

Abstract
Herbaria are an invaluable source of plant material that can be used in a variety of biological studies. The use of herbarium specimens is associated with a number of challenges including sample preservation quality, degraded DNA, and destructive sampling of rare specimens. In order to more effectively use herbarium material in large sequencing projects, a dependable and scalable method of DNA isolation and library preparation is needed. This paper demonstrates a robust, beginning-to-end protocol for DNA isolation and high-throughput library construction from herbarium specimens that does not require modification for individual samples. This protocol is tailored for low quality dried plant material and takes advantage of existing methods by optimizing tissue grinding, modifying library size selection, and introducing an optional reamplification step for low yield libraries. Reamplification of low yield DNA libraries can rescue samples derived from irreplaceable and potentially valuable herbarium specimens, negating the need for additional destructive sampling and without introducing discernible sequencing bias for common phylogenetic applications. The protocol has been tested on hundreds of grass species, but is expected to be adaptable for use in other plant lineages after verification. This protocol can be limited by extremely degraded DNA, where fragments do not exist in the desired size range, and by secondary metabolites present in some plant material that inhibit clean DNA isolation. Overall, this protocol introduces a fast and comprehensive method that allows for DNA isolation and library preparation of 24 samples in less than 13 h, with only 8 h of active hands-on time with minimal modifications.
Keywords: Genetics, Issue 133, Herbarium, DNA isolation, high-throughput sequencing, museomics, phylogenomics, grasses
Introduction
Herbarium collections are a potentially valuable source of both species and genomic diversity for studies including phylogenetics1,2,3, population genetics4,5, conservation biology6, invasive species biology7, and trait evolution8. The ability to obtain a rich diversity of species, populations, geographical locations, and time points highlights the "treasure chest"9 that is the herbarium. Historically, the degraded nature of herbarium-derived DNA has hindered PCR-based projects, often relegating researchers to using only markers found in high copy, such as regions of the chloroplast genome or the internal transcribed spacer (ITS) of the ribosomal RNA. Quality of specimens and DNA vary extensively based on methods of preservation9,10, with double-stranded breaks and fragmentation from heat used in the drying process being the most common forms of damage, creating the so-called 90% DNA lock-up that has encumbered PCR-based studies11. Aside from fragmentation, the second most prevalent issue in herbarium genomics is contamination, such as that derived from endophytic fungi13 or fungi acquired postmortem after collection but before mounting in the herbarium12, though this problem can be solved bioinformatically given the right fungal database (see below). A third, and less common, problem is sequence modification through cytosine deamination (C/G→T/A)14, although it is estimated to be low (~0.03%) in herbarium specimens11. With the advent of high-throughput sequencing (HTS), the issue of fragmentation can be overcome with short reads and sequencing depth12,15, allowing genomic-level data acquisition from numerous specimens with low quality DNA, and even sometimes permitting whole genome sequencing15.
Herbarium samples are becoming more frequently used and are a larger component of phylogenetic projects16. A current challenge of using herbarium specimens for HTS is consistently obtaining sufficient double stranded DNA, a necessary prerequisite for sequencing protocols, from numerous species in a timely manner, without needing to optimize methods for individual specimens. In this paper, a protocol for DNA extraction and library preparation of herbarium specimens is demonstrated that takes advantage of existing methods and modifies them to allow for fast and replicable results. This method allows for complete processing from specimen to a library of 24 samples in 13 h, with 8 h hands-on time, or 16 h, with 9 h hands-on time, when the optional reamplification step is required. Simultaneous processing of more samples is achievable, though the limiting factor is centrifuge capacity and technical skill. The protocol is designed to require only typical laboratory equipment (thermocycler, centrifuge, and magnetic stands) instead of specialized equipment, such as a nebulizer or sonicator, for shearing DNA.
DNA quality, fragment size, and quantity are limiting factors for the use of herbarium specimens in high-throughput sequencing experiments. Other methods for isolating herbarium DNA and creating high-throughput sequencing libraries have demonstrated the utility of using as little as 10 ng of DNA16; however they require experimentally determining the optimum number of PCR cycles required for library preparation. This becomes impractical when dealing with exceedingly small amounts of viable double stranded DNA (dsDNA), as some herbarium specimens produce only enough DNA for a single library preparation. The method presented here uses a single number of cycles regardless of sample quality, so no DNA is lost in library optimization steps. Instead, a reamplification step is invoked when libraries do not meet the minimum amounts needed for sequencing. Many herbarium samples are rare and possess little material making it difficult to justify destructive sampling in many cases. To counter this, the presented protocol allows dsDNA input sizes less than 1.25 ng into the library preparation process, expanding the scope of viable samples for high-throughput sequencing and minimizing the need for destructive sampling of specimens.
The following protocol has been optimized for grasses and tested on hundreds of different species from herbarium samples, although we expect that the protocol can be applied to many other plant groups. It includes an optional recovery step that can be used to save low quality and/or rare specimens. Based on over two hundred herbarium specimens tested, this protocol works on specimens with low tissue input and quality, allowing for the preservation of rare specimens through minimal destructive sampling. Here it is shown that this protocol can provide high quality libraries that can be sequenced for phylogenomics-based projects.
Protocol
1. Prior to Start
Make fresh cetyl trimethylammonium bromide (CTAB) buffer17 by adding 20 g of CTAB, 10 g of polyvinylpyrrolidone (PVP) 40, 100.0 mL 1 M Tris pH 8.0, 40 mL of 0.5 M ethylenediaminetetraacetic acid (EDTA) pH 8.0, 280.0 mL of 5 M NaCl, and 400.0 mL of reagent water together, and bring the total volume to 1 L using reagent-grade water. Adjust the pH to 8.0. NOTE: Additional reagents can be added to CTAB depending on secondary compounds in individual taxa. See Allen et al.18 for a thorough list of additive reagents.
- Add 10 µL of β-mercaptoethanol per 5 mL of CTAB buffer. NOTE: this can be prepared in batches of 50 mL and stored at room temperature for 3–4 weeks.
- Heat the CTAB solution in a 65 °C water bath.
Chill mortars and pestles in -20 °C for at least 20 min.
Label 4 sets of 2 x n 2-mL tubes (where n = the number of samples). Put 1 set of the labeled tubes on ice.
Chill isopropanol on ice or in a -20 °C freezer.
Remove solid phase reversible immobilization (SPRI) beads (see Table of Materials) from the fridge, and allow them to equilibrate to room temperature (at least 30 min).
Prepare 80% ethanol.
Select herbarium specimens for extraction and retrieve ~1 cm2, or 10 mg, of tissue per specimen, preferably leaf material.
2. DNA Extraction
Grind ~1 cm2 of preselected herbarium tissue using prechilled mortars and pestles. Add liquid nitrogen and 30–50 mg sterilized sand. Grind until tissue turns into fine powder. NOTE: 10 mg or more tissue is desirable, but less has also worked in some cases. Once the liquid nitrogen evaporates, add more as needed until tissue is fully ground. Another common method for disrupting cells and tissues is the use of a bead beater. However, this method was found to not work well for the specimens used in these experiments.
Transfer the frozen powder into two 2 mL tubes (add no more than half of the tube's volume). Add 600 µL of warm CTAB solution to each tube and mix the tubes thoroughly by inversion and vortexing. NOTE: Since the quantity and quality of herbarium material is often low, performing DNA isolation in two repetitions helps to obtain higher yields.
Incubate the samples in a 65 °C water bath for 1–1.5 h, vortexing every 15 min.
Centrifuge samples at 10,000 x g for 5 min. Transfer the supernatant to a fresh set of labeled tubes (~500 µL). Discard the pellet using a standard non-chlorinated disposal procedure.
Add 4 µL of RNase-A (10 mg/mL) to each tube and mix by inverting or pipetting. Incubate the samples at 37 °C in a heat block or water bath for 15 min.
Add an equal volume (~500 µL) of a 25:24:1 phenol:chloroform:isoamyl alcohol mixture once the tubes have reached room temperature. Mix thoroughly by pipetting up and down and/or with inversion. Centrifuge the tubes at 12,000 x g for 15 min. Transfer the aqueous layer (upper layer) to a fresh set of labeled tubes (~400 µL). Discard the organic layer into a chlorinated waste container. NOTE: Step 2.6 may be repeated if large quantities of secondary compounds are expected in plant material.
Add an equal volume (~400 µL) of a 24:1 chloroform:isoamyl alcohol mixture. Mix thoroughly by pipetting up and down and/or with inversion. Centrifuge the tubes at 12,000 x g for 15 min. Transfer the aqueous layer (upper layer) to a fresh set of prechilled, labeled tubes (~300 µL). Discard the organic layer into a chlorinated waste container.
Add an equal volume (~300 µL) of prechilled isopropanol and 12 µL of 2.5 M sodium acetate to each tube. Incubate samples at -20 °C for 30–60 min. NOTE: The incubation times can be extended (up to overnight incubation), but DNA quality will decrease the longer the samples incubate.
Take the samples out of the freezer and centrifuge the tubes at 12,000 x g for 15 min. Remove and discard the supernatant gently without disturbing the pellet. Wash the pellet by suspension with fresh 70% ethanol (approximately 300–500 µL). For each doubled sample, consolidate the two individual pellets into one with accompanying ethanol. NOTE: Samples should be consolidated into one tube using ethanol first and then proceed. It is not necessary to wash each sample with ethanol separately.
Centrifuge the tubes at 12,000 x g for 10 min. Remove and discard the supernatant gently without disturbing the pellet. Air dry the pellets. NOTE: Samples can be dried faster using a dry heat block (do not exceed 65 °C). Make sure that the samples do not over-dry, as this can decrease the final yield of DNA.
Suspend the isolated DNA in 50 µL of 1x TE. Store in the -20 °C freezer for long term storage or 4 °C for use in the following week.
3. Quality Control (QC)
- Run an agarose gel for quality check.
- Prepare a 1x Tris/Borate/EDTA (TBE) buffer by adding 54 g Tris base, 27.5 g boric acid, and 3.75 g EDTA disodium salt, bringing the total volume to 5 L using reagent grade water.
- Prepare a 1% agarose gel by adding 1 g agarose to 100 mL of 1x TBE. Microwave the solution until no agarose is visible. Add 0.01% nucleic acid gel stain (see Table of Materials). Let the flask cool until it is warm to the touch. Mix well by stirring. Pour agarose in a gel tray and let it sit until it solidifies.
- Mix 3 µL of sample, 2 µL of reagent grade water, and 1 µL of 6x loading dye. Load the samples in the gel matrix, noting their order.
- Run the samples for 60–70 min at 60–70 V. Image the gel under UV light with correct exposure and focus. NOTE: Presence of a clear high molecular weight band is a sign of high quality DNA, while smears usually indicate DNA degradation. Most herbarium specimens are degraded.
- Run a dsDNA quantification analysis (see Table of Materials) to determine the quantity of double stranded DNA.
- Use 2 µL of sample for analysis. NOTE: Dilutions are not needed for the quantification analysis of herbarium material as they tend to be in minimal amounts. Successful libraries have been made from as little as 1.26 ng total dsDNA from this step.
4. DNA Shearing
NOTE: This is an optimized version of a commercial double-stranded fragmentase protocol (see Table of Materials).
Place dsDNA fragmentation enzyme on ice after vortexing for 3 s.
In a sterile 0.2 mL polymerase chain reaction (PCR) tube, mix 1–16 µL isolated DNA with 2 µL of accompanying fragmentation reaction buffer. Bring the total volume to 18 µL by adding nuclease free water. Add 2 µL of dsDNA fragmentation enzyme and vortex the mixture for 3 s. NOTE: The amount of DNA needed varies depending on DNA concentration (aim for 200 ng total in the tube).
Incubate the samples at 37 °C for 8.5 min. Then add 5 µL of 0.5 M EDTA to the tubes. NOTE: This step needs to be performed as soon as the incubation period is over to terminate the reaction and prevent DNA samples from over-shearing.
5. Bead Clean-up
Homogenize the SPRI beads by vortexing.
Bring the total volume of the sheared DNA to 50 µL by adding 25 µL of nuclease free water. Add 45 µL of room temperature SPRI beads (90% volume) to 50 µL of sheared DNA and mix thoroughly by pipetting up and down. NOTE: Adding beads at 90% of total sample volume is done to remove the smallest of DNA fragments, often below 200 base pairs.
Let the samples incubate for 5 min. Put the tubes on a magnetic plate and let them sit for 5 min. Carefully remove and discard the supernatant. NOTE: Be careful not to disturb the beads, as they contain the desired DNA targets.
Add 200 µL of fresh 80% ethanol to the tubes while on the magnetic stand. Incubate at room temperature for 30 s and then carefully remove and discard the supernatant. Repeat this step once. Air dry the beads for 5 min while the tube is on the magnetic stand with its lid open. NOTE: Avoid over-drying the beads. This can result in lower recovery of DNA.
Remove the tubes from the magnet. Elute the DNA from the beads into 55 µL of 0.1x TE and mix thoroughly by pipetting up and down. Incubate at room temperature for 5 min. Place tubes on the magnetic stand and wait for the solution to turn clear (~2 min).
Pull off 52 µL of the supernatant. Run a DNA quantification analysis on the samples to check the recovery and the initial concentration that goes into library prep. NOTE: Libraries have been made with total dsDNA estimated to be less than 1.25 ng, though in each case reamplification was required.
6. Library Preparation
NOTE: This is a modified version of a commercially available library kit (see Table of Materials protocol).
- End Prep
- Add 3 µL of endonuclease and phosphate tailing enzymes, and 7 µL of accompanying reaction buffer to 50 µL of cleaned, sheared DNA. Mix thoroughly by pipetting up and down. Spin the tubes to remove bubbles. NOTE: The total volume should be 60 µL.
- Place the samples in a thermocycler with the following program: 30 min at 20 °C, 30 min at 65 °C, then hold at 4 °C. NOTE: Heated lid was set to ≥75 °C
- Adaptor Ligation
- Dilute the adaptor 25–50 fold (working adaptor concentration of 0.6–0.3 µM). Add 30 µL of ligation master mix, 1 µL of ligation enhancer, and 2.5 µL of adaptor for high-throughput short read sequencing to the tubes. NOTE: The total volume should be 93.5 µL.
- Mix thoroughly by pipetting up and down. Spin the tubes to remove bubbles. Incubate the tubes at 20 °C for 15 min.
- Add 3 µL of commercial mixture of uracil DNA glycosylase (UDG) and the DNA glycosylase-lyase Endonuclease VIII (see Table of Materials) to the tubes. Ensure that the total volume is 96.5 µL. Mix thoroughly and incubate at 37 °C for 15 min using a thermocycler. NOTE: The lid should be set to ≥47 °C. The original version of the commercial protocol has size selection after the adaptor ligation step, followed by a bead cleanup as the final step. This protocol, which achieves higher yields, switches the order of these steps and implements size selection as a final step.
- Cleanup to Remove Enzymes and Small Fragments
- Homogenize magnetic beads by vortexing.
- Add 78 µL of SPRI magnetic beads (80% volume) and mix thoroughly by pipetting up and down. NOTE: Adding beads at 80% of total sample volume is done to remove the smallest DNA fragments, which are often shorter than 250 base pairs. The more stringent removal of small DNA fragments is to (i) remove surplus adaptors and (ii) emphasize amplification of larger fragments in the following steps.
- Let the samples incubate for 5 min. Put the tubes on a magnetic plate for 5 min. Carefully remove and discard the supernatant that contains the DNA outside the desired size range. NOTE: Be careful not to disturb the beads that contain the desired DNA targets.
- Add 200 µL of fresh 80% ethanol to the tubes while on the magnetic stand. Incubate at room temperature for 30 s and then carefully remove and discard the supernatant. Repeat this step once. Air dry the beads for 5 min while the tube is on the magnetic stand with lid open. NOTE: Avoid over drying the beads. This can result in lower recovery of DNA.
- Remove the tubes from the magnet. Elute the DNA target from the beads by adding 17 µL of 0.1x TE and mix thoroughly by pipetting up and down.
- Incubate at room temperature for 5 min. Place tubes on the magnetic stand and wait for the solution to turn clear (~2 min).
- Pull off 15 µL of the supernatant.
- PCR amplification
- Add 25 µL of high fidelity PCR master mix, 5 µL of high-throughput short read sequencing library prep 5' primer, and 5 µL of high-throughput short read sequencing library prep 3' primer, to 15 µL of the cleaned adaptor-ligated DNA. NOTE: Total volume should be 50 µL.
- Mix well by vortexing. Place the samples into a thermocycler using the settings found in Table 1: Thermocycler amplification setting. NOTE: A large number of cycles is needed due to the low quantity of input DNA.
| Cycle Step | Temp. | Time | Cycles |
| Initial Denaturation | 98 °C | 30 s | 1 |
| Denaturation | 98 °C | 10 s | 12 |
| Annealing/Extension | 65 °C | 75 s | 12 |
| Final Extension | 65 °C | 5 min | 1 |
| Hold | 4 °C |
Table 1: PCR protocol denaturation, annealing, and extension times and temperatures. Temperature and times were optimized for the reagents presented in this protocol. If reagents are altered, temperatures and times should be optimized again.
- Size Selection for Desired Library Size NOTE: This bead step will remove fragments both above and below the target range.
- Homogenize SPRI beads by vortexing.
- Add 25 µL (50% volume) of room temperature magnetic beads and mix thoroughly by pipetting up and down. Let the samples incubate for 5 min. Put the tubes on a magnetic plate and let them sit for 5 min. Carefully remove and transfer the supernatant to a new set of labeled tubes. NOTE: This volume can be adjusted based on desired library size. The supernatant contains DNA fragments of the desired size. In the first bead incubation, the beads are binding larger library fragments. These are removed to focus on those in the 400–600 base pair range. The supernatant contains smaller fragments.
- Add 6 µL of room temperature SPRI beads to the supernatant and mix thoroughly by pipetting up and down. Let the samples incubate for 5 min. Put the tubes on a magnetic plate and let them sit for 5 min. NOTE: This volume can be adjusted based on desired library size in accordance with step 6.5.2.
- Carefully remove and discard the supernatant. NOTE: Be careful not to disturb the beads that contain the desired DNA. In the second bead incubation, the beads are binding to the fragments left after the initial removal of the largest DNA fragments. This set of fragments is usually in the desired size range.
- Add 200 µL of fresh 80% ethanol to the tubes while on the magnetic stand. Incubate at room temperature for 30 s, then carefully remove and discard the supernatant. Repeat. Air dry the beads for 5 min while the tube is on the magnetic stand with lid open. NOTE: Avoid over-drying the beads. This can result in lower recovery of DNA.
- Remove the tubes from the magnet. Elute the DNA target from the beads into 33 µL of 0.1x TE and mix thoroughly by pipetting up and down.
- Incubate at room temperature for 5 min. Place tubes on the magnetic stand and wait for the solution to turn clear (~2 min). Pull off 30 µL of the supernatant and transfer to 2 mL tubes (see Table of Materials) for storage. NOTE: The libraries can be kept at -20 ˚C for long-term storage.
- Quality control
- Run a quality control test on the DNA Libraries. Refer to steps 3.1 and 3.2. NOTE: For DNA libraries, run the gel for ~45 min at 96 V.
- Library reamplification: Optional if library quantity is not sufficient. NOTE: Samples with library concentrations below 10 nM can be reamplified using the following steps. Reamplification of low concentration libraries can achieve workable results for sequencing, but reamplification may cause a modest shift in base composition diversity, though gathered data (Table 3) suggest that this is negligible for certain metrics.
- Dilute the universal reamplification primers 10-fold using 0.1x TE.
- Add 25 µL of high fidelity PCR master mix, 5 µL of diluted universal reamplification primer 1 (AATGATACGGCGACCACCGA), and 5 µL of diluted universal reamplification primer 2 (CAAGCAGAAGACGGCATACGA) to 15 µL of low concentration libraries. NOTE: Total volume should be at 50 µL.
- Mix well by vortexing. Place the samples into a thermocycler using the settings found in Table 1: Thermocycler amplification setting NOTE: The large number of cycles is needed due to low quantity of input DNA.
- Bead clean-up
- Homogenize SPRI beads by vortexing. Add 45 µL of room temperature SPRI beads (90% volume) and mix thoroughly by pipetting up and down.
- Let the samples incubate for 5 min. Put the tubes on a magnetic plate for 5 min.
- Carefully remove and discard the supernatant that contains the unwanted DNA. NOTE: Be careful not to disturb the beads that contain the desired DNA targets.
- Add 200 µL of fresh 80% ethanol to the tubes while on the magnetic stand. Incubate at room temperature for 30 s, and then carefully remove and discard the supernatant. Repeat this step once.
- Air dry the beads for 5 min while the tube is on the magnetic stand with its lid open. NOTE: Avoid over-drying the beads. This can result in lower recovery of the DNA target.
- Remove the tubes from the magnet. Elute the DNA target from the beads into 33 µL of 0.1x TE and mix thoroughly by pipetting up and down.
- Incubate at room temperature for 5 min. Place tubes on the magnetic stand and wait for the solution to turn clear (~2 min).
- Pull off 30 µL of the supernatant. The libraries can be stored at -20 °C for long-term storage.
- Quality control
- Run a quality control test on the DNA Libraries. Refer to steps 3.1 and 3.2. For DNA libraries, run the gel for ~45 min at 96 V. NOTE: If a double band is seen in the gel, this is likely a consequence of primer exhaustion from the reamplification step. The bands can be removed by repeating 6.9, but using only one cycle in the PCR program depicted in Table 1.
Representative Results
DNA Isolation and Final Library Yield In this study, the efficacy of the protocol for the isolation of herbarium DNA and the recovery of high quality sequencing libraries was demonstrated using fifty different samples with the oldest from 1920 and the youngest from 2012 (Table 2). For each sample, approximately 10 mg of leaf tissue was used for DNA isolation. Greener leaf tissue was favored if available, and no tissue with obvious fungal contamination was selected. Successful isolations can be made using yellow or brown tissue, though yield should be expected to be lower. Total double stranded DNA (dsDNA) from the initial isolation ranged from 3.56 ng to 2,610 ng. As expected, DNA obtained from herbarium specimens was highly degraded (Figure 1A, Supplemental Figure 1). A portion of these isolations were used for enzymatic shearing (1.26–464 ng). Though herbarium DNA is already sheared through the preservation process, optimization of the protocol requires additional shearing to improve overall library yield. The total recovery of dsDNA post-shearing ranged from <1% to 51% of input dsDNA, resulting in a minimum of less than 1.25 ng of starting DNA for library preparation and a maximum of 328 ng. The extreme loss of DNA in some samples can be attributed to the already small fragment size of much of the DNA prior to enzymatic shearing (Figure 1A, Supplemental Figure 1). The use of a 90% volume bead cleanup on the sheared DNA purposely removed the smallest fragments of DNA to enrich for larger, more desirable fragment sizes. These small fragments were especially seen in samples TK463, TK657, and TK694, as denoted by an intense signal at the 100 base pair mark (Figure 1A).
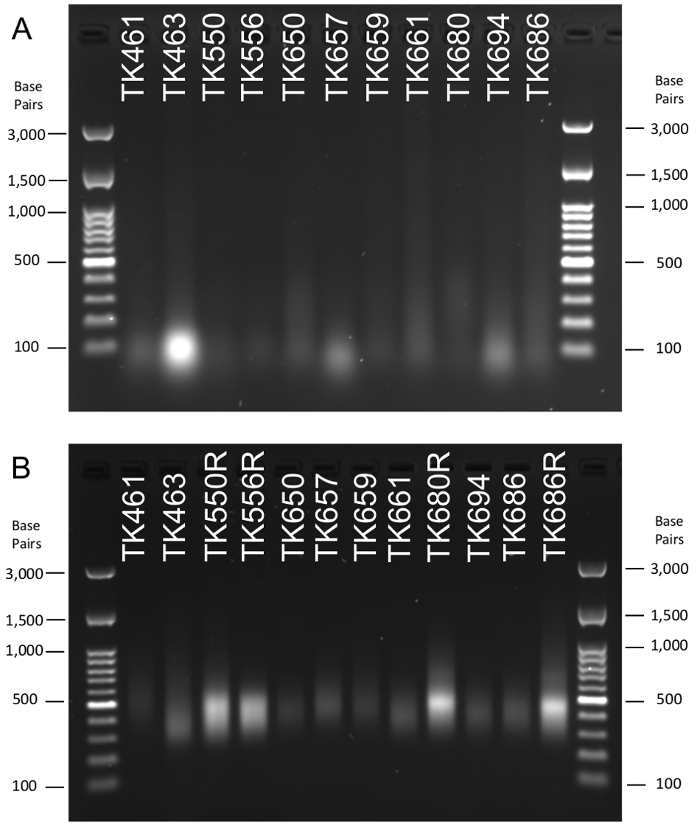
The total quantity of the library post size selection ranged from 1.425 ng to 942.5 ng (Table 2, Supplemental Table 1). For 23 of the samples, the initial extraction and library preparation did not yield an adequate amount of library (<10 nM; Table 2, Supplemental Table 1), so these samples were subjected to the reamplification and recovery steps of the protocol, resulting in a 14–680x increase in total library (Table 2, Supplemental Table 1). Final libraries resulted in a band between 350 and 500 base pairs (Figure 1B, Supplemental Figure 2). At times, a second band that was larger than the expected library size was seen (Figure 1B, Supplemental Figure 2). This occurred when the reamplification PCR exhausted available primers and began annealing library adaptors of non-homologous DNA fragments. This creates a molecule where the ends (the adaptors) were properly annealing, but the DNA insert did not. This "bubbled" molecule appeared larger on a gel, as it moved more slowly through the gel matrix. These annealing errors were fixed by preparing another reamplification reaction from the already reamplified library and running it for a single cycle. This single cycle provided primers for proper annealing and amplification, removing the second band (Supplemental Figure 3).
Reamplification of libraries facilitated final library concentrations of at least 10 nM. These concentrations allowed libraries to be diluted to equal molarity and pooled in equal representation, helping to negate issues that would have arisen with unequal sample quality and sequencing library yield. If the goal of a project is chloroplast genome sequencing, then the total amount of sequencing needed will vary as different lineages and tissues differ in the total percent of reads that originate from chloroplast DNA19. Typically, 50–100x projected coverage of the chloroplast genome is sufficient for assembly, and sequencing runs can be pooled to include as many as 70 individuals depending on species and sequencing method.
Testing for Contamination, Bias and Variation Caused by Reamplification A notable concern for HTS sequencing is introduction of bias into libraries through extensive PCR amplification20. To test the effects of reamplification and identify potential bias in common phylogenetic applications of herbarium material, we compared a successfully sequenced library (TK686) with the same library diluted 1:5 and reamplified (designated TK686-R). Both TK686 and TK686-R were sequenced on an Illumina HiSeq4000 at the University of Illinois Roy J. Carver Biotechnology Center and the Michigan State University Research Technology Support Facility, respectively, using paired end 150 base pair reads (see Table 3 for sequencing details). Raw reads have been deposited in the NCBI SRA (SRP128083). Reads were cleaned using Trimmomatic v.0.3621 including adaptor trimming using NEB adaptor sequence, quality filtering for an average phred score of 20 for a 10 base pair sliding window, and a minimum cut off size of 40 base pairs. As one of the primary issues with herbarium specimens is fungal contamination, contamination was estimated by mapping reads against a portion of the JGI MycoCosm fungal genome database22 (312 nuclear genomes and 79 mitochondrial genomes) using bowtie2 v. 2.2.923 using the "very-sensitive-local" parameter set. TK686 and TK686-R libraries were indistinguishable in nuclear fungal contamination (9.24% and 9.68%, respectively) and mitochondrial fungal contamination (0.94% and 0.8%) (Table 3). Though this is only one example, it does suggest that fungal contamination of herbarium samples is not negligible and should be removed prior to using herbarium-derived sequence data. The database and commands used to identify and remove fungal contamination can be found in M. R. McKain's GitHub repository Herbarium Genomics24.
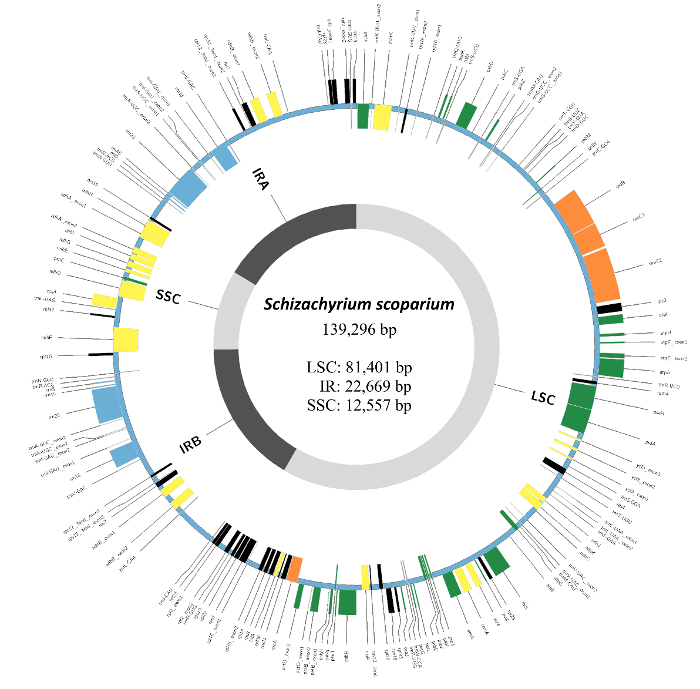
Chloroplast genome sequencing is commonly used for phylogenetic analyses, and herbarium specimens are increasingly used as source material12. In order to test the fidelity of reamplification in the chloroplast genome assembly, chloroplast genomes for both TK686 and TK686-R were assembled using Fast-Plast v.1.2.525 under default settings with the bowtie index set to Poales. A full chloroplast genome was obtained for TK686-R, but the TK686 chloroplast genome was assembled into seven contigs due to lower read depth. The TK686 contigs were assembled manually following McKain et al.26 Fully assembled chloroplast genomes were aligned to each other in a GUI-based alignment software (see Table of Materials) and variation between assemblies was assessed. A total of 12 SNPs and one indel were identified between chloroplast assemblies for TK686 and TK686-R. For each variant, coverage was assessed in read sets from both TK686 and TK686-R. In all cases, the most common variant was the same between the two libraries. TK686 demonstrated one T→C, one G→A, one G→T, one G→-, one C→A, one T→A, and four C→A variants. Five of these variants occurred within a homopolymer string, suggesting their incorporation into the assembly may have been the result of sequencing error and low overall coverage. The others may have been the result of either chloroplast haplotype variation, sequencing error, or cytosine deamination, or some combination of these factors. TK686-R had one C→T, one G→T, and one A→G. The C→T and G→T variants were found in a homopolymer as above. Ultimately, identical complete chloroplast genomes were identified from both read sets. A single chloroplast genome from TK686 was annotated using Verdant21 and compared to other members of the tribe Andropogoneae. All standard chloroplast features were annotated: 8 rRNAs, 38 tRNAs, and 84 protein coding genes. The complete chloroplast genome is available from GenBank (MF170217) and Verdant27.
Potential bias from the reamplification of the libraries was also determined through estimation of percent GC and total estimated transposon content. Percent GC was estimated using a custom script24. Differences in GC content of the two samples were negligible, with 49.7% GC in TK686 and 51.2% GC in TK686-R. Transposon composition was estimated using Transposome28. For both libraries, 100,000 reads were randomly subsampled and transposon composition was estimated using a percent identity of 90, a fraction coverage of 0.55, a cluster size of 100, and the RepBase 21.10 grass repeat reference set29. This was repeated 100 times to perform bootstrapping on transposon estimation from these datasets. Total genome percentages for major subfamilies of transposons were extracted from Transposome output, and the mean and standard deviation of these for the 100 replicates were estimated. All scripts used to generate these outputs can be found in M. R. McKain's Github repository Transposons30, and all results have been deposited in Dryad (doi:10.5061/dryad.r8t2m). Using the two most prevalent transposon subfamilies as indicators (Copia and Gypsy long terminal repeat retrotransposons), the results were nearly identical with Copia at 33.52 ± 4.00% and 31.68 ± 2.94% and Gypsy at 24.83 ± 2.72% and 24.00 ± 2.35% in TK686 and TK686-R, respectively (Table 3). These test results suggest that the reamplification step of this protocol did not create meaningful sequencing bias for high-level genome metrics. However, it should be noted that this single example may not be fully representative of all reamplified libraries. This single test demonstrates that the reamplification step was not inherently biasing genome metrics in TK686/TK686-R. Introduced bias would not affect the assembly of a chloroplast genome given sufficient coverage of sequencing, but it is recommended that experiments, as presented in this study with TK686/TK686-R, are conducted on target lineages to verify that bias is not occurring during studies investigating transposable element diversity.
Figure 1: Agarose gel images of A) DNA isolation and B) final sequencing libraries from ten herbarium specimens. For each lane, 3 µL of DNA or library was used. (A) DNA was degraded in all herbarium isolations as seen by the general smear. (B) Final sequencing libraries depict a primary band of 300-500 base pairs with a wider distribution of 200-1,000 base pairs; the latter is more prevalent in reamplified libraries. Lanes for both (A) and (B) were identified by sample and can be compared to results in Table 2. Ladder size was depicted in base pairs (bp). Please click here to view a larger version of this figure.
Figure 2: Circular plot of Schizachyrium scoparium (TK686) chloroplast genome with annotation. The fully assembled genome from shotgun sequencing of herbarium-derived DNA exhibited a total length of 139,296 base pairs (bp), a large single copy region (LSC) of 81,401 bp, an inverted repeat region (IR) of 22,669 bp, and a small single copy region (SSC) of 12,557 bp. All standard protein-coding genes, tRNAs, and rRNAs for members of the Andropogoneae tribe were identified in the annotation. Please click here to view a larger version of this figure.
Table 2: DNA extraction and library preparation results for ten herbarium samples and four reamplified libraries. Thetotal double stranded DNA at various steps in the protocol demonstrated how variable quality can be, especially when filtered for size. Please click here to download this table.
Table 3: Sequencing statistics for TK686 and the reamplified TK686R. Reamplification does not affect the overall incidence of fungal genome contamination, GC content estimation, transposon composition estimation, or the ability to assemble whole chloroplast genomes. Please click here to download this table.
Supplemental Table 1: DNA extraction and library preparation results for forty additional herbarium samples, including twenty reamplified libraries.Total double stranded DNA at various steps in the protocol demonstrated how variable quality can be, especially when filtered for size. Please click here to download this table.
Supplemental Figure 1: Agarose gel images of forty additional DNA isolations from herbarium specimens. Both (A) and (B) depict twenty separate DNA isolations and demonstrate the characteristic degradation of herbarium-derived DNA. For each lane, 3 µL of DNA was used. Lanes for both (A) and (B) were identified by sample and can be compared to results in Supplemental Table 1. Ladder size is depicted in base pairs (bp). White flecks on the image are due to artefacts in the gel imager that could not be removed with cleaning. Please click here to download this figure.
Supplemental Figure 2: Agarose gel images of forty additional sequencing libraries from herbarium-derived DNA. For each lane, 3 µL of library was used. Final sequencing libraries are found in both (A) and (B) with an average size of 300-500 base pairs. Secondary bands seen in some amplified samples suggest "bubbling" of libraries. Lanes for both (A) and (B) are identified by sample and can be compared to results in Supplemental Table 1. Ladder size is depicted in base pairs (bp). Please click here to download this figure.
Supplemental Figure 3: Agarose gel image of secondary band removal with an additional single cycle PCR step. Please click here to download this figure.
Discussion
The protocol presented here is a comprehensive and robust method for DNA isolation and sequencing library preparation from dried plant specimens. The consistency of the method and minimal need to alter it based on specimen quality make it scalable for large herbarium-based sequencing projects. The inclusion of an optional reamplification step for low yield libraries allows the inclusion of low quality, low quantity, rare, or historically important samples that would otherwise not be suitable for sequencing.
Importance of Initial DNA Yield Herbarium-derived DNA is often degraded as a consequence of initial specimen preservation11, with DNA of specimens less than 300 years old being as degraded as DNA isolated from animal remains that are several hundred to thousands of years old31,32. Consequently, optimization of initial DNA yield is vital in obtaining enough high-quality dsDNA for successful sequencing library preparation. For grass species, an optimal yield is achieved through the combined use of sterilized sand and liquid nitrogen in the initial grinding step, providing a more thorough destruction of cell walls and release of nucleic acids. This approach increases both desirable larger dsDNA and undesirable smaller fragments (Figure 1, Supplemental Figure 1, Table 2, Supplemental Table 1). Subsequent bead cleaning steps isolate and enrich for fragments of a size appropriate for sequencing (300–500 base pairs), greatly reducing recovery but also enriching for longer fragments (Table 2, Supplemental Table 1). Alterations to the initial DNA isolation steps may be necessary based on the lineage being sampled in order to reduce the effects of secondary metabolites on downstream processing18.
Optimization of Library Adaptors The concentration of adaptors used for ligation has a direct effect on the amount of adaptor dimer in finished libraries. Adaptor dimers result from adaptor self-ligation when insufficient sample is present, and contaminate sequencing runs33. The relatively low total dsDNA available from herbarium specimens necessitates dilution of adaptors prior to ligation. Adaptors can be diluted 50-fold from the stock concentration of 15 µM (see Protocol Section) facilitating high-throughput library preparation without the need to individually measure and dilute adaptor for each sample (Table 2, Supplemental Table 1). Though saturation of adaptors could in principle decrease overall library yield, it is unlikely that herbarium specimens will yield dsDNA in such excess of adaptor.
Variation in Bead Cleaning Steps for Higher Yields Size selection in preparation of sequencing libraries is usually done after adaptor ligation, allowing for amplification of fragments primarily within the desired size range; this is done by removing fragments that are both larger and smaller than the target size. The low amount of herbarium-derived dsDNA for library preparation is exacerbated after size selection at this step, resulting in unworkably low total dsDNA and ultimately low yield in the final library. By conducting a standard bead-cleaning step using 90% volume beads after ligation, more total dsDNA remains for enrichment in the amplification step. Extremely small DNA fragments are preferentially removed using 90% volume beads. Size selection is conducted in the final step on the amplified library, which ensures enrichment of desired fragment sizes. Total volumes of beads can be adjusted to select the desired range, though the two-step volumes of 25 µL and 6 µL of beads are optimized to retrieve libraries of 400-500 base pair inserts within this protocol (Figure 1B, Supplementary Figure 2).
Sample Rescue through Reamplification of Libraries Despite best practices in DNA isolation and library preparation, final concentrations of sequencing libraries may be inadequate for further sequencing. The destructive nature of sampling and often limited expendable material from herbarium specimens does not always permit repeating DNA isolation. By reamplifying the library up to 12 additional PCR cycles, even exceptionally poor libraries can be saved. A standard primer pair is used for amplification, which is compatible with either dual or single indexed library protocols. A primary concern for reamplification is the introduction of bias, often through reduction in GC-rich portions of the genome20. By using a high-fidelity polymerase (see Table of Materials), these potential biases are potentially avoided. This is demonstrated through the minimal variation of GC content of the sequenced libraries TK686 and TK686-R (Table 3). As a second verification, the transposon content of both TK686 and TK686-R was estimated and showed no discernible differences (Table 3). Finally, the whole chloroplast genome of this accession was assembled from TK686 and TK686-R, which resulted in identical sequences after close inspection of SNP variation between the two assemblies (Figure 2, Table 3). These tests suggest that standard genomic metrics, such as GC content and transposon composition, and the ability to assemble complete chloroplast genomes, may not be affected by reamplification. This opens up the possibility of incorporating herbarium specimens thought to be too degraded or lacking in material into phylogenomic studies without concern for introduced bias through PCR. These reamplified libraries might also be used for sequence capture34, although it would be necessary to test whether SNP calling is biased. It is recommended that small scale tests of bias be conducted with each project to verify that bias is not introduced into sequencing libraries.
Limitations and Possible Modifications Even though this protocol has worked on hundreds of herbarium specimens, poorly preserved tissues may still fail at any step. It is, however, exceedingly rare for libraries to fail from tissues with successful DNA extractions, especially after library rescue through reamplification. The size selection steps can be modified to target different sized fragments or to reduce the wide range of fragments seen in some final libraries. As with all plant-based extraction protocols, steps may be needed to remove lineage-specific secondary compounds that can impede the overall protocol. As presented, this protocol provides a standard method for DNA isolation and high-throughput library preparation for grass herbarium specimens, and through verification and experimentation is likely to be amendable to other plant lineages.
Disclosures
The authors declare they have no competing interests.
Acknowledgments
We thank Taylor AuBuchon-Elder, Jordan Teisher, and Kristina Zudock for assistance in sampling herbarium specimens, and the Missouri Botanical Garden for access to herbarium specimens for destructive sampling. This work was support by a grant from the National Science Foundation (DEB-1457748).
References
- Savolainen V, Cuénoud P, Spichiger R, Martinez MDP, Crèvecoeur M, Manen JF. The use of herbarium specimens in DNA phylogenetics: Evaluation and improvement. Plant Syst Evo. 1995;197(1-4):87–98. [Google Scholar]
- Zedane L, Hong-Wa C, Murienne J, Jeziorski C, Baldwin BG, Besnard G. Museomics illuminate the history of an extinct, paleoendemic plant lineage (Hesperelaea, Oleaceae) known from an 1875 collection from Guadalupe Island, Mexico. Bio J Linn Soc. 2016;117(1):44–57. [Google Scholar]
- Teisher JK, McKain MR, Schaal BA, Kellogg EA. Polyphyly of Arundinoideae (Poaceae) and Evolution of the Twisted Geniculate Lemma Awn. Ann Bot. 2017. [DOI] [PMC free article] [PubMed]
- Cozzolino S, Cafasso D, Pellegrino G, Musacchio A, Widmer A. Genetic variation in time and space: the use of herbarium specimens to reconstruct patterns of genetic variation in the endangered orchid Anacamptis palustris. Conserv Gen. 2007;8(3):629–639. [Google Scholar]
- Wandeler P, Hoeck PEA, Keller LF. Back to the future: museum specimens in population genetics. Tre Eco & Evo. 2007;22(12):634–642. doi: 10.1016/j.tree.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Rivers MC, Taylor L, Brummitt NA, Meagher TR, Roberts DL, Lughadha EN. How many herbarium specimens are needed to detect threatened species? Bio Conserv. 2011;144(10):2541–2547. [Google Scholar]
- Saltonstall K. Cryptic invasion by a non-native genotype of the common reed, Phragmites australis, into North America. PNAS USA. 2002;99(4):2445–2449. doi: 10.1073/pnas.032477999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard G, et al. From museums to genomics: old herbarium specimens shed light on a C3 to C4 transition. J Exp Bot. 2014;65(22):6711–6721. doi: 10.1093/jxb/eru395. [DOI] [PubMed] [Google Scholar]
- Särkinen T, Staats M, Richardson JE, Cowan RS, Bakker FT. How to open the treasure chest? Optimising DNA extraction from herbarium specimens. PLoS ONE. 2012;7(8):e43808. doi: 10.1371/journal.pone.0043808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SA. DNA analysis of tropical plant species: An assessment of different drying methods. Plant Syst Evo. 1994;188(1-2):57–64. [Google Scholar]
- Staats M, et al. DNA damage in plant herbarium tissue. PLoS ONE. 2011;6(12):e28448. doi: 10.1371/journal.pone.0028448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker FT, et al. Herbarium genomics: plastome sequence assembly from a range of herbarium specimens using an Iterative Organelle Genome Assembly pipeline. Bio J of the Linn Soc. 2016;117(1):33–43. [Google Scholar]
- Camacho FJ, Gernandt DS, Liston A, Stone JK, Klein AS. Endophytic fungal DNA, the source of contamination in spruce needle DNA. Mol Eco. 1997;6(10):983–987. [Google Scholar]
- Hofreiter M, Jaenicke V, Serre D, Von Haeseler A, Pääbo S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucl Acids Res. 2001;29(23):4793–4799. doi: 10.1093/nar/29.23.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staats M, et al. Genomic treasure troves: Complete genome sequencing of herbarium and insect museum specimens. PLoS ONE. 2013;8(7):e69189. doi: 10.1371/journal.pone.0069189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker FT. Herbarium genomics: skimming and plastomics from archival specimens. Webbia. 2017;72(1):35–45. [Google Scholar]
- Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bul. 1987;19:11–15. [Google Scholar]
- Allen GC, Flores-Vergara MA, Krasynanski S, Kumar S, Thompson WF. A modified protocol for rapid DNA isolation from plant tissue using cetryltrimethylammonium bromide. Nat Prot. 2006;1:2320–2325. doi: 10.1038/nprot.2006.384. [DOI] [PubMed] [Google Scholar]
- Twyford AD, Ness RD. Strategies for complete plastid genome seqeuncing. Mol Eco Resour. 2016. [DOI] [PMC free article] [PubMed]
- Aird D, et al. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Bio. 2011;12(2):R18. doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinf. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev IV, et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucl Acids Res. 2014;42(1):D699–D704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Meth. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKain MR. Herbarium Genomics. 2017. Github Repository https://github.com/mrmckain/
- McKain MR, Wilson MA. Fast-Plast: Rapid de novo assembly and finishing for whole chloroplast genomes. 2017. Github Repository https://github.com/mrmckain/
- McKain MR, McNeal JR, Kellar PR, Eguiarte LE, Pires JC, Leebens-Mack J. Timing of rapid diversification and convergent origins of active pollination within Agavoideae (Asparagaceae) Am J Bot. 2016;103(10):1717–1729. doi: 10.3732/ajb.1600198. [DOI] [PubMed] [Google Scholar]
- McKain MR, Hartsock RH, Wohl MM, Kellogg EA. Verdant: automated annotation, alignment, and phylogenetic analysis of whole chloroplast genomes. Bioinf. 2016. [DOI] [PMC free article] [PubMed]
- Staton SE, Burke JM. Transposome: A toolkit for annotation of transposable element families from unassembled sequence reads. Bioinf. 2015;31(11):1827–1829. doi: 10.1093/bioinformatics/btv059. [DOI] [PubMed] [Google Scholar]
- Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA. 2015;6(1):11. doi: 10.1186/s13100-015-0041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKain MR. Transposons. 2017. Github Repository https://github.com/mrmckain/
- Weiß CL, et al. Temporal patterns of damage and decay kinetics of DNA retrieved from plant herbarium specimens. Royal Soc Open Sci. 2016;3(6):160239. doi: 10.1098/rsos.160239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer S, Krause J, Guschanski K, Savolainen V, Pääbo S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS ONE. 2012;7(3):e34131. doi: 10.1371/journal.pone.0034131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head SR, et al. Library construction for next-generation sequencing: overviews and challenges. BioTechniques. 2014;56(2):61–64. doi: 10.2144/000114133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover CE, Salmon A, Wendel JF. Targeted sequence capture as a powerful tool for evolutionary analysis. Am J Bot. 2012;99:312–319. doi: 10.3732/ajb.1100323. [DOI] [PubMed] [Google Scholar]