

Abstract
The growing deficit in suitable tissues for patients awaiting organ transplants demonstrates the clinical need for engineered tissues as alternative graft sources. Demonstrating safety and efficacy by tracking the migration and fate of implanted cells is a key consideration required for approval of promising engineered tissues. Cells from transgenic animals that express green fluorescent protein (GFP) are commonly used for this purpose. However, GFP can create difficulties in practice due to high levels of green autofluorescence in many musculoskeletal tissues. Tandem-dimer tomato (tdTomato) is a stable, robust red fluorescent protein that is nearly threefold brighter than GFP. Our objective was to create a line of transgenic rats that ubiquitously express tdTomato in all cells, driven by the human ubiquitin C promoter. We sought to determine the rats' utility in tissue engineering applications by fabricating engineered skeletal muscle units (SMUs) from isolated muscle-derived tdTomato cells. These tdTomato SMUs were implanted into a volumetric muscle loss (VML) defect of the tibialis anterior muscle in a rat ubiquitously expressing GFP. We also evaluated a novel method for modularly combining individual SMUs to create a larger engineered tissue. Following a recovery period of 28 days, we found that implantation of the modular SMU led to a significant decrease in the size of the remaining VML deficit. Histological analysis of explanted tissues demonstrated both tdTomato and GFP expression in the repair site, indicating involvement of both implanted and host cells in the regeneration process. These results demonstrate the successful generation of a tdTomato transgenic rat, and the use of these rats in tissue engineering and cell migration applications. Furthermore, this study successfully validated a method for scaling engineered tissues to larger sizes, a factor that will be important for repairing volumetric injuries in more clinically relevant models.
Keywords: : cell migration, cell tracking, tissue engineering, tdTomato
Introduction
Tissue engineering and regenerative medicine (TE/RM) products for repair or regeneration of failing organs present a promising approach to address the growing deficit in tissues suitable for implantation.1–5 Despite considerable advances in the field in recent years, only a fraction of potential TE/RM products have reached the clinic.3,6 To realize the full potential of TE/RM in addressing the need for alternative transplant tissues and organs, it will be essential for researchers to navigate the complex regulatory processes required for clinical approval. Recent regulatory guidance has indicated that TE/RM products in preclinical development should focus on testing the in vivo stability of implanted cellular and scaffold-based constructs.1,7 Stability refers both to the potential of cells to migrate out of the implanted construct into the host tissue and to the alteration of the implanted cells and matrix as the host tissue remodels and resorbs the construct. As a result, development of assays for measuring and tracking the interactions between the host and implanted tissue are imperative for novel TE/RM products in development.
Fortunately for tissue engineering researchers, a wide variety of such cell tracking tools is available. Fluorescence-based reporters are the most commonly used tool for identifying individual cells and structures due to the wide range of available labeling techniques, from transfection to genetic recombination to higher resolution techniques, such as Forster resonance energy transfer (FRET) and fluorescence in situ hybridization.8–11 The number of fluorophores available for cell tracking studies has expanded to meet increasing demands for multicolor labeling strategies. Green fluorescent protein (GFP) from the Aequorea jellyfish sees the most widespread use for tracking gene expression, protein localization, and FRET.8,12–14 For in vivo imaging, however, the shorter wavelength GFP signal is limited by low tissue penetration and high background due to tissue autofluorescence.15,16 In contrast, longer wavelength red fluorescent protein derived from Discosoma coral (DsRed) presents an alternative to GFP with distinct spectral characteristics.17 In particular, tandem-dimer Tomato (tdTomato), a variant of DsRed, has demonstrated increased stability and fluorescence intensity in comparison to enhanced GFP and is among the brightest of the fluorescent proteins.18,19 Compared with the other DsRed derivatives, tdTomato is both the brightest and most resistant to photobleaching.19 Thus, tdTomato may ultimately be best suited for detection of cell migration in implanted engineered tissues and resulting histological sections.13
Kobayashi et al. first generated a ubiquitous tdTomato rat in 2012 through germline transmission of one Rosa26-tdTomato knockin embryonic stem cell line.20 This was again accomplished by Igarashi et al. in 2016 through the injection of intracytoplasmic Cre mRNA into fertilized rat ova, yielding their FLAME line of transgenic rats.21 However, none of the articles that have referenced these works has applied the technology to cell migration or tissue engineering applications. Rather, their technologies have been widely applied to genetic modification studies, including the generation of a ubiquitous mCherry rat.22 As these exciting research tools were not immediately available due to cost and accessibility for use in our tissue engineering applications, we decided to have the University of Michigan's Transgenic Animal Model Core develop a rat line for our tissue engineering needs. In an effort to increase accessibility to this technology, we will be making our animals available through the Rat Resource & Research Center (RRRC).
Our previous work has studied the native skeletal muscle repair mechanism,23,24 involving the design and fabrication of scaffoldless tissue-engineered skeletal muscle units (SMUs), and demonstrating potential for repairing damage in a rat model of volumetric muscle loss (VML).25,26 Implanted SMUs have integrated with the host tissue by forming vascular and neural interfaces, regenerated muscle fibers in the defect area, and restored a portion of the VML force deficit following 28 days in vivo.25 Based on our previous study, we hypothesized that implanting larger engineered tissues to match the size of the VML defect more closely would result in a reduced VML deficit after a 28-day recovery period. However, scaling-up engineered tissues is accompanied by a set of challenges. Tissue-engineered constructs are typically avascular, so they rely on diffusion alone to allow adequate nutrient delivery and waste removal. A necrotic core will form if nutrients cannot adequately penetrate the entire thickness. Thus, in this study, we have taken a modular approach to scale-up, in which smaller tissues are fabricated as per our standard protocol, and then combined just before implantation. By combining the engineered tissues just before implantation, we can avoid necrosis through host-driven vascularization, which previous work has shown to occur within a 28-day implantation for SMUs up to 2 mm in diameter.25
Additionally, we were curious to determine to what degree the implanted tissue would be infiltrated by host cells and whether SMU-derived cells would migrate out of the implant into the surrounding host tissue. Previous cell tracking experiments studied GFP as a fluorophore for labeling engineered tissue cell migration following implantation into wild-type animals and concluded that a second fluorophore was needed. Thus, this study was designed with a dual purpose: (1) use cells isolated from a tdTomato rat to fabricate SMUs and track the cell migration when implanted into a GFP host, and (2) demonstrate a technique for modular combination of multiple SMUs to create a larger engineered tissue with greater potential for regeneration of VML.
Methods
Animal care
All animal care procedures followed The Guide for Care and Use of Laboratory Animals,27 according to a protocol approved by the University Committee for the Use and Care of Animals. SMUs for implantation were engineered using soleus muscles and bone marrow from tdTomato rats, generated as described below. Control SMUs were fabricated from Fischer 344 rats (strain no. 403; Charles River Laboratories, Wilmington, MA) or from F344-Tg(UBC-EGFP)F455Rrrc rats (strain no. 307; Rat Resource & Research Center, Columbia, MO).
Generation of ubiquitous tdTomato rat
To generate transgenic tdTomato rats, an ∼3.9 kb fragment of DNA was isolated from pAAV-UbC-tdTomato28 by digestion with PciI and RsrII restriction enzymes (cat. no. R0655S and R0501S; New England BioLabs, Ipswitch, MA). This isolated fragment contained the human ubiquitin C promoter and β-globin intron enhancer upstream of the red fluorescent protein, tdTomato, followed by a human growth hormone poly-adenylation sequence (Fig. 1A). The fragment was purified and injected into the pronuclei of fertilized rat eggs, which were then surgically implanted into recipient rats as described by Filipiak and Saunders.29 The genotype of the offspring was determined by polymerase chain reaction (PCR) analysis of DNA obtained from tail biopsies (Fig. 1B) using the Qiagen DNeasy Kit (cat. no. 69504; Qiagen, Valencia, CA) and Platinum Taq Polymerase (cat. no. 13000-012; Thermo Fisher Scientific, Waltham, MA). Primers for PCR are listed in Table 1, and rat beta-2-microglobulin used as a control.
FIG. 1.

Genetic modification strategy. (A) A DNA fragment encoding tdTomato preceded by a human UbC promoter and a β-globin intron enhancer and followed by a hGH pA sequence was inserted into recipient rats through injection into the pronuclei of fertilized eggs. (B) PCR confirmed the presence of tdTomato and human UbC in transgenic animals, with rat β-2-microglobulin as a control. hGH pA, human growth hormone poly-adenylation; PCR, polymerase chain reaction; tdTomato, tandem-dimer tomato; UbC, ubiquitin C.
Table 1.
Primers Used for Genotyping
| Gene | Fwd. primer (5′-3′) | Rev. primer (5′-3′) | Exp. size (bp) |
|---|---|---|---|
| hUbC | CACCCGTTCTGTTGGCTTAT | CCAAAAACGGCCAGAATTTA | 703 |
| tdT | CTCCGAGGACAACAACATGG | CTTGTACAGCTCGTCCATGC | 349 |
| B2 M | GTGCTTGTCTCTCTGGCCGTCG | AACGCCCACTCCTTTCCCGAGA | 144 |
PCR confirmed the presence of hUbC and tdTomato in transgenic rats. Rat B2 M was used as a control.
B2 M, beta-2-microglobulin; hUbC, human ubiquitin C; PCR, polymerase chain reaction; tdT, tandem-dimer tomato.
Tissue fluorescence validation
Animals were anesthetized using isoflurane (Piramal, Bethlehem, PA, NADA no. 200-237) before imaging with an IVIS Spectrum (cat. no. 124262; PerkinElmer, Waltham, MA) in vivo imaging system. Animals were placed in an upright position and the dorsal side was viewed from above. The GFP signal was acquired at an excitation of 465 nm and an emission of 520 nm, whereas the tdTomato signal was acquired using an excitation of 535 nm and a 580 nm emission. Captured images were analyzed using the Living Image software associated with the IVIS system. Background fluorescence and autofluorescence from the fur of the animals were removed so that the average radiance could be quantified.
Tissue dissection and muscle-derived cell isolation
Before dissection, intraperitoneal injections of sodium pentobarbital (65 mg/kg; Merck Animal Health, Madison, NJ, NADA no. 119-807) were used to induce a deep plane of anesthesia. Supplemental pentobarbital doses were administered as required to maintain adequate anesthesia depth. Once under anesthesia, tissues were dissected as described in previous studies.25,26,30,31 Both soleus muscles were removed under aseptic conditions and sanitized in 70% ethanol. After tissue dissection, animals were subsequently euthanized through the administration of a bilateral pneumothorax. Muscles were minced using a razor blade, placed under ultraviolet light for 15 min, and added to a dissociation solution consisting of 32 U dispase (1.8 U/mg, cat. no. 17105-041; Thermo Fisher Scientific) and 2390 U type IV collagenase (239 U/mg, cat. no. 17104-019; Thermo Fisher Scientific). The mixture was maintained at 37°C with agitation for 90 min. The resulting suspension was then filtered with a 100 μm mesh filter (cat. no. 22-363-549; Thermo Fisher Scientific) and centrifuged. The supernatant dissociation solution was aspirated away, and the cells were resuspended in a growth medium.
Cell fluorescence validation
To confirm the presence of fluorescence signals, muscle isolates from a tdTomato, GFP, and wild-type animal were individually seeded for imaging. Additionally, these three cell populations were combined at equal densities and seeded for simultaneous visualization. On day 6 of SMU fabrication, shortly before reaching confluence, developing cell monolayers were imaged. Bright field, green, and red fluorescence images were captured using an EVOS FL Color Imaging System (cat. no. AMEFC4300; Thermo Fisher Scientific) with the exposure maintained at 500 ms for all fluorescence images. The mean fluorescence intensity for each image was measured using ImageJ.
SMU formation
In this study, all cells used for SMU formation were isolated from the transgenic tdTomato rats described above. As detailed in previous studies, bone and tendon constructs were fabricated to act as anchors onto which developing muscle monolayers could attach and fuse.32,33 Bone marrow from both femurs was removed under aseptic conditions. Isolated bone marrow cells were plated in 100-mm tissue culture plates (cat. no. 353003; Corning, Corning, NY). Following four passages to drive cells to the bone or tendon lineage, cell monolayers were delaminated from the tissue culture plate and were pinned in cylindrical form on a Sylgard 184 substrate (cat. no. 4019862; Dow Chemical Corp., Midland, MI) using minutien pins (cat. no. 26002-20; Fine Science Tools, Foster City, CA). The resulting tissue constructs were cut into 2.5 mm sections to be used as engineered bone–tendon anchors for construct implantation.
SMUs were engineered in 60-mm polystyrene plates (cat. no. 353002; Corning) as described previously.25,34 Isolated muscle-derived cells in muscle growth medium (MGM) were seeded onto a PDMS substrate coated with laminin (cat. no. 23017-015; Thermo Fisher Scientific). After initial plating for 4 days to allow attachment, cells were subsequently fed MGM every 2 days until becoming fully confluent and forming a network of elongating myotubes on day 7. At this point, bone–tendon anchors were pinned onto the cell monolayers at a spacing of 2.5 cm, and the medium was switched to muscle differentiation medium. Monolayers delaminated from the plates on day 14, rolling into cylindrical muscle constructs, held at length by the engineered bone–tendon anchors.
To create engineered tissues more closely matching the VML deficit, three individual SMUs were combined as pictured in Figure 2A–C. Each SMU was allowed to fuse for 24 h in 3D cylindrical form (Fig. 2A) before being transferred to a dish with two other SMUs on day 15 (Fig. 2B). Using minutien pins, the bone–tendon anchors were superimposed and pinned together (Fig. 2C). The combined SMUs were maintained in this form for 48 h to allow fusion before implantation on day 17. This modular approach to scale-up allowed us to create a larger tissue for implantation while avoiding the potential necrosis that occurs in larger, avascular tissues with prolonged time in vitro.
FIG. 2.

SMU fabrication and surgical procedures. To create larger engineered skeletal muscle tissues more closely matching the VML defect, (A) individual tdT SMUs composed of skeletal muscle supported by bone–tendon anchors, (B) were transferred in sets of three to a common substrate, and (C) were combined by pinning the bone–tendon anchors together. After creating a VML injury in GFP rats by removing ∼30% of the TA muscle, either (D) the defect was left unrepaired except for a redirected nerve branch and its supporting vasculature, or (E) a combined SMU was implanted to fill the defect. Black arrowheads indicate the redirected nerve. Following 28 days of recovery, a representative TA muscle is pictured from (F) the VML Only group, and (G) the VML+SMU group. Graft and contralateral TAs were subsequently explanted, weighed, and preserved for histology. GFP, green fluorescent protein; SMU, skeletal muscle unit; TA, tibialis anterior; VML, volumetric muscle loss.
Surgical procedures
All host rats in this study expressed ubiquitous GFP fluorescence. Each animal was anesthetized using isoflurane. An incision along the lower left hindlimb exposed the tibialis anterior (TA) muscle, and a longitudinal cut removed ∼30% of the TA volume. The resected muscle was blotted and weighed to quantify the mass of the induced VML defect. The implantation procedure followed previously established techniques,25 with animals randomly assigned to a VML Only or VML+SMU group. In both groups, the peroneal nerve distal to the innervation of the extensor digitorum longus was transected along with its associated vasculature and routed to the area of VML. A 0.9 mm diameter tunnel was drilled into the proximal tendon insertion site on the tibia. In the VML Only group, transected nerve branch was sutured into the defect, but no additional tissue was added. In the VML+SMU group, an engineered tissue composed of three combined tdTomato SMUs was placed in the repair site, with one bone–tendon end placed in tibial bone tunnel and sutured to periosteum, and the other bone–tendon anchor sutured to the distal TA tendon. The transected nerve branch was then sutured to the SMU using 9-0 suture. The surgical site was then closed with surgical staples, and carprofen (Zoetis, Parsippany, NJ, NADA no. 141-199) was administered following each procedure at a dose of 5 mg/kg every 24 h for 48 h postsurgery.
Following 28 days of recovery, rats were anesthetized with an injection of sodium pentobarbital (65 mg/kg). The TA muscles were removed, blotted, and weighed. Immediately after muscle mass was measured, muscles were coated in tissue freezing medium (cat. no. 15146-019; VWR, Radnor, PA), frozen in 2-methylbutane (cat. no. 03551-4; Thermo Fisher Scientific) cooled by dry ice, and stored at −80°C until analyzed for histology.
Histochemical and immunohistochemical analysis
Cross-sections of the repaired and contralateral TA muscles at a thickness of 12 μm were mounted on Superfrost Plus microscopy slides (cat. no. 12-550-15; Thermo Fisher Scientific) and used for histological analysis. General morphology was observed with Hematoxylin and Eosin (H&E).35 For immunohistochemical (IHC) analysis, frozen sections were fixed in methanol at −20°C for 10 min. The sections were submerged for 15 min in 0.05% Triton X-100 (Sigma-Aldrich) in DPBS (PBST) and blocked with PBST containing 3% bovine serum albumin (cat. no. A2153-10g; Sigma-Aldrich) at room temperature. Sections were then incubated overnight at 4°C with primary antibodies listed below. Immunofluorescent staining with specific antibodies was performed to detect the presence of myosin heavy chains (1:100 dilution, cat. no. MF20; Developmental Studies Hybridoma Bank, Iowa City, IA) and laminin (1:200 dilution, cat. no. ab7463; Abcam, Cambridge, MA). Following three washes in PBST, the sections were incubated in 1:500 dilutions of AlexaFluor 647 anti-mouse and 488 anti-rabbit secondary antibodies to target myosin heavy chains and laminin, respectively (cat. no. A31571 and A11034; Thermo Fisher Scientific). Following three PBST washes, sections were fixed in ProLong Gold with DAPI (cat. no. P36935; Thermo Fisher Scientific) and coverslipped. The sections were examined and photographed with an Olympus FluoView 1000 microscope, and images were analyzed using ImageJ.
Statistical analysis
Values are presented as mean ± standard error. Measurements of significant differences between mean were performed using GraphPad Prism 7 (GraphPad Software, Inc., La Jolla, CA). Mean was compared using either a Student's t-test or one-way ANOVA with Tukey's post hoc comparisons. Differences were considered significant at p < 0.05.
Results
tdTomato fluorescence was greater than GFP fluorescence in both transgenic animals in vivo and isolated cells in vitro
Upon analysis of in vivo fluorescence images from wild-type, GFP, and tdTomato animals (n = 3 each), several important differences were evident. The raw fluorescence signal from the wild-type animal indicated a distinct signal in the green channel localized to the fur (data not shown). After removing this autofluorescence baseline, green fluorescence was readily detectable in the ears, feet, and tails of GFP animals (Fig. 3C), as was red fluorescence in the tdTomato rats (Fig. 3D). Quantification of the average radiance indicated that the GFP signal (1.67 × 108 ± 1.22 × 107 photons/sec/cm2) was significantly weaker than the tdTomato (2.65 × 108 ± 3.35 × 107 photons/sec/cm2, p = 0.020).
FIG. 3.
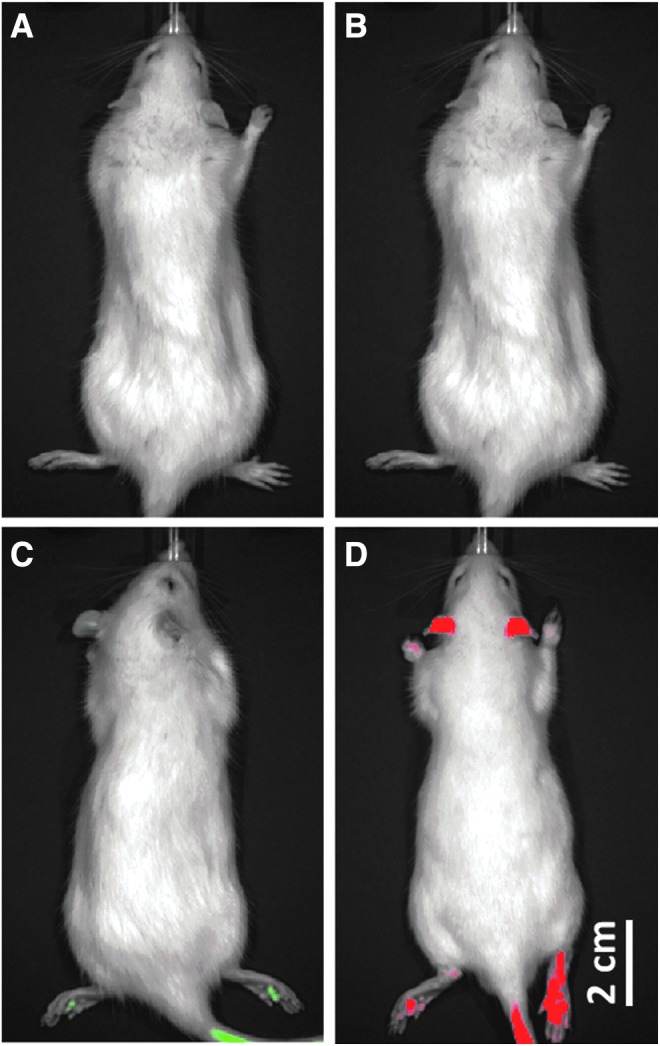
In vivo fluorescence signals. Representative images of the red and green fluorescent signals demonstrate the relative intensity of GFP and tdT from in vivo imaging. After removing background tissue and fur autofluorescence, minimal fluorescence was present in wild-type animals in (A) the green fluorescent channel or (B) the red channel. In the ears, feet, and tail, (C) the GFP signal was observed to be significantly weaker (1.67 × 108 ± 1.22 × 107 photons/sec/cm2) than (D) the tdT signal (2.65 × 108 ± 3.35 × 107 photons/sec/cm2, p = 0.020).
On the cellular level, microscopy was used to validate fluorescence expression. Following growth for 6 days in vitro, isolated cells maintained their respective fluorescence (Fig. 4). Ubiquitous green fluorescence was observed in cells isolated from GFP animals, and ubiquitous red fluorescence was present in cells from tdTomato rats. Quantification of these fluorescence intensities indicated that the tdTomato signal was significantly greater than the GFP signal (8.83 ± 1.51 vs. 3.22 ± 0.55, p = 0.008, n = 6). When cells from a GFP, tdTomato, and wild-type rat were seeded evenly and cultured together, these three populations were evident from their fluorescence signals when analyzed on day 6. In many cases, fusion of myoblasts into multinucleated myotubes resulted in muscle cells expressing only one type of fluorescence. However, several chimeric myotubes expressing both red and green fluorescence were observed, indicating myoblasts from these different animals were able to interact with each other and fuse normally.
FIG. 4.

Cell fluorescence in muscle isolates. Following 6 days in culture, fluorescence was observed in proliferating cells. (Top Row) Triculture of equal populations of tdTomato, GFP, and wild-type cells exhibited cells expressing either red, green, or no fluorescence. Fusion of red and green myoblasts into a chimeric myotube is indicated by the white arrowhead. (2nd Row) Ubiquitous red fluorescence alone was observed in cells isolated from tdTomato rats, (3rd Row) whereas only green fluorescence was present in cells from the GFP animal. Quantification of these fluorescence intensities indicated the tdTomato signal was significantly greater than the GFP signal (8.83 ± 1.51 vs. 3.22 ± 0.55, p = 0.008, n = 6). (Bottom Row) Cells isolated from wild-type animals exhibited no red or green fluorescence above background levels.
Cell tracking indicated cell migration both into and out of the implanted engineered muscle
Following 28 days of implantation, cross-sections of the surgical TA muscles were examined for migration of GFP cells from the host into the engineered tissue and for the remaining presence of tdTomato cells in the implanted engineered tissue or for their migration into the host. The location of the VML defect in the VML Only group (Fig. 5A) and the surgically repaired area in the VML+SMU group (Fig. 5B) was evident from the presence of the suture landmarks. Examination of the GFP signal indicated widespread migration of host cells into the defect site, such that distinguishing host and implant was difficult based on relative GFP intensity alone. From analysis of the tdTomato signal, it was evident that the implanted tissue maintained some level of tdTomato expression despite the infiltration of host GFP cells. Additionally, engraftment of tdTomato cells from the engineered tissue with host fibers was visible (Fig. 5B–D).
FIG. 5.

Cell migration following SMU implantation. Examination of tdT and GFP signals in explanted TA cross-sections (A) confirmed the absence of tdT signal in VML Only animals and (B) demonstrated the presence of both persistent tdT signal and infiltrating GFP signal in the implanted tdT SMU. White arrows indicate the suture landmarks used to locate the VML defect or SMU implant. (C, D) show higher magnification images of the implanted tissue from other animals.
Implantation of the combined SMU restored a portion of the VML deficit and regenerated small muscle fibers in the defect site
To evaluate muscle regeneration, the pre- and postimplantation masses of the VML defect were compared. Before implantation, the average VML deficit was 101 ± 10 mg in the VML Only group (n = 7 for each group). This value was not significantly different (p = 0.44) from the VML deficit of 91 ± 8.0 mg in the repaired VML+SMU group before tissue implantation (Fig. 6I). The average mass of the combined SMU placed into the VML defect was 53 ± 1.1 mg. After 28 days of recovery, implantation of the combined SMU led to a significant recovery of the remaining VML deficit. The VML Only group exhibited a mass deficit of 105 ± 14 mg, whereas the VML+SMU group had a significantly smaller deficit of 60 ± 14 mg (p = 0.026, Fig. 6J).
FIG. 6.

Muscle recovery with 28-day implantation of combined SMU. H&E images of explanted TA muscles indicated (A) minimal regeneration in animals subjected to a VML deficit alone, in comparison to (C) those receiving an engineered tdT SMU implant. IHC for myosin heavy chains (MF20, recolored in red), laminin (green), and DAPI (blue) (B) demonstrated substantial extracellular matrix deposition in the VML Only group and (D) indicated some formation of distinct muscle fibers in the VML+SMU group. It is important to note that the MF20 was labeled with a far red antibody to avoid potential overlap with the tdT signal. Arrows indicate the suture landmarks used to locate the VML defect or SMU implant and differ in color only to provide visible contrast. (I) There was no significant difference in the size of VML defect between VML Only (101 ± 10 mg) and VML+SMU (91 ± 8.0 mg) subjects. (E–H) Show higher magnification H&E and IHC images of the implanted tissue from two other animals. All images are serial sections to those in Figure 5. (J) Implantation of the combined SMU did lead to a significant decrease in the size of the VML deficit following 28 days of recovery, with the VML Only deficit remaining largely unchanged at 105 ± 14 mg and the VML+SMU deficit falling to 60 ± 14 mg. This decrease can partially be attributed to the added mass of the engineered tissue itself, but the formation of muscle fibers in the implant area suggests some degree of myogenesis due to the SMU. (K) Quantification of CSA indicated that regenerating fibers in the implant area (166 ± 16 μm2) were ∼9.5% of the average CSA of control native fibers, 1931 ± 216 μm2. # indicates significant different from VML Only or Native control (p < 0.05). CSA, cross-sectional area; H&E, Hematoxylin and Eosin; IHC, immunohistochemical.
Examination of the explanted TA cross-sections exhibited similarly improved regeneration of the VML defect with SMU implantation. H&E images were used to identify the VML defect in the VML Only group (Fig. 6A) and the surgically repaired area in the VML+SMU group (Fig. 6C). These regions were characterized by hypercellularity and an increased amount of connective tissue compared with native skeletal muscle morphology, and we used this altered morphology of the tissue to identify the surgical sites during histological analysis. The VML+SMU group exhibited a greater degree of hypercellularity than the VML Only group, with minimal collagen crimping, suggesting that the repaired muscles were undergoing remodeling. IHC for myosin heavy chains and laminin identified muscle fibers surrounded by extracellular matrix within the implant area (Fig. 6D, F, H). The average cross-sectional area (CSA) of these fibers, 166 ± 16 μm2, was ∼9.5% of the average CSA of control native fibers, 1931 ± 216 μm2 (Fig. 6K). These values match the findings from our previous study of VML repair,25 suggesting both that tdTomato cells possess similar regenerative potential to wild-type cells and that the process of combining individual SMUs to engineer a larger tissue did not adversely affect myogenesis.
Discussion
One of the primary purposes of this study was to develop a ubiquitous tdTomato rat for use in combination with GFP markers to enable tracking of cell migration in implantation studies. Although the enhanced form of GFP certainly has applicability for cell tracking, it is especially limited in vivo by its relatively short wavelength and overlap with tissue autofluorescence. Thus, we used tdTomato as an alternative fluorophore in our engineered tissues and used it in combination with GFP to identify cellular origin and observe cell migration in implantation studies. After generating a ubiquitous tdTomato rat, validating the tdTomato fluorescence expression, and demonstrating intensity superior to GFP at the cell and tissue level, we studied the localization of tdTomato protein following 28 days of implantation in a model of VML. Although host GFP cells clearly infiltrated the implanted tdTomato tissue over this recovery period, distinct areas expressing tdTomato fluorescence were still evident in the defect area. Additionally, migration of tdTomato cells from the SMU implant into the host tissue was consistently observed. Because cell migration was observed both into and away from the implant, it was difficult to determine the source of regenerating muscle fibers. The ability to track cell migration in both directions, however, highlights the potential utility of this approach for a wide variety of cell tracking applications.
The presence of regenerating muscle fibers, accompanied by a decrease in size of the VML defect, following implantation of the engineered skeletal muscle construct demonstrates the potential of our engineered SMUs for VML repair. In the VML Only negative control group, animals subjected to a VML injury without any implant confirmed the inability of the host to repair this defect naturally. In contrast, the engineered tissue implant successfully integrated with the host tissue and could not be distinguished by gross observation alone, as well as significantly decreased the size of the VML defect. Small but distinct regenerating muscle fibers, comparable in size to past studies at the same time point, were present in the implanted SMU following the 28 days of recovery as well. These findings address the second primary purpose of this study, investigating whether implanting larger engineered tissues leads to greater recovery of the volumetric deficit. At the time of implantation, the larger, combined SMU weighed ∼50% of the muscle removed. After the 28 days of recovery, this larger engineered tissue had restored nearly half of the mass of the VML deficit. Based on these promising results, we expect it will be necessary to match the size of our engineered SMUs to the original deficit for full recovery of muscle mass at this time point.
Ultimately, this study successfully accomplished its two goals. A tdTomato rat was generated through genetic modification, and the isolated tdTomato muscle progenitor cells were used to fabricate tissue-engineered skeletal muscle. These single SMUs were successfully combined in a modular fashion to form SMUs closer in size to induced VML deficits. This method of modularly combining SMUs to create a larger tissue for implantation was implemented to help avoid the diffusion limitations of larger, avascular tissues which typically develop a necrotic core after extended time in vitro. Importantly, this fabrication method maintained the regenerative potential of the engineered SMUs, as evidenced by the presence of small, regenerating muscle fibers accompanied by both resilient tdTomato and infiltrating GFP fluorescence within the surgical site after the 28-day implantation.
While these results demonstrate the applicability of our tdTomato rat for cell tracking studies, further examination at greater resolution will likely be needed to define the specific mechanisms behind migration into and away from the implanted engineered tissue. Specifically, we are curious to study cell migration at more acute time points to relate this process to essential steps in the native repair mechanism. Additional questions relating to the scaling of engineered tissues remain as well. Our results suggest that engineering even larger tissues to match the size of the VML deficit may be necessary for full recovery of mass, and this finding will be important as researchers seek to repair volumetric injuries in more clinically relevant models. Fortunately, our study successfully validated a modular approach for scaling engineered tissues to a larger geometry. We hope researchers in the field can use this scale-up model for tissue fabrication for advancing their technologies toward clinical relevance and can use the combination of two fluorescent markers in cell tracking experiments for both tissue engineering therapies and basic biological studies.
Acknowledgments
The authors would like to acknowledge Thomas Saunders from the University of Michigan Transgenic Animal Model Core. Peter C. Macpherson, Rachel Armstrong, Matthew Nguyen, and Arthur Su provided essential technical assistance. They would also like to acknowledge the financial support of the NIH through R01 AR067744-011.
Disclosure Statement
No competing financial interests exist.
References
- 1.Lee M.H., Arcidiacono J.A., Bilek A.M., et al. Considerations for tissue-engineered and regenerative medicine product development prior to clinical trials in the United States. Tissue Eng Part B Rev 16, 41, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Orlando G., Wood K.J., Stratta R.J., et al. Regenerative medicine and organ transplantation: past, present, and future. Transplantation 91, 1310, 2011 [DOI] [PubMed] [Google Scholar]
- 3.Orlando G., Soker S., Stratta R.J., and Atala A. Will regenerative medicine replace transplantation? Cold Spring Harb Perspect Med 3, a015693, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abouna G.M. Organ shortage crisis: problems and possible solutions. Transplant Proc 40, 34, 2008 [DOI] [PubMed] [Google Scholar]
- 5.Heidary Rouchi A., and Mahdavi-Mazdeh M. Regenerative medicine in organ and tissue transplantation: shortly and practically achievable? Int J Organ Transplant Med 6, 93, 2015 [PMC free article] [PubMed] [Google Scholar]
- 6.Butler D. Translational research: crossing the valley of death. Nature 453, 840, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Omstead D.R., Baird L.G., Christenson L., et al. Voluntary guidance for the development of tissue-engineered products. Tissue Eng 4, 239, 1998 [DOI] [PubMed] [Google Scholar]
- 8.Progatzky F., Dallman M.J., and Lo Celso C. From seeing to believing: labelling strategies for in vivo cell-tracking experiments. Interface Focus 3, 20130001, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kretzschmar K., and Watt F.M. Lineage tracing. Cell 148, 33, 2012 [DOI] [PubMed] [Google Scholar]
- 10.Luche H., Weber O., Nageswara Rao T., Blum C., and Fehling H.J. Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. Eur J Immunol 37, 43 2007 [DOI] [PubMed] [Google Scholar]
- 11.Moter A., and Göbel U.B. Fluorescence in situ hybridization (FISH) for direct visualization of microorganisms. J Microbiol Methods 41, 85, 2000 [DOI] [PubMed] [Google Scholar]
- 12.Tao W., Evans B.G., Yao J., et al. Enhanced green fluorescent protein is a nearly ideal long-term expression tracer for hematopoietic stem cells, whereas DsRed-express fluorescent protein is not. Stem Cells 25, 670, 2007 [DOI] [PubMed] [Google Scholar]
- 13.Morris L.M., Klanke C.A., Lang S.A., Lim F.Y., and Crombleholme T.M. TdTomato and EGFP identification in histological sections: insight and alternatives. Biotech Histochem 85, 379, 2010 [DOI] [PubMed] [Google Scholar]
- 14.Tsien R.Y. The green fluorescent protein. Annu Rev Biochem 67, 509, 1998 [DOI] [PubMed] [Google Scholar]
- 15.Skylaki S., Hilsenbeck O., and Schroeder T. Challenges in long-term imaging and quantification of single-cell dynamics. Nat Biotechnol 34, 1137, 2016 [DOI] [PubMed] [Google Scholar]
- 16.Spronken M.I., Short K., Herfst S., et al. Optimisations and challenges involved in the creation of various bioluminescent and fluorescent influenza A virus strains for in vitro and in vivo applications. PLoS One 10, e0133888, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell R.E., Tour O., Palmer A.E., et al. A monomeric red fluorescent protein. Proc Natl Acad Sci U S A 99, 7877, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaner N.C., Patterson G.H., and Davidson M.W. Advances in fluorescent protein technology. J Cell Sci 120, 4247, 2007 [DOI] [PubMed] [Google Scholar]
- 19.Shaner N.C., Campbell R.E., Steinbach P.A., et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22, 1567, 2004 [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi T., Kato-Itoh M., Yomaguchi T., et al. Identification of rat Rosa26 locus enables generation of knock-in rat lines ubiquitously expressing tdTomato. Stem Cells Dev 21, 2981, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Igarashi H., Koizumi K., Kaneko R., et al. A novel reporter rat strain that conditionally expresses the bright red fluorescent protein tdTomato. PLoS One 11, e0155687, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Y., Yu L., Pan S., et al. CRISPR/Cas9-mediated targeting of the Rosa26 locus produces Cre reporter rat strains for monitoring Cre-loxP-mediated lineage tracing. FEBS J 284, 3262, 2017 [DOI] [PubMed] [Google Scholar]
- 23.Gumucio J.P., Flood M.D., Bedi A., et al. Inhibition of prolyl 4-hydroxylase decreases muscle fibrosis following chronic rotator cuff tear. Bone Joint Res 6, 57, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gumucio J.P., Flood M.D., Roche S.M., et al. Stromal vascular stem cell treatment decreases muscle fibrosis following chronic rotator cuff tear. Int Orthop 40, 759, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.VanDusen K.W., Syverud B.C., Williams M.L., Lee J.D., and Larkin L.M. Engineered skeletal muscle units for repair of volumetric muscle loss in the tibialis anterior muscle of a rat. Tissue Eng Part A 20, 2920, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams M.L., Kostrominova T.Y., Arruda E.M., and Larkin L.M. Effect of implantation on engineered skeletal muscle constructs. J Tissue Eng Regen Med 7, 434, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Committee for the Update of the Guide for the Care and Use of Laboratory Animals, eigth edition. Washington, DC: National Academies Press, 2010 [Google Scholar]
- 28.Soderblom C., Lee D.H., Dawood A., et al. 3D imaging of axons in transparent spinal cords from rodents and nonhuman primates. eNeuro 2, 1, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Filipiak W.E., and Saunders T.L. Advances in transgenic rat production. Transgenic Res 15, 673, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Mertens J.P., Sugg K.B., Lee J.D., and Larkin L.M. Engineering muscle constructs for the creation of functional engineered musculoskeletal tissue. Regen Med 9, 89, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weist M.R., Wellington M.S., Bermudez J.E., et al. TGF-β1 enhances contractility in engineered skeletal muscle. J Tissue Eng Regen Med 7, 562, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Syed-Picard F.N., Larkin L.M., Shaw C.M., and Arruda E.M. Three-dimensional engineered bone from bone marrow stromal cells and their autogenous extracellular matrix. Tissue Eng Part A 15, 187, 2009 [DOI] [PubMed] [Google Scholar]
- 33.Ma J., Smietana M., Kostrominova T.Y., et al. Three-dimensional engineered bone-ligament-bone constructs for anterior cruciate ligament replacement. Tissue Eng Part A 18, 103, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Syverud B.C., VanDusen K.W., and Larkin L.M. Effects of dexamethasone on satellite cells and tissue engineered skeletal muscle units. Tissue Eng Part A 22, 480, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luna L.G. Manual of Histologic Staining Methods of the Armed Forces Institute of Pathology, third edition. New York: McGraw-Hill, Inc., 1960 [Google Scholar]