment with one or more prior regimens that included a VEGF inhibitor. Eligible patients were ≥ 18 years of age, had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 1, and had adequate bone marrow (absolute neutrophil count ≥ 1.5 × 109/L; hemoglobin ≥ 9 g/dL; platelets ≥ 1 × 105/μL), hepatic (AST/ALT ≤ 2.5 × upper limit of normal; total bilirubin ≤ 2.0 mg/dL), and renal (serum creatinine < 2 × upper limit of normal or creatinine clearance ≥ 50 mL/min) function. Patients in the expansion cohort were required to have measurable disease per Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1).14 Patients with untreated brain metastases, uncontrolled or poorly controlled hypertension, a major cardiac event in the prior 6 months, and malabsorption as a result of prior gastrointestinal surgery or gastrointestinal disease, and those requiring warfarin anticoagulation were excluded.
All patients provided written informed consent. The study protocol was approved by institutional review boards at all participating institutions. The study (NCT02293980) was conducted in accordance with good clinical practice and the Declaration of Helsinki.
Study Design and Treatment
The primary objective of this phase I, open-label, multicenter, dose-escalation trial was to identify the maximum tolerated dose (MTD) and/or the recommended phase II dose (RP2D) of PT2385 in patients with advanced ccRCC. Secondary objectives were to evaluate the safety, pharmacokinetic (PK), and pharmacodynamic (PD) profiles, and to assess the antitumor activity of PT2385 in this patient population.
In a standard 3 + 3 design,15 cohorts of three to six patients were treated with oral PT2385 twice per day. A starting dose of 100 mg twice a day was selected on the basis of results of preclinical toxicology studies (data not shown). Dose levels were increased by 100% until the first drug-related grade ≥ 2 toxicity was reported. Thereafter, dose increases were by ≤ 50%. Patients at each dose level had to be treated for a minimum of 3 weeks before enrollment occurred at the next dose level. Dose-limiting toxicity (DLT) was defined as febrile neutropenia; grade 4 neutropenia lasting for > 5 days; grade 4 thrombocytopenia (or grade 3 with bleeding); grade 4 anemia unexplained by underlying disease; grade 3 or 4 nausea, vomiting, or diarrhea persisting for > 72 hours despite optimal treatment; grade ≥ 3 increased transaminase levels; any other grade ≥ 3 nonhematologic toxicity; any other grade ≥ 3 laboratory toxicity not resolving within 72 hours and considered clinically significant by the investigator; or any PT2385-related toxicity resulting in discontinuation before 21 days of treatment. After determination of the MTD or RP2D, ≤ 25 patients were to be enrolled in the expansion portion.
Assessments
Blood samples for PK and PD assessment were drawn in patients under fasted conditions 1 hour before and 1, 1.5, 2, 4, 6, 8, 12, and 24 hours after PT2385 administration on day 1 of weeks 1 and 3, and before drug administration on day 1 of weeks 2, 4, 5, 6, 7, 10, 13, and 16, and every 12 weeks thereafter. The evening doses on day 1 of weeks 1 and 3 were withheld to allow for full 24-hour PK assessments. Area under the concentration-time curve (AUC), maximum concentration (Cmax), time of maximum concentration (Tmax), and terminal half-life (T1/2) were determined. The plasma concentrations of PT2385 were determined using a validated liquid chromatography-tandem mass spectrometry method. Plasma Epo was measured with the Epo Access Immunoassay System (Beckman Coulter, Brea, CA).
Adverse events (AEs) were coded using Medical Dictionary for Regulatory Activities version 16 terminology. AE severity was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03 or later. Tumors were assessed by contrast or noncontrast computed tomography or magnetic resonance imaging and/or physical examination at baseline within 7 days before the week 7 visit and every 9 weeks thereafter. Tumor responses were evaluated using RECIST 1.1.14 Patients were considered to have stable disease (SD) if they had SD at their first restaging scan.
Statistical Analyses
The sample size was based on the need to establish the MTD and/or RP2D (≤ 48 patients), and ≤ 25 additional patients were to be enrolled in the expansion cohort. All statistical analyses were performed using SAS version 9.3 or higher (SAS Institute, Cary, NC). PK analyses were performed using Phoenix WinNonlin software (Certara, Princeton, NJ). Best response by RECIST and progression-free survival (PFS) time were investigator determined. Baseline patient and disease characteristics, efficacy, PK, and safety data were summarized. Cox proportional hazards regression models were used to identify factors potentially related to PFS (Appendix, online only).
RESULTS
Patient Characteristics
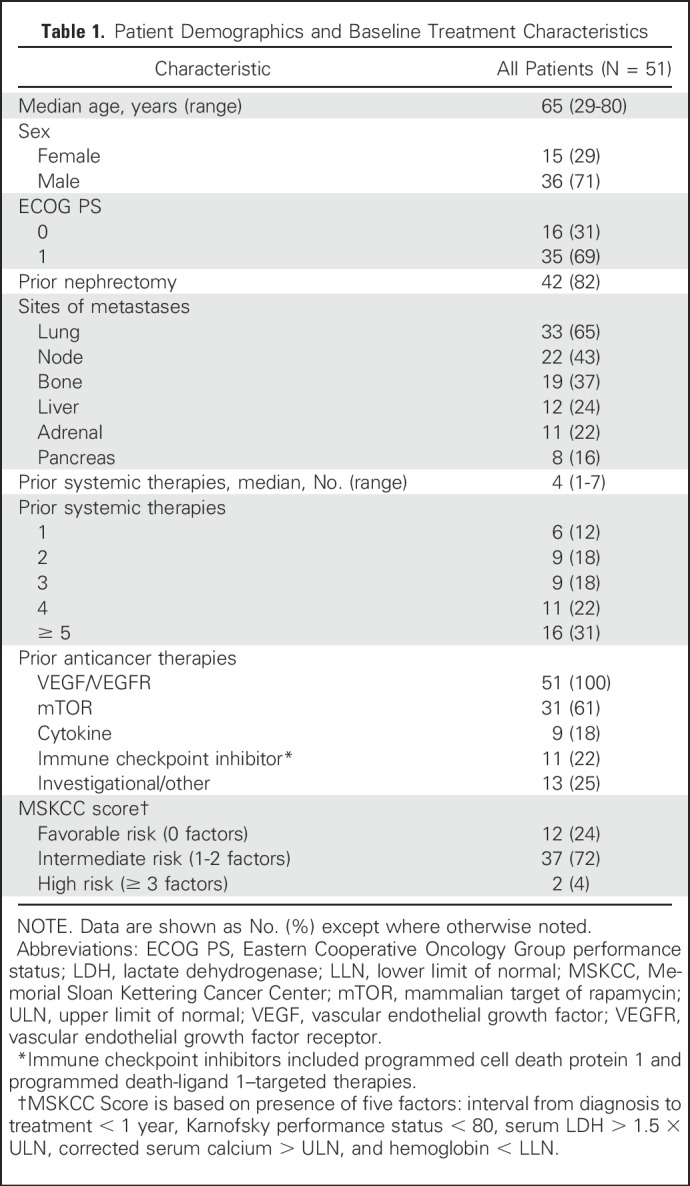
Twenty-six patients were enrolled in the dose-escalation portion, and 25 were enrolled in the expansion portion of the study. The majority of patients (71%) were male (Table 1). Most patients (69%) had an Eastern Cooperative Oncology Group performance status of 1. The median number of prior systemic therapies was four. All patients had been previously treated with VEGF-targeted therapy. The majority (61%) had also been treated with mammalian target of rapamycin inhibitors, and 39% had received prior immune checkpoint inhibitors (22%) and/or cytokines (18%).
Table 1.
Patient Demographics and Baseline Treatment Characteristics
Dose Escalation and MTD
No DLT was observed at the initial dose of 100 mg twice a day (n = 3) or at twice-a-day doses of 200 mg (n = 3), 400 mg (n = 4), 800 mg (n = 7), 1,200 mg (n = 6), or 1,800 mg (n = 3). Two days after the end of the 21-day DLT period, one patient in the 800-mg group experienced a grade 4 pulmonary embolism that was considered possibly related to PT2385 by the investigator in the context of disease progression. Following the consensus recommendation of the investigators, four more patients were treated at 800 mg twice a day and three at 1,200 mg twice a day to obtain additional safety, PK, and PD data. No patients experienced a DLT at any dose ≤ 1,800 mg twice a day; thus, an MTD was not identified.
PK and PD
Exposure to PT2385 was variable and increased up to 800 mg twice a day (Fig 2A). No additional increases were seen up to the highest dose level of 1,800 mg twice a day. At day 15, PT2385 (800 mg) was rapidly absorbed (median Tmax, 2 hours) with a mean Cmax of 3.1 µg/mL and a mean T1/2 of 17 hours. A 2.5-fold accumulation in AUC was observed from day 1 to day 15. Similar values were observed when all available data at 800 mg twice a day were combined (dose escalation plus expansion cohort; Fig 2A). Rapid reductions in plasma Epo were observed at all dosing levels (Fig 2B). Doses > 800 mg twice a day did not result in greater reductions in Epo. Because the MTD was not reached, the RP2D was based on the observed safety, PK, and PD data, and was determined to be 800 mg twice a day.
Fig 2.

Pharmacokinetics of PT2385. (A) Mean PT2385 plasma concentration, day 15, in all cohorts in the dose escalation phase. (Bottom) Pharmacokinetics in all patients (dose escalation and expansion cohorts; n = 32) treated with PT2385 at the recommended phase II dose (800 mg twice per day). (B) Pharmacodynamic response as assessed by decreases in the hypoxia-inducible factor-2α (HIF-2α) target erythropoietin. Plasminogen activator inhibitor-1, insulin-like growth factor-binding protein 3, and vascular endothelial growth factor A also were assessed as potential pharmacodynamic markers but no significant relationships with PT2385 exposure were observed. Abbreviations: AUClast,, area under the concentration-time curve from time 0 to the last quantifiable time point; Cmax, maximum concentration; PO, orally; SD, standard deviation; T1/2, terminal half-life; Tmax, time of maximal concentration.
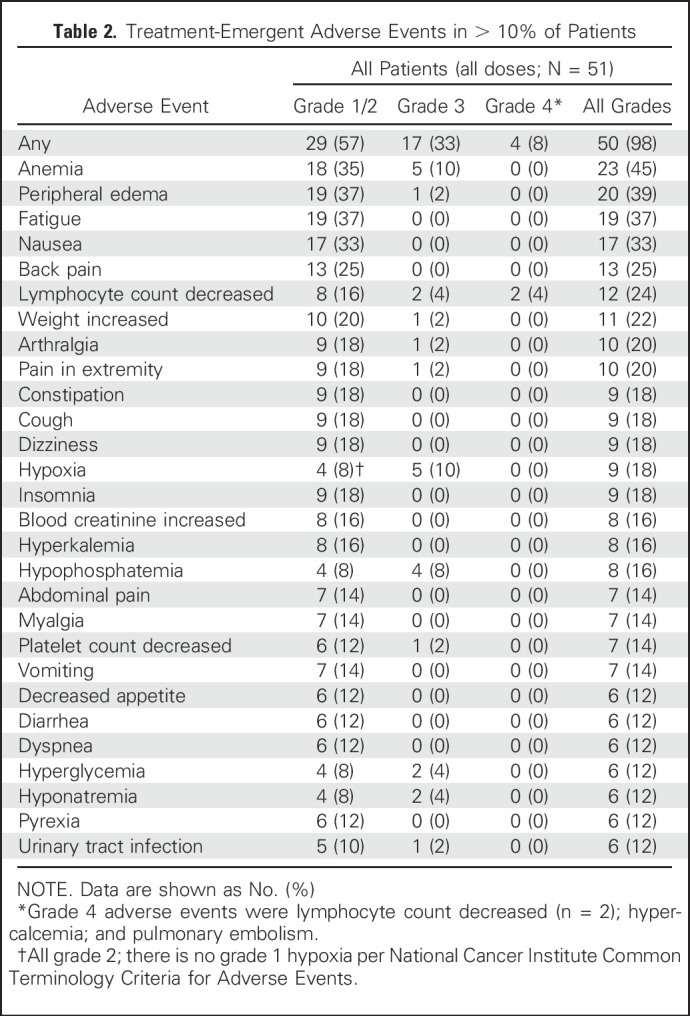
Safety
Treatment-emergent AEs occurring in > 10% of patients are listed in Table 2. The majority of AEs were grade 1 or 2 (Table 2). The most common all-grade AEs were anemia (45%), peripheral edema (39%), and fatigue (37%). Peripheral edema was not associated with thromboembolic events or cardiovascular toxicity. The most common grade ≥ 3 AEs were anemia (10%, all grade 3), hypoxia (10%, all grade 3), lymphopenia (8%; 4%, grade 3, 4%, grade 4), and hypophosphatemia (8%, all grade 3). Anemia was asymptomatic. Two patients each required a single prophylactic transfusion of packed red blood cells. Lymphopenia was not associated with infections. The only serious AE occurring in > 1 patient was hypoxia, which occurred in two patients treated at the 1,200-mg twice-a-day dose level.
Table 2.
Treatment-Emergent Adverse Events in > 10% of Patients
There were five dose interruptions (dose range, 400 to 1,800 mg twice a day) and two dose reductions (one each at 800 mg twice a day and 1,200 mg twice a day) for AEs. No patients discontinued treatment because of AEs, and there were no deaths during the study. Reasons for treatment discontinuation (n = 43) were progressive disease (41 patients; 95%), patient decision (one patient; 2%), and palliative radiotherapy (one patient; 2%).
Efficacy
Fifty-one patients were treated, and 50 were evaluable for response. As best response, one patient (2%) had a complete response (CR), six (12%) had a partial response (PR), 26 (52%) had SD, and 17 (34%) had tumor progression. The disease control rate (CR plus PR plus SD) was 66%. Twenty-one patients (42%) had SD or better for ≥ 4 months. The CR occurred in a patient receiving the 200-mg twice-a-day dose (Fig 3). PRs occurred in one patient receiving the 1,200-mg twice-a-day dose, four patients receiving the 800-mg twice-a-day dose, and one patient receiving the 400-mg twice-a-day dose. Thirteen patients remained in the study for ≥ 1 year (Fig 4). At a median follow-up of 17.5 months, 25% of patients had PFS > 14 months.
Fig 3.

Maximum radiographic response (n = 49). Note that one patient discontinued the study because of clinical progression before the first scheduled radiologic assessment. Abbreviations: CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease; SLD, sum of the longest diameter.
Fig 4.

Treatment duration in all patients evaluable for response (n = 50). (*)The case of patient 11 was previously referenced.16
An analysis of the relationship between PK and Memorial Sloan Kettering Cancer Center Score, a risk score that incorporates prognostic factors associated with survival in patients with previously treated metastatic RCC,17 with PFS was performed (Appendix). The variable with the most significant effect on PFS was the trough PT2385 concentration at day 15, 12 hours postdose (P = .002). Notably, 16 of 26 patients (62%) with a PT2385 trough concentration ≥ 0.5 µg/mL had SD or better for ≥ 4 months, whereas only four of 22 patients (18%) with a PT2385 trough concentration of < 0.5 µg/mL had SD or better for ≥ 4 months (Appendix Fig A1, online only).
DISCUSSION
There is a growing body of evidence that HIF-2α is a major oncogenic driver in VHL-deficient ccRCC.4,6-8 In contrast to many currently available therapies, which inhibit a single signaling pathway, direct inhibition of the transcriptional activity of HIF-2α has the potential to inhibit a number of pathways that contribute to tumor growth in ccRCC. However, to date, there have been no viable HIF-2α inhibitors for clinical use.
Targeting protein–protein interactions with small molecules for therapeutic applications has proven to be challenging.18 Crystallographic studies identified a unique 290Å cavity in the PAS-B domain of HIF-2α that could accommodate a small molecule, which might disrupt heterodimerization with ARNT.11,12 Iterative structure-based drug design was used to optimize candidate compounds, leading to the discovery of PT2385.13 Occupancy of the HIF-2α PAS-B domain pocket results in a conformation change in HIF-2α and destabilization of its interaction with ARNT. As such, the mechanism of action of PT2385 involves an allosteric disruption of a protein–protein interaction between HIF-2α and ARNT. Recent data have shown that PT2385 is highly selective in its disruption of the HIF-2α/ARNT heterodimer and has no effect on the HIF-1α/ARNT heterodimer.13 PT2385 has shown efficacy in ccRCC xenograft models, including one derived from a tumor refractory to sunitinib and everolimus.13
PT2385 is the first HIF-2α inhibitor to be tested in humans and to show activity in patients with ccRCC. PT2385 treatment resulted in rapid and pronounced decreases in Epo (known to be regulated by HIF-2α9) at all doses, demonstrating target engagement and biologic activity. PT2385 did not cause DLT at any dose, including those as high as 1,800 mg twice a day. Both PK and PD profiling indicated maximum compound exposure at 800 mg twice a day, establishing this as the RP2D.
A limitation of this study is the lack of a priori statistical calculation in selecting the size of the expansion cohort. However, in a heavily pretreated population with few treatment options, a response rate > 5% would be reasonable hypothesis-generating evidence that a compound warrants additional investigation. A sample of ≥ 25 patients provides a 95% lower confidence limit for overall response that excludes 5% if the observed response rate is ≥ 10%. Thus, the observed 14% response rate supports additional investigation of PT2385 efficacy in a larger patient population.
Drug exposure variability was observed in patients treated with PT2385, and greater exposure seemed to be correlated with longer PFS. The effect of administered dose and plasma trough levels of PT2385 on clinical activity should be confirmed in ongoing studies of PT2385. Preliminary data with a structurally related compound in clinical development do not show this variable drug exposure (data not shown).
In this study, PT2385 had a favorable safety profile, with most treatment-emergent AEs being grade 1 or 2, and no patients discontinuing treatment owing to AEs. Anemia, the most frequently reported treatment-emergent AE in this study, may be the result of on-target effects of HIF-2α antagonism as reflected in reduced Epo, which functioned as a PD biomarker. Anemia was predominantly low-grade (grade 1 or 2, 35%; grade 3, 10%; no grade 4). During the course of the study, hypoxia observed by routine pulse oximetry monitoring emerged as a likely treatment-emergent AE (nine of 51 patients, 18%) and, along with anemia, was the most common grade ≥ 3 AE (n = 5 for each). All nine instances occurred in patients treated at doses ≥ 800 mg twice a day. Seven of the nine events occurred during the first 5 weeks of therapy. PT2385 dosing was interrupted in two patients; one resumed the study drug within 1 week with no additional episodes of hypoxia, and one was taken out of the study for progressive disease. It should be noted that pulse oximetry was not a required measurement per the study protocol; thus, the incidence of hypoxia may be under-reported. A possible mechanistic link to PT2385 is suggested by a recent study showing that HIF-2α plays a role in ventilatory control and carotid body biology in rodents.19 Alternatively, HIF-2α inhibition may lead to an impaired pulmonary vascular response to hypoxia.20-22
In this phase I dose-escalation study, PT2385 showed promising efficacy in a highly pretreated patient population (31% of patients received five or more prior therapies). Although many caveats necessarily apply to cross-trial comparisons, other phase I studies of targeted agents in heavily pretreated ccRCC populations, where drugs were dose-reduced or discontinued in a large number of patients, may be helpful in providing a context for these findings.23,24 In this study, there were relatively few dose interruptions, two dose reductions, and no treatment discontinuations as a result of AEs. Interestingly, unlike VEGF signaling inhibitors,25 PT2385 was not observed to cause hypertension or apparent cardiac toxicity. The favorable tolerability profile of PT2385 over time was evident in that 21 patients with SD or better remained in the study for ≥ 4 months, and 13 patients remained in the study for ≥ 1 year without discontinuation as a result of drug intolerability. Also, one patient previously treated with sunitinib and temsirolimus achieved a CR on single-agent PT2385, continuing to receive treatment for nearly 2 years at the time of data cutoff.
The favorable tolerability profile and activity of PT2385 monotherapy provide a rationale for exploring PT2385 in combination with other active agents for ccRCC. The second part of this study, therefore, investigates PT2385 plus the anti–programmed death ligand 1 monoclonal antibody nivolumab.26,27 A third part assesses PT2385 plus the multitargeted tyrosine kinase inhibitor cabozantinib.25,28 We anticipate these combination studies will build on the encouraging results from this first-in-human trial of the HIF-2α inhibitor PT2385.
ACKNOWLEDGMENT
We thank the patients, their family members, nursing staff, study personnel, and investigators. Twist Medical provided medical writing support, funded by Peloton Therapeutics. The authors directed development of the manuscript and are fully responsible for all content and editorial decisions.
Appendix
Analyses of the Relationship Between Pharmacokinetics and Memorial Sloan Kettering Cancer Center Score with Progression-Free Survival
Univariable Cox regression models were used to evaluate the relationship between standard pharmacokinetic parameters and Memorial Sloan Kettering Cancer Center Score17 with progression-free survival (PFS). The pharmacokinetic parameters investigated were maximum concentration (Cmax), Cmax normalized for dose, terminal half-life, area under the concentration-time curve extrapolated to infinity (AUCinf), area under the concentration-time curve from time 0 to the last quantifiable time point (AUClast), AUCinf and AUClast normalized for dose, median trough concentration, and time of maximum concentration at days 1 and 15, predose levels of PT2385 at days 8, 15, 22, 29, 36, 43, 64, 85, and 106, and 12 hr postdose levels at days 1 and 15 (Appendix Table A1, online only). The variables selected for inclusion in the multiple regression model (defined as P < .10, indicated in boldin Appendix Table A1) were Cmax and normalized Cmax at days 1 and 15, AUClast and normalized AUClast at day 15, predose and 12-hour postdose concentration at day 15, concentration at days 29 and 36, and Memorial Sloan Kettering Cancer Center Score. In the stepwise Cox model, the subset of variables that best predicted PFS (P < .05) was trough PT2385 concentration at day 15, 12 hours postdose, and predose PT2385 concentration at day 29. The variable with the most significant effect on PFS was the trough PT2385 concentration at day 15, 12 hours postdose (P = .002). The value of postdose concentration at day 15 that maximized the difference in PFS between patients with levels below the value versus at or above the value was determined to be 0.5 µg/mL. Specifically, 16 of 26 patients (62%) with a PT2385 trough concentration ≥ 0.5 µg/mL had SD or better for ≥ 4 months, whereas only four of 22 patients (18%) with a PT2385 trough concentration of < 0.5 µg/mL had SD or better for ≥ 4 months (Appendix Fig. A1)
Fig. A1.

Relationship between PT2385 exposure and progression-free survival in patients experiencing steady-state exposure trough concentrations < 0.5 µg/mL versus ≥ 0.5 µg/mL (all evaluable patients in dose escalation phase and expansion cohort; n = 48). Abbreviation: h, hours
Table A1.
Univariable and Multivariable Analyses

Footnotes
Supported by Peloton Therapeutics.
Presented in part at the 51st Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, June 3-7, 2016.
Clinical trial information: NCT02293980.
See accompanying article on page 908
AUTHOR CONTRIBUTIONS
Conception and design: Kevin D. Courtney, Jeffrey R. Infante, Robert A. Figlin, James Brugarolas, Ann M. Lowe, Keshi Wang, Eli M. Wallace, John A. Josey, Toni K. Choueiri
Administrative support: Toni K. Choueiri
Provision of study materials or patients: Kevin D. Courtney, Jeffrey R. Infante, Elaine T. Lam, Robert A. Figlin, Brian I. Rini, James Brugarolas, Toni K. Choueiri
Collection and assembly of data: Kevin D. Courtney, Jeffrey R. Infante, Elaine T. Lam, Brian I. Rini, James Brugarolas, Naseem J. Zojwalla, Ann M. Lowe, Keshi Wang, Eli M. Wallace, John A. Josey
Data analysis and interpretation: Kevin D. Courtney, Jeffrey R. Infante, Elaine T. Lam, Robert A. Figlin, Brian I. Rini, James Brugarolas, Naseem J. Zojwalla, Keshi Wang, Eli M. Wallace, John A. Josey, Toni K. Choueiri
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2α Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Kevin D. Courtney
Stock or Other Ownership: Regeneron (I)
Consulting or Advisory Role: Sanofi
Research Funding: Astellas Pharma (Inst), Eli Lilly (Inst), Medivation (Inst), Aragon Pharmaceuticals, Astellas Pharma, PSMA Development Company, Stemline Therapeutics (Inst), Peloton Therapeutics (Inst), Merck Sharp & Dohme (Inst), Corvus Pharmaceuticals (Inst), Clovis Oncology (Inst), Hoffmann-La Roche (Inst)
Patents, Royalties, Other Intellectual Property: Spouse receives patent royalties from Athena Diagnostics (I)
Jeffrey R. Infante
Employment: Johnson & Johnson
Research Funding: Celldex (Inst), ARMO BioSciences (Inst), BioMed Valley Discoveries (Inst), Novartis (Inst), Janssen Oncology (Inst), GlaxoSmithKline (Inst), Immunocore (Inst), Calithera Biosciences (Inst), Calithera Biosciences (Inst), Phosplatin Therapeutics (Inst), Genentech/Roche (Inst), Roche (Inst), Aileron Therapeutics (Inst), AstraZeneca (Inst), eFFECTOR Therapeutics (Inst), MedImmune (Inst), Pfizer (Inst), Bristol-Myers Squibb (Inst), Tesaro (Inst), Celldex (Inst), Merck (Inst)
Elaine T. Lam
Research Funding: Peloton Therapeutics (Inst)
Robert A. Figlin
Consulting or Advisory Role: Pfizer, Cerulean, Calithera
Research Funding: Pfizer, Novartis, Cerulean, Peloton Therapeutics
Brian I. Rini
Consulting or Advisory Role: Company: Pfizer,
Research Funding: Pfizer (Inst), Roche/Genentech (Inst), Bristol-Myers Squibb (Inst)
Travel, Accommodations, Expenses: Pfizer
James Brugarolas
Consulting or Advisory Role: Nektar
Research Funding: Genentech/Roche, Peloton Therapeutics
Patents, Royalties, Other Intellectual Property: Patent WO 2017/053192 biomarkers of response to HIF-2-alpha inhibition in cancer and methods for the use thereof
Travel, Accommodations, Expenses: Nektar
Other Relationship: Nektar, National Institutes of Health/National Cancer Institute SPORE (P50CA196516)
Naseem J. Zojwalla
Employment: Peloton Therapeutics
Leadership: Peloton Therapeutics
Stock or Other Ownership: Peloton Therapeutics
Ann M. Lowe
Employment: Peloton Therapeutics, Rigel Pharmaceuticals, ArmaGen, Bellicum Pharmaceuticals, Aeglea Biotherapeutics
Leadership: Aeglea Biotherapeutics, Aeglea Biotherapeutics (I)
Stock or Other Ownership: Aeglea Biotherapeutics (I), Rigel Pharmaceuticals (I), Shattuck Laboratories (I), Rigel Pharmaceuticals
Consulting or Advisory Role: Peloton Therapeutics, Rigel Pharmaceuticals, Aeglea Biotherapeutics, Bellicum Pharmaceuticals, ArmaGen
Keshi Wang
Employment: Peloton Therapeutics, Peloton Therapeutics (I)
Stock or Other Ownership: Peloton Therapeutics, Peloton Therapeutics (I)
Research Funding: Peloton Therapeutics
Eli M. Wallace
Employment: Peloton Therapeutics
Leadership: Peloton Therapeutics
Stock or Other Ownership: Peloton Therapeutics
John A. Josey
Employment: Peloton Therapeutics, Peloton Therapeutics (I)
Leadership: Peloton Therapeutics
Stock or Other Ownership: Peloton Therapeutics, Peloton Therapeutics (I)
Patents, Royalties, Other Intellectual Property: Peloton Therapeutics (Inst)
Toni K. Choueiri
Honoraria: NCCN, UpToDate
Consulting or Advisory Role: Pfizer, Bayer, Novartis, GlaxoSmithKline, Merck, Bristol-Myers Squibb, Roche/Genentech, Eisai, Foundation Medicine, Cerulean Pharma, AstraZeneca, Peloton Therapeutics, Exelixis, Prometheus, Alligent
Research Funding: Pfizer (Inst), Novartis (Inst), Merck (Inst), Exelixis (Inst), Company: TRACON Pharma (Inst), GlaxoSmithKline (Inst), Bristol-Myers Squibb (Inst), AstraZeneca (Inst), Peloton Therapeutics (Inst), Roche/Genentech (Inst), Celldex (Inst), Agensys (Inst)
Travel, Accommodations, Expenses: Advisory boards/consultancy
UT Southwestern Medical Center owns stock in Peloton Therapeutics and has a financial interest in the clinical trial described in the article.
REFERENCES
- 1.Choueiri TK, Motzer RJ: Systemic therapy for metastatic renal-cell carcinoma. N Engl J Med 376:354-366, 2017 [DOI] [PubMed] [Google Scholar]
- 2. American Cancer Society: Kidney cancer (adult)—renal cell carcinoma. http://www.cancer.org/ cancer/kidneycancer/detailedguide/
- 3. National Cancer Institute: Cancer stat facts: Kidney and renal pelvis cancer. http:// seer.cancer.gov/statfacts/html/kidrp.html.
- 4.Kondo K, Kim WY, Lechpammer M, et al. : Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol 1:E83, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nickerson ML, Jaeger E, Shi Y, et al. : Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res 14:4726-4734, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cowey CL, Rathmell WK: VHL gene mutations in renal cell carcinoma: Role as a biomarker of disease outcome and drug efficacy. Curr Oncol Rep 11:94-101, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato Y, Yoshizato T, Shiraishi Y, et al. : Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet 45:860-867, 2013 [DOI] [PubMed] [Google Scholar]
- 8.Shen C, Kaelin WG, Jr: The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol 23:18-25, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haase VH: Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev 27:41-53, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao J, Du F, Shen G, et al. : The role of hypoxia-inducible factor-2 in digestive system cancers. Cell Death Dis 6:e1600, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheuermann TH, Tomchick DR, Machius M, et al. : Artificial ligand binding within the HIF2alpha PAS-B domain of the HIF2 transcription factor. Proc Natl Acad Sci USA 106:450-455, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogers JL, Bayeh L, Scheuermann TH, et al. : Development of inhibitors of the PAS-B domain of the HIF-2α transcription factor. J Med Chem 56:1739-1747, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallace EM, Rizzi JP, Han G, et al. : A small-molecule antagonist of HIF2α is efficacious in preclinical models of renal cell carcinoma. Cancer Res 76:5491-5500, 2016 [DOI] [PubMed] [Google Scholar]
- 14.Eisenhauer EA, Therasse P, Bogaerts J, et al. : New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 45:228-247, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Le Tourneau C, Lee JJ, Siu LL: Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst 101:708-720, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen W, Hill H, Christie A, et al. : Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 539:112-117, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Motzer RJ, Bacik J, Schwartz LH, et al. : Prognostic factors for survival in previously treated patients with metastatic renal cell carcinoma. J Clin Oncol 22:454-463, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Koehler AN: A complex task? Direct modulation of transcription factors with small molecules. Curr Opin Chem Biol 14:331-340, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodson EJ, Nicholls LG, Turner PJ, et al. : Regulation of ventilatory sensitivity and carotid body proliferation in hypoxia by the PHD2/HIF-2 pathway. J Physiol 594:1179-1195, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith TG, Brooks JT, Balanos GM, et al. : Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med 3:e290, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. doi: 10.1152/japplphysiol.00535.2013. Petousi N, Croft QP, Cavalleri GL, et al: Tibetans living at sea level have a hyporesponsive hypoxia-inducible factor system and blunted physiological responses to hypoxia. J Appl Physiol (1985) 116:893-904, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frise MC, Robbins PA: The pulmonary vasculature--lessons from Tibetans and from rare diseases of oxygen sensing. Exp Physiol 100:1233-1241, 2015 [DOI] [PubMed] [Google Scholar]
- 23.Angevin E, Lopez-Martin JA, Lin CC, et al. : Phase I study of dovitinib (TKI258), an oral FGFR, VEGFR, and PDGFR inhibitor, in advanced or metastatic renal cell carcinoma. Clin Cancer Res 19:1257-1268, 2013 [DOI] [PubMed] [Google Scholar]
- 24.Choueiri TK, Pal SK, McDermott DF, et al. : A phase I study of cabozantinib (XL184) in patients with renal cell cancer. Ann Oncol 25:1603-1608, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moslehi JJ: Cardiovascular toxic effects of targeted cancer therapies. N Engl J Med 375:1457-1467, 2016 [DOI] [PubMed] [Google Scholar]
- 26.Motzer RJ, Escudier B, McDermott DF, et al. : Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 373:1803-1813, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Motzer RJ, Rini BI, McDermott DF, et al. : Nivolumab for metastatic renal cell carcinoma: Results of a randomized phase II trial. J Clin Oncol 33:1430-1437, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. doi: 10.2147/DDDT.S104225. Procopio G, Grassi P, Verzoni E, et al: Cabozantinib in the treatment of advanced renal cell carcinoma: design, development, and potential place in the therapy. Drug Des Devel Ther 10:2167-2172, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]