

ABSTRACT
Introduction: This study sought to estimate the global prevalence of transthyretin familial amyloid polyneuropathy (ATTR‐FAP). Methods: Prevalence estimates and information supporting prevalence calculations was extracted from records yielded by reference‐database searches (2005–2016), conference proceedings, and nonpeer reviewed sources. Prevalence was calculated as prevalence rate multiplied by general population size, then extrapolated to countries without prevalence estimates but with reported cases. Results: Searches returned 3,006 records; 1,001 were fully assessed and 10 retained, yielding prevalence for 10 “core” countries, then extrapolated to 32 additional countries. ATTR‐FAP prevalence in core countries, extrapolated countries, and globally was 3,762 (range 3639–3884), 6424 (range, 1,887–34,584), and 10,186 (range, 5,526–38,468) persons, respectively. Discussion: The mid global prevalence estimate (10,186) approximates the maximum commonly accepted estimate (5,000–10,000). The upper limit (38,468) implies potentially higher prevalence. These estimates should be interpreted carefully because contributing evidence was heterogeneous and carried an overall moderate risk of bias. This highlights the requirement for increasing rare‐disease epidemiological assessment and clinician awareness. Muscle Nerve 57: 829–837, 2018
Keywords: amyloidosis, ATTR‐FAP, epidemiology, prevalence, transthyretin familial amyloid polyneuropathy
Abbreviations
- 1M
million
- ATTR‐FAP
transthyretin familial amyloid polyneuropathy
- EMA
European Medicines Agency
- MeSH
Medical Subject Heading
- PRISMA
Preferred Reporting Items for Systematic Reviews and Meta‐Analyses
- THAOS
Transthyretin Amyloidosis Outcomes Survey
Transthyretin familial amyloid polyneuropathy (ATTR‐FAP) is a rare autosomal dominant disease characterized by polyneuropathy due to amyloid deposition in the peripheral nerves and major organs.1 Associated axonal degeneration of myelinated fibers and systemic amyloid deposition leads to a mixed clinical phenotype consisting of sensory and motor impairment and multiple organ failure. Left untreated, inexorable loss of function ensues, with an average survival of 10–15 years after symptom onset.2, 3, 4, 5
Endemic to Portugal, Sweden, and specific regions of Japan,1 ATTR‐FAP has been reported in 36 countries worldwide,6 with an estimated global prevalence of 5,000–10,000 persons.7, 8 This commonly accepted prevalence range, however, does not appear to have been formally or transparently determined. Prevalence outside of the 3 endemic areas noted above has been sparsely reported; when given, these estimates pertain primarily to subnational geographic foci. Epidemiological estimates for ATTR‐FAP are also hindered by underdiagnosis and misdiagnosis resulting from insufficient disease awareness and a clinical presentation involving nonspecific clinical features that are hallmarks of more common diseases.9 Thus, there is a strong basis on which to assume that the commonly held range of 5,000–10,000 persons with ATTR‐FAP worldwide does not reflect the true global prevalence. This study was conducted to address this knowledge gap by estimating global ATTR‐FAP prevalence comprehensively.
MATERIALS AND METHODS
Global ATTR‐FAP prevalence was assessed through a synthesis of epidemiological evidence for reported endemic countries and foci extracted from a comprehensive literature review. In addition, that evidence was extrapolated to countries with reported ATTR‐FAP cases but without published prevalence estimates.
Literature Review
A literature review was undertaken according to modified Cochrane10 and PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses)11 guidelines, which were reflected in a protocol developed specifically for this study. This protocol specified the outcomes of interest as (1) the prevalence of persons with symptomatic ATTR‐FAP globally and (2) the prevalence of persons with symptomatic ATTR‐FAP in each country for which data were available. Aspects of these guidelines were modified to reflect the review of observational, noninterventional studies.
The content and composition of the search terms were developed iteratively against a records standard published by the European Medicines Agency (EMA).12 The EMA report used 14 published studies to determine ATTR‐FAP epidemiology in Europe. The search string for the present review was revised until it yielded all 14 studies. The final search strategy was composed of 2 parts:
Part 1: ((“Amyloid Neuropathies, Familial/epidemiology” [MeSH] OR “Amyloidosis, Familial”[MeSH] OR “Amyloid Neuropathies, Familial/ethnology”[MeSH] OR “Amyloid Neuropathies, Familial/etiology”[MeSH] OR “Amyloid Neuropathies, Familial/genetics”[MeSH] OR “Amyloid Neuropathies, Familial/statistics and numerical data” [Mesh]) AND (incidence[MeSH] OR prevalence[MeSH] OR penetrance[MeSH])) OR ((transthyretin AND (familial OR hereditary OR inherited))
OR
Part 2: ((“fap ttr” OR “ttr fap” OR “attr”) OR (“ttr” AND “fap”)) AND amyloid* AND (neuropath* OR polyneuropath*) AND (inciden* OR prevalen* OR epidemiol* OR ((penetrance OR genotyp* OR phenotyp*) OR (genetic AND (variat* OR varian* OR variab*))) OR epidemiol* OR “rate per” OR (country OR region OR population))).
Part 1 was a string of Medical Subject Heading (MeSH) and structured terms that oriented the search toward properly categorized studies of ATTR‐FAP epidemiology specifically in PubMed. Structured search terms were modified according to each of the databases being searched. Part 2 was a string of unstructured terms and phrases derived from keywords included in relevant studies.
These searches were applied to Embase, PubMed, Scopus, and Web of Science, and were limited to records published from 2005 to 2015 (inclusive) without limits on other record characteristics. Additional records to be assessed for potential inclusion were sourced in 3 ways. First, all abstracts, working papers, and posters reported in the proceedings of the following 5 conferences were reviewed: (a) First European Congress on Hereditary ATTR Amyloidosis (ATTR 2015); (b) International Society of Amyloidosis (ISA 2010, 2012, and 2014); (c) International Symposium on Familial Amyloidotic Polyneuropathy (ISFAP 2013). Second, commonly cited sources of prevalence estimates published prior to 2005 were assessed in full text for inclusion in this review. Third, country‐specific health statistics and relevant disease websites reporting prevalence data or data that could be used to calculate prevalence in the given country were included, as warranted. These website sources were identified through an unstructured search of online sources.
Record Selection, Eligibility, and Data Processing
Records yielded by the search were first deduplicated and then assessed for inclusion via 3 iterative rounds of review (title, abstract, and full text) according to a prespecified rubric. Assessment was completed independently by 3 reviewers, and inconsistencies were adjudicated by consensus among the reviewers. Records were retained if they reported the prevalence of ATTR‐FAP within a specific geographic area or if they reported details that would allow the calculation of ATTR‐FAP prevalence in a specific geographic area.
Evidentiary strength and risk of bias of retained studies (including gray literature) was determined by application of the American Academy of Neurology's clinical practice process for classifying population screening studies.13 Evidence classifications range from class I (strongest evidence, low risk of bias) to class IV (weakest evidence, very high risk of bias). The primary focus of the evidence classification is to determine the objectivity of the retained sources. For the purpose of the present review, an objective source was one in which the determination of the outcome (i.e., the diagnosis of persons with ATTR‐FAP and the determination of prevalence) was unlikely to be affected by observer expectations.
In the absence of available guidelines for grading epidemiological studies of rare diseases, this process was chosen for its simplicity and applicability to the field of neurology. However, it is important to note that not all of the criteria are applicable to a review focusing on a noninterventional topic nor are all of the criteria applicable to a rare disease. For example, to determine the national prevalence of a rare disease, it would not be feasible for a study to statistically sample the general population for assessment and then apply diagnostic strategies to that sample. Instead, a more feasible approach (which we expected to observe in this review) would be for the collected studies to have determined prevalence based on the number of patients diagnosed, treated, or registered at specialty treatment centers within a given country. Because of such considerations, we applied the evidence classification process to the extent possible in an effort to assess the validity of our findings and of the contributing evidence.
Data Synthesis and Analysis
Extracted prevalence estimates were converted, when not originally reported as such, to the common denominator of 1 million persons. The prevalence estimates were categorized in 1 of 4 mutually exclusive categories pertaining to the geographic locus: (A) endemic country, national estimate; (B) endemic country, regional estimate; (C) nonendemic country, national estimate; (D) nonendemic country, regional estimate. The distinction of endemic versus nonendemic countries was made with the commonly cited assumption that Portugal, Sweden, and Japan are endemic,7 and all others were considered nonendemic. Estimates noted in the records pertaining to a country as a whole were designated national, and those noted as pertaining to areas within a country were designated regional.
For countries with reported prevalence estimates, the total number of persons with ATTR‐FAP was calculated as the prevalence rate per million (1M) multiplied by the general population expressed in millions. With the exception of Northern Ireland14 and Luso‐Brazilians in Portugal,15 general population sizes were drawn from the World Bank database.16 When multiple prevalence rates were reported for a country, we first constructed a range consisting of a low prevalence estimate and a high prevalence estimate. In other words, the lowest prevalence rate would be used to estimate the minimum (i.e., low) prevalence, and the highest prevalence rate would be used to estimate the maximum (i.e., high) prevalence. Unless specified otherwise, the mid estimate for a given country was equal to the average value (i.e., [high + low]/2).
Extracted prevalence estimates were extrapolated to countries in which ATTR‐FAP cases have been reported but prevalence has not. These countries were identified from (1) a recently presented case series of 532 ATTR‐FAP cases published between 2005 and 2015,6 (2) a list of countries with at least 1 clinic site enrolling patients into the THAOS (Transthyretin Amyloidosis Outcomes Survey) disease registry,17 and (3) countries that are known to the authors to have ATTR‐FAP patients. Unless additional information was available, the low, mid, and high prevalence estimates for these extrapolated countries were calculated as the lowest, middle, and highest nonendemic national prevalence rates extracted from the literature multiplied by general population size.
RESULTS
The search strategy yielded 3,006 unique records, of which 1,001 were assessed in full and 10 were retained as sources of ATTR‐FAP prevalence estimates (Fig. 1). One of the 10 retained records, Parman et al.,18 was published while our research was ongoing and was included retroactively. The search was rerun to include the year 2016, but no additional relevant records were identified. Two19, 20 of the 10 records provided information that we used to generate prevalence estimates for 2 countries; Benson19 reported ATTR‐FAP prevalence among the Caucasian population in the United States, and the Center for Studies of Paramyloidosis Antonio Rodrigues de Mello20 reported an estimate for the number of persons in Brazil with ATTR‐FAP. Also included in Table 1 are ratings for evidentiary strength and risk of bias, which indicated an overall moderate evidentiary strength and moderate risk of bias: class I (n = 5), class II (n = 1), and class IV (n = 4).
Figure 1.
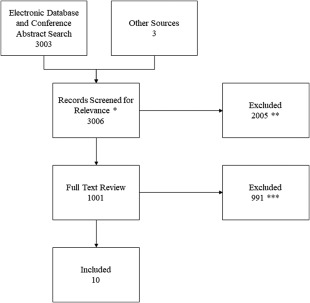
Record adjudication diagram shows the numbers of records adjudicated at each step of the comprehensive review. A total of 3,006 records potentially containing prevalence information were evaluated and 10 were ultimately retained. *Included record title and abstract review. **Records (2,005) excluded for not pertaining to amyloidosis and/or not expected to have prevalence information for ATTR‐FAP. ***Records (643) excluded for pertaining to other types of amyloidosis, records (206) excluded for containing ATTR‐FAP cases but no prevalence information, and records (142) excluded for being observational or interventional studies for ATTR‐FAP but without prevalence information. ATTR‐FAP, transthyretin familial amyloid polyneuropathy.
Table 1.
Characteristics of retained sources of prevalence information
| Reference | Country | Prevalence area | Evidence required for definitive diagnosis | Source(s) for diagnosed persons used to estimate prevalence | Evidence class, risk of biasa |
|---|---|---|---|---|---|
| Dardiotis et al.21 | Cyprus | National | Genetic or pathologicb | Persons enrolled with the clinical and neurogenetic data bank of the Cyprus Institute of Neurology and Genetics | 1, Low |
| Ines et al.22 | Portugal | Northern | Diagnoses recorded via claims database, objectivity is questionablec | Persons receiving prescription medication or liver transplantation treatment for ATTR‐FAP as identified in the electronic prescription data, provided by the Central Health Administration of Portugal | 4, Very high |
| Kato‐Motozaki et al.23 | Japan | Nagano, Kumamoto, Ishikawa (national)d | Genetic or pathologice | Persons with reported clinical data C registered in a database of patients with amyloidosis for 2003–2005 maintained by the Japanese Ministry of Health, Labor, and Welfare | 2, Moderate |
| Mazzeo et al.24 | Italy | Sicily | Genetic and pathologicf | Persons followed at the Neuromuscular Center of the AOU Policlinico Hospital from 1995 to 2015 | 1, Low |
| Munar‐Ques et al.25 | Spain | Majorca, Minorca | Genetic and pathologicg | Persons at risk were initially identified through a survey of physicians and were subsequently followed up for definitive diagnosis | 1, Low |
| Parman et al.18 | Multipleh | National | Not reported (expert self‐report, survey based) | Estimates were based on a self‐report survey of clinicians and epidemiologists in each country | 4, Very high |
| Sousa et al.26 | Sweden | Pitea, Skelleftea | Genetici | Persons maintained in a centralized national registry | 1, Low |
| Sousa et al.27 | Portugal | Northern | Genetici | Persons registered with the CEPARM noted as being diagnosed with ATTR‐FAP | 1, Low |
| Benson19 | USA | National | None reported (anecdotal) | Not reported (anecdotal) | 4, Very high |
| CEPARM20 | Brazil | National | Diagnoses recorded via registry without information for means of diagnosis, objectivity is questionableb | Persons registered with the CEPARM noted as being diagnosed with ATTR‐FAP. | 4, Very high |
ATTR‐FAP, transthyretin familial amyloid polyneuropathy; CEPARM, Centro de Estudos em Paramiloidose Antônio Rodrigues de Mello (Brazilian Association of Paramyloidosis.
Evidence classes range from 1 (strongest evidence, low risk of bias) to 4 (weakest evidence, very high risk of bias).
All individuals underwent genetic testing via polymerase chain reaction amplified TTR exons 1‐4; products sequenced bidirectionally with Beckman Coulter kit; sequencing products automatically compared with normal TTR sequence listed in GenBank. Pathologic diagnosis was established by using sural nerve or rectal biopsy. All patients were examined by 1 author of record.
Persons with ATTR‐FAP were identified from a central electronic prescription monitoring database by the presence of a diagnostic and treatment codes for ATTR‐FAP. More information regarding the diagnostic and treatment codes used was not reported in the record.
The record's authors extrapolated ATTR‐FAP prevalence estimates for these 3 subnational regions to a national perspective.
Persons with ATTR‐FAP were identified from a compulsory amyloidosis database maintained by the Japanese Ministry of Health, Labor, and Welfare. Available information with which to identify persons with ATTR‐FAP in the database included age, sex, present address, birth place, age at disease onset, family history, clinical manifestations at the time of onset and at present, laboratory findings, genetic analysis of the TTR gene, and histology and immunohistochemistry of biopsy specimens. It was not clear from this record whether all persons in this registry diagnosed with ATTR‐FAP had both genetic and pathologic diagnosis, but here it is presumed as such. This ambiguity was cause to categorize this evidence as Class 2.
All included persons with ATTR‐FAP had received genetic confirmation of disease. The study was retrospective and observational and the procedures for genetic testing were not reported; it was clear whether the investigators conducted the genetic testing. Pathologic diagnosis was performed via sural nerve biopsy was confirmed for 16 of the 76 individuals. It is unclear whether pathologic diagnosis was conducted on the other individuals.
Confirmation of ATTR‐FAP was established in 19 of 104 patients by detection of amyloid in biopsies stained with Congo red and in 83 of 104 by the presence of the biochemical marker TTRVal30Met in serum and/or the G for A substitution in the TTR gene.
Bulgaria, Cyprus, France, Germany, Italy, Netherlands, Portugal, Spain, Sweden, and Turkey.
Diagnosis confirmed by genetic testing in all individuals. Pathologic diagnosis was not reported. Prevalence estimates were based only on individuals reported to have a positive genetic diagnosis and symptomatic disease characterized by polyneuropathy.
Among the 10 retained records, 8 records18, 21, 22, 23, 24, 25, 26, 27 directly reported 25 prevalence estimates pertaining to 11 countries (for pertinent details of the 10 retained records, see Table 1). National level prevalence estimates were reported for 10 of the countries, and these 10 constituted the core group of countries for which ATTR‐FAP population sizes were directly estimable. The 25 prevalence estimates were placed into 1 of 4 geographic categories for which the numbers of contributing records and prevalence estimate ranges were (A) endemic country, national estimate (n = 4; range, 0.99–204.00); (B) endemic country, regional estimate (n = 7; range, 3.80–1,631.20); (C) nonendemic country, national estimate (n = 8; range, 0.32–43.34); and (D) nonendemic country, regional estimate (n = 6; range, 0.20–50.00).
Higher prevalence estimates were reported in endemic countries (Portugal, Sweden, and Japan) and even higher in specific regions within those endemic countries (Fig. 2). The highest prevalence estimates were reported for Northern Portugal (1,631.20/1M) and Northern Sweden (1,040.00/1M). Findings in Japan were noteworthy in that the national prevalence estimate (0.99/1M) was less than the national prevalence in many nonendemic countries, namely Italy, France, Bulgaria, Netherlands, and Germany. Regional prevalences in Japan's Nagano, Kumamoto, and Ishikawa prefectures, however, were among the moderate to high values, which is consistent with the contention that Japan's designation as an endemic country is driven by a few isolated regions with high prevalence,1 as opposed to Portugal and Sweden, where the geographic distribution of ATTR‐FAP—although still somewhat confined to subnational foci—is more homogeneous.
Figure 2.
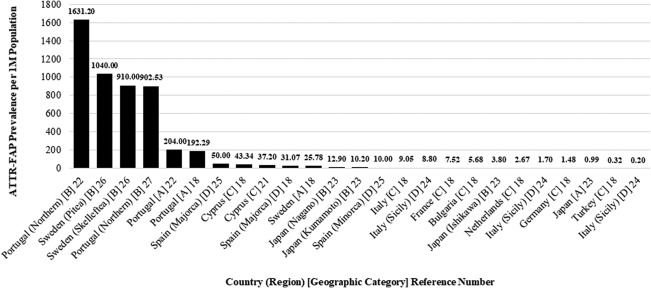
Extracted ATTR‐FAP prevalence values per 1 million general population (1M). The highest and lowest prevalence values reported in the literature were for subnational regions. The highest prevalence was reported in Northern Portugal (1,631.20/1M), and the lowest was reported in Sicily (0.20/1M). Values are categorized as A (endemic country, national estimate), B (endemic country, regional estimate), C (nonendemic country, national estimate), and D (nonendemic country, regional estimate). ATTR‐FAP, transthyretin familial amyloid polyneuropathy
The low, mid, and high nonendemic national prevalence estimates applied to the extrapolated countries were 0.32/1M (Turkey), 1.48/1M (Germany), and 7.52/1M (France), respectively. These values were derived from Parman et al.,18 which was a survey of ATTR‐FAP clinician and epidemiologist members of the European Network for ATTR‐FAP and reported the total number of known persons with ATTR‐FAP in their respective countries in the year 2014. Those values were combined with general population sizes to derive prevalence per 1 million general population. Note that Parman and colleagues included a prevalence estimate for Italy (9.05/1M) that was greater than the value chosen for the high estimate (France, 7.52/1M). The former was not used because of the uncertainty with which it was originally reported.
Low, mid, and high estimated ATTR‐FAP population sizes for each country and the cumulative global ATTR‐FAP population size in each scenario are presented in Figure 3, which was populated with data reported in Table 2. Among the 10 core countries, a cumulative population of 3,762 persons (range, 3,639–3,884) with ATTR‐FAP was estimated; Portugal was the largest contributor with 2,051 persons (range, 1,990–2,111). Again, ATTR‐FAP population sizes were directly estimable in the 10 core countries by using prevalence estimates reported in the literature. Across the 32 countries to which Parman and colleagues' estimates were extrapolated, the estimated ATTR‐FAP population size was 6,424 persons (range, 1,887–34,584). Thus, the cumulative estimated number of persons with ATTR‐FAP across the 42 countries considered was 10,186 persons (range, 5,526–38,468).
Figure 3.
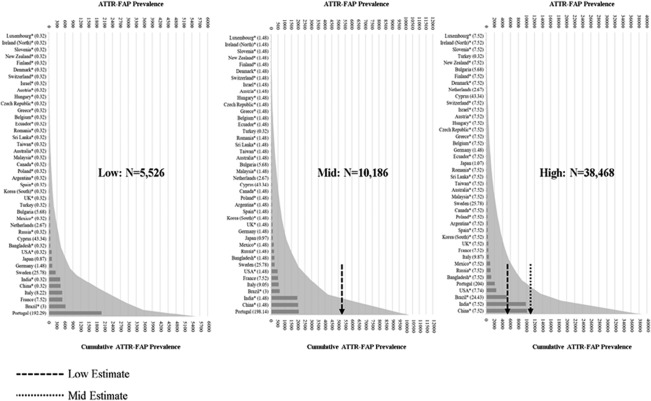
Low, mid, and high country‐specific and global cumulative ATTR‐FAP prevalence. Countries are listed from top to bottom in order of lower to higher ATTR‐FAP population size. Prevalence values (per 1 million population) used in each calculation are listed in parentheses, and global cumulative prevalence is depicted as a curve in gray. *Countries for which prevalence estimates were based on extrapolation. ATTR‐FAP, transthyretin familial amyloid polyneuropathy.
Table 2.
Prevalence estimates by country
| Country | General population, M | Prevalence low | Prevalence mid | Prevalence high |
|---|---|---|---|---|
| Totals | 4,582.3 | 5,526 | 10,186 | 38,468 |
| Core | 460.1 | 3,639 | 3,762 | 3884 |
| Turkey | 78.7 | 25 | 25 | 25 |
| Bulgaria | 7.2 | 41 | 41 | 41 |
| Netherlands | 16.9 | 45 | 45 | 45 |
| Cyprus | 1.2 | 51 | 51 | 51 |
| Germany | 81.4 | 121 | 121 | 121 |
| Japan | 127.0 | 111 | 123 | 135 |
| Sweden | 9.8 | 253 | 253 | 253 |
| France | 66.8 | 502 | 502 | 502 |
| Italy | 60.8 | 500 | 550 | 600 |
| Portugal | 10.3 | 1,990 | 2,051 | 2111 |
| Extrapolated | 4,122.2 | 1,887 | 6,424 | 3,4584 |
| Luxembourg | 0.6 | 0 | 1 | 4 |
| Slovenia | 2.1 | 1 | 3 | 16 |
| Ireland (North) | 1.9 | 1 | 3 | 14 |
| New Zealand | 4.6 | 1 | 7 | 35 |
| Finland | 5.5 | 2 | 8 | 41 |
| Denmark | 5.7 | 2 | 8 | 43 |
| Israel | 8.4 | 3 | 12 | 63 |
| Switzerland | 8.3 | 3 | 12 | 62 |
| Austria | 8.6 | 3 | 13 | 65 |
| Hungary | 9.8 | 3 | 15 | 74 |
| Czech Republic | 10.6 | 3 | 16 | 79 |
| Greece | 10.8 | 3 | 16 | 81 |
| Belgium | 11.3 | 4 | 17 | 85 |
| Ecuador | 16.1 | 5 | 24 | 121 |
| Romania | 19.8 | 6 | 29 | 149 |
| Sri Lanka | 21.0 | 7 | 31 | 158 |
| Taiwan | 23.5 | 8 | 35 | 177 |
| Australia | 23.8 | 8 | 35 | 179 |
| Korea (South) | 50.6 | 16 | 75 | 381 |
| Malaysia | 30.3 | 10 | 45 | 228 |
| Canada | 35.9 | 12 | 53 | 270 |
| Poland | 38.0 | 12 | 56 | 286 |
| Argentina | 43.4 | 14 | 64 | 326 |
| Spain | 46.4 | 15 | 69 | 349 |
| UK | 65.1 | 21 | 97 | 490 |
| Mexico | 127.0 | 41 | 188 | 955 |
| Russia | 144.1 | 46 | 214 | 1,084 |
| Bangladesh | 161.0 | 52 | 239 | 1,211 |
| Brazila | 207.8 | 623 | 623 | 5,078 |
| USAb | 321.4 | 104 | 476 | 2,488 |
| India | 1,311.1 | 423 | 1,943 | 9,858 |
| China | 1,347.7 | 435 | 1,997 | 10,134 |
ATTR‐FAP, transthyretin familial amyloid polyneuropathy; M, million; UK, United Kingdom.
For Brazil, 623 patients from Centro de Estudos em Paramiloidose Antônio Rodrigues de Mello were used for the low/middle estimate per author judgment. The high estimate was derived as the summed prevalence from 2 population subgroups, Luso‐Brazilians and Portuguese non‐Luso‐Brazilians. ATTR‐FAP prevalence for Luso‐Brazilians was equal to the product of the subgroup population size (i.e., 25M15 and the prevalence rate given in Parman et al.18 (i.e., 192.29/1M). ATTR‐FAP prevalence for Portuguese non‐Luso‐Brazilians was equal to the product of the subgroup population size (i.e., 182.8M16 and the mid prevalence rate applied elsewhere in this analysis (i.e., 1.48/1M18.
DISCUSSION
The global prevalence of ATTR‐FAP was estimated to be 10,186 persons (range, 5,526–38,468). Both evidentiary strength and risk of bias for the studies contributing to our estimates were moderate overall (5 class I, 1 class II, and 4 class IV records). ATTR‐FAP population sizes reported for the extrapolated countries should be interpreted more cautiously because these estimates were subject to a greater degree of author judgment compared with core country estimates. Additionally, we relied heavily on 1 class IV study (Parman et al.18, and methodological options for deriving estimates and for assessing underlying uncertainty were limited by the heterogeneity and scarcity of data. The evidentiary strength of our estimates might therefore also be considered moderate. However, this determination and the classifications of contributing studies should be interpreted with the understanding that the evidence grading criteria could not be strictly applied to this review for reasons described in the Materials and Methods section, so this should be considered a comprehensive rather than a systematic review.
Low, mid, and high country‐specific and global ATTR‐FAP population sizes were estimated by the varying of assumptions for the underlying risk of ATTR‐FAP within populations. The low and mid global estimates (5,526 and 10,186, respectively) were generated under more conservative assumptions, and these were consistent with the range reported anecdotally in the literature from separate sources (5,000–10,000).7, 8 Under the most conservative scenario, traditionally endemic countries (Portugal, Sweden, and Japan)18 together contributed 43% of prevalent persons globally even though their general populations constitute only 3% of the total population considered in this analysis. Their relative combined contribution decreased to 24% in the mid estimate and 6% in the high estimate primarily because of large contributions by nonendemic high‐population countries (e.g., India and China). The total “at‐risk” population in this analysis (4.6 billion persons from 42 countries) is only 60% of the total global population (7.4 billion). Additional ATTR‐FAP cases might be found in the 40% of the global population living in areas not considered here, particularly in former Portuguese colonies such as Angola, Cape Verde, Equatorial Guinea, and Mozambique.
One might reasonably question the present prevalence estimates and, in particular, extrapolations beyond the 10 core countries. Addressing the core countries first, we applied country‐specific prevalence rates to the most recent general population statistics. This method of multiplying the rate of disease for a given population by the size of that population has precedence in the epidemiology literature, for example in Alzheimer disease,29 but is not without its limitations. However, to have reported ATTR‐FAP prevalence for only those 10 core countries belies the reality of this disease, which, we know from published cases, is found in at least 32 additional countries.6 For these extrapolated estimates, we applied the lowest prevalence rates to generate conservative values in the base case, and we explored realistic ranges by applying other appropriate prevalence multipliers. Mediating population‐level factors were also considered. For example, in Brazil, the cumulative national prevalence was derived based on Portuguese‐descended and non–Portuguese‐descended subpopulations separately.
This synthesis relied heavily on the country‐specific prevalence rates reported in Parman et al.18 These estimates were based on a survey of European ATTR‐FAP clinicians and researchers that reported their impressions, but not necessarily direct observations, of ATTR‐FAP prevalence in their respective countries. Because of the indirect estimation of ATTR‐FAP prevalence through experts' self‐report by Parman and colleagues, we rated the strength of their evidence as class IV. Nevertheless, for multiple reasons, these estimates were indispensable to deriving our original estimates for extrapolated countries. First, the prevalence rates reported in Parman et al.18 were generated from the responses of 15 ATTR‐FAP clinical and epidemiological experts practicing in Europe. Each expert (identified by name and affiliation in the report) is affiliated with a national ATTR‐FAP registry or national ATTR‐FAP specialized treatment center. Our interpretation is that the experts' self‐reports of the number of persons with ATTR‐FAP are likely to have been accurate. Second, many of the country‐level prevalence rates reported by Parman and colleagues (e.g., Germany, Italy, Turkey, and The Netherlands) are not available elsewhere.
Our approach has some additional limitations. Because of a general lack of prevalence data outside traditionally endemic areas, it was necessary to impute prevalence for many of the countries considered here based on a range of plausible values. With the heterogeneity among the retained studies and the general scarcity of data, it was also not possible to formally assess precision of our estimates, which might have otherwise been achieved by deriving confidence intervals around the estimates through meta‐analysis. These findings should also be interpreted with the understanding that clinical definitions of ATTR‐FAP evolved over the time horizon of the literature search, so the ATTR‐FAP populations forming the bases of the extracted original prevalence estimates may not have been entirely consistent. However, the impact of this was mitigated by retaining only estimates that were derived from populations with noted peripheral neuropathy, which is a constellation of symptoms that is consistently represented across the literature. Perhaps more important in its implications for these findings, the emergence of survival‐extending ATTR‐FAP treatments has potentially caused an increase in prevalence over time. Thus, inclusion of older prevalence estimates here may have skewed estimates downward. Similarly, the number of ATTR‐FAP diagnoses and the number of regions where ATTR‐FAP has been observed have been increasing because of improved diagnostic processes and clinician knowledge.30, 31 Presumably this also would have resulted in underestimated prevalence in some areas, especially nonendemic ones.
It is hoped that these findings will encourage clinicians to consider ATTR‐FAP as part of the differential diagnosis for patients presenting with otherwise idiopathic polyneuropathy and systemic features such as cardiomyopathy and autonomic or gastrointestinal dysfunction.32 From public health and policy perspectives, these findings highlight the requirement to allocate more resources toward rare disease surveillance, epidemiological assessment, diagnostics, specialty care, and increasing clinician knowledge, especially for very rare conditions such as ATTR‐FAP. This is particularly important in countries such as Brazil and the United States, where sizeable populations of persons descended from traditionally endemic areas may equate to higher rates of underdiagnosis of ATTR‐FAP.
Ethical Publication Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Funding: This research was supported by Pfizer Inc.
Conflicts of Interest: M. Stewart, M. Hopps, S. Fallet, and L. Amass are employees of Pfizer Inc. M. F. Botteman and A. S. Chopra are employees of Pharmerit International, which received funding from Pfizer Inc. for study design, execution, analysis, and manuscript development. J. A. Carter is an independent scientific consultant who was funded by Pharmerit International. H. H. Schmidt and M. Waddington‐Cruz were investigators for the study and were not financially compensated for collaborative efforts on publication‐related activities. H. H. Schmidt has received support from FoldRx Pharmaceuticals (acquired by Pfizer Inc. in October 2010), Pfizer, IONIS, and Alnylam as a clinical investigator. M. Waddington‐Cruz has received support from FoldRx Pharmaceuticals as a clinical investigator, has served on the scientific advisory board of Pfizer Inc., has received funding from Pfizer Inc. for scientific meeting expenses (travel, accommodations, and registration), has received research support from the National Institutes of Health, and currently serves on the THAOS (Transthyretin Amyloidosis Outcomes Survey) scientific advisory board.
REFERENCES
- 1. Yamashita T, Ando Y, Okamoto S, Misumi Y, Hirahara T, Ueda M. Long‐term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology 2012;78(9):637–643. [DOI] [PubMed] [Google Scholar]
- 2. Reinés JB, Vera TR, Martín MU, Serra HA, Campins MM, Millán J. Epidemiology of transthyretin‐associated familial amyloid polyneuropathy in the Majorcan area: Son Llàtzer Hospital descriptive study. Orphanet J Rare Dis 2014;9:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Planté‐Bordeneuve V, Lozeron P. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012;79(8):785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bekircan‐Kurt C, Gunes N, Yilmaz A, Erdem‐Ozdamar S, Tan E. Three Turkish families with different transthyretin mutations. Neuromuscul Disord 2015;25:686–692. [DOI] [PubMed] [Google Scholar]
- 5. Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A 2012;109(24):9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Waddington‐Cruz M, Schmidt HH, Botteman MF, Carter JA, Chopra AS, Stewart M. Epidemiological and clinical characteristics of persons with transthyretin familial amyloid polyneuropathy: a global synthesis of 532 cases. Amyloid 2017;24(Suppl):109–110. [DOI] [PubMed] [Google Scholar]
- 7. Plante‐Bordeneuve V. Update in the diagnosis and management of transthyretin familial amyloid polyneuropathy. J Neurol 2014;261(6):1227–1233. [DOI] [PubMed] [Google Scholar]
- 8. Misu KI, Hattori N, Nagamatsu M, Ikeda SI, Ando Y, Nakazato M. Late‐onset familial amyloid polyneuropathy type I (transthyretin Met30‐associated familial amyloid polyneuropathy) unrelated to endemic focus in Japan. Clinicopathological and genetic features. Brain 1999;122(1):1951–1962. [DOI] [PubMed] [Google Scholar]
- 9. Hawkins PN, Ando Y, Dispenzeri A, Gonzalez‐Duarte A, Adams D, Suhr OB. Evolving landscape in the management of transthyretin amyloidosis. Ann Med 2015;47(8):625–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Higgins JPT, Green S, eds. Cochrane handbook for systematic reviews of interventions version 5.1.0. Updated March 2011. Available at http://handbook-5-1.cochrane.org/. Accessed December 7, 2017.
- 11. Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. Int J Surg 2010;8(5):336–341. [DOI] [PubMed] [Google Scholar]
- 12. European Medicines Agency . Examples of prevalence sources previously considered in orphan medicinal product designation procedures. Published 2014. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Other/2014/12/WC500179298.pdf. Accessed December 7, 2017.
- 13. Gronseth GS, Woodroffe LM, Getchius TSD, eds. Clinical practice guideline process manual. St Paul, MN: American Academy of Neurology; 2011. Available at http://tools.aan.com/globals/axon/assets/9023.pdf. Accessed December 7, 2017. [Google Scholar]
- 14. Northern Ireland Statistics and Research Agency . The population of Northern Ireland. http://www.nisra.gov.uk/statistics/population.html. Accessed December 13, 2016.
- 15. Council of the Luso‐Brazilian Community of the State of Sao Paulo. Portuguese Immigration Day: News [in Portuguese]. http://www.cclb.org.br/noticias/jul08/jul02_02.htm. Published 2008. Accessed December 7, 2017.
- 16. The World Bank . World DataBank. http://databank.worldbank.org/data/home.aspx. Accessed December 7, 2017.
- 17. Plante‐Bordeneuve V, Suhr OB, Maurer MS, White B, Grogan DR, Coelho T. The Transthyretin Amyloidosis Outcomes Survey (THAOS) registry: design and methodology. Curr Med Res Opin 2013;29(1):77–84. [DOI] [PubMed] [Google Scholar]
- 18. Parman Y, Adams D, Obici L, Galan L, Guergueltcheva V, Suhr OB. Sixty years of transthyretin familial amyloid polyneuropathy (TTR‐FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR‐FAP. Curr Opin Neurol 2016;29(Suppl 1):S3–S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Benson MD. Hereditary amyloidosis In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, et al, eds. The online metabolic and molecular bases of inherited disease. New York, NY: McGraw‐Hill Medical; 2007. Available at https://ommbid.mhmedical.com/book.aspx?bookID=971. Accessed December 7, 2017. [Google Scholar]
- 20. CEPARM . Recent findings. http://ceparm.com/en/ceparm/recent-findings/. Published 2015. Accessed December 7, 2017.
- 21. Dardiotis E, Koutsou P, Papanicolaou EZ, Vonta I, Kladi A, Vassilopoulos D. Epidemiological, clinical and genetic study of familial amyloidotic polyneuropathy in Cyprus. Amyloid 2009;16(1):32–37. [DOI] [PubMed] [Google Scholar]
- 22. Inês M, Coelho T, Conceição I, #Duarte‐Ramos F, de Carvalho M, Costa J. Epidemiology of transthyretin familial amyloid polyneuropathy in Portugal. Orphanet J Rare Dis 2015;10(Suppl 1):P21–P21. [DOI] [PubMed] [Google Scholar]
- 23. Kato‐Motozaki Y, Ono K, Shima K, Morinaga A, Machiya T, Nozaki I. Epidemiology of familial amyloid polyneuropathy in Japan: identification of a novel endemic focus. J Neurol Sci 2008;270(1–2):133–140. [DOI] [PubMed] [Google Scholar]
- 24. Mazzeo A, Russo M, Di Bella G, Minutoli F, Stancenelli C, Gentile L. TTR‐FAP: a single‐center experience in Sicily, an Italian endemic area. Orphanet J Rare Dis 2015;10(Suppl 1):O1–O1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Munar‐Ques M, Saraiva MJM, Viader‐Farre C, Zabay‐Becerril JM, Mulet‐Ferrer J. Genetic epidemiology of familial amyloid polyneuropathy in the Balearic Islands (Spain). Amyloid 2005;12(1):54–61. [DOI] [PubMed] [Google Scholar]
- 26. Sousa A, Andersson R, Drugge U, Holmgren G, Sandgren O. Familial amyloidotic polyneuropathy in Sweden: geographical distribution, age of onset, and prevalence. Hum Hered 1993;43(5):288–294. [DOI] [PubMed] [Google Scholar]
- 27. Sousa A, Coelho T, Barros J, Sequeiros J. Genetic epidemiology of familial amyloidotic polyneuropathy (FAP)‐type I in Povoa do Varzim and Vila do Conde (north of Portugal). Am J Med Genet 1995;60(6):512–521. [DOI] [PubMed] [Google Scholar]
- 28. United States Census Bureau . Population quick facts. https://www.census.gov/quickfacts/fact/table/US/PST045216. Published 2016. Accessed December 7, 2017.
- 29. Brookmeyer R, Evans DA, Hebert L, Langa KM, Heeringa SG, Plassman BL. National estimates of the prevalence of Alzheimer's disease in the United States. Alzheimers Dement 2011;7(1):61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yukio A, Yoshiki S, Konen O, Taro Y, Mitsuharu U, Yohei M. Effects of tafamidis treatment on transthyretin (TTR) stabilization, efficacy, and safety in Japanese patients with familial amyloid polyneuropathy (TTR‐FAP) with Val30Met and non‐Varl30Met: a phase III, open‐label study. J Neurol Sci 2016;362:266–271. [DOI] [PubMed] [Google Scholar]
- 31. Ando Y, Nakamura M, Araki S. Transthyretin‐related familial amyloidotic polyneuropathy. Arch Neurol 2005;62(7):1057–1062. [DOI] [PubMed] [Google Scholar]
- 32. Conceicao I, Gonzalez‐Duarte A, Obici L, Schmidt HH, Simoneau D, Ong ML. “Red‐flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst 2016;21(1):5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]