

Abstract
Deficiency in ATP binding cassette A3 (ABCA3) causes neonatal respiratory distress, hypoxemic respiratory failure, and interstitial lung disease. ABCA3 transports phospholipids into the lamellar bodies of type II alveolar cells, a critical step in alveolar surfactant production. We report a term infant with ABCA3 surfactant deficiency syndrome with the E292V (c.875A>T; p.Glu292Val) mutation in trans with a novel C‐terminal frame shift mutation (c.4938delC; p.Met1647fs). This mutation removes the final 58 amino acids and substitutes 33 incorrect amino acids. The frame shift spares membrane spanning and nucleotide binding domains, but disrupts a highly conserved C‐terminal domain, which includes sequence motifs necessary for the function of human paralogs ABCA1, ABCA4, and the bacterial homolog DrrA. This observation suggests the C‐terminal domain is also required for normal function of ABCA3.
Keywords: ABCA3, childhood, genetics, interstitial lung disease (ILD), surfactant biology and pathophysiology
1. CASE REPORT
A 2438 g male infant was born at 37 weeks gestation by spontaneous vaginal delivery following a healthy pregnancy. He developed immediate respiratory distress and hypoxemia, and the initial chest radiograph showed diffuse ground glass opacities (Figure 1A). Blood, urine, and cerebrospinal fluid cultures were negative. Tachypnea, a grunting cough, and abnormal chest radiography persisted despite 7 days treatment with intravenous ampicillin and gentamicin in a neonatal intensive care unit. Following discharge, his tachypnea persisted and he failed to gain adequate weight despite supplemental 22 kcal/oz formula. He was readmitted to a local hospital at 1 month of age with respiratory distress and hypoxemia. Nasal PCR testing was positive for adenovirus. He received supplemental oxygen and was discharged home after his respiratory distress improved. At 2 months, he was admitted to the pediatric intensive care unit with respiratory distress, perioral cyanosis, and worsening failure to thrive. Chest radiography continued to show diffuse ground glass opacities (Figure 1B). The SpO2 decreased below 80%, and he received 4 L/min of oxygen by nasal cannula. The hypoxemia was unresponsive to azithromycin, vancomycin, and ceftriaxone.
Figure 1.
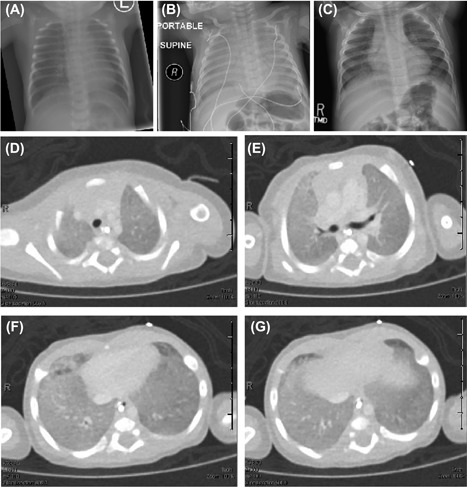
A‐C) Chest radiographs on day of life 1 (A) and upon readmission at 2 months of age (B) show persistent diffuse ground glass appearance. At 12 months of life (C), we observed marked improvement. D‐G, Axial sections from a chest CT with contrast at 2 months of life reveal diffuse ground glass opacities with minimal thickening of the interlobular septum. No airway or vascular abnormalities were detected
A chest CT at 2 months of life (Figure 1D‐G) revealed diffuse ground glass opacities with minimal thickening of the interlobular septum. No airway or vascular abnormalities were detected. A complete blood count was normal except for elevated eosinophils (1310 cells/mm3). The C‐reactive protein, basic metabolic panel, thyroid studies, fluoroscopic swallow study, and echocardiogram were normal. Infectious disease testing included viral respiratory panel, chlamydia PCR, and mycoplasma PCR, all of which were negative. Due to the persistent and diffuse lung parenchymal abnormalities, we suspected childhood interstitial lung disease, including disorders of surfactant protein metabolism (SP‐C, ABCA3, TTF‐1/NKX2.1), pulmonary interstitial glycogenosis (PIG), and neuroendocrine hyperplasia of infancy (NEHI). Primary ciliary dyskinesia was considered as a possible cause of neonatal respiratory distress, although the imaging appeared more consistent with an alveolar or interstitial disorder. Focused genetic testing for surfactant disorders (SFTPC, ABCA3, NKX2.1) revealed two mutations in the ABCA3 (Reference sequence GRCh37/hg19: ENSG00000167972; ENST00000301732). Parental testing confirmed that a known pathogenic mutation (c.875A>T; p.Glu292Val) was inherited maternally and a novel frame shift (c.4938delC; p.Met1647fs) was inherited paternally.
His treatment focused on nutritional rehabilitation. Growth improved dramatically with gastrostomy supplementation. There were no objective changes following 10 days on 10 mg/kg of oral prednisolone or 3 days on 10 mg/kg of intravenous methylprednisolone. He has received no further corticosteroids. In consultation with other experts, we prescribed azithromycin 10 mg/kg three times weekly and hydroxychloroquine 50 mg/kg daily. At age 1, his oxygen requirement has decreased from 4 to 3 L/min by nasal cannula, and we observed interval improvement by chest radiograph (Figure 1C).
2. DISCUSSION
In ABCA3 deficiency, there is a recognized correlation between genotype and phenotype. Patients with two null mutations, defined by premature stop codon or frame shift, have earlier symptoms and greater mortality by 1 year of life.1 The prognosis for patients with other mutations is variable, and may depend on residual protein function. In heterologous cells, p.Glu292Val is synthesized and trafficked to vesicles, but has defective ATPase activity.2 The frame shift mutation c.4938delC has not been described in literature, the NHLBI GO Exome Sequencing Project (ESP), or the Exome Aggregate Consortium database. This mutation spares the membrane spanning and nucleotide binding domains. To understand how c.4938delC disrupts ABCA3 function, we compared this mutation to disruptions of the C‐terminus in related ABC class A family members. The C‐terminal sequence is conserved in ABC class A members through evolution (Figure 2). Similar deletions of the C‐terminus have been implicated in Tangier disease (ABCA1) and Stargardt disease (ABCA4).3, 4 Deletion of the C‐terminal domain of ABCA1 and ABCA4 permits protein expression in heterologous cells, but impairs function. ABCA1 lacking the C‐terminus is trafficked to the cell membrane but lacks ATPase activity and cholesterol efflux.3 ABCA4 lacking the C‐terminus is retained in the endoplasmic reticulum and lacks ATPase activity.4 Both proteins require a VFVNFA motif for normal function (Figure 2). In Streptomyces, the ABC class A protein DrrA is the catalytic subunit of the DrrAB complex. This complex functions an ATP‐dependent transporter of doxorubicin and other antibiotics. Chemical cross‐linking studies revealed that the LDEVFL motif of DrrA interacts with the pore‐forming subunit DrrB.5 The LDEVFL motif overlaps with the VFVNFA motif of ABCA1 and 4 (Figure 2). Mutations to the LDEVFL motif abolish the ATPase activity of DrrA and dimerization with DrrB, resulting in susceptibility of bacteria to doxorubicin.5 Thus, three other related ABC class A family members require the C‐terminal domain for normal function. It remains unclear whether C‐terminal truncations of ABCA3 impair protein localization, ATPase activity (as in ABCA1), or both (as in ABCA4). Thus, future studies analyzing the functional consequences of this and other ABCA3 mutations remain needed. Based on the patient's phenotype of chronic respiratory failure, we conclude that c.4938delC disrupts the function of ABCA3 in vivo.
Figure 2.

The C‐terminal sequence of ABCA3 is highly conserved. The predicted protein sequence of c.4938delC starting at p.Met1647 is compared to ABCA3 protein sequences from other vertebrates and other human ABC class A family members. Sequences known to be critical for the function of ABCA1 (Tangier Disease) and ABCA4 (Stargardt Disease) are highlighted. At bottom is the C‐terminal sequence of the Streptomyces peucetius homolog DrrA with a highlighted sequence critical for protein interaction and ATPase activity
ETHICS STATEMENT
The patient's parents have signed written informed consent for publication of this case.
Akil N, Fischer AJ. Surfactant deficiency syndrome in an infant with a C‐terminal frame shift in ABCA3: A case report. Pediatric Pulmonology. 2018;53:E12–E14. https://doi.org/10.1002/ppul.23994
REFERENCES
- 1. Wambach JA, Casey AM, Fishman MP, et al. Genotype‐phenotype correlations for infants and children with ABCA3 deficiency. Am J Respir Crit Care Med. 2014; 189:1538–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wambach JA, Yang P, Wegner DJ, et al. Functional characterization of ATP‐binding cassette transporter A3 mutations from infants with respiratory distress syndrome. Am J Respir Cell Mol Biol. 2016; 55:716–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fitzgerald ML, Okuhira K, Short GF, 3rd , Manning JJ, Bell SA, Freeman MW. ATP‐binding cassette transporter A1 contains a novel C‐terminal VFVNFA motif that is required for its cholesterol efflux and ApoA‐I binding activities. J Biol Chem. 2004; 279:48477–48485. [DOI] [PubMed] [Google Scholar]
- 4. Zhong M, Molday LL, Molday RS. Role of the C terminus of the photoreceptor ABCA4 transporter in protein folding, function, and retinal degenerative diseases. J Biol Chem. 2009; 284:3640–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang H, Pradhan P, Kaur P. The extreme C terminus of the ABC protein DrrA contains unique motifs involved in function and assembly of the DrrAB complex. J Biol Chem. 2010; 285:38324–38336. [DOI] [PMC free article] [PubMed] [Google Scholar]