

Abstract
Objective
EMBODY 1 (ClinicalTrials.gov identifier: NCT01262365) and EMBODY 2 (ClinicalTrials.gov identifier: NCT01261793) investigated the efficacy and safety of epratuzumab, a CD22‐targeted humanized monoclonal IgG antibody, in patients with systemic lupus erythematosus (SLE). The studies showed no significant difference from placebo in primary or secondary clinical outcome measures but did demonstrate B cell–specific immunologic activity. The aim of this post hoc analysis was to determine whether epratuzumab had a different clinical efficacy profile in SLE patients with versus those without an associated diagnosis of Sjögren's syndrome (SS).
Methods
The efficacy and safety of epratuzumab were compared between 2 patient subpopulations randomized in EMBODY 1 and 2: SLE patients with and those without a diagnosis of associated SS. British Isles Lupus Assessment Group (BILAG) total score, BILAG‐based Combined Lupus Assessment (BICLA) clinical response to treatment, biologic markers (including B cells, IgG, IgM, and IgA), and safety were assessed.
Results
A total of 1,584 patients were randomized in the EMBODY 1 and EMBODY 2 trials; 113 patients were anti‐SSA positive and had a diagnosis of associated SS, and 1,375 patients (86.8%) had no diagnosis of associated SS (918 patients were randomized to receive epratuzumab and 457 to receive placebo). For patients with associated SS, but not those without associated SS, a higher proportion of patients receiving epratuzumab achieved a BICLA response and a reduction from baseline in BILAG total score. B cell reduction was faster in patients with associated SS. The sensitivity of B cells to epratuzumab as measured by the mean concentration producing 50% of the maximum B cell count depletion was lower for patients with associated SS (9.5 μg/ml) versus the total EMBODY population (87.1 μg/ml). No difference in the frequency of adverse events in those receiving placebo was reported.
Conclusion
Patients with SLE and associated SS treated with epratuzumab showed improvement in SLE disease activity, which was associated with bioactivity, such as decreases in B cell number and IgM level.
Sjögren's syndrome (SS) is a progressive autoimmune disease that primarily affects the exocrine glands, with systemic complications. The disease is characterized by reduced mucosal secretions resulting in dryness of mucosal surfaces, which typically manifests as dryness of the eyes and mouth 1. Histopathologic analyses of affected tissue samples reveal lymphocyte infiltration, the formation of ectopic germinal centers, and cell death. Analyses of the lymphocytes implicate a central role of B cells in the pathogenesis of the disease 2, 3, 4, 5.
SS is described as either primary or associated. Primary SS develops independently of any other condition, while associated SS (also known as secondary SS) is linked to a coexisting autoimmune disease such as systemic lupus erythematosus (SLE) 6, 7. SLE is a chronic autoimmune disease predominantly affecting the musculoskeletal, mucocutaneous, and renal systems. Between 6.5% and 19% of patients with SLE develop associated SS 8. The pathogenesis of SLE and associated SS is associated with elevated B cell activation and subsequent tissue infiltration and destruction 9, 10; thus, targeting B cells has been widely seen as a potential therapeutic option for both SLE and associated SS 11, 12, 13.
Epratuzumab (developed by Immunomedics) is a humanized monoclonal antibody that targets the B cell–specific protein CD22, resulting in a reduction in both B cell activity and the number of B cells in peripheral blood. CD22 is an inhibitory co‐receptor of the B cell receptor (BCR); following ligation to epratuzumab, CD22 is rapidly internalized and phosphorylated 14, 15, leading to the removal of BCR complexes from the cell surface. This down‐modulates BCR‐activated signaling, functionally inhibiting B cells; for example, there is reduced proliferation and maturation of B cells and a diminished production of proinflammatory cytokines 16, 17, 18.
In the phase IIb EMBLEM trial 19, patients with moderate‐to‐severe SLE treated with epratuzumab demonstrated clinically relevant improvements in disease activity, which were maintained during the 3.2‐year open‐label extension study 20. Following the EMBLEM trial, 2 phase III, randomized, placebo‐controlled, double‐blind, multicenter trials (EMBODY 1 and 2) assessed the efficacy and safety of epratuzumab in the treatment of moderately to severely active SLE 21. In those trials there was no statistically significant difference in British Isles Lupus Assessment Group (BILAG)–based Combined Lupus Assessment (BICLA) 22 response rates at week 48 between epratuzumab‐treated and placebo‐treated patients (the primary end point). Changes in immunologic parameters were observed in epratuzumab‐treated patients, including a 30–40% reduction in the number of peripheral B cells, consistent with previous studies with epratuzumab.
The efficacy of epratuzumab in primary SS was evaluated in 2006 in a phase I/II open‐label study, in which 16 patients were enrolled and received 4 infusions of epratuzumab 23. Similar to the EMBLEM trial in SLE patients, epratuzumab reduced peripheral B cell counts and was associated with improved clinical markers of disease activity.
A subset of SLE patients enrolled into the EMBODY trial were diagnosed as having associated SS according to their medical history. Since B cells have been implicated in the pathogenesis of SS, and epratuzumab has shown efficacy in a small open trial of patients with primary SS, the objective of this post hoc analysis was to determine whether epratuzumab demonstrated a different clinical efficacy profile in SLE patients with a diagnosis of associated SS.
Patients and methods
The analyses reported here represent post hoc analyses of the EMBODY trials (ClinicalTrials.gov identifiers: NCT01262365 and NCT01261793). The EMBODY trial inclusion and exclusion criteria, and the study design, have been reported previously 21 and are described briefly below.
Patients. To be eligible for inclusion in the EMBODY trials, patients had to be ≥18 years of age and have a diagnosis of moderately to severely active SLE fulfilling ≥4 of 11 of the revised American College of Rheumatology criteria (24) (if patients had a neurologic disorder, ≥5 of 11 criteria were required). All patients had disease activity in the musculoskeletal, mucocutaneous, or cardiorespiratory systems, as defined by the BILAG Index (2004 version) 25, 26. Patients must have had BILAG grade A activity in ≥1 of these systems, or BILAG grade B activity in ≥2 of these systems. Additionally, all patients had an SLE Disease Activity Index 2000 (SLEDAI‐2K) (27) score of ≥6, and had to be positive at screening for antinuclear antibodies (ANAs; titer ≥1:80) and/or anti–double‐stranded DNA (defined as a positive result on either a multiplex immunoassay or a Farr assay). Key exclusion criteria included SLE disease activity involving the renal system (defined by BILAG level A renal activity); serum creatinine level of >2.5 mg/dl, a clinically significant increase in serum creatinine level within 4 weeks prior to screening, or proteinuria >3.5 gm/day; and severe neuropsychiatric SLE disease.
Study design. EMBODY 1 and EMBODY 2 were identical phase III, randomized, double‐blind, placebo‐controlled, multicenter studies 21. The studies consisted of a 2‐week screening period, followed by a 48‐week double‐blind treatment period, and a 4‐week safety follow‐up (13 weeks for patients who discontinued prior to week 48). Patients were randomized 1:1:1 to receive placebo, epratuzumab 600 mg every week, or epratuzumab 1,200 mg every other week. Infusions of active drug or placebo were administered over a 4‐week dosing period at the beginning of each 12‐week treatment cycle. This dosing pattern was repeated for 4 cycles (48 weeks total). In all cases, the study drug was given in addition to the patients' existing standard therapy, consisting of corticosteroids, antimalarials, and/or immunosuppressants.
Post hoc analyses. The efficacy and safety of epratuzumab compared to placebo were assessed in 2 subpopulations of randomized patients. The first subpopulation (patients with associated SS) consisted of SLE patients with a diagnosis of associated SS according to their medical history (provided by patients at screening) who were positive for anti‐SSA (also called anti‐Ro). The second subpopulation (patients without associated SS) consisted of all other SLE patients (i.e., those without a medical history of associated SS). Two sets of sensitivity analyses were performed for all outcomes, one on SLE patients positive for anti‐SSA and the other on SLE patients negative for anti‐SSA. Unexpectedly, some patients were positive for antidrug antibody at baseline before receiving epratuzumab or placebo. Because the significance of baseline antidrug antibody is uncertain, patients positive for antidrug antibody at baseline were excluded from all analyses.
The proportion of patients with a BICLA response was recorded at each study visit. All of the following were required to achieve a BICLA response: all BILAG As at study entry improved to B, C, or D, all BILAG Bs improved to C or D, no new BILAG As, no more than 1 new BILAG B, no worsening (no increase) in SLEDAI‐2K total score compared to study entry, no worsening in physician's global assessment of disease activity (PGA) compared to study entry (<10‐mm increase on a 100‐mm visual analog scale [VAS]), and no disallowed changes in concomitant medications, including increase in or addition of immunosuppressants or antimalarials compared to baseline, increase of >25% in corticosteroids compared to baseline from week 0 to 8, or any increase in corticosteroids compared to baseline after week 8 for an SLE‐related indication. Other clinical efficacy variables included BILAG total score (generated by converting A, B, C, D, and E to 12, 8, 1, 0, and 0, respectively 28), PGA (on a 100‐mm VAS), and SLEDAI total score at each visit. Pharmacodynamic (PD) and immunologic variables included levels of CD19+ B cells, immunoglobulins, autoantibodies, extractable nuclear antigen antibodies, and complement proteins. Measurements of immunologic and lupus‐associated laboratory parameters, including anti‐SSA autoantibodies, were conducted by a central laboratory.
The pharmacokinetics (PK) of epratuzumab were characterized using nonlinear mixed‐effects modeling (population approach). The following epratuzumab exposure measurements were determined: the average plasma concentration at steady state (Cssav) for each 12‐week treatment period, and the predicted plasma concentration at each study visit (Cvisit). Cssav was derived from patient‐specific estimates of epratuzumab clearance (CL; liter/week) and the total dose over the 12‐week treatment period (i.e., 2,400 mg; dose 12w) using the following equation:
Cvisit was the patient‐specific prediction of epratuzumab plasma concentration at the time of each study visit based on the established population PK model.
The total B cell count was modeled as a continuous PD variable that was allowed to change from baseline over the study duration for each individual. The effect of epratuzumab exposure on the B cell count, corrected for placebo effects, was described using an Emax model, where the effect increases in a nonlinear manner to a maximum value (Emax). The model comprised 2 parameters: the concentration of epratuzumab producing 50% of the maximum response (EC50), and the maximum effect produced (Emax). The population PK/PD model for B cell count was first developed with the complete data set, and then applied to the associated SS subpopulation to assess placebo and epratuzumab exposure‐response for each population.
Treatment‐emergent adverse events (AEs) and serious treatment‐emergent AEs were monitored throughout the study.
Statistical analysis. Efficacy variables were analyzed using the full analysis set, which consisted of all patients with a valid baseline and post‐baseline measurement who received at least one partial dose of study medication. For BICLA response, missing data were imputed using modified nonresponder imputation, with P values calculated using logistic regression with factors for treatment, pooled region, and baseline disease severity.
For change from baseline in total BILAG score, PGA, and total SLEDAI score, missing values were imputed using last observation carried forward, and P values were calculated (at weeks 24 and 48 only) using analysis of covariance adjusted for baseline, pooled region, and baseline disease severity. Observed values were used for all immunologic and serologic measurements. Safety variables for this analysis were analyzed using the full analysis set.
Results
Patient disposition and baseline characteristics. A total of 1,584 patients were randomized in EMBODY 1 and EMBODY 2 (21). Of these, 113 patients were anti‐SSA positive and had a diagnosis of associated SS (7.1%; 73 patients were randomized to the epratuzumab group and 40 to placebo; 33 patients [29.5%] were diagnosed as having SS before their SLE diagnosis); 1,375 patients (86.8%) had no diagnosis of associated SS (918 patients were randomized to the epratuzumab group and 457 to placebo).
Of all the patients randomized to the EMBODY 1 and 2 trials, 96 (6.1%) were excluded from the analysis because they did not meet the subset definitions or were positive for antidrug antibody at baseline. These patients were either antidrug antibody positive at baseline (48 patients), had a diagnosis of associated SS but were not anti‐SSA positive (39 patients), or were both antidrug antibody positive at baseline and had a diagnosis of associated SS but were not anti‐SSA positive (9 patients). In total, 1,329 patients without associated SS and 112 patients with associated SS received at least 1 dose of study drug and had a post‐baseline assessment and were included in the full analysis set.
Overall, 26 patients with associated SS (35.6%) and 298 patients without associated SS (32.5%) receiving epratuzumab, and 16 patients with associated SS (40.0%) and 150 patients without associated SS (32.8%) receiving placebo discontinued before the end of the study (data are available upon request from the corresponding author). The most common reasons for discontinuation were AEs and lack of efficacy. Patient characteristics, patient clinical features, and the proportion of patients receiving controlled medications at baseline were similar for both patients with associated SS and patients without associated SS (Table 1), although small differences were apparent in sex, age, and serologic profiles.
Table 1.
Baseline demographic and disease characteristics of patients with SLE with and those without associated SSa
| Non‐SS patients | SS patientsb | |||||
|---|---|---|---|---|---|---|
| Placebo (n = 443) | Epratuzumab 600 mg (n = 450) | Epratuzumab 1,200 mg (n = 436) | Placebo (n = 40) | Epratuzumab 600 mg (n = 31) | Epratuzumab 1,200 mg (n = 41) | |
| Age, mean ± SD years | 40.7 ± 12.4 | 41.4 ± 12.0 | 41.0 ± 11.5 | 44.3 ± 11.9 | 46.4 ± 12.3 | 47.2 ± 11.0 |
| Sex, no. (%) female | 414 (93.5) | 412 (91.6) | 406 (93.1) | 40 (100) | 30 (96.8) | 41 (100) |
| Weight, mean ± SD kg | 72.3 ± 19.9 | 71.6 ± 18.7 | 73.2 ± 19.5 | 68.2 ± 16.7 | 72.2 ± 26.6 | 69.1 ± 15.3 |
| BMI, mean ± SD kg/m2 | 27.1 ± 7.3 | 26.7 ± 6.9 | 27.2 ± 7.0 | 25.5 ± 6.0 | 26.8 ± 8.8 | 25.9 ± 5.0 |
| Time since SS diagnosis, median (range) years | – | – | – | 7.5 (0–2.6) | 5.1 (0–34) | 5.3 (0–32) |
| Time since SLE diagnosis, median (range) years | 5.7 (0–37) | 5.5 (0–43) | 6.0 (0–34) | 7.3 (0–33) | 7.7 (0–40) | 6.7 (0–29) |
| SLEDAI total score, median (range) | 10 (4–28) | 10 (4–25) | 10 (4–27) | 10 (4–22) | 10 (4–22) | 9 (6–21) |
| Physician's global assessment of disease activity, median (range) | 55 (15–87) | 57 (12–100) | 56 (13–92) | 54 (15–79) | 58 (25–99) | 58 (27–91) |
| Patient's global assessment of disease activity, median (range) | 60 (3–100) | 62 (5–100) | 62 (0–100) | 60 (5–83) | 56 (3–86) | 59 (0–95) |
| BILAG total score, median (range)c | 20 (2–60) | 19.5 (9–45) | 20 (9–60) | 20 (12–37) | 20 (13–42) | 20 (12–56) |
| Medication use at baseline, no. (%) | ||||||
| Immunosuppressants | 204 (46.0) | 208 (46.2) | 206 (47.2) | 18 (45.0) | 18 (58.1) | 21 (51.2) |
| Antimalarials | 296 (66.8) | 317 (70.4) | 294 (67.4) | 23 (57.5) | 20 (64.5) | 25 (61.0) |
| Antimalarials and immunosuppressants | 134 (30.2) | 142 (31.6) | 140 (32.1) | 10 (25.0) | 11 (35.5) | 17 (41.5) |
| Corticosteroids | 411 (92.8) | 434 (96.4) | 417 (95.6) | 35 (87.5) | 29 (93.5) | 37 (90.2) |
| ANA titer >1:80, no. (%) | 382 (86.2) | 395 (87.8) | 382 (87.6) | 40 (100) | 29 (93.5) | 39 (95.1) |
| Anti‐dsDNA positive, no. (%) | 245 (55.3) | 229 (50.9) | 226 (51.8) | 21 (52.5) | 20 (64.5) | 12 (29.3) |
| Low complement, no. (%)d | 207 (46.7) | 212 (47.1) | 196 (45.0) | 24 (60.0) | 14 (45.2) | 19 (46.3) |
| Anti‐Sm positive, no. (%) | 115 (26.1) | 107 (23.9) | 119 (27.5) | 12 (30.0) | 10 (32.3) | 6 (14.6) |
| Anti‐RNP positive, no. (%) | 123 (28.0) | 129 (28.9) | 133 (30.7) | 13 (32.5) | 11 (35.5) | 8 (19.5) |
| Anti‐SSA positive, no. (%)e | 200 (45.5) | 198 (44.3) | 203 (46.9) | 40 (100) | 31 (100) | 41 (100) |
| Anti‐SSB positive, no. (%) | 78 (17.7) | 86 (19.2) | 77 (17.8) | 23 (57.5) | 19 (61.3) | 22 (53.7) |
| CRP, mean ± SD mg/liter | 8.6 ± 12.9 | 8.0 ± 10.6 | 8.4 ± 15.7 | 3.9 ± 4.0 | 8.5 ± 13.2 | 5.5 ± 6.7 |
Analyses were conducted using the full analysis set. Data were not available for all patients. Epratuzumab 600 mg was administered weekly, and epratuzumab 1,200 mg was administered every other week. Both dosing regimens were repeated for 4 weeks every 12 weeks. BMI = body mass index; SLEDAI = Systemic Lupus Erythematosus Disease Activity Index; BILAG = British Isles Lupus Assessment Group; ANA = antinuclear antibody; anti‐dsDNA = anti–double‐stranded DNA; CRP = C‐reactive protein.
Systemic lupus erythematosus (SLE) patients with a diagnosis of Sjögren's syndrome (SS) according to their medical history who were also anti‐SSA positive.
Generated by converting A, B, C, D, and E to 12, 8, 1, 0, and 0, respectively.
Defined as a C3 level of <0.9 gm/liter or a C4 level of <180 mg/liter.
Defined according to the rules of the central laboratory, which may have varied across treatment regions.
Clinical response . The primary analysis of the EMBODY trials revealed no significant difference between the epratuzumab and placebo treatment groups in BICLA response rate, change from baseline in BILAG total score, or PGA at week 48 21.
Consistent with these results, SLE patients without associated SS receiving epratuzumab failed to demonstrate a BICLA response compared with placebo for the primary end point at week 24. Differences in BICLA response compared with placebo were only observed at weeks 12, 16, and 20 for patients receiving 600 mg every week, and no difference was observed at any time point for patients receiving 1,200 mg every other week (Figure 1A). In contrast, a rapid BICLA response was observed for SLE patients with associated SS who were anti‐SSA positive. By week 16, BICLA response was greater in patients receiving epratuzumab than in those receiving placebo (P < 0.05). This response was maintained through week 48 (Figure 1E).
Figure 1.
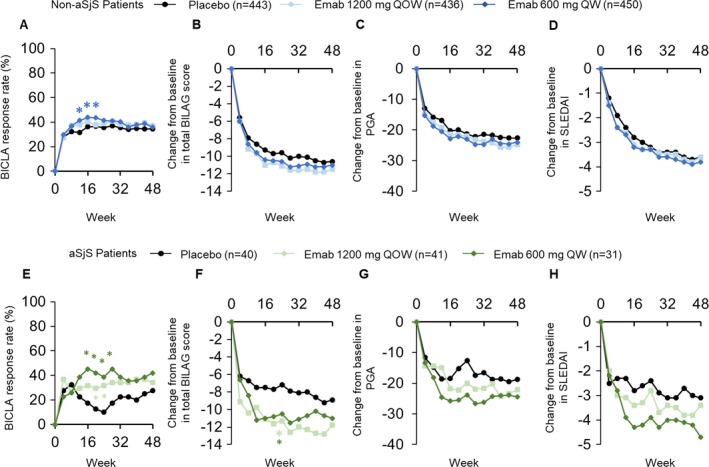
A–D, British Isles Lupus Assessment Group (BILAG)–based Combined Lupus Assessment (BICLA) response rate (A), change in total BILAG score from baseline (B), change in physician's global assessment (PGA) from baseline (C), and change in Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) from baseline (D) in SLE patients without associated Sjögren's syndrome (SS; non‐aSjS patients) receiving placebo or epratuzumab (Emab). E–H, BICLA response rate (E), change in total BILAG score from baseline (F), change in PGA from baseline (G), and change in SLEDAI from baseline (H) in SLE patients with associated SS receiving placebo or epratuzumab. Analyses were conducted using the full analysis set. Values are the median percent change from baseline. For total BILAG score, PGA, and SLEDAI, statistical testing was conducted only at weeks 24 and 48. Nonresponder imputation was used for the BICLA response. The last observation carried forward method was used to impute missing values for the total BILAG score, PGA, and SLEDAI. Light asterisk = P < 0.05 for epratuzumab 1,200 mg every other week (QOW) versus placebo; dark asterisk = P < 0.05 for epratuzumab 600 mg every week (QW) versus placebo.
Similar to the BICLA response rate, only a small difference was observed in change in total BILAG score from baseline in patients without associated SS receiving epratuzumab versus those receiving placebo (Figure 1B). However, differences were observed in SLE patients with associated SS. By week 24, patients receiving either epratuzumab dosing regimen reported greater reductions in total BILAG score compared with placebo (P < 0.05) (Figure 1F). No significant differences, but a trend in favor of epratuzumab response, were observed for both PGA response rate and SLEDAI change from baseline in both SLE patients without associated SS and those with associated SS receiving epratuzumab versus placebo (Figures 1C, D, G, and H).
At baseline, serum C‐reactive protein (CRP) concentration was higher in the patients without associated SS (Table 1). To assess whether baseline CRP levels had an effect on the clinical efficacy measurements, the total EMBODY population was reanalyzed according to CRP levels at baseline, using the median value (3.0 mg/liter [range 0.12–182.09]) as a cutoff (data available upon request from the corresponding author). Compared to placebo, changes from baseline in total BILAG score, BICLA response, PGA, and SLEDAI were greater for patients with a CRP concentration higher than the median value.
Immunologic response. By week 48 in the EMBODY trials, a reduction in B cell count from baseline of between 30% and 40% was observed in SLE patients receiving epratuzumab. Consistent with these results, a reduction in CD19+ B cell count of between 30% and 40% was observed in both patients without associated SS and patients with associated SS receiving epratuzumab, but not those receiving placebo (Figures 2A and B). No difference was seen between patients with associated SS and those without associated SS.
Figure 2.
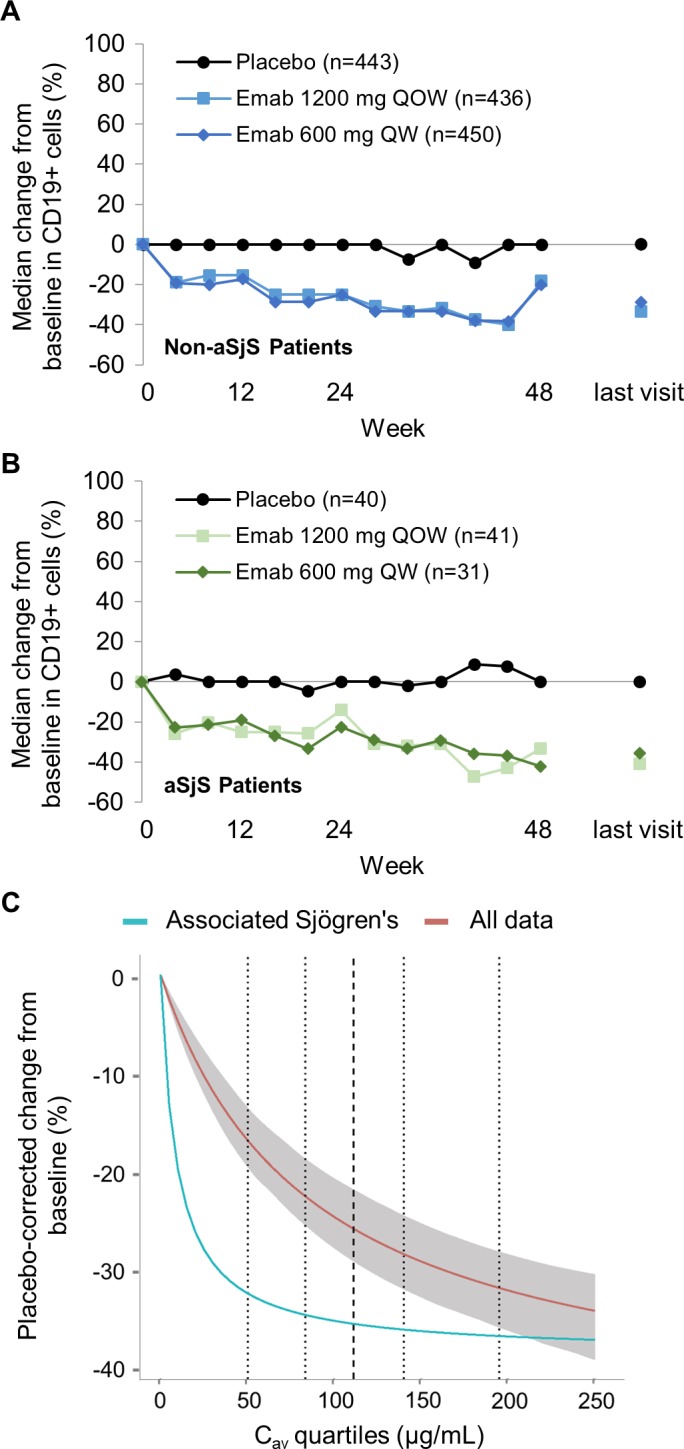
A and B, Percentage change from baseline in CD19+ cells from patients with systemic lupus erythematosus (SLE) without associated Sjögren's syndrome (SS; non‐aSjS patients) (A) and patients with SLE with associated SS (B) receiving placebo or epratuzumab (Emab). C, Pharmacokinetics/pharmacodynamics modeling of B cell reduction following intravenous epratuzumab. Dotted lines represent the 5th, 25th, 75th, and 95th percentiles, respectively, of the observed epratuzumab average steady‐state plasma drug concentration during multiple‐dose administration; broken line represents the 50th percentile. The shaded area shows the 95% confidence interval based on 500 bootstrap sampling with replacement available for all the “All data” model only. QOW = every other week; QW = every week. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.40425/abstract.
PK/PD modeling was used to compare the effect of the serum epratuzumab concentration on CD19+ B cell count at week 48 (Figure 2C). B cell counts were reduced by ~30% of baseline values in both the entire SLE population and the patients with associated SS. However, the reduction was faster in the population with associated SS, and the sensitivity to epratuzumab, as measured by the EC50, was substantially lower for patients with associated SS receiving epratuzumab (9.5 μg/ml) compared with the total EMBODY population (87.1 μg/ml).
Reductions in IgM levels were greater in patients receiving epratuzumab compared with those receiving placebo and were similar for patients with and those without associated SS (Figure 3). By week 48 in both groups, median percentage changes from baseline of ~20% were observed for patients receiving epratuzumab compared with no change in patients receiving placebo (Figures 3A and C). No changes in either IgG (Figures 3B and D) or IgA (data not shown) were observed over the 48 weeks in any group.
Figure 3.
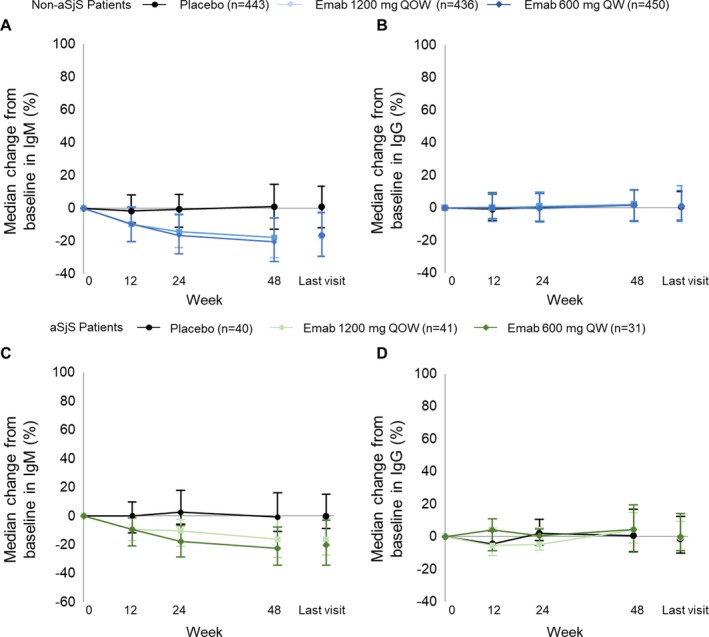
A and B, Median percent change from baseline in IgM (A) and IgG (B) serum concentrations in patients with systemic lupus erythematosus (SLE) without associated Sjögren's syndrome (SS; non‐aSjS) receiving placebo or epratuzumab (Emab). C and D, Median percent change from baseline in IgM (C) and IgG (D) serum concentrations in patients with SLE with associated SS receiving placebo or epratuzumab. Analyses were conducted using the full analysis set and observed data. Values are the median (25th–75th percentile) percent change from baseline. QOW = every other week; QW = every week. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.40425/abstract.
Serum concentration of anti‐SSA decreased from baseline through week 48 in patients with associated SS and was greater in patients receiving either dose of epratuzumab (−51.24 units/ml and −84.45 units/ml) versus placebo (−26.83 units/ml). However, no changes were observed in patients without associated SS (Figures 4A and C) (data are available upon request from the corresponding author). No significant differences in anti‐SSB titers between treatment groups were observed for either population (Figures 4B and D).
Figure 4.
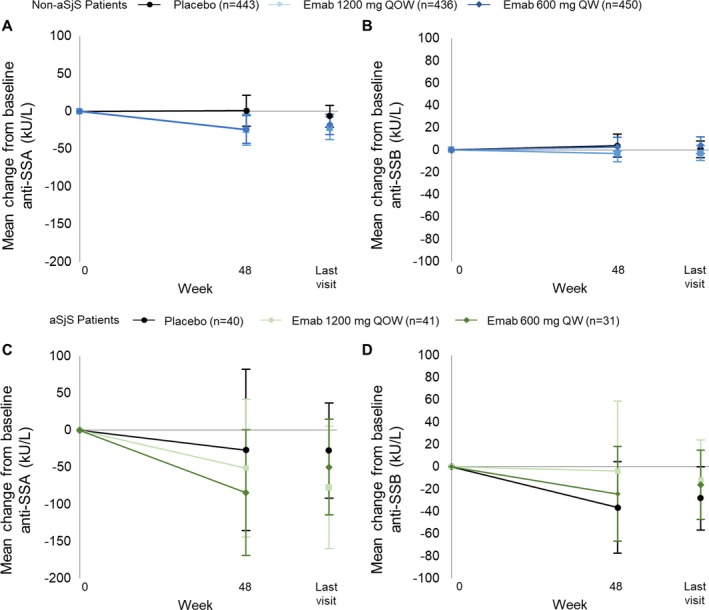
A and B, Mean change from baseline in anti‐SSA (A) and anti‐SSB (B) in patients with systemic lupus erythematosus (SLE) without associated Sjögren's syndrome (SS; non‐aSjS) receiving placebo or epratuzumab (Emab). C and D, Mean change from baseline in anti‐SSA (C) and anti‐SSB (D) in patients with SLE with associated SS receiving placebo or epratuzumab. Analyses were conducted using the full analysis set and observed data. Values are the mean (95% confidence interval) change from baseline. QOW = every other week; QW = every week. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.40425/abstract.
Subgroup analyses. To discern whether anti‐SSA positivity contributed to the clinical responses observed above, all analyses performed for this study were repeated for anti‐SSA–positive patients without associated SS and anti‐SSA–negative patients without associated SS. At baseline, demographic and disease characteristics were similar between the groups (data are available upon request from the corresponding author).
Changes from baseline in total BILAG score, PGA, SLEDAI, CD19+ cell count, IgM, IgG, and anti‐SSB at each study visit were similar for both patient populations and consistent with the results reported above (data are available upon request from the corresponding author). Differences between the 2 patient populations were only observed for BICLA response rate, where there was a greater difference between the epratuzumab and the placebo arms for anti‐SSA–positive patients without associated SS than anti‐SSA–negative patients without associated SS. However, the magnitude of the difference between the treatment and placebo arms was similar to the difference observed in the original population of patients without associated SS.
Safety. In this subpopulation of SLE patients with a diagnosis of associated SS, the incidence of AEs was comparable between the placebo and epratuzumab treatment groups, and was similar to the incidence of treatment‐emergent AEs reported by non‐SS patients (Table 2). Treatment‐emergent AEs occurred in 61 patients receiving epratuzumab (84.7%) and 33 patients receiving placebo (82.5%). Serious AEs occurred in 15 patients receiving epratuzumab (20.8%) and 7 patients receiving placebo (17.5%). There was 1 death in the associated SS subpopulation; this was in the placebo treatment group, and therefore unrelated to the study drug. The death was caused by pneumonia.
Table 2.
Summary of treatment‐emergent AEsa
| Non‐SS patients | SS patients | |||||
|---|---|---|---|---|---|---|
| Placebo (n = 443) | Epratuzumab 600 mg (n = 450) | Epratuzumab 1,200 mg (n = 436) | Placebo (n = 40) | Epratuzumab 600 mg (n = 31) | Epratuzumab 1,200 mg (n = 41) | |
| Any treatment‐emergent AE | 374 (84.4) | 374 (83.1) | 377 (86.5) | 33 (82.5) | 28 (90.3) | 33 (80.5) |
| Serious treatment‐emergent AEs | 75 (16.9) | 77 (17.1) | 75 (17.2) | 7 (17.5) | 7 (22.6) | 8 (19.5) |
| Discontinuation due to treatment‐emergent AEs | 33 (7.4) | 22 (4.9) | 35 (8.0) | 3 (7.5) | 4 (12.9) | 1 (2.4) |
| Infusion reaction treatment‐emergent AEs | 32 (7.2) | 31 (6.9) | 49 (11.2) | 2 (5.0) | 4 (12.9) | 5 (12.2) |
| Drug‐related treatment‐emergent AEs | 126 (28.4) | 131 (29.1) | 152 (34.9) | 13 (32.5) | 9 (29.0) | 13 (31.7) |
| Severe treatment‐emergent AEs | 63 (14.2) | 62 (13.8) | 61 (14.0) | 5 (12.5) | 5 (16.1) | 8 (19.5) |
| All deaths | 3 (0.7) | 1 (0.2) | 3 (0.7) | 1 (2.5) | 0 (0.0) | 0 (0.0) |
Analyses were conducted using the full analysis set. Values are the number (%). AEs = adverse events; SS = Sjögren's syndrome.
Discussion
In this post hoc analysis of the EMBODY phase III trials, treatment with epratuzumab in addition to standard therapies resulted in improved SLE‐specific clinical outcomes at week 48 compared with placebo in patients with a diagnosis of associated SS. Similar to the published EMBODY results 21, no improvements in SLE‐specific clinical outcomes at week 48 were observed in patients without a diagnosis of associated SS.
The mechanism of action driving the different responses in the 2 study populations is not known. However, since epratuzumab reduces B cell activity and the number of B cells in peripheral blood targets, differences in B cell response to epratuzumab may lead to different SLE‐specific clinical outcomes at week 48. To investigate this, changes in B cell numbers and immunoglobulin (IgM, IgG, and IgA) concentrations were compared. Interestingly, despite the different responses observed in SLE‐specific clinical outcomes, no differences were observed in changes from baseline in either immunoglobulin concentration or B cell count. Exposure‐response analysis for B cell reduction modeling over placebo was used to further compare the effect of epratuzumab on B cells in the 2 patient populations. Differences were observed that suggested that B cells from patients with associated SS respond more quickly to epratuzumab than B cells from patients without associated SS.
The majority of the baseline demographics of the patients with associated SS were similar to those of the wider SLE population enrolled in the trial. However, there were differences. Mean patient age and the proportion of women were both higher in the group with associated SS. Serologic differences were also observed; CRP was lower and anti‐SSB was higher in the associated SS population, while anti‐ANA and anti‐Sm were lower. Analyses investigating whether CRP level correlates with treatment outcomes were performed by grouping patients according to high and low CRP levels using the median value as a cutoff. These analyses suggested that the efficacy of epratuzumab may be higher in patients with high CRP levels at baseline in the original study. However, although baseline CRP values were higher in the patients without associated SS than in the patients with associated SS, measures of epratuzumab efficacy were lower in these patients without SS. These results suggest that CRP does not represent a helpful biomarker in identifying patients who may respond to epratuzumab.
There is published evidence of differences in impaired B cell regulation in Sjögren's patients compared with SLE patients which may contribute to the clinical response achieved by patients with associated SS. Although SLE and associated SS are both characterized by hyperactivated B cells, in SS, exocrine glands are the primary target for the disease, which is thought to be mediated by an antigen‐driven, germinal center–type B cell response 29, 30. Taken together, these data suggest that the pathophysiology of associated SS may differ from that in the wider SLE population. If these differences provide a better target for epratuzumab, this may explain why clinical efficacy and the exposure‐response analysis for B cell reduction modeling over placebo were only observed in patients with associated SS, despite similar B cell reductions being observed in both populations.
To date, no clinical trials have prospectively assessed the efficacy of B cell modulators in patients with SLE and an associated diagnosis of SS. The few trials that have investigated B cell modulators as a potential therapy for SS have assessed their use in patients with primary SS.
The efficacy of rituximab, an anti‐CD20 monoclonal antibody, in patients with primary SS has been assessed in several trials. In 2 phase II trials, rituximab reduced the signs and symptoms of the disease, improving salivary production, health‐related quality of life, and measures of fatigue 31, 32. Similar results were observed in the Tolerance and Efficacy of Rituximab in Primary Sjögren's Syndrome (TEARS) trial 33. This trial enrolled 120 patients with primary SS. Although it failed its primary end point, the trial demonstrated early clinical improvements and improvements in patient‐reported outcomes. The Trial of Anti–B Cell Therapy in Patients with Primary Sjögren's Syndrome (TRACTISS), a multicenter, randomized, double‐blind, parallel‐group placebo‐controlled trial, was designed to further assess the potential benefits of rituximab in SS patients 34. This study also failed to meet its clinical end point; the only outcome measure with a significant improvement was unstimulated saliva flow. This might be related to the incomplete depletion of salivary autoreactive B cells, the absence of targeting of pathogenic long‐lived plasma cells, and the effect of rituximab on regulatory (protective) B cells.
The Belimumab in Sjögren's Syndrome (BELISS) study was an open‐label trial assessing the efficacy and safety of belimumab, an anti–B lymphocyte stimulator monoclonal antibody, in primary SS. The trial enrolled 30 patients and achieved its week 28 primary end point of improvement in 2 of 5 items: reduction of ≥30% in dryness score on a VAS, ≥30% in fatigue VAS score, ≥30% in VAS pain score, ≥30% in systemic activity VAS assessed by the physician and/or >25% improvement in any B cell activation biomarker values 35. Based on these criteria, 60% of patients receiving belimumab achieved a clinical response.
Similar effects were observed in the only open trial to date assessing the efficacy of epratuzumab in primary SS, although caution is required when interpreting the results because that study included only 16 patients 23. A reduction of <30% in total B cell count was maintained throughout the 32‐week trial, and 53% of the patients achieved at least a 20% improvement in 2 of the following 4 domains: dryness of the eyes (Schirmer I test), dryness of the mouth, fatigue (VAS), and erythrocyte sedimentation rate and/or IgG level. The results of that study, in conjunction with the results from the BELISS and TEARS trials, suggest that anti–B cell therapies may be effective in the treatment of associated SS or SLE patients with anti‐SSA antibodies.
There were several limitations to this study. The EMBODY program was designed to assess the clinical efficacy and safety of epratuzumab in SLE patients, and so no associated SS–specific clinical data were collected. Patients with associated SS were identified from a clinician's identification and diagnosis of the disease, and so may not be accurate (since no formal objective assessment of tear, salivary flow, or salivary gland biopsy was performed or assessed according to the American‐European Consensus Group proposed definition for associated SS 36). Consequently, patients may have been incorrectly categorized as not having associated SS. This could explain why BICLA response was higher in anti‐SSA–positive patients without associated SS, since some patients might have been diagnosed as having associated SS if they had been fully assessed; however, the additional selection criterion of a positive anti‐SSA titer was included for a more defined SS patient population. The population size assessed, although relatively large for a Sjögren's study, included only 112 patients in the full analysis set. The efficacy and safety of epratuzumab in a larger patient population with SS‐specific outcome measures now needs to be assessed to determine if epratuzumab would be helpful for treating patients with SS. In addition, other studies could be considered to determine whether lupus patients with anti‐SSA (and/or anti‐SSB) antibodies are more likely to respond to epratuzumab and other B cell–targeted therapies than those with other serologic phenotypes.
In conclusion, patients with SLE and associated SS treated with epratuzumab showed improvements in SLE disease activity compared with patients treated with placebo, which was not observed in patients without associated SS. B cell numbers, IgM levels, and anti‐SSA levels were consistently reduced in all patient populations receiving epratuzumab; however, B cell reduction was faster in the associated SS patient population. These data suggest that epratuzumab may have clinical benefits in certain subsets of SLE patients and so stratification of SLE patients may be appropriate. Further trials examining the effectiveness of epratuzumab in primary SS are needed to confirm the effectiveness of epratuzumab in this population.
Author contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Gottenberg had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Gottenberg, Dörner, Bootsma, Devauchelle‐Pensec, Bowman, Mariette, Bartz, Oortgiesen, Shock, Koetse, Galateanu, Bongardt, Wegener, Goldenberg, Meno‐Tetang, Kosutic, Gordon.
Acquisition of data
Gottenberg, Dörner, Bootsma, Devauchelle‐Pensec, Mariette, Gordon.
Analysis and interpretation of data
Gottenberg, Dörner, Bootsma, Devauchelle‐Pensec, Bowman, Mariette, Bartz, Oortgiesen, Shock, Koetse, Galateanu, Bongardt, Wegener, Goldenberg, Meno‐Tetang, Kosutic, Gordon.
Role of the study sponsor
UCB Pharma designed and sponsored the randomized trials from which data were extracted for the present post hoc analysis. UCB Pharma also supported the analysis and interpretation of the data. UCB Pharma had no role in the writing of the manuscript or the decision to submit the manuscript for publication. Medical writing and editorial assistance was provided by Simon Foulcer, PhD (Costello Medical, Cambridge, UK), based on the authors' input and direction. Publication of this article was not contingent upon approval by UCB Pharma.
Additional disclosures
Authors Wegener and Goldenberg are employees of Immunomedics.
Acknowledgments
The authors thank the patients and their caregivers in addition to the investigators and their teams who contributed to this study.
ClinicalTrials.gov identifiers: NCT01262365; NCT01261793.
Supported by UCB Pharma.
Dr. Dörner has received research support from UCB Pharma. Dr. Mariette has received consulting fees from Biogen, Bristol‐Myers Squibb, GlaxoSmithKline, LFB, Pfizer, and UCB Pharma (less than $10,000 each) and research support from Biogen, Pfizer, and UCB Pharma. Mr. Koetse and Dr. Kosutic own stock or stock options in UCB Pharma. Dr. Gordon has received consulting fees, speaking fees, and/or honoraria from GlaxoSmithKline, MedImmune, Merck Serono, Parexel, and UCB Pharma (less than $10,000 each).
References
- 1. Fox RI. Sjogren's syndrome. Lancet 2005;366:321–31. [DOI] [PubMed] [Google Scholar]
- 2. Youinou P, Devauchelle‐Pensec V, Pers JO. Significance of B cells and B cell clonality in Sjögren's syndrome. Arthritis Rheum 2010;62:2605–10. [DOI] [PubMed] [Google Scholar]
- 3. Salomonsson S, Jonsson MV, Skarstein K, Brokstad KA, Hjelmström P, Wahren‐Herlenius M, et al. Cellular basis of ectopic germinal center formation and autoantibody production in the target organ of patients with Sjögren's syndrome. Arthritis Rheum 2003;48:3187–201. [DOI] [PubMed] [Google Scholar]
- 4. Risselada AP, Looije MF, Kruize AA, Bijlsma JW, van Roon JA. The role of ectopic germinal centers in the immunopathology of primary Sjögren's syndrome: a systematic review. Semin Arthritis Rheum 2013;42:368–76. [DOI] [PubMed] [Google Scholar]
- 5. Ambrus JL, Suresh L, Peck A. Multiple roles for B‐lymphocytes in Sjögren's syndrome. J Clin Med 2016;5:E87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Manoussakis MN, Georgopoulou C, Zintzaras E, Spyropoulou M, Stavropoulou A, Skopouli FN, et al. Sjögren's syndrome associated with systemic lupus erythematosus: clinical and laboratory profiles and comparison with primary Sjögren's syndrome. Arthritis Rheum 2004;50:882–91. [DOI] [PubMed] [Google Scholar]
- 7. Fox PC. Autoimmune diseases and Sjögren's syndrome. Ann N Y Acad Sci 2007;1098:15–21. [DOI] [PubMed] [Google Scholar]
- 8. Patel R, Shahane A. The epidemiology of Sjögren's syndrome. Clin Epidemiol 2014;6:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choi J, Kim ST, Craft J. The pathogenesis of systemic lupus erythematosus: an update. Curr Opin Immunol 2012;24:651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mariette X, Gottenberg JE. Pathogenesis of Sjögren's syndrome and therapeutic consequences. Curr Opin Rheumatol 2010;22:471–7. [DOI] [PubMed] [Google Scholar]
- 11. Looney RJ, Anolik JH, Campbell D, Felgar RE, Young F, Arend LJ, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose‐escalation trial of rituximab. Arthritis Rheum 2004;50:2580–9. [DOI] [PubMed] [Google Scholar]
- 12. Shirota Y, Yarboro C, Fischer R, Pham TH, Lipsky P, Illei GG. Impact of anti‐interleukin‐6 receptor blockade on circulating T and B cell subsets in patients with systemic lupus erythematosus. Ann Rheum Dis 2013;72:118–28. [DOI] [PubMed] [Google Scholar]
- 13. Stohl W, Hiepe F, Latinis KM, Thomas M, Scheinberg MA, Clarke A, et al. Belimumab reduces autoantibodies, normalizes low complement levels, and reduces select B cell populations in patients with systemic lupus erythematosus. Arthritis Rheum 2012;64:2328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lumb S, Fleischer SJ, Wiedemann A, Daridon C, Maloney A, Shock A, et al. Engagement of CD22 on B cells with the monoclonal antibody epratuzumab stimulates the phosphorylation of upstream inhibitory signals of the B cell receptor. J Cell Commun Signal 2016;10:143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carnahan J, Wang P, Kendall R, Chen C, Hu S, Boone T, et al. Epratuzumab, a humanized monoclonal antibody targeting CD22: characterization of in vitro properties. Clin Cancer Res 2003;9:3982s–90s. [PubMed] [Google Scholar]
- 16. Rossi D, Spina V, Bomben R, Rasi S, Dal‐Bo M, Bruscaggin A, et al. Association between molecular lesions and specific B‐cell receptor subsets in chronic lymphocytic leukemia. Blood 2013;121:4902–5. [DOI] [PubMed] [Google Scholar]
- 17. Fleischer V, Sieber J, Fleischer SJ, Shock A, Heine G, Daridon C, et al. Epratuzumab inhibits the production of the proinflammatory cytokines IL‐6 and TNF‐α, but not the regulatory cytokine IL‐10, by B cells from healthy donors and SLE patients. Arthritis Res Ther 2015;17:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jacobi AM, Goldenberg DM, Hiepe F, Radbruch A, Burmester GR, Dorner T. Differential effects of epratuzumab on peripheral blood B cells of patients with systemic lupus erythematosus versus normal controls. Ann Rheum Dis 2008;67:450–7. [DOI] [PubMed] [Google Scholar]
- 19. Wallace DJ, Kalunian K, Petri MA, Strand V, Houssiau FA, Pike M, et al. Efficacy and safety of epratuzumab in patients with moderate/severe active systemic lupus erythematosus: results from EMBLEM, a phase IIb, randomised, double‐blind, placebo‐controlled, multicentre study. Ann Rheum Dis 2014;73:183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wallace DJ, Hobbs K, Clowse ME, Petri M, Strand V, Pike M, et al. Long‐term safety and efficacy of epratuzumab in the treatment of moderate‐to‐severe systemic lupus erythematosus: results from an open‐label extension study. Arthritis Care Res (Hoboken) 2016;68:534–43. [DOI] [PubMed] [Google Scholar]
- 21. Clowse ME, Wallace DJ, Furie RA, Petri MA, Pike MC, Leszczyński P, et al. Efficacy and safety of epratuzumab in moderately to severely active systemic lupus erythematosus: results from two phase III, randomized, double‐blind, placebo‐controlled trials. Arthritis Rheumatol 2017;69:362–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wallace DJ, Strand V, Furie R, Petri M, Kalunian K, Pike M, et al. Evaluation of treatment success in systemic lupus erythematosus clinical trials: development of the British Isles Lupus Assessment Group‐Based Composite Lupus Assessment endpoint [abstract]. Arthritis Rheum 2011;63 Suppl:S885. [Google Scholar]
- 23. Steinfeld SD, Tant L, Burmester GR, Teoh NK, Wegener WA, Goldenberg DM, et al. Epratuzumab (humanised anti‐CD22 antibody) in primary Sjogren's syndrome: an open‐label phase I/II study. Arthritis Res Ther 2006;8:R129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hochberg MC, for the Diagnostic and Therapeutic Criteria Committee of the American College of Rheumatology . Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997;40:1725. [DOI] [PubMed] [Google Scholar]
- 25. Yee CS, Farewell V, Isenberg DA, Griffiths B, Teh LS, Bruce IN, et al. The BILAG‐2004 index is sensitive to change for assessment of SLE disease activity. Rheumatology (Oxford) 2009;48:691–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Isenberg DA, Rahman A, Allen E, Farewell V, Akil M, Bruce IN, et al. BILAG 2004: development and initial validation of an updated version of the British Isles Lupus Assessment Group's disease activity index for patients with systemic lupus erythematosus. Rheumatology (Oxford) 2005;44:902–6. [DOI] [PubMed] [Google Scholar]
- 27. Gladman DD, Ibanez D, Urowitz MB. Systemic Lupus Erythematosus Disease Activity Index 2000. J Rheumatol 2002;29:288–91. [PubMed] [Google Scholar]
- 28. Yee CS, Cresswell L, Farewell V, Rahman A, Teh LS, Griffiths B, et al. Numerical scoring for the BILAG‐2004 index. Rheumatology (Oxford) 2010;49:1665–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hansen A, Lipsky PE, Dorner T. B cells in Sjogren's syndrome: indications for disturbed selection and differentiation in ectopic lymphoid tissue. Arthritis Res Ther 2007;9:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ambrosi A, Wahren‐Herlenius M. Update on the immunobiology of Sjogren's syndrome. Curr Opin Rheumatol 2015;27:468–75. [DOI] [PubMed] [Google Scholar]
- 31. Dass S, Bowman SJ, Vital EM, Ikeda K, Pease CT, Hamburger J, et al. Reduction of fatigue in Sjogren syndrome with rituximab: results of a randomised, double‐blind, placebo‐controlled pilot study. Ann Rheum Dis 2008;67:1541–4. [DOI] [PubMed] [Google Scholar]
- 32. Pijpe J, van Imhoff GW, Spijkervet FK, Roodenburg JL, Wolbink GJ, Mansour K, et al. Rituximab treatment in patients with primary Sjögren's syndrome: an open‐label phase II study. Arthritis Rheum 2005;52:2740–50. [DOI] [PubMed] [Google Scholar]
- 33. Devauchelle‐Pensec V, Mariette X, Jousse‐Joulin S, Berthelot JM, Perdriger A, Puechal X, et al. Treatment of primary Sjogren syndrome with rituximab: a randomized trial. Ann Intern Med 2014;160:233–42. [DOI] [PubMed] [Google Scholar]
- 34. Bowman SJ, Everett CC, O'Dwyer JL, Emery P, Pitzalis C, Ng WF, et al. Randomized controlled trial of rituximab and cost‐effectiveness analysis in treating fatigue and oral dryness in primary Sjögren's syndrome. Arthritis Rheumatol 2017;69:1440–50. [DOI] [PubMed] [Google Scholar]
- 35. Mariette X, Seror R, Quartuccio L, Baron G, Salvin S, Fabris M, et al. Efficacy and safety of belimumab in primary Sjogren's syndrome: results of the BELISS open‐label phase II study. Ann Rheum Dis 2015;74:526–31. [DOI] [PubMed] [Google Scholar]
- 36. Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, et al, and the European Study Group on Classification Criteria for Sjögren's Syndrome . Classification criteria for Sjögren's syndrome: a revised version of the European criteria proposed by the American‐European Consensus Group. Ann Rheum Dis 2002;61:554–8. [DOI] [PMC free article] [PubMed] [Google Scholar]