

Abstract
Motivation
Structural variation (SV) detection from short-read whole genome sequencing is error prone, presenting significant challenges for population or family-based studies of disease.
Results
Here, we describe SV2, a machine-learning algorithm for genotyping deletions and duplications from paired-end sequencing data. SV2 can rapidly integrate variant calls from multiple structural variant discovery algorithms into a unified call set with high genotyping accuracy and capability to detect de novo mutations.
Availability and implementation
SV2 is freely available on GitHub (https://github.com/dantaki/SV2).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Structural variation (SV) is defined as a change of the structure of a chromosome larger than 50 bp. SV is a major contributor to human genetic variation and is implicated in a variety of human diseases (Conrad et al., 2010; Sudmant et al., 2015). In particular, de novo structural mutations (those in offspring and not in parents) contribute significant risk for idiopathic autism and intellectual disability (Brandler et al., 2016). Accurate detection of de novo SVs is challenging due to genotyping errors, such as false-positives in offspring and false-negatives in parents. Additionally, genotyping errors bias the transmission of variants from parent to offspring, confounding analysis of pedigrees.
Characterizing SV from next generation sequencing is a difficult task due to the broad range of SV sizes (50 bp–50 Mb) and types. Hence, to fully capture the diversity of SVs a multitude of tools is required, each designed as a standalone solution (Francioli et al., 2014; Sudmant et al., 2015). Methods for harmonizing variant calls and confidence scores from multiple SV calling methods into a unified set of SV genotypes are lacking.
2 Input and methods
SV2 (support-vector structural-variant genotyper) is an open source application written in Python that requires a BAM file, a single nucleotide variant (SNV) VCF file, and either a BED or VCF file of deletions and duplications as input. SV2 operates in three stages: preprocessing, feature extraction, followed by genotyping (Fig. 1). Genotyping utilizes four informative features: depth of coverage, discordant paired-ends, split-reads and heterozygous allele ratio (HAR). Genotyping is performed with supervised support vector machine classifiers trained on whole genome sequences (WGS) from the 1000 Genomes Project (1KGP). The resulting VCF file of genotypes contains annotations for genes, repeat elements and variant identifiers of common SVs recorded by the 1KGP (Sudmant et al., 2015).
Fig.1.
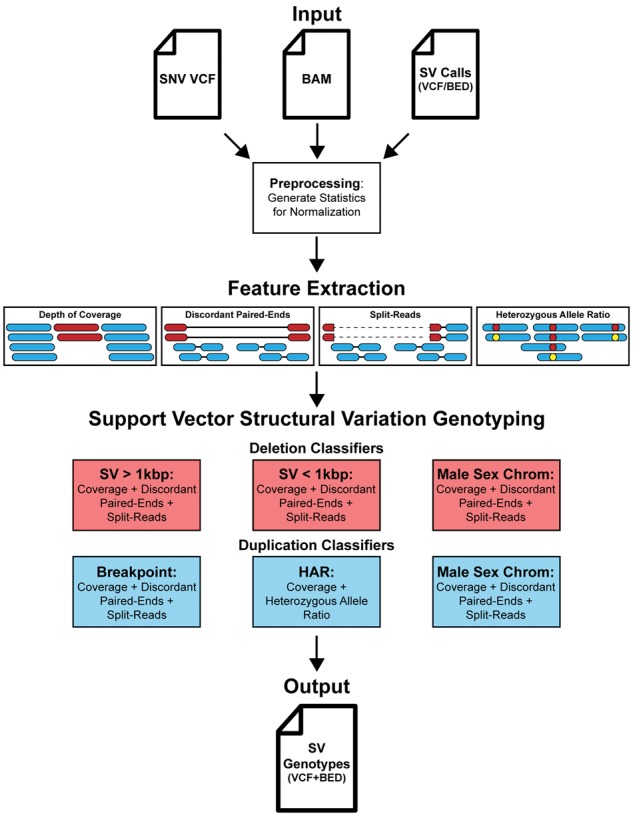
SV2 workflow. SV2 requires a VCF file of SNVs, a BAM file, and a set of SVs to genotype as input. Before genotyping, preprocessing is performed where the median coverage, insert size, and read length is recorded for feature normalization. Features for genotyping, which include depth of coverage, discordant paired-ends, split-reads, and HAR, are measured for each SV. SVs are then genotyped with an ensemble of support vector machine classifiers. SV2 produces two output files, a BED file and a VCF, containing annotations for RefSeq genic elements, RepeatMasker repeats, segmental duplications, short tandem repeats, and common SVs from the 1000 Genomes phase 3 call set
3 Genotyping classifiers
Our training set included high coverage (48×, N = 27) and low coverage (7×, N = 2, 494) WGS from the 1KGP. Training features were collected from a gold-standard SV call set on the above individuals with an estimated false discovery rate (FDR) of 1–4% (Sudmant et al., 2015), totaling 297 131 genotypes from 11 747 unique loci (Supplementary Table S1).
An ensemble of SV detection methods is required to capture a wide diversity of SVs, resulting in a set of SVs with variable characteristics. Deletions have a very broad size range (50 bp–>10 Mb), and as deletions become very small, the signal from discordant paired-ends diminishes and signal from split-reads increases. Duplication calls, in contrast, tend to be larger (>3 kb Sudmant et al., 2015) with less precise breakpoints. In addition, genotyping methods must account for the ploidy of sex chromosomes in males and females. Consequently, a one-size-fits-all genotyping method performs poorly on the combined calls.
With our goal of combining SV calls from multiple methods and generating a uniform set of genotypes, we developed a genotyper consisting of multiple classifiers, each designed to genotype a different category of SV. To contend with the variable characteristics described earlier, deletions were stratified by size and duplications by the availability of breakpoint features (discordant paired-ends or split-reads). Deletions were trained in three classifiers: (i) deletions greater than 1 kb, (ii) deletions less than or equal to 1 kb and (iii) deletions on male sex chromosomes. All three deletion classifiers implemented coverage, discordant paired-end and split-read features extracted from high coverage WGS. Duplications were trained in three additional classifiers: (iv) duplications with breakpoint features, (v) duplications without breakpoint features and (vi) duplications on male sex chromosomes. Classifier v was unique in that it included heterozygous SNV allele ratios as a feature (‘HAR’ Fig. 1). The deletion male sex chromosomes classifier was not split by size due to the sparse number of training examples (N = 191, Supplementary Table S1).
4 Performance results
For this study, we implemented three independent WGS datasets for training, evaluation and validation (Supplementary Table S2). Parameter selection for classifiers was based on a 7-fold cross-validation of the training set (Supplementary Fig. S1) and further based on classifier performance in an independent dataset consisting of 42× WGS of 57 subjects (Supplementary Table S3), a subset of samples from our previous study of autism (‘evaluation dataset’, Brandler et al., 2017). Genotyping accuracy was evaluated further based on rates of SV transmission in families from an additional 1827 genomes from Brandler et al. Finally, classifier performance was independently evaluated in a third dataset consisting of 72× Illumina WGS of nine subjects from the 1KGP (‘validation dataset’). We confirmed SV2 genotypes in the evaluation and validation datasets using two orthogonal platforms: Illumina 2.5 M microarrays and Pacific Biosciences (PacBio) single molecule sequencing, respectively.
Performance was evaluated at two levels of SV filtering stringency: a ‘standard’ level and a stricter level for calling de novo mutations, which are enriched for false-positive calls. Our formulation of filters applied the evaluation set of 57 subjects with SV2 genotypes from SV calls from ForestSV (v0.3.3; Michaelson and Sebat 2012), LUMPY (v0.2.13; Layer et al., 2014) and Manta (v1.1.1; Chen et al., 2015). We implemented Illumina 2.5 M arrays to calculate the FDR using SVToolkit (v2.0 sourceforge.net/projects/svtoolkit), which performs an in silico validation by ranking microarray probes intensities, defining the FDR as two times the fraction of variants with P-value >0.5 (Sudmant et al., 2015). The final thresholds for filters for each stringency level considered variant length, feature availability and FDR (Supplementary Table S4).
Evaluation: Using the aforementioned evaluation set, we found a FDR of 40% for both unfiltered deletions (N = 5344) and duplications (N = 776) (Fig. 2A). Filtering at the standard level of stringency reduced the FDR to 1.24% for deletions and 4.41% for duplications (Supplementary Fig. S2). We then ascertained the FDR of unfiltered de novo variants to be 60% for deletions and 86% for duplications. Applying the de novo filters reduced the FDR to 0.54% for deletions and 0% for duplications (Supplementary Fig. S2). We extended our evaluation of SV2 genotyping with an additional 1827 individuals to the previous evaluation set of 57 subjects, totaling 619 families (N = 1884). Validation of SV2 genotypes implemented the group-wise transmission disequilibrium test (Chen et al., 2015), which tests for deviations from the expected variant transmission rate (50%). An under-transmission of variants indicates either an enrichment of either false positives in parents or false negatives in the offspring. As expected, unfiltered SV calls exhibited a significant under-transmission bias: transmission rates of 39.8% for deletions and 35.08% for duplications (deletions: P = 9.61 × 10–51, N = 105 023; duplications P = 7.8 × 10–18, N = 346 173) (Fig. 2B). Applying standard genotype likelihood filters reduced the transmission bias to 48.2% (P = 1.32 × 10-2, N = 40 587) for deletions and 47.3% (P = 3.39 × 10-3, N = 3863) for duplications. Applying more stringent de novo filters further reduced under-transmission bias to 49.1% (P = 1.32 × 10-2, N = 21 772) for deletions and 49.3% (P = 1.0, N = 2847) for duplications (Supplementary Fig. S3).
Fig. 2.

SV2 genotyping performance. (A) False discovery rate across SV2 genotype likelihoods estimated from Illumina 2.5 M arrays (N = 57) and PacBio long reads (N = 9). Black dotted line indicates 5% FDR. (B) Group-wise transmission disequilibrium tests across SV2 genotype likelihoods in 630 offspring with shaded regions representing one standard deviation. (C) ROC curves of WGS genotyping calculated from Illumina 2.5 M arrays for SV2, SVTyper, and Manta in 57 individuals. (D) ROC curves of WGS genotyping calculated from supporting PacBio long-reads for SV2, SVTyper and Manta for SVs in nine individuals
Performance of SV2 was then compared to that of two widely used SV genotyping software SVTyper (v0.0.4; Chiang et al., 2015) and Manta. For this comparison, SVTyper genotyped SV predictions using the companion tool LUMPY, Manta produced genotypes for its predictions, and SV2 genotyped the union of LUMPY and Manta calls for the previous evaluation set of 57 subjects with Illumina 2.5 M arrays. Receiver operating characteristic (ROC) curves for each genotyping method were generated, specifying true and false positives with SVToolkit. SV2 achieved the best genotyping accuracy with an AUC of 0.92 for deletions and 0.8 for duplications, in contrast to Manta (deletion AUC = 0.84, duplication AUC = 0.69) and SVTyper (deletion AUC = 0.9, duplications AUC = 0.78) (Fig. 2C).
Validation: Further assessment of SV2’s genotype likelihood filters leveraged PacBio long-read WGS (26×) on 9 subjects from the 1KGP. This validation set is independent from the training set since SV2 genotypes were generated using a separate deep (72×) Illumina WGS library with SV predictions from LUMPY and Manta, both of which were not implemented in the training call set (Sudmant et al., 2015). To comply with the data release requirements for these data, only variants on chromosome 1 were analyzed. As a precaution for overfitting, we excluded SVs that overlapped with > =80% reciprocal overlap to SVs in our training set. Additionally, we omitted variants with less than three PacBio reads within 1 kbp flanking regions. Valid WGS genotypes required at least one supporting breakpoint with 50% reciprocal overlap to a PacBio split-read or CIGAR string. The FDR was 6.53% (N = 3121) and 17.72% (N = 413) for unfiltered deletions and duplications respectively (Fig. 2A). SV2 standard filters, lowered the FDR for deletions to 0.85% (de novo filters: 0.62%) and for duplications to 0% (de novo filters: 0%). With these data, we then compared SV2 genotyping performance to the aforementioned genotyping methods. Likewise, we found that SV2 produced the optimal performance with AUCs of 0.98 for deletions and duplications. Conversely, Manta performance resulted in an AUC of 0.9 for deletions and 0.81 for duplications, and SVTyper producing AUCs of 0.97 for deletions and 0.91 for duplications (Fig. 2D).
5 Conclusions
SV2 is unique from other genotyping methods in its use of machine learning, specifically a radial basis function kernel which is able to distinguish classes among nonlinear distributions (Supplementary Fig. S4). SV2 can rapidly genotype a wide variety of deletions and duplications (Supplementary Fig. S5). Exceptions include variants that completely overlap segmental duplications, short tandem repeats, centromeres, telomeres or other unsequenceable regions due to complications with unique-mappability from short-read technology. SV2 is designed for genotyping SVs in population genetic studies, including studies of complex traits or disease in pedigrees or case-control samples. SV2 could be further applied to any comparison of SVs across samples, e.g. in identifying somatic variants from multiple genomes derived from individual cells or clones of one individual (Abyzov et al., 2012). SV2 aids in variant post-processing by recording overlap to common filtering criteria and gene elements, and provides the option to merge divergent breakpoints according to the optimal genotype likelihood. Ultimately, SV2's strength is its ability to harmonize SV predictions from multiple callers, simplifying genotyping, likelihood estimation, analysis of SV association, and providing a much-needed tool for accurately detecting de novo mutations.
Supplementary Material
Acknowledgements
We would like to thank the 1000 Genomes Project and the Human Genome Structural Variation Consortium for providing PacBio SMRT alignments and complementary high coverage PCR-free Illumina short-read alignments for chromosome 1.
Funding
This project was supported by grants to JS from the National Institutes of Health (NIH grants HG007497, MH076431 and MH113715). D.A. was supported by a T32 predoctoral training grant from the NIH (GM008666). Analysis of low coverage genomes was funded by a research grant through Amazon Web Services.
Conflict of Interest: none declared.
References
- Abyzov A. et al. (2012) Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature, 492, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandler W.M. et al. (2016) Frequency and complexity of de novo structural mutation in autism. Am J Hum Genet, 98, 667–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandler W.M. et al. (2017) Paternally inherited noncoding structural variants contribute to autism. bioRxiv, 102327. [Google Scholar]
- Chen R. et al. (2015) A haplotype-based framework for group-wise transmission/disequilibrium tests for rare variant association analysis. Bioinformatics, 31, 1452–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. et al. (2015) Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics, 32, 1220–1222. [DOI] [PubMed] [Google Scholar]
- Chiang C. et al. (2015) SpeedSeq: ultra-fast personal genome analysis and interpretation. Nature Methods, 12, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad D.F. et al. (2010) Origins and functional impact of copy number variation in the human genome. Nature, 464, 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francioli L.C. et al. (2014) Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet., 46, 818–825. [DOI] [PubMed] [Google Scholar]
- Layer,R.M. et al. (2014). LUMPY: a probabilistic framework for structural variant discovery. Genome Biol., 15, R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson,J.J., and Sebat,J. (2012). forestSV: structural variant discovery through statistical learning. Nature Methods, 9, 819–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant P.H. et al. (2015) An integrated map of structural variation in 2, 504 human genomes. Nature, 526, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.