

Abstract
Cystatin C (CysC) is a major protein component of Bunina bodies, which are a pathological hallmark observed in the remaining motor neurons of patients with amyotrophic lateral sclerosis (ALS). Dominant mutations in the SOD1 gene, encoding Cu/Zn superoxide dismutase (SOD1), are causative for a subset of inherited ALS cases. Our previous study showed that CysC exerts a neuroprotective effect against mutant SOD1‐mediated toxicity in vitro; however, in vivo evidence of the beneficial effects mediated by CysC remains obscure. Here we examined the therapeutic potential of recombinant human CysC in vivo using a mouse model of ALS in which the ALS‐linked mutated SOD1 gene is expressed (SOD1G93A mice). Intracerebroventricular administration of CysC during the early symptomatic SOD1G93A mice extended their survival times. Administered CysC was predominantly distributed in ventral horn neurons including motor neurons, and induced autophagy through AMP‐activated kinase activation to reduce the amount of insoluble mutant SOD1 species. Moreover, PGC‐1α, a disease modifier of ALS, was restored by CysC through AMP‐activated kinase activation. Finally, the administration of CysC also promoted aggregation of CysC in motor neurons, which is similar to Bunina bodies. Taken together, our findings suggest that CysC represents a promising therapeutic candidate for ALS.
Keywords: Amyotrophic lateral sclerosis (ALS), Cu/Zn superoxide dismutase (SOD1), Cystatin C (CysC), AMP‐activated kinase (AMPK)
Abbreviations used
- ALS
amyotrophic lateral sclerosis
- AMPK
AMP‐activated kinase
- CC
compound C
- CysC
cystatin C
- DMEM
Dulbecco's modified Eagle's medium
- FBS
fetal bovine serum
- GFAP
glial fibrillary acidic protein
- HA‐tag
influenza hemagglutinin‐tag
- HSP70
heat‐shock protein 70
- PBS
phosphate‐buffered saline
- PGC‐1α
peroxisome proliferator‐activated receptor gamma coactivator 1‐α
- poly‐Ubi
poly‐ubiquitin
- SIRT1
sirtuin 1
- SNP
single‐nucleotide polymorphism
- SOD1
Cu/Zn superoxide dismutase
- TDP‐43
TAR DNA binding protein‐43
Impairment of protein quality control systems is deeply involved in the pathology of neurodegenerative diseases including amyotrophic lateral sclerosis (ALS). ALS is an adult‐onset, fatal neurodegenerative disease that is characterized by the selective loss of upper and lower motor neurons. Although 90% of ALS cases are of sporadic etiology, approximately 10% are inherited and dominant mutations in the gene encoding Cu/Zn superoxide dismutase (SOD1) are frequent causes of the inherited form of ALS (Cleveland and Rothstein 2001). Transgenic rodents expressing the human SOD1 gene with ALS‐linked mutations have been widely used as animal models of the disease (Bendotti and Carri 2004; Bruijn et al. 2004). Mutant SOD1 proteins induce neurodegeneration through gain‐of‐toxicity mechanisms. Misfolded and consequently aggregated mutant SOD1 proteins have been tightly linked to the toxicity. Some studies also suggested involvement of accumulated, misfolded SOD1 species in sporadic ALS (Bosco et al. 2010; Gauthier et al. 2011; Guareschi et al. 2012). Therefore, a reduction in toxic SOD1 proteins is one of the viable therapeutic strategies for ALS.
Cystatin C (CysC) is an endogenous protease inhibitor expressed in various tissues (Abrahamson et al. 1986). CysC is also well characterized as a marker of the glomerular filtration rate to evaluate renal function, whereas some studies have also revealed its unique neuroprotective role in neurodegenerative diseases (Gauthier et al. 2011). In a mouse model of Alzheimer's disease, over‐expressed human CysC reduced deposition of amyloidal‐β fibrils (Mi et al. 2007). CysC also prolonged survival of dopaminergic neurons in a rat model of Parkinson's disease (Xu et al. 2005). In ALS, CysC is a major protein component of Bunina bodies, protein inclusions specifically observed in the remaining motor neurons of sporadic ALS (Okamoto et al. 1993), and a reduced concentration of CysC in cerebrospinal fluid is negatively correlated with the survival time of ALS patients (Wilson et al. 2010). We previously examined the potential neuroprotective properties of CysC against mutant SOD1‐linked neurotoxicity using cultured cells, and found that CysC exhibited neuroprotection via a combination of AMP‐activated kinase (AMPK) activation and cathepsin inhibition (Watanabe et al. 2014b). AMPK is a master regulator of intracellular energy metabolism, and regulates autophagic flux by phosphorylation of mammalian target of rapamycin. CysC promoted mutant SOD1 degradation through the activation of autophagy by AMPK, and at the same time, suppressed aberrant proteolysis by lysosomal cathepsins. CysC increased viability of mutant SOD1‐expressing primary motor neurons as well as neuroblastoma cell lines in vitro. These studies suggest that CysC is a promising neuroprotective agent for ALS. However, it is still uncertain whether CysC confers the beneficial effects in SOD1‐linked ALS models in vivo.
In this study, we aimed to examine the beneficial effects of CysC in vivo using transgenic mice carrying the ALS‐linked, mutated SOD1 gene (SOD1G93A mice). We found that intracerebroventricular administration of recombinant human CysC extended the lifespan of SOD1G93A mice, and that CysC conferred the beneficial effects through AMPK activation to induce autophagy and peroxisome proliferator‐activated receptor gamma coactivator 1‐α (PGC‐1α) expression. In addition, we also found that administered CysC formed detergent‐insoluble tetramers to form aggregates in motor neurons, suggesting that generation of Bunina bodies may be triggered by CysC tetramer formation.
Materials and methods
Animals
Transgenic mice expressing SOD1G93A (B6.Cg‐Tg(SOD1*G93A)1Gur/J) (RRID: IMSR_JAX:004435) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Genotyping for SOD1G93A mice was performed as previously described (Watanabe et al. 2014a). The SOD1G93A mice weighing more than 30g (in male) or 20g (in female) were used for the surgery described below. The times for disease end‐stage were determined as previously described (Watanabe et al. 2016). Briefly, the disease end‐stage was determined as the time when animals in a lateral position were unable to right themselves within 20 s, an endpoint commonly used for mutant SOD1 mice that is in compliance with the requirements of the Animal Use and Care Committee of Nagoya University. The mice were housed in the specific pathogen free environment (with a 12 h light‐dark cycle at 23 ± 1°C and 50 ± 5% humidity) and treated in compliance with the requirements of the Animal Care and Use Committee. The experiments using genetically modified animals and organisms were approved by the Animal Care and Use committee and the recombinant DNA experiment committee of Nagoya University (approval numbers #17240 and #143‐11, respectively). This study was not pre‐registered.
Antibodies
Antibodies used were as follows: anti‐phosphorylated AMPK (Thr172) (1:250, #sc‐33524, RRID: AB_2169714, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐LC3 (1:500, #NB100‐2331, RRID: AB_10001955, Novus Biologicals LLC, Littleton, CO, USA), anti‐CysC (1 : 500 for immunoblotting, 1 : 100 for immunohistochemistry, #ab109508, RRID: AB_10888303, Abcam, Cambridge, UK), anti‐β‐actin (1 : 5000, #5441, RRID: AB_476744, Sigma‐Aldrich Co LLC, St. Louis, MO, USA), anti‐influenza hemagglutinin‐tag (HA‐tag) (1 : 500, #11‐867‐423‐001, RRID: AB_10094468, Roche, Basel, Switzerland), anti‐p62 (for immunoblotting, 1 : 1000, #PM045, RRID: AB_2169714, Medical and Biological Laboratories (MBL), Nagoya, Japan) (for immunohistochemistry, 1 : 100, #03‐GP62‐C, RRID: AB_1542690, American Research Products Inc., Belmont, MA, USA), anti‐poly‐ubiquitin (poly‐Ubi) (1 : 1000 for immunoblotting, 1:500 for immunohistochemistry, #D058‐3, RRID: AB_592937, MBL), anti‐glial fibrillary acidic protein (1 : 250, #G3893, RRID: AB_477010, Sigma‐Aldrich), and anti‐Iba‐1 (1:500, #019‐10741, Wako Pure Chemical Industries Ltd., Osaka, Japan). Rabbit anti‐human SOD1 antibody was raised in our laboratory against a recombinant human SOD1 peptide (aa 24‐36), and purified with protein A (Bruijn et al. 1997).
Intracerebroventricular administration of CysC
Recombinant human CysC with or without an HA‐tag at its N‐terminus was prepared from Escherichia Coli strain Rosetta (DE3) (EMD Millipore Corp., Billerica, MA, USA) as described previously (Watanabe et al. 2014b). The purified CysC was sterilized by passing through a 0.22 μm filter and stored in sterile phosphate‐buffered saline (PBS) at −80°C until use. Intracerebroventricular administration of CysC was performed with an Alzet® osmotic pump (model #1004; Alzet Corp, Palo Alto, CA, USA) with a brain infusion kit (Alzet). To minimize the animals’ suffering and pain, the mice were anesthetized with three types of mixed anesthetic agents; medetomidine chloride (0.75 mg/kg) (ZENOAQ, Fukushima, Japan), midazolam (4 mg/kg) (SANDOZ, Tokyo, Japan), and butorphanol tartrate (5 mg/kg) (Meiji Seika Pharma Co., Ltd., Tokyo, Japan) during the surgery. The osmotic pump, filled with sterile PBS (control) or CysC (25 ng/μL in sterile PBS), was subcutaneously implanted in 100‐day‐old SOD1G93A mice to allow continuous infusion of CysC or PBS into the lateral ventricle of the brain. CysC was continuously infused at 0.11 μL/h for 4 weeks. The implanted pump was remained on the back of the SOD1G93A mice until the end‐stage. No randomization and blinding were performed in this study. The mice failed to recover their body weights after the surgery were excluded from this study.
Immunoblotting
Mice were killed with intraperitoneal injection of pentobarbital (5 mg/mL), and perfused with PBS for 10 min to remove the blood. The spinal cords and the brains were immediately dissected out and frozen in liquid nitrogen, and stored at −80°C until use. Mouse spinal cords were sonicated on ice in lysis buffer [50 mM Tris‐HCl (pH 7.4), 150 mM NaCl, 1 mM ethylenediamine‐tetraacetic acid, 1% Triton X‐100, and protease inhibitor cocktail (Roche)]. The lysates were centrifuged at 15 000 g for 10 min at 4°C, and the supernatant was collected. For the analyses of insoluble protein species, the pellets were re‐solubilized in the equivalent volume of the lysis buffer supplemented with 2% sodium dodecyl‐sulfate. All the protein concentrations were measured using the Bio‐Rad protein assay kit (Bio‐Rad, Hercules, CA, USA), and 20 μg of total protein was analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis with a reducing agent, 2‐mercaptoethanol. Densitometric analyses were performed after the chemiluminescence detection (Luminata Crescendo; EMD Millipore) using an image analyzer LAS‐4000 mini (Fuji Film, Tokyo, Japan) with the equipped software (Multi Gauge; Fuji Film).
Immunohistochemistry
Immunohistochemical analyses were performed as described elsewhere (Yamashita et al. 2007; Watanabe et al. 2014b). In brief, mice at the disease end‐stage were deeply anesthetized with pentobarbital, and sequentially perfused transcardially with PBS and 4%(w/v) paraformaldehyde in 0.1 M phosphate buffer for 10 min. After the incubation with 30%(w/v) sucrose in PBS, the dissected lumbar spinal cords were embedded in Tissue‐Tek OCT compound medium (Sakura Finetek, Tokyo, Japan), and frozen at −80°C until use. After blocking, the 12 μm‐sliced lumbar spinal cord sections were incubated with the primary antibodies for overnight at 4°C. Bound antibodies were detected with Alexa Fluor 488/564‐conjugated secondary antibodies (1 : 500, Thermo Fisher Scientific Inc., Waltham, MA, USA). Immunostained images were obtained by confocal laser scanning microscopy (LSM700; Carl Zeiss AG, Oberkochen, Germany) and the equipped software (Zen; Carl Zeiss, AG, USA).
Cell culture, and total RNA isolation
Mouse neuroblastoma Neuro2a cells (RRID: CVCL_0470) were maintained in Dulbecco's modified Eagle's medium containing 4.5 g/L glucose and supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (all from Thermo Fisher Scientific) in a humidified atmosphere containing 5% CO2 at 37°C. The cells were seeded on 6 well plate at 4.0 × 105/well, and treated with 1 μM CysC in the absence or presence of 10 μM Compound C (EMD Millipore) for 24 h. Total RNA was isolated from the cells using the NucleoSpin RNA Plus kit (Macherey‐Nagel, Düren, Germany). Total RNA of mouse brains was extracted with TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. The concentration of total RNA was determined using a spectrophotometer (NanoDrop ND‐2000; Thermo Fisher Scientific).
Quantitative RT‐PCR
cDNA was synthesized from 1 μg of total RNA using PrimeScript Reverse Transcriptase (Takara Bio, Shiga, Japan). Quantitative RT‐PCR was performed using SYBR Premix Ex Taq II (Takara), with gene‐specific primers in the Thermal Cycler Dice Real Time System II (Takara). Relative mRNA expression was calculated by a standard curve method normalizing to β‐actin (Actb) and relative to the control samples. The gene‐specific primes that were used are listed as follows: Pargc1a (5′‐CCGTCTCTACTTAAGAAGCTC‐3′ and 5′‐ GTTCTGAGTGCTAAGACCG‐3′), Vegfa (5′‐GATCAAACCTCACCAAAGCC‐3′ and 5′‐TCTTTCTTTGGTCTGCATTCAC‐3′), Tfeb (5′‐GCTAACAGATGCTGAGAGC‐3′ and 5′‐GTTGAACCTGCGTCTTCTC‐3′), Actb (5′‐GCTATGTTGCTCTAGACTTCG‐3′ and 5′‐GGATTCCATACCCAAGAAGG‐3′).
Statistical analyses
Survival times were analyzed with a log‐rank test, and no sample size calculation was performed in this study. All the data from semi‐quantitative immunoblotting and quantitative RT‐PCR were analyzed by an unpaired t‐test or one‐way anova followed by the post hoc Tukey's multiple comparison t‐test. All the statistical analyses were carried out using GraphPad Prism software (GraphPad Software, La Jolla, CA, USA).
Results
CysC administration extends the survival times of SOD1G93A mice
In this study, we administered CysC into the lateral ventricles of the mouse brain, since the molecular size of CysC, a 13.2 kDa protein, was too large to penetrate the blood‐brain barrier. Recombinant human CysC was purified from Escherichia coli as described in the Materials and Methods section, and the purity of CysC used for an administration was confirmed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Fig. 1a). To confirm a delivery of administered CysC to the lumbar spinal cords, where the lower motor neurons innervating the hindlimbs are located, we tested influenza hemagglutinin (HA)‐tagged CysC administration in wild‐type mice. After 1 week of continuous HA‐tagged CysC administration using an osmotic pump, the CysC was successfully delivered to the lumbar spinal cord and was predominantly distributed in the ventral horn neurons (Fig. 1b and c), whereas CysC was rarely found in neurons of the dorsal horn. These data indicate that intracerebroventricular administration was sufficient to deliver CysC to lower motor neurons in the lumbar spinal cord.
Figure 1.
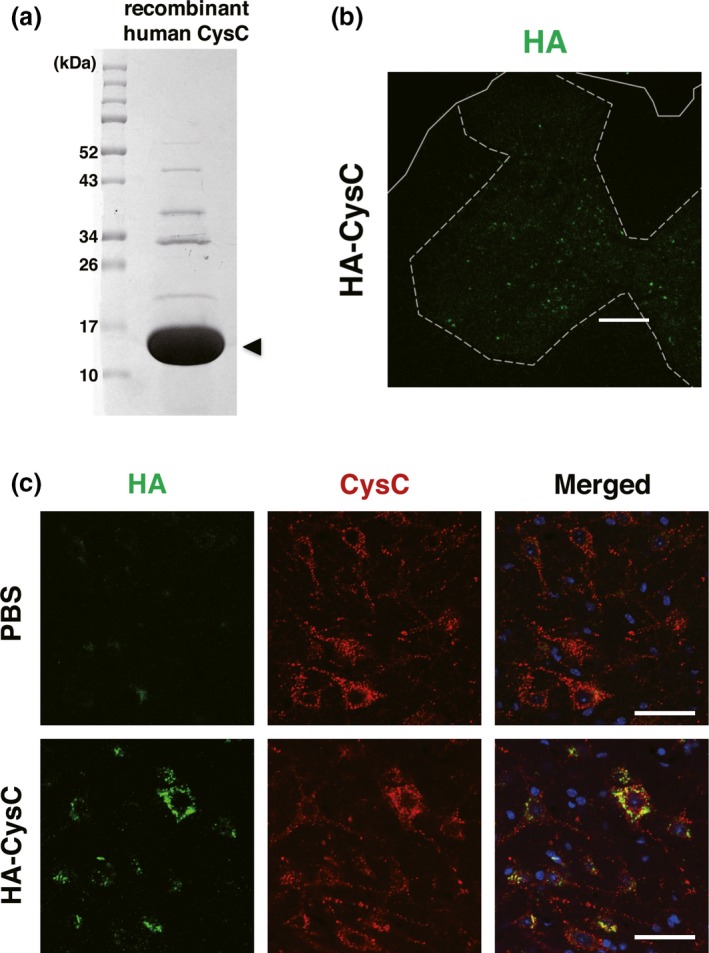
Intracerebroventricularly administered Cystatin C (CysC) is delivered to lumbar ventral horn neurons. (a) sodium dodecyl sulfate–polyacrylamide gel electrophoresis analysis of purified recombinant human CysC. The protein sample was separated on a 15% sodium dodecyl sulfate‐polyacrylamide gel, and stained with Coomassie Brilliant Blue. Molecular size markers are shown on the left (in kDa). The arrowhead indicates recombinant human CysC (13.2 kDa). (b and c) The uptake of HA‐tagged CysC by lumbar ventral horn neurons. CysC was administered for 1 week to 100‐day‐old non‐transgenic mice using an osmotic pump. After the administration, the delivered CysC in the lumbar spinal cords was detected by immunofluorescence using an anti‐HA antibody. Note that the anti‐CysC antibody labeled both the endogenous murine and exogenous recombinant human CysC in the lumbar ventral horn neurons. Scale bars: 50 μm.
Then, to examine the protective effect of CysC in ALS model mice, we administered non‐tagged recombinant human CysC into SOD1G93A mice at the early symptomatic phase, from 100 days old, around the time at which SOD1G93A mice typically reach disease onset (Fig. 2a) (Watanabe et al. 2014a; Endo et al. 2015). The continuous infusion of CysC extended the survival time of SOD1G93A mice for an average of 9.6 days (6.48% of the total survival time, Fig. 2b). This result indicates that CysC administration ameliorates disease in SOD1G93A mice.
Figure 2.
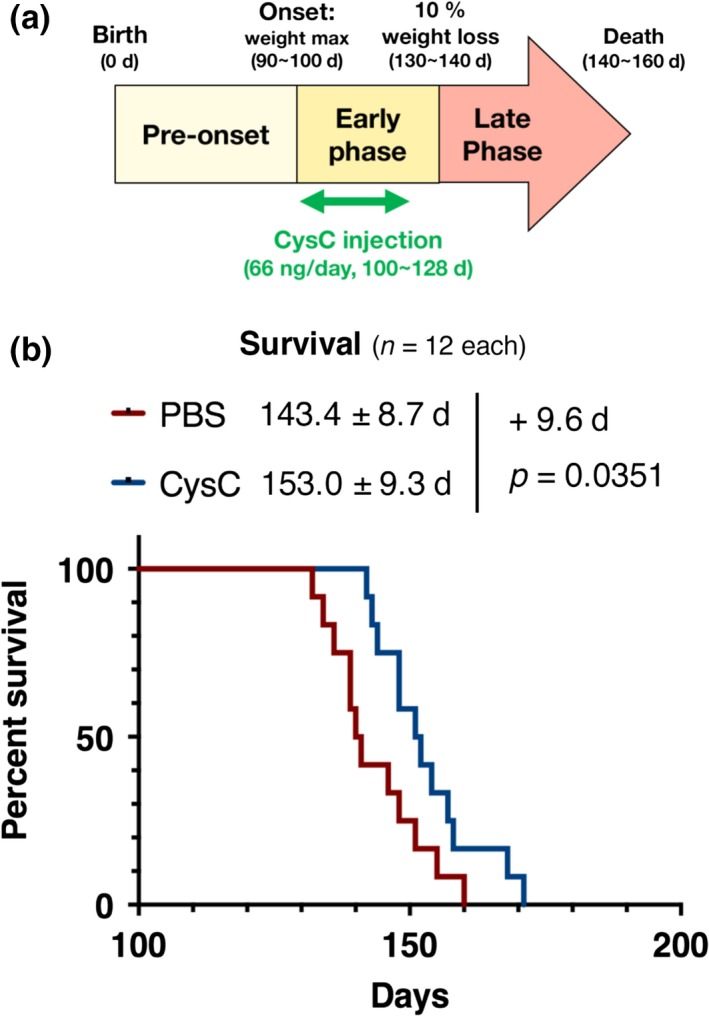
Administration of Cystatin C (CysC) extends the survival time of SOD1G93A mice. (a) Schematic diagram of CysC administration. The osmotic pump was implanted at 100 days of age (around the time of onset of disease). CysC was continuously delivered at 66 ng/day for 4 weeks during the early phase of the disease. (b) Survival curves of the phosphate‐buffered saline (PBS)‐ or CysC‐infused SOD1G93A mouse cohort plotted over time (n = 12 in total, n = 5 in male and n = 7 in female for both groups). The survival data were analyzed by the log‐rank test, and the average survival times are shown with SD.
AMPK activation plays a key role in the CysC‐mediated autophagic clearance of mutant SOD1 in vivo
We previously showed that autophagy induction through AMPK activation is essential for the neuroprotection by CysC in vitro (Watanabe et al. 2014b). Therefore, we next examined whether AMPK activation was involved in the CysC‐mediated survival extension in SOD1G93A mice. Consistent with the in vitro cell model, the AMPK phosphorylation level was restored (Fig. 3a and b), and the insoluble mutant SOD1 protein levels were decreased in the lumbar spinal cord of SOD1G93A mice with administration of the CysC (Fig. 3a and c). Accumulation of LC3‐II, an autophagosome marker, was also decreased in the CysC‐administered mice (Fig. 3a, d, and e). Taken together, these results indicate that autophagic turnover was increased by CysC administration to degrade insoluble mutant SOD1 species. Moreover, human SOD1 immunoreactivity was inversely correlated with that of CysC (Fig. 3f–h), supporting the notion that CysC promoted degradation of mutant SOD1 species.
Figure 3.
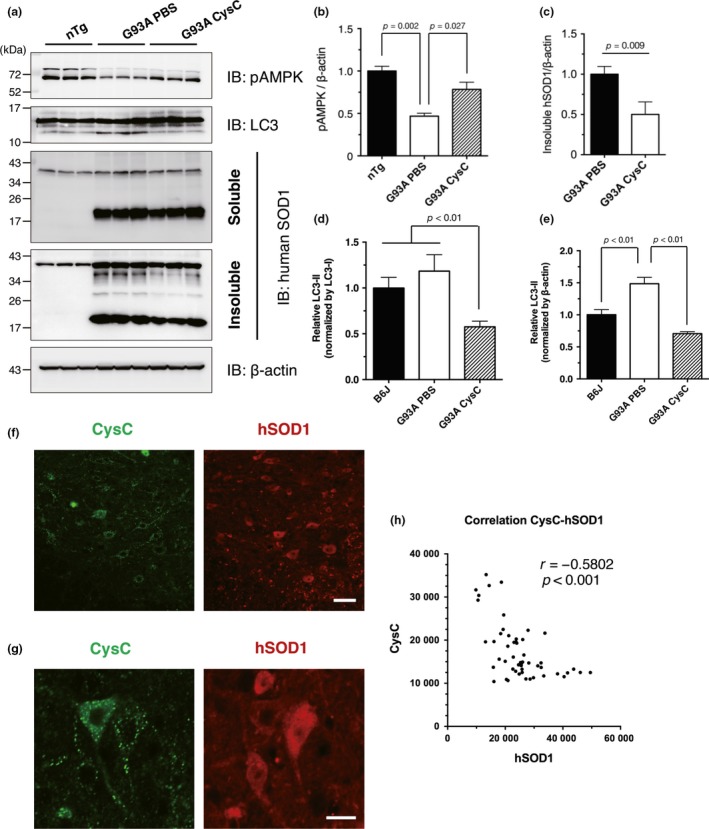
Cystatin C (CysC) activates the AMP‐activated kinase (AMPK) pathway to reduce the insoluble mutant SOD1 species. (a–e) Immunoblotting analyses of phosphorylated AMPK (pAMPK) (a and b), soluble or insoluble mutant SOD1 species (a and c), and LC3 (a, d and e) in the lumbar spinal cords of 5‐month‐old non‐transgenic control mice (nTg), phosphate‐buffered saline (PBS)‐administered SOD1G93A mice (G93A PBS), and CysC‐administered SOD1G93A mice (G93A CysC) at end‐stage. The level of LC3‐II was normalized by two distinct internal controls; LC3‐I (the pre‐modified form of LC3‐II; (d) or β‐actin (e). (f–h) The amount of SOD1 protein was negatively correlated with that of CysC in the ventral horn neurons. The lumbar spinal cord sections of end‐stage SOD1G93A mice administered with CysC were immunostained for SOD1 and CysC (F and G). The fluorescent intensities were quantified and plotted for at least 50 ventral horn neurons of CysC‐infused SOD1G93A mice (h). Representative fluorescence images used for quantification are shown (f and g). Similar staining was also obtained in three independent experiments. Scale bars: 50 μm (f) and 20 μm (g).
However, the levels of other autophagic markers, p62 and poly‐ubiquitin, were not changed by CysC administration at the disease end‐stage (Fig. 4a). Immunofluorescence analysis at the disease end‐stage revealed that p62 and poly‐ubiquitin was predominantly accumulated in non‐neuronal cells or debris, in which CysC was not transduced (Fig. 4b). To examine the effect of CysC on proteostasis during the early symptomatic stage, we evaluated amounts of the autophagic markers above after 2 weeks of CysC administration (4‐month‐old; about 1–1.5 month prior to the disease end‐stage) (Fig. 4c). Similar to the disease end‐stage, CysC successively activated AMPK. In contrast to the end‐stage, the levels of insoluble p62 and poly‐ubiquitinated proteins were decreased by CysC administration. On the other hand, activation of glial cells, astrocytes and microglia, was not altered by the CysC (Fig. 4d), suggesting that CysC selectively targeted neurons in the lumbar spinal cords of SOD1G93A mice.
Figure 4.
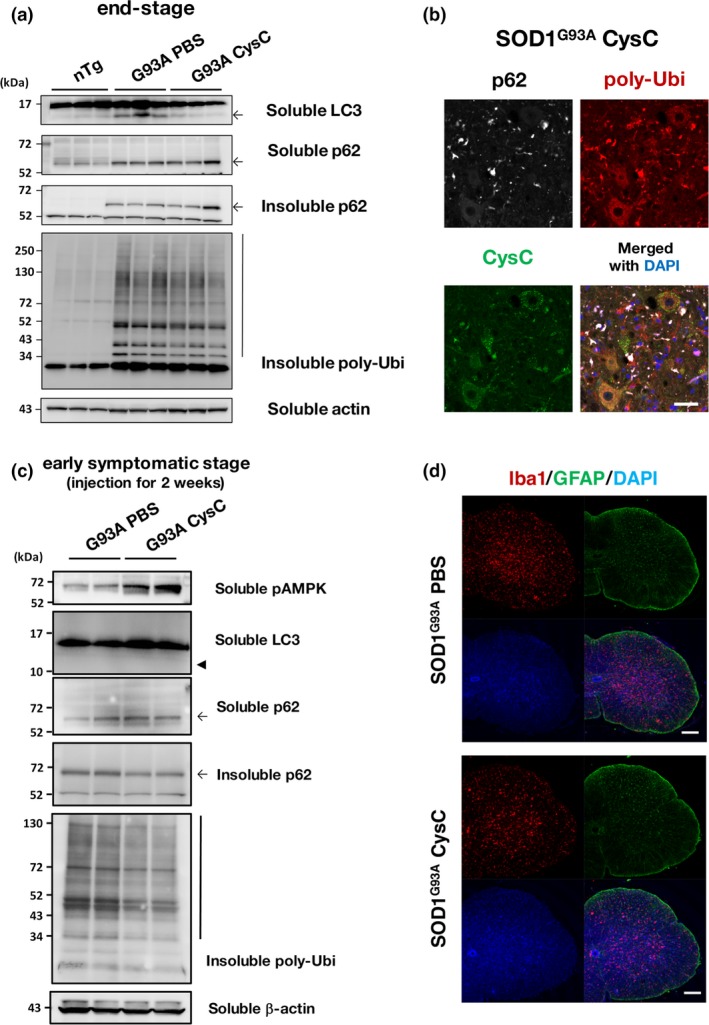
Cystatin C (CysC) administration reduces insoluble p62 and poly‐ubiquitin accumulation in the lumbar spinal cords of early symptomatic SOD1G93A mice. (a) Immunoblotting analyses of p62 and poly‐ubiquitin (poly‐Ubi). Note that detergent (1% Triton X‐100)‐insoluble p62 and poly‐Ubi was accumulated in SOD1G93A mice, but they were not affected by CysC administration. (b) Co‐localization of p62 and poly‐Ubi aggregates were predominantly observed in outside of neurons. A representative image for the lumbar spinal cord section of the end‐stage SOD1G93A mice with CysC administration stained for p62, poly‐ubiquitin, and CysC, along with a merged image with DAPI. (c) Immunoblotting analyses of spinal cords derived from the early symptomatic stage (4 months old) SOD1G93A mice administered with phosphate‐buffered saline (PBS) or CysC for 2 weeks. In contrast to the end‐stage samples (a), the amounts of insoluble p62 and poly‐ubiqutinated proteins were reduced by CysC administration, suggesting the improved protein degradation. It should be also noted that bands corresponding to LC3‐II were hardly detected (arrowhead). (d) Astrocytes and microglia in the lumbar spinal cords of end‐stage SOD1G93A mice, with or without administration of CysC, were immunostained with antibodies against glial fibrillary acidic protein (GFAP) and Iba‐1 with DAPI. Scale bars: 50 μm (b), and 100 μm (d).
CysC activates the PGC‐1α pathway through AMPK activation
To explore the mechanism of the beneficial effects of CysC in SOD1G93A mice, we focused on the PGC‐1α pathway, a male‐dominant disease modifier of ALS (Eschbach et al. 2013). PGC‐1α is a transcription coactivator that regulates genes involved in energy metabolism as well as in mitochondrial biogenesis. A recent study showed that CysC up‐regulated Vegfa, a gene regulated by PGC‐1α, to prevent neuronal cell death in Parkinson's diseases model mice (Zou et al. 2017), suggesting the involvement of the PGC‐1α pathway in the CysC‐mediated survival extension. First, we confirmed that CysC treatment induced PGC‐1α expression in mouse neuroblastoma Neuro2a cells (Fig. 5a). Compound C, an AMPK inhibitor, prevented the PGC‐1α induction by CysC, indicating that CysC regulated PGC‐1α through AMPK. In the brains of SOD1G93A male mice, PGC‐1α mRNA was down‐regulated, as previously reported (Bayer et al. 2017); however, the level of PGC‐1α mRNA was partially restored in the brains of CysC‐infused SOD1G93A mice (Fig. 5b). Moreover, the mRNA level of Vegfa was also restored in the brains of CysC‐infused SOD1G93A mice (Fig. 5c). Tfeb, another PGC‐1α responsive gene (Bayer et al. 2017), also showed a trend toward restoration of its levels, which was similar to Vegfa in the CysC‐infused SOD1G93A mice (Fig. 5d). These results suggest that activation of the PGC‐1α pathway through AMPK activation by CysC contributes to the extended survival times in SOD1‐ALS mice.
Figure 5.
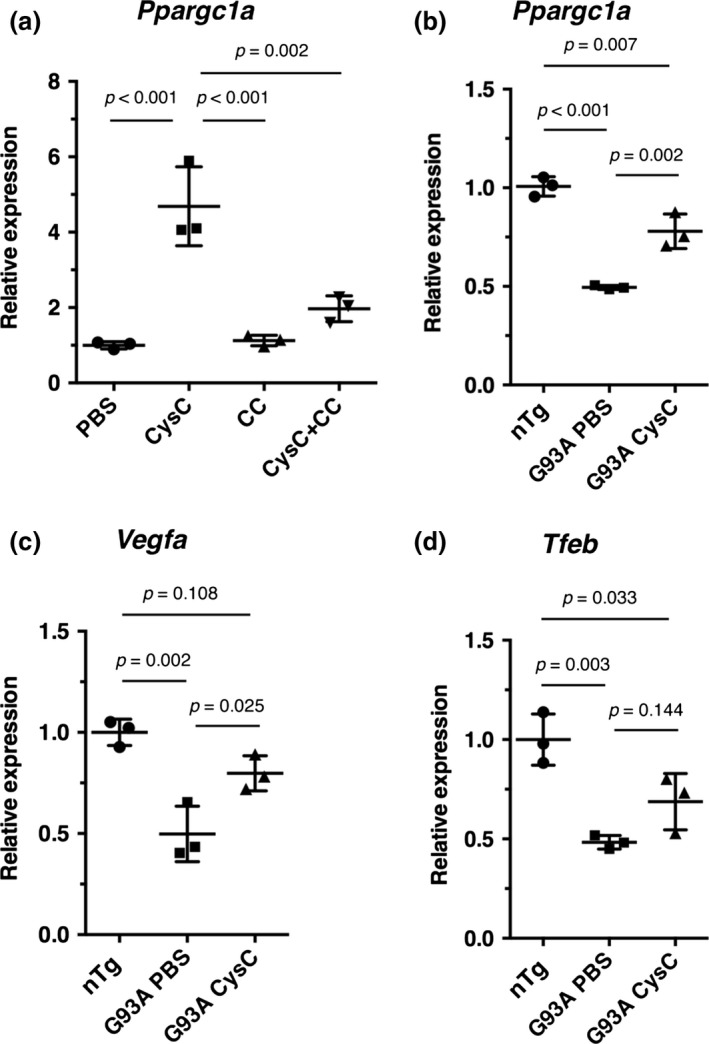
Cystatin C (CysC) up‐regulates PGC‐1α and its responsive genes in the SOD1G93A mice. (a) mRNA levels of Ppargc1a (murine PGC‐1α coding gene) in Neuro2a cells treated with phosphate‐buffered saline (PBS) or CysC (1 μM) for 24 h. The cells were also treated with CysC in the presence of compound C (CC), a selective inhibitor of AMP‐activated kinase (AMPK). (b‐d) mRNA levels of Ppargc1a (b), Vegfa (c), and Tfeb (d) in the brains of non‐transgenic control (nTg), PBS‐ (G93A PBS) or CysC‐infused (G93A CysC) SOD1G93A male mice. The expression levels relative to PBS‐treated control cells (a), or nTg controls (b–d) were measured by quantitative RT‐PCR, and expressed as mean ± SEM (n = 3).
Administration of CysC induces inclusion bodies in the motor neurons
We have also reported that CysC forms aggregates in neuronal cells under oxidative stress (Watanabe et al. 2014b). Similarly, in this study, we found that the administration of CysC induced CysC aggregation in motor neurons (Fig. 6a). No CysC aggregates were observed in the PBS‐infused control mice. The CysC aggregates were predominantly 3–5 μm in diameter (Fig. 6b), and only 1–2 motor neurons with CysC aggregates were found per a ventral horn section. The aggregated CysC did not co‐localize with poly‐ubiquitin or p62, which are general markers of protein aggregates (Fig. 6c and d). In addition, we found eosinophilic inclusion bodies, which were similar in size to the CysC aggregates, in the ventral horn neurons of the CysC‐infused SOD1G93A mice (Fig. 6e). Immunoblotting revealed that the soluble form of CysC was decreased in the spinal cords of CysC‐infused SOD1G93A mice (Fig. 6f and g), and conversely, that the Triton X‐100‐insoluble tetramer of CysC was increased (Fig. 6f and h). These data imply that the CysC tetramer is a core structure of CysC aggregates in motor neurons.
Figure 6.
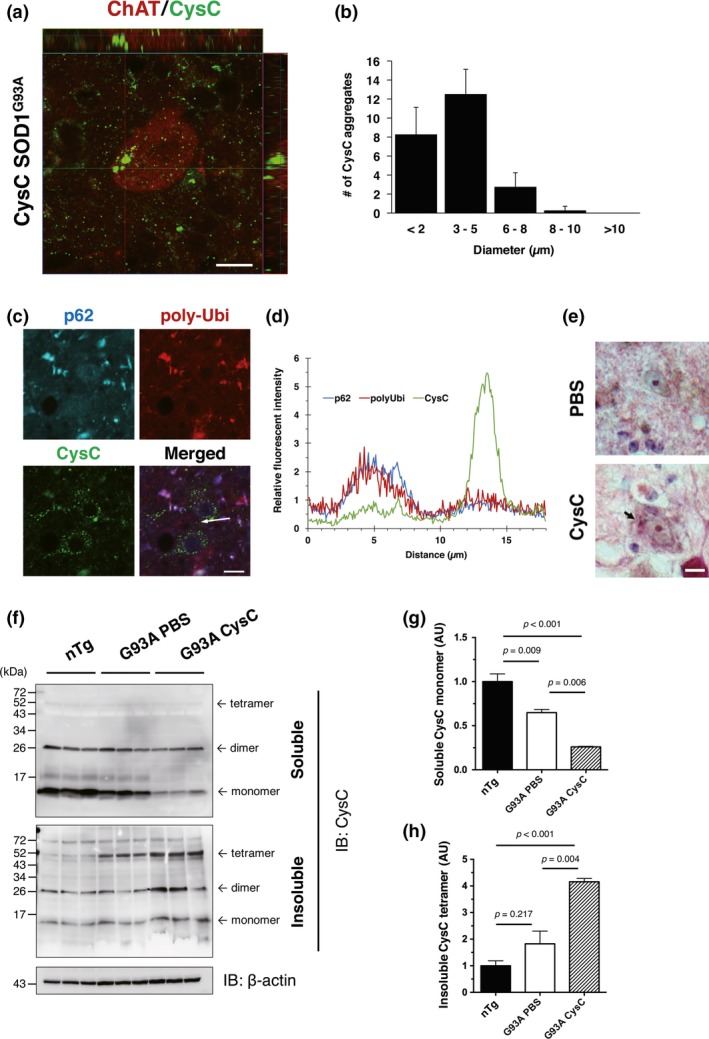
Administration of Cystatin C (CysC) induces its aggregation in lumbar motor neurons. (a and b) CysC aggregation in the motor neurons of CysC‐administered SOD1G93A mice. CysC aggregates were found in the motor neurons of CysC‐administered mice. The motor neurons were immunostained by the antibodies for ChAT (red) and CysC (green) (a). Averaged number of CysC aggregates in ChAT‐positive motor neurons was plotted against the aggregate diameter (b). A total of 30 aggregates each in the lumbar spinal cords from three CysC‐administered SOD1G93A mice were measured. (c and d) CysC aggregates did not co‐localize with p62 or poly‐ubiquitin (poly‐Ubi). The lack of co‐localization was confirmed by a fluorescence intensity profile (d), where indicated by an arrow in (c). (e) Eosinophilic inclusions found in the ventral horn neurons of the CysC‐administered mice. (f–h) Immunoblotting analyses of CysC aggregation. In the CysC‐administered SOD1G93A mouse spinal cords, detergent (Triton X‐100)‐soluble monomeric CysC was decreased (g), whereas insoluble tetrameric CysC was accumulated (h). The amounts of CysC were normalized to β‐actin, and are shown as mean ± SEM. Scale bars: 20 μm.
Discussion
In this study, we demonstrated that continuous administration of CysC extended survival times of SOD1G93A ALS model mice presumably through activation of an AMPK‐dependent autophagic pathway as we previously demonstrated in cultured cells. AMPK activation by CysC was also beneficial likely through PGC‐1α up‐regulation. Furthermore, CysC formed Bunina body‐like aggregates in the CysC‐infused mouse motor neurons, suggesting that Bunina bodies are byproducts of the beneficial effects of CysC.
Autophagy, as well as ubiquitin–proteasome system, is one of the major degradation machinery of misfolded proteins (Klionsky and Emr 2000). Impairment of protein degradation machinery is deeply involved in ALS pathology (Webster et al. 2017). Misfolded SOD1 and/or TAR DNA binding protein 43 are highly accumulated in motor neurons of ALS patients; therefore, facilitating degradation of these misfolded proteins is a promising therapeutic strategy for ALS. There are several strategies for eliminating disease‐causing proteins. Efficacy of down‐regulation of SOD1 mRNA by the intrathecal administration of anti‐sense oligonucleotides was evaluated in a clinical trial in SOD1‐ALS patients (Miller et al. 2013; Schoch and Miller 2017). Alternatively, promotion of protein degradation machineries or induction of molecular chaperones is other viable strategies. In this context, we recently reported that sirtuin 1 (SIRT1) over‐expression in the central nervous system slowed the disease progression of mutant SOD1 mice. This was because of the reduction in toxic SOD1 species through enhancement of the expression of the molecular chaperone, heat‐shock protein 70 (Watanabe et al. 2014a). Inducers of autophagy, such as trehalose or rapamycin, also extend the survival of ALS model mice by degrading abnormal toxic protein species (Wang et al. 2012; Castillo et al. 2013). In this study, we demonstrated that the administration of CysC ameliorated the disease in SOD1G93A mice. CysC also activated AMPK in vivo as it did in cultured cell lines. Furthermore, insoluble SOD1 species, which are toxic and degraded partially by autophagy (Kabuta et al. 2006), were also reduced by the CysC administration. This indicates that autophagy induced by CysC is also beneficial in an in vivo model of ALS. Contrary to the increased level of an autophagosome marker, LC3‐II, in CysC treated cells (Watanabe et al. 2014b), the level of LC3‐II was decreased by the administration of CysC in ALS mice at the disease end‐stage. LC3‐II rapidly accumulates during autophagosome formation; however, it is degraded when autophagosomes are fused with lysosomes. Indeed, during an early symptomatic stage, LC3‐II level was not detectable in SOD1G93A mouse spinal cords. Impairment of autophagy flux is generally observed in ALS (Webster et al. 2017), and LC3‐II accumulation is also promoted by the reduced turnover of autophagosomes (Mizushima and Yoshimori 2007). Therefore, the reduction in LC3‐II at the disease end‐stage by CysC administration probably represents an increase in autophagy turnover rather than impairment of autophagy. Moreover, inverse correlation between the levels of mutant SOD1 and CysC in ventral horn neurons, as well as reduction in p62 or poly‐ubiquitinated proteins in the early symptomatic CysC‐administered SOD1G93A mice, supports the notion that CysC promotes degradation of toxic SOD1 species through autophagy. Taken together, all these results indicate that enhancing the CysC‐mediated autophagy is a promising therapeutic strategy against SOD1‐mediated neurodegeneration.
Decreased expression of PGC‐1α was observed in the spinal cords of SOD1‐ALS mice (Bayer et al. 2017). In addition, a single‐nucleotide polymorphism in the PGC‐1α promoter region (rs11737023) that resulted in the decreased expression of PGC‐1α, correlated with the shortened survival times of male ALS patients (Eschbach et al. 2013). Moreover, PGC‐1α deficiency shortened the survival times of male SOD1G93A mice (Eschbach et al. 2013). These results suggest that PGC‐1α is a disease modifier with a predominant effect in male, and that up‐regulation of PGC‐1α may contribute to the extended survival of male ALS patients. PGC‐1α is mainly regulated by two distinct pathways; AMPK and SIRT1 (Ruderman et al. 2010). CysC induced PGC‐1α through AMPK activation in this study. Similarly, we also previously reported that SIRT1 over‐expression extended the survival times of SOD1G93A mice (Watanabe et al. 2014a). It should be noted that SIRT1 only extended the survival times of the low expression line of SOD1G93A mice, and failed to show any beneficial effects in the high expression SOD1G93A line as used in this study. In contrast, CysC was apparently beneficial in the high expression SOD1G93A line. This implies that AMPK is a dominant regulator of PGC‐1α in ALS, and therefore, that targeting the AMPK/PGC‐1α pathway may represent a novel therapeutic strategy. Further studies focused on the role of the AMPK/PGC‐1α axis in ALS are required.
A Bunina body is an ALS‐specific inclusion body which is found in the remaining motor neurons of sporadic ALS patients (Okamoto et al. 2008). CysC was shown to be a major protein component of Bunina bodies, but the mechanism of Bunina body formation has remained uncovered (Okamoto et al. 1993). In this study, we found that exogenous CysC triggered formation of CysC aggregates in motor neurons for the first time. The CysC aggregates induced by the exogenous administration of CysC well shared features with Bunina bodies, including size, cell‐type specificity, and absence of co‐localization with p62 or ubiquitin, and eosinophilic aggregates. Therefore, the CysC aggregates observed in our study were possibly formed by a mechanism similar to that of Bunina bodies. Furthermore, our finding that CysC tetramers are rapidly increased in the CysC‐administered mice is well consistent with a previous study showing that chicken cystatin forms a tetramer prior to its amyloidogenesis (Sanders et al. 2004). Tetrameric units of human CysC formed by amyloid‐like intermolecular β‐sheets interactions were also observed in its crystal structure (Janowski et al. 2005). Taken together, these observations suggest that the CysC tetramer is the core structure of CysC aggregates. While, Bunina bodies are not found in ALS patients with an inherited SOD1 mutation. The reduced level of endogenous CysC in SOD1‐ALS mice, which we reported in our previous study (Watanabe et al. 2014b), may explain the absence of Bunina bodies in SOD1‐ALS cases. This finding suggests that a certain amount of endogenous CysC is required for the formation of Bunina bodies. Although the detailed mechanisms of Bunina body formation need more investigation, these results suggest that Bunina bodies are a byproduct of the beneficial effects of CysC. Moreover, we also previously demonstrated that oxidative stress, instead of CysC uptake itself, was a trigger of CysC aggregation in cultured neuronal cells (Watanabe et al. 2014b). In the cited study, we also showed that CysC was still enzymatically active under the oxidative stress condition, in which CysC aggregates were formed. These observations suggest that CysC aggregation does not represent a complete loss of its enzymatic function; however, further investigation to reveal the detailed enzymatic roles of CysC in vivo is required.
In conclusion, we demonstrated that the intraventricular administration of CysC extended the survival times of mutant SOD1‐linked ALS mice. CysC conferred the beneficial effects in vivo via the AMPK/PGC‐1α pathway. Moreover, we showed that the CysC administration induced the formation of CysC aggregates that were similar to Bunina bodies in spinal motor neurons for the first time. All of our findings demonstrate the neuroprotective role of CysC in ALS pathology, and suggest that CysC may represent a novel therapeutic candidate for ALS.
Author contributions
SW and KY designed the study with a support from KW. SW conducted the biochemical, histological, behavioral analyses with supports from OK, FE under the supervision of KY. SW and KY interpreted the data and wrote the manuscript. All authors read and approved the manuscript.
Acknowledgments and conflict of interest disclosure
This work was supported by Grants‐in‐Aid for Scientific Research 26293208, 16H01336 (to KY), and 17H0496, 25860252 (to SW) from the Ministry for Education, Culture, Sports, Science and Technology, Japan, Grants‐in‐Aid for Research on Rare and Intractable Diseases, the Research Committee on Establishment of Novel Treatments for ALS from Japan Agency for Medical Research and Development (AMED), Uehara Memorial Foundation, Takeda Science Foundation (to KY), and Grants‐in‐Aid to SW from Hori foundation, Japan. We also thank our laboratory members, and Center for Animal Research and Education, Nagoya University for technical supports. The authors declare that they have no conflict of interest.
All experiments were conducted in compliance with the ARRIVE guidelines.
References
- Abrahamson M., Barrett A. J., Salvesen G. and Grubb A. (1986) Isolation of six cysteine proteinase inhibitors from human urine. Their physicochemical and enzyme kinetic properties and concentrations in biological fluids. J. Biol. Chem. 261, 11282–11289. [PubMed] [Google Scholar]
- Bayer H., Lang K., Buck E. et al (2017) ALS‐causing mutations differentially affect PGC‐1alpha expression and function in the brain vs. peripheral tissues. Neurobiol. Dis. 97, 36–45. [DOI] [PubMed] [Google Scholar]
- Bendotti C. and Carri M. T. (2004) Lessons from models of SOD1‐linked familial ALS. Trends Mol. Med. 10, 393–400. [DOI] [PubMed] [Google Scholar]
- Bosco D. A., Morfini G., Karabacak N. M. et al (2010) Wild‐type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn L. I., Becher M. W., Lee M. K. et al (1997) ALS‐linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1‐containing inclusions. Neuron 18, 327–338. [DOI] [PubMed] [Google Scholar]
- Bruijn L. I., Miller T. M. and Cleveland D. W. (2004) Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 27, 723–749. [DOI] [PubMed] [Google Scholar]
- Castillo K., Nassif M., Valenzuela V., Rojas F., Matus S., Mercado G., Court F. A., van Zundert B. and Hetz C. (2013) Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 9, 1308–1320. [DOI] [PubMed] [Google Scholar]
- Cleveland D. W. and Rothstein J. D. (2001) From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2, 806–819. [DOI] [PubMed] [Google Scholar]
- Endo F., Komine O., Fujimori‐Tonou N. et al (2015) Astrocyte‐derived TGF‐beta1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep. 11, 592–604. [DOI] [PubMed] [Google Scholar]
- Eschbach J., Schwalenstocker B., Soyal S. M. et al (2013) PGC‐1alpha is a male‐specific disease modifier of human and experimental amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 3477–3484. [DOI] [PubMed] [Google Scholar]
- Gauthier S., Kaur G., Mi W., Tizon B. and Levy E. (2011) Protective mechanisms by cystatin C in neurodegenerative diseases. Front. Biosci. (Scholar edition) 3, 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guareschi S., Cova E., Cereda C., Ceroni M., Donetti E., Bosco D. A., Trotti D. and Pasinelli P. (2012) An over‐oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl Acad. Sci. USA 109, 5074–5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski R., Kozak M., Abrahamson M., Grubb A. and Jaskolski M. (2005) 3D domain‐swapped human cystatin C with amyloidlike intermolecular beta‐sheets. Proteins 61, 570–578. [DOI] [PubMed] [Google Scholar]
- Kabuta T., Suzuki Y. and Wada K. (2006) Degradation of amyotrophic lateral sclerosis‐linked mutant Cu, Zn‐superoxide dismutase proteins by macroautophagy and the proteasome. J. Biol. Chem. 281, 30524–30533. [DOI] [PubMed] [Google Scholar]
- Klionsky D. J. and Emr S. D. (2000) Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi W., Pawlik M., Sastre M. et al (2007) Cystatin C inhibits amyloid‐beta deposition in Alzheimer's disease mouse models. Nat. Genet. 39, 1440–1442. [DOI] [PubMed] [Google Scholar]
- Miller T. M., Pestronk A., David W. et al (2013) An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first‐in‐man study. Lancet Neurol. 12, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. and Yoshimori T. (2007) How to interpret LC3 immunoblotting. Autophagy 3, 542–545. [DOI] [PubMed] [Google Scholar]
- Okamoto K., Hirai S., Amari M., Watanabe M. and Sakurai A. (1993) Bunina bodies in amyotrophic lateral sclerosis immunostained with rabbit anti‐cystatin C serum. Neurosci. Lett. 162, 125–128. [DOI] [PubMed] [Google Scholar]
- Okamoto K., Mizuno Y. and Fujita Y. (2008) Bunina bodies in amyotrophic lateral sclerosis. Neuropathol. 28, 109–115. [DOI] [PubMed] [Google Scholar]
- Ruderman N. B., Xu X. J., Nelson L., Cacicedo J. M., Saha A. K., Lan F. and Ido Y. (2010) AMPK and SIRT1: a long‐standing partnership? Am. J. Physiol. Endocrinol. Metab. 298, E751–E760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders A., Jeremy Craven C., Higgins L. D., Giannini S., Conroy M. J., Hounslow A. M., Waltho J. P. and Staniforth R.A. (2004) Cystatin forms a tetramer through structural rearrangement of domain‐swapped dimers prior to amyloidogenesis. J. Mol. Biol. 336, 165–178. [DOI] [PubMed] [Google Scholar]
- Schoch K. M. and Miller T. M. (2017) Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron 94, 1056–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang I. F., Guo B. S., Liu Y. C., Wu C. C., Yang C. H., Tsai K. J. and Shen C. K. (2012) Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA‐binding protein 43. Proc. Natl Acad. Sci. USA 109, 15024–15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S., Ageta‐Ishihara N., Nagatsu S. et al (2014a) SIRT1 overexpression ameliorates a mouse model of SOD1‐linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system. Mol. Brain 7, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S., Hayakawa T., Wakasugi K. and Yamanaka K. (2014b) Cystatin C protects neuronal cells against mutant copper‐zinc superoxide dismutase‐mediated toxicity. Cell Death Dis. 5, e1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S., Ilieva H., Tamada H. et al (2016) Mitochondria‐associated membrane collapse is a common pathomechanism in SIGMAR1‐ and SOD1‐linked ALS. EMBO Mol. Med. 8, 1421–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster C. P., Smith E. F., Shaw P. J. and De Vos K. J. (2017) Protein homeostasis in amyotrophic lateral sclerosis: therapeutic opportunities? Front. Mol. Neurosci. 10, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M. E., Boumaza I., Lacomis D. and Bowser R. (2010) Cystatin C: a candidate biomarker for amyotrophic lateral sclerosis. PLoS ONE 5, e15133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Sheng J., Tang Z. et al (2005) Cystatin C prevents degeneration of rat nigral dopaminergic neurons: in vitro and in vivo studies. Neurobiol. Dis. 18, 152–165. [DOI] [PubMed] [Google Scholar]
- Yamashita H., Kawamata J., Okawa K., Kanki R., Nakamizo T., Hatayama T., Yamanaka K., Takahashi R. and Shimohama S. (2007) Heat‐shock protein 105 interacts with and suppresses aggregation of mutant Cu/Zn superoxide dismutase: clues to a possible strategy for treating ALS. J. Neurochem. 102, 1497–1505. [DOI] [PubMed] [Google Scholar]
- Zou J., Chen Z., Wei X. et al (2017) Cystatin C as a potential therapeutic mediator against Parkinson's disease via VEGF‐induced angiogenesis and enhanced neuronal autophagy in neurovascular units. Cell Death Dis. 8, e2854. [DOI] [PMC free article] [PubMed] [Google Scholar]