
Fig. 1.
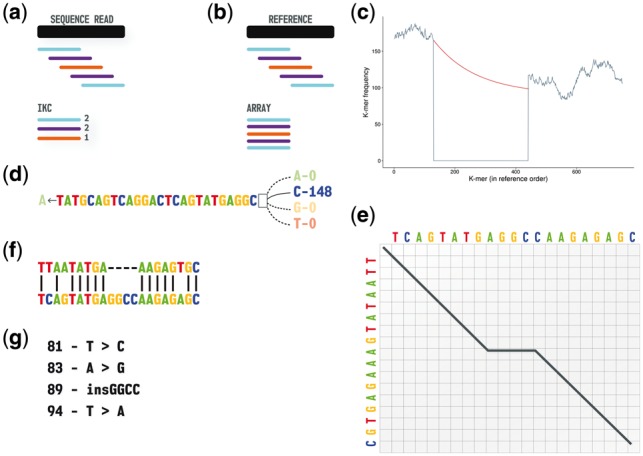
Overview of the Kestrel process from sequence data to variant call. (a) The sequence reads are converted to an IKC file. (b) The reference sequence is converted into an array of k-mers and left in reference order. (c) K-mer frequencies from the sequence reads (vertical axis) are assigned to the ordered k-mers of the reference (horizontal axis). A decline and recovery of the frequencies bound an active region where one or more variants are present. The recovery threshold is degraded with an exponential decay function (red) to allow for declining read coverage. (d) Starting from the left anchor k-mer (last k-mer with a high frequency), the first base is removed, each possible base is appended, and the base that recovers the k-mer frequency is appended to the haplotype. (e) A modified alignment algorithm tracks haplotype reconstruction and terminates the process when an optimal alignment is reached. (f) This algorithm yields an alignment of the reference sequence and haplotype within the active region. (g) Variant calls are extracted from the alignment (Color version of this figure is available at Bioinformatics online.)