

Summary
Barley (Hordeum vulgare) is an established model to study domestication of the Fertile Crescent cereals. Recent molecular data suggested that domesticated barley genomes consist of the ancestral blocks descending from multiple wild barley populations. However, the relationship between the mosaic ancestry patterns and the process of domestication itself remained unclear.
To address this knowledge gap, we identified candidate domestication genes using selection scans based on targeted resequencing of 433 wild and domesticated barley accessions. We conducted phylogenetic, population structure, and ancestry analyses to investigate the origin of the domesticated barley haplotypes separately at the neutral and candidate domestication loci.
We discovered multiple selective sweeps that occurred on all barley chromosomes during domestication in the background of several ancestral wild populations. The ancestry analyses demonstrated that, although the ancestral blocks of the domesticated barley genomes were descended from all over the Fertile Crescent, the candidate domestication loci originated specifically in its eastern and western parts.
These findings provided the first molecular evidence implicating multiple wild or protodomesticated lineages in the process of barley domestication initiated in the Levantine and Zagros clusters of the origin of agriculture.
Keywords: ancestry analysis, barley, cereals, domestication, selection scan, targeted resequencing
Introduction
Domesticated barley (Hordeum vulgare L. ssp. vulgare) is one of the Neolithic founder crops, which facilitated establishment of the early agricultural societies (Lev‐Yadun et al., 2000). Owing to its striking environmental plasticity, barley is an important staple crop in a wide range of agricultural environments (Dawson et al., 2015). The first traces of barley cultivation were found at archaeological sites in the Fertile Crescent, which dated back to c. 10 000 bc (Zohary et al., 2012). The Fertile Crescent is the primary habitat of the crop progenitor wild barley (H. vulgare ssp. spontaneum). However, its isolated populations have spread as far as North African and European shores of the Mediterranean Basin and East Asia (Harlan & Zohary, 1966). Wild barley is a rich but under‐utilized reservoir of novel alleles for breeding of barley cultivars better adapted to predicted future climatic perturbations.
In contrast to some other crops, the visible phenotype of domesticated barley did not diverge dramatically from its wild form. So far, the spike rachis brittleness has remained the only well‐characterized domestication trait that exhibits a clear dimorphism between the wild and domesticated subgroups, which are characterized by the brittle and nonbrittle spikes, respectively (Abbo et al., 2014; Pourkheirandish et al., 2015). Other traits differentiated between the modern‐day wild and domesticated genotypes and underlying genes that define the barley domestication syndrome, as a complex of all characters that characterize the domesticated phenotype, are as yet undiscovered (Hammer, 1984; Meyer & Purugganan, 2013). When adaptive phenotypes are not clearly defined, the so‐called bottom‐up approach, which starts with the identification of genome‐wide signatures of selection, has proven instrumental in reconstructing the genetic architecture of the domestication syndrome (Ross‐Ibarra et al., 2007; Shi & Lai, 2015). In other crops, the selection scans detected multiple selective sweep regions associated with domestication, which comprised hundreds of candidate domestication genes (Huang et al., 2012; Hufford et al., 2012; Lin et al., 2014; Schmutz et al., 2014; Zhou et al., 2015).
The circumstances of barley domestication are debatable and its genome‐wide effects on the domesticated barley genomes remain poorly understood (Pankin & von Korff, 2017). The early models based on diversity analyses of isolated genes and neutral DNA markers proposed the Israel‐Jordan area as a primary center of cultivated barley origin and proposed the eastern Fertile Crescent, the Horn of Africa, Morocco and Tibet as the alternative centers of domestication (Negassa, 1985; Molina‐Cano et al., 1999; Badr et al., 2000; Morrell & Clegg, 2007; Dai et al., 2012). As regards the number and the timescale of domestication events, one school of thought maintains that Neolithic domestication in the Near East has been a rapid centric innovation (Abbo et al., 2010; Heun et al., 2012; Zohary et al., 2012). Conversely, the archeobotanical evidence and simulation studies prompted development of the so‐called protracted domestication model, which postulates that domestication of the Near Eastern crops has been a slow polyphyletic process dispersed over large territories (Allaby et al., 2008; Fuller et al., 2011, 2012; Purugganan & Fuller, 2011). The diphyletic origin of the nonbrittle spike phenotype of cultivated barley and the heterogeneous (mosaic) ancestry of cultivated barley genomes, which consisted of ancestral fragments originating from several wild barley populations, supported the protracted model (Allaby, 2015; Poets et al., 2015; Pourkheirandish et al., 2015). The heterogeneous origin of domesticated barley genomes hints at the existence of several founder lineages of barley cultivation. However, the link between the mosaic ancestry patterns and the process of domestication remained unclear.
Here, we partitioned the domesticated barley genomes into the neutral (in relation to domestication) and domestication sweep regions identified by selection scans and separately reconstructed the phylogeographic history of their origin, focusing on the hypothesis of the Fertile Crescent origins of domesticated barley. To this end, we resequenced a diversity panel comprising 344 wild barley accessions from the Fertile Crescent and 89 domesticated genotypes using a custom genome‐wide enrichment assay (c. 544 000 single nucleotide polymorphisms (SNPs)). The selection scans identified multiple domestication sweep regions on every barley chromosome. Analysis of the top candidate genes within the domestication sweeps suggested cases of parallelism in targets of selection during domestication of barley and other crop species. The patterns of ancestry at the neutral loci revealed signatures of abundant continuous gene flow, which hindered identification of lineages descending from the independent founder events. Nevertheless, heterogeneous ancestry of the domestication sweep loci provided the first molecular evidence that multiple domestication sweeps occurred in the background of several progenitor wild barley populations residing in the eastern and western clusters of the Fertile Crescent.
Materials and Methods
Plant material and Btr genotyping assay
A panel consisting of 344 wild and 89 domesticated lines was selected to maximize genetic diversity and to cover the entire range of the wild and landrace barley habitats in the Fertile Crescent (Supporting Information Table S1). The elite barley cultivars were sampled to represent northern European, East Asian, North American and Australian breeding programs. The largest part of the germplasm set, 98% of wild and 40% of domesticated barley genotypes, originated from the area of the Fertile Crescent. The selection of domesticated barley originated from various breeding programs and represented the whole variety of cultivated barley life forms, namely two‐ (71%) and six‐row (29%) genotypes with winter (45%) and spring (55%) growth habits based on the passport data. All material was purified by single‐seed descent to eliminate accession heterogeneity.
Leaf samples for DNA extraction were collected from single 3‐wk‐old plants. The DNA was extracted using the DNeasy Plant Mini kit (Qiagen) and quantified using the NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and electrophoresis in the 0.8% agarose gel.
The DNA samples of domesticated barley were genotyped using PCR markers distinguishing loss‐of‐function alleles of the brittleness genes Btr1 and Btr2. The markers were amplified using allele‐specific primer pairs Btr1f 5′‐CCGCAATGGAAGGCGATG‐3′/Btr1r 5′‐CTATGAAACCGGAGAGGC‐3′ (c. 200 bp fragment, presence – Btr1/absence – btr1) and Btr2f 5′‐AATACGACTCACTATAGGGTTCGTCGAGCTCGCTATC‐3′/Btr2r 5′‐GTGGAGTTGCCACCTGTG‐3′ (c. 160 bp fragment, 11 bp deletion in the btr2 allele). PCR reactions (1 × PCR buffer, 0.1 M primers, 1 U Taq polymerase, 100 ng DNA) were incubated in the PTC DNA Engine thermocycler (Bio‐Rad, Hercules, CA, USA) under the following conditions: 95°C for 3 min; 30 cycles of 95°C for 20 s, 60°C for 30 s, 72°C for 1 min; 72°C for 5 min.
Design of the enrichment assay and SNP calling
To resequence barley genotypes, we designed a custom target enrichment assay, which included 666 loci implicated in the candidate domestication and environmental adaptation pathways in barley and other species and 1000 neutral loci covering all barley chromosomes to attenuate effects of the biased selection (Fig. S1; Table S2). Among the selected loci were known barley genes implicated in the regulation of flowering time, development of meristem and inflorescences, tillering, seed dormancy, and carbohydrate metabolism; barley homologs of flowering genes from the other grass species, such as Brachypodium and rice (Higgins et al., 2010); and barley homologs of 259 Arabidopsis genes characterized by the development‐related gene ontology (GO) terms that have been confirmed experimentally (Table S3). The barley homologs were extracted from the following sources: the NCBI UniGene dataset (ftp://ftp.ncbi.nih.gov/repository/UniGene/Hordeum_vulgare), IBGSC High and Low confidence genes (IBGSC, 2012), and the HarvEST unigene assembly 35 (http://harvest.ucr.edu). See Methods S1 for the detailed description of the gene selection and design of the capture baits.
The SNP calling pipeline consisted of three modules: quality control and filtering of Illumina read libraries; mapping the reads to the custom reference (Methods S2; Table S4); and extracting and filtering both variant (SNP) and invariant sites (Fig. S2), implemented in a series of bash scripts using standard bioinformatics tools (Methods S3). Genetic positions of the reference contigs were determined according to two versions of the POPSEQ maps, which are hereafter termed Mascher_2013 (Morex × Barke map; Mascher et al., 2013b) and Beier_2017 (Beier et al., 2017). Homology between the selected target loci and HORVU genes (Mascher et al., 2017) was determined using reciprocal blastn algorithm.
To determine the ancestral status, the SNPs were genotyped in silico in two Hordeum species, H. bulbosum and H. pubiflorum, using the Hordeum exome Illumina datasets and the aforementioned bioinformatics pipeline (Mascher et al., 2013a). Alleles that were identical in both species were assigned as ancestral.
De facto captured regions were defined as those with the depth of coverage ≥ 8 in at least one of the samples, which was determined using bedtools v.2.16.2, vcftools v.0.1.11 and R (Danecek et al., 2011; Quinlan, 2014). Functional effects of the SNPs were predicted using snpeff 3.6b with the custom CDS coordinates (Cingolani et al., 2012). The coordinates were determined on the target genomic contigs based on the spidey predictions (Wheelan et al., 2001).
Population structure
The population structure was explored using Bayesian clustering algorithms (structure and instruct), maximum‐likelihood (ML) and Neighbor‐Net phylogenetic analyses and principal component analysis (PCA). For all the population structure analyses, the subset of putatively neutral SNPs based on the snpeff flags with minor allele frequency > 0.05 and missing data frequency < 0.5 was selected. The vcf files were converted into the ped format using the tabix utility of samtools and plink 1.9 (Chang et al., 2015). The SNPs in very high linkage disequilibrium (r 2 > 0.99) were pruned using plink. The PCA was performed using the smartpca utility of the eigensoft software 5.0.2 (Patterson et al., 2006).
The faststructure software (Raj et al., 2014) was applied with 20 iterations for a predefined number of populations (K). The optimal K for wild barley was chosen to represent the model with maximum marginal likelihood tested for K from 2 to 25. The output matrices were summarized using clumpak (Kopelman et al., 2015), reordered and plotted using an in‐house R script. The instruct tools, which extends the structure model to include selfing (Gao et al., 2007), owing to very high computational intensity, was run on 10 randomly drawn subsamples of 1000 SNP markers for five independent chains.
The geographic centers of the populations were calculated as a median of the latitude and longitude of the genotypes comprising the populations. The vector geographic map dataset was downloaded from Natural Earth repository and manipulated in R (http:/www.naturalearthdata.com).
The ML phylogeny rooted to H. bulbosum and H. pubiflorum outgroup species was estimated from the genome‐wide SNP dataset using the GTRCAT model with Lewis's ascertainment bias correction to account for the absence of invariant sites in the alignment and the rapid bootstrap search, which stopped after 200 replicates according to the majority‐rule tree‐based criteria for bootstopping (autoMRE_IGN) implemented in RAxML 8.2.8 (Stamatakis, 2014). The admixed wild genotypes (faststructure single population ancestry component < 95%) were excluded from the input dataset as gene flow between the genotypes may lead to inaccurate placement of the admixed accession on the bifurcating phylogenetic tree. The trees were manipulated using dendroscope 3.5.7 (Huson & Scornavacca, 2012). The Neighbor‐Net phylogenetic network was constructed for all the wild and domesticated accessions using splitstree 4.14.6 (Huson & Bryant, 2006).
Identification of domestication sweeps
The putative signatures of selection related to domestication were identified using several complementary tests – the diversity reduction index (π wild/π domesticated, DRI), Fay&Wu's H norm (Zeng et al., 2006) and the composite likelihood ratio (CLR) test. The π and H norm statistics were calculated for the individual loci and sliding 10 cM windows (step 1 cM) using mstatspop software, which account for missing genotypes in the data, with 1000 permutations (release 0.1b 20150803; http://bioinformatics.cragenomica.es/numgenomics/people/sebas/software/software.html). A sum of segregating and invariant sites was used to normalize the π‐values. The software sweeD 3.3.2 (Pavlidis et al., 2013) was used to calculate the CLR test of Kim & Stephan (2002) as expanded by Nielsen et al. (2005). sweeD calculates the ratio of the maximum composite likelihoods under a neutral model, which uses the genome‐wide site frequency spectrum (SFS) as a reference, to the maximum composite likelihoods under a selective sweep model. The CLR tests were calculated separately for wild and domesticated subsets from the unfolded SFS of individual reference contigs containing at least four SNPs at two grid points across each contig. The genome‐wide reference SFS was calculated using sweeD's ‘‐osfs’ flag and provided for the CLR calculations for the individual loci.
To estimate statistical thresholds of the CLR neutral distribution, following Nielsen et al. (2005), we simulated 1000 datasets assuming a standard neutral model without recombination using the coalescent simulation software ms with the number of segregating sites (S) and the number of samples (n) as the input parameters describing the wild and domesticated barley populations (Hudson, 2002). Variation of the CLR and in the simulated neutral datasets was assessed using sweeD with the threshold to reject neutrality at the 99th percentile of the neutral CLR values. The variation of H norm in the same neutral datasets was assessed using the msstats software (https://github.com/molpopgen/msstats) and the threshold was chosen as the 99.9th percentile to minimize the number of false positives as a result of the likely deviation of barley demographic history from the standard model. For the DRI, the top 95th percentile was used as a cutoff value following Liu et al. (2017). Significance of the overlaps between the tests was estimated using hypergeometric test in R.
Ancestry of domesticated barley genomes
To estimate ancestry of the domesticated barley loci, we calculated pairwise ML distances between each wild and domesticated genotype separately for each locus (i.e. individual contig in the mapping reference; in total 39.6 million comparisons) using the GTRGAMMA model in RAxML 8.2.8 (Stamatakis, 2014). If an allele in a domesticated genotype had a smallest ML distance with a population‐specific wild allele, this wild allele was deemed ancestral for this locus in this domesticated genotype. The cases where a domesticated barley allele was equally distant to wild barley alleles found in several populations represent the instances of incomplete lineage sorting and therefore were not accounted for in a cumulative ancestry of a genotype. A sum of all loci with assigned origin in a single domesticated accession sorted by the locus name we termed an ‘ancestry palette’. The ancestry palettes of the individual accessions were pairwise‐correlated using the Jaccard index‐based similarity measure (J) implemented in R:
where X and Y are vectors of individual elements of the ancestry palettes, i.e. concatenated locus name and corresponding ancestral population (e.g. locus1_population1) in a pair of genotypes. The ancestry similarity of 1 means identical ancestry palettes and that of 0 means that no loci originate from the same population in a pair of accessions. Ancestry similarity heatmaps were visualized using ‘heatmap.2’ function of the ‘gplots’ R package.
The kernel density estimates were plotted using the ‘geom_density’ function of the ggplot2 R package with the values binned into the 0.1× and 2× of the default bin size for the distributions of the longitudes and the similarity indices, respectively.
In this study, we used the term ‘protodomesticated lineage’ to describe descendants of an event when barley cultivation has been initiated from a wild barley population, in which at least one of the selective sweeps found in the modern‐day cultivated barley genomes (detected in this study) occurred. The scripts and the accompanying files used for the analyses are available in an online repository at https://github.com/artempankin/korffgroup.
Results
More than 500 000 SNPs discovered by the targeted resequencing assay
A total of 433 barley accessions, including 344 wild and 89 domesticated barley genotypes, were analyzed in this study. To maximize diversity, the barley genotypes were selected to cover the entire range of wild barley habitats in the Fertile Crescent and to represent the whole variety of domesticated barley life forms from various breeding programs (Table S1). Additionally, domesticated barley is classified into the btr1 (btr1Btr2) and btr2 (Btr1btr2) types based on the allelic status of the spike brittleness genes Btr1 and 2; independent mutations in either of these genes convert the wild‐type brittle spikes into the nonbrittle spikes of the domesticated forms (Pourkheirandish et al., 2015). To further verify representativeness of the selected genotypes, we screened for the Btr mutations using allele‐specific markers. In our genotype set, the btr1 and btr2 types were represented by 71% and 29% of the domesticated accessions, respectively (Table S1).
Illumina enrichment resequencing of 433 barley genotypes yielded c. 8 billion reads (0.56 Tb of data; deposited at NCBI SRA BioProject PRJNA329198; Notes S1). Cumulatively, the captured regions comprised c. 13.8 Mbp (Table S5), 1.33 Mbp of which resided in the coding regions (CDS). Per‐sample analysis of the coverage revealed that c. 87% of the captured regions were covered above the SNP calling threshold and that the between‐sample variation was relatively low, with the median depth of coverage varying from 45 to 130 (Fig. S3). The SNP calling pipeline identified 544 318 high‐quality SNPs, including c. 190 000 singletons (Table S5). Of all the SNPs, 37 870 resided in CDS and c. 43% of them were nonneutral based on the Snpeff predictions. The CDS were more conserved than the noncoding regions with the average SNP density of 29 and 41 SNPs per kbp, respectively; 45% of the SNPs could be located on the barley genetic map, whereas for 37% of the SNPs, only the chromosome could be assigned (Fig. S4).
Patterns of recent admixture between wild and domesticated barley
In domestication studies, where patterns of genetic variation are contrasted between wild and domesticated genotypes, it is critical to distinguish these subgroups and exclude genotypes of unclear provenance. The PCA based on the SNP markers revealed two distinct clusters corresponding to the domesticated and wild subspecies, with multiple genotypes scattered between these clusters (Fig. 1a). faststructure analysis (K = 2) revealed patterns of recent admixture between wild and domesticated subspecies in 36% and 12% of the domesticated and wild genotypes, respectively. These admixed accessions corresponded to the genotypes intermediate between wild and cultivated barley clusters in the PCA (Fig. 1b). Both faststructure and instruct models produced matching admixture patterns (r 2 > 0.99) (Fig. S5). The admixed domesticates did not originate from any specific locality and the admixed wild barley were spread all over the Fertile Crescent, indicating that the admixture was not restricted to any particular geographical area (Table S1). These admixed genotypes of ambiguous provenance were removed from further analyses.
Figure 1.
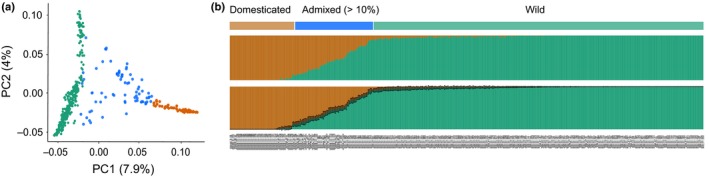
Genome‐wide analysis of admixture between wild and domesticated barley. (a) Principal component (PC) analysis of 433 barley genotypes. The first two PCs discern subgroups of wild (green) and domesticated (orange) barley and admixed (blue) genotypes. A percentage of the total variation explained by the PCs is shown in parentheses. (b) Global genetic ancestry of the wild and domesticated barley genotypes as determined by the population structure analysis using faststructure (c. 315 000 SNPs) and instruct (10 random samples of 1000 SNPs) models – upper and lower panels, respectively. Proportions of wild and domesticated ancestral clusters are shown as green and orange vertical bars, respectively. The standard deviations on the instruct plots are shown as whiskers.
Footprints of domestication‐related selection
Selection acting on a beneficial mutation affects various aspects of genetic variation such as allele frequency, nucleotide diversity and linkage disequilibrium in the neighboring regions in a process called selective sweep. To scan for signatures of selective sweeps, which occurred during domestication (hereafter domestication sweep), we performed genome scans using several statistics – the CLR, Fay & Wu's H norm, and the DRI (π wild/π domesticated). These statistics explore different patterns of molecular variation and therefore presumably reveal signatures left by selection under different scenarios (Innan and Kim 2004). Both the CLR and H norm statistics are site frequency‐based and describe variation of the SFS at the tested loci. The H norm statistics detects enrichment of the high‐frequency derived alleles (the right tail of an unfolded SFS), whereas sweeD CLR takes advantage of the patterns of variability in the complete SFS scanned along the sequence length. Strong deviations of these statistics from the expected genome‐wide values, as tested by coalescent simulations under the neutral scenario, indicate selection. The DRI statistics reveals a severe depletion of nucleotide diversity in the domesticated genotypes at certain loci detected as statistical outliers. All three statistics have frequently been used in the selection scans to reveal domestication sweeps and candidate domestication genes in other crop species (Huang et al., 2012; Lin et al., 2014; Wang et al., 2014; Civáň et al., 2015; Velasco et al., 2016; Zhong et al., 2017).
Altogether the scans identified 137 outlier contigs carrying signatures of a selective sweep – 91 of them could be located on the map and covered all barley chromosomes (Table S6). Of those, 20 contigs (16 and 14 mapped locations on Mascher_2013b and Beier_2017 maps, respectively) were outliers in at least two of the scans (Figs 2, S6; Table S6). The overlap between the CLR and H norm scans was relatively high, 38% (P‐value < 1.0e–07), and the overlaps between the DRI scan and the other two tests were significant but less prominent (8–10%; P‐value < 0.05) consistent with the previously reported values (Liu et al., 2017).
Figure 2.
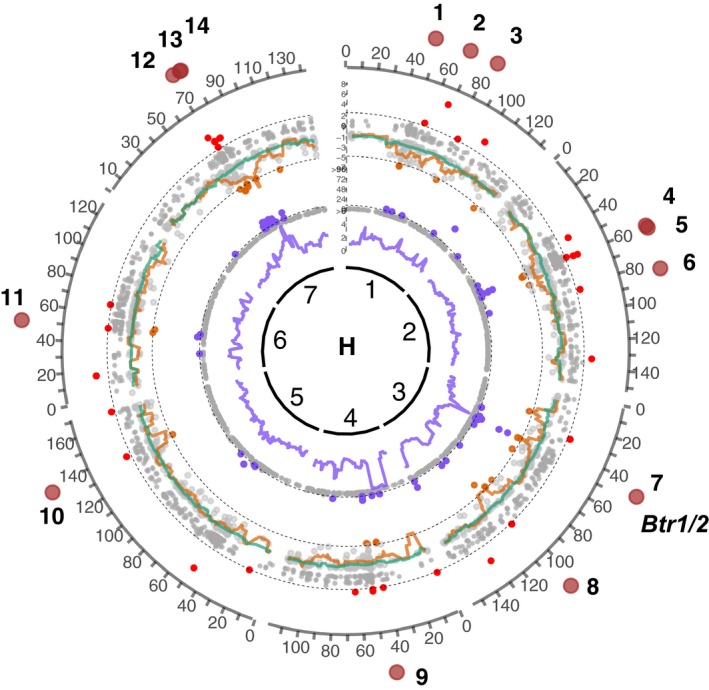
Genomic signatures of domestication selective sweeps (Beier_2017 map). Genome scans for signatures of selection associated with domestication. The sliding‐window and individual‐target values are shown as lines and points, respectively. The innermost circle represents barley linkage groups (H) followed by the diversity reduction index (π wild/π dom) (violet); the normalized Fay&Wu's H norm statistics for the wild (green) and domesticated (orange) groups; and the composite likelihood ratio statistics (sweeD CLR) for the domesticated group (red). The outlier thresholds are shown by dashed lines and the nonoutlier loci are shown as gray dots for all the tests. Fourteen candidate selected regions supported by at least two of the statistics are shown as brown circles on the outermost layer. Btr1/2, brittle rachis domestication genes (Pourkheirandish et al., 2015).
Among the top outliers including the loci in the overlaps between the tests were homologs of genes implicated in the modulation of the light signaling, circadian clock, and carbohydrate metabolism pathways (Table S6). None of the candidate domestication genes identified in this study have been functionally characterized in barley; however, putative function can often be inferred from homology.
In our scans, the barley homolog of the Arabidopsis gene EMPFINDLICHER IM DUNKELROTEN LICHT 1 – like 3 (EDL3) of the EID1 gene family had the strongest CLR signal among all the genes (seq375; CLR = 7.13). In Arabidopsis, EDL3 has been implicated in regulation of the photoperiod pathway and ABA signaling, whereas in tobacco, the EDL3 homolog encodes a key element of the circadian clock (Koops et al., 2011; Xu and Johnson, 2001). In tomato, a circadian clock gene homologous to EID1 has been implicated in domestication (Müller et al., 2016). The homologs of the Arabidopsis CULLIN4 (CUL4; seq442, AK371672; H norm = −5.1, CLR = 2.7) and SUPRESSOR OF PHYA 2 (SPA2; seq108, MLOC_52815; DRI = 53), encoding two members of the COP1‐CUL4‐SPA protein complex implicated in regulation of the light signaling pathway, are other examples where strong signatures of selection have been found in our scans and in another crop species (Zhu et al., 2008). In the common bean, the homologs of CUL4 and CONSTITUTIVELY PHOTOMORPHOGENIC 1 (COP1) – a gene encoding another member of the COP1‐CUL4‐SPA complex – have been independently targeted by selection in two separate domestication events (Schmutz et al., 2014). Two genes of the starch metabolism pathway – the homologs of the alpha‐ and beta‐amylases (AMY, seq669, DRI = 23; BAM1, seq345, H norm = −5.32) – were strong outliers in the DRI and H norm scans. Previous studies discovered reduced variation at the alpha‐amylase locus in barley domesticates compared with wild genotypes and hinted at the functional divergence of the wild and domesticated alpha‐amylase alleles (Kilian et al., 2006; Cu et al., 2013).
The location of the only known barley domestication locus Btr1/2 coincided with the domestication sweep 6 on the chromosome 3 and thus the Btr1/2 genes were probably direct targets of selection within the sweep 6 (Fig. 2). The location of selective sweeps 13–16 presumably corresponded to the region of depleted diversity on the chromosome 7 discovered by Russell et al. (2016), who speculated that the NUD gene, controlling the naked (hulless) grain phenotype (Taketa et al., 2004), might have been a direct target of domestication in this region. In our scans, the NUD gene itself did not carry a selection signature and thus apparently was not the target of selection at this locus. Indeed, both hulless and hulled genotypes are ubiquitously present in the domesticated barley gene pool and thus the naked grain phenotype represents an improvement but not a domestication trait (Saisho & Purugganan, 2007).
Population structure and ancestry analyses
Next, we investigated whether the local ancestry patterns in cultivated barley genomes could shed light on the phylogeographic history of barley domestication. To this end, we first explored the population structure of wild barley genotypes using Bayesian clustering, PCA and phylogenetic analyses and then searched within the wild gene pool for putative ancestral alleles for each locus of the domesticated genotypes using the ML approach (see the Materials and Methods section; Fig. S7). Following a widely held assumption, we assumed that any individual genomic locus in a domesticated barley accession descended from a wild population that carried the phylogenetically closest allele and that the habitat of this wild population indicates a place of origin of the cultivated barley allele (Civáň et al., 2015; Pourkheirandish et al., 2015). The cases where a putative ancestral allele was not specific to a single wild population were excluded from the analysis to alleviate adverse effects of the incomplete lineage sorting on the ancestry estimates.
Nine wild barley populations were suggested by faststructure, which corresponded to the clearly defined clusters on the ML phylogenetic tree and the Neighbor‐Net network (Figs 3abc, S8, S9, S10; Notes S2). The domesticated barley genotypes branched off as a monophyletic cluster on the ML phylogram at a sister position to the cluster of wild barley genotypes (Fig. S9). This apparently inaccurate position of the domesticated barley cluster on the ML phylogeny indicated a complex reticulate genealogy of the domesticated barley genomes that could not be reliably described by a bifurcating tree.
Six wild populations (Carmel and Galilee (CG), Golan Heights (GH), Hula Valley and Galilee (HG), Judean Desert and Jordan Valley (JJ), Negev Mountains (NM), Sharon, Coastal Plain and Judean Lowlands (SCJ)) were concentrated in the South Levant and the other three (Lower Mesopotamia (LM), North Levant (NL) and Upper Mesopotamia (UM)) occupied large areas of the northern and eastern Fertile Crescent. Habitats of the wild populations were distinct, with very few immigrants and genotypes of mixed ancestry occurring mostly in the borders of overlapping areas (Figs S11, S12). Only 23 wild accessions had a highly admixed ancestry and could not be attributed to any of the nine populations (Fig. 3a).
Figure 3.
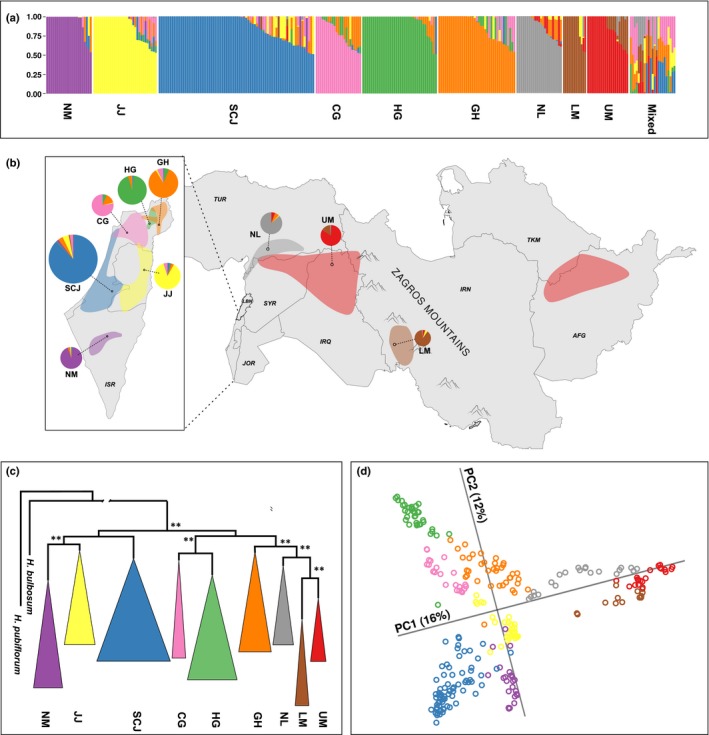
Geographic distribution, structure and phylogeny of nine wild barley (Hordeum vulgare ssp. spontaneum) populations. The colors correspond to the nine wild barley populations: Carmel & Galilee (CG, pink); Golan Heights (GH, orange); Hula Valley & Galilee (HG, green); Judean Desert & Jordan Valley (JJ, yellow); Lower Mesopotamia (LM, brown); Negev Mountains (NM, magenta); North Levant (NL, gray); Sharon, Coastal Plain & Judean Lowlands (SCJ, blue); and Upper Mesopotamia (UM, red). (a) Population structure of wild barley as determined by faststructure for K = 9. Vertical bars correspond to individual genotypes and colors indicate their membership in the nine subpopulations. (b) Distribution of the wild barley populations within the Fertile Crescent. The pie charts represent the ancestral composition of the populations as determined by faststructure and are connected to the geographic centers of population distributions by dashed lines. The size of the pie charts reflects the number of genotypes in the populations. The country codes (ISO 3166) are shown in italics. (c) The maximum likelihood phylogeny of wild barley accessions (faststructure single population ancestry component > 95%). The clusters were collapsed based on the population assignment. Hordeum bulbosum and Hordeum pubiflorum were used as distant outgroup species and the length of the outgroup branch was artificially shortened. The branches with the bootstrap support values > 0.6 are labeled by double asterisks. (d) Principal component (PC) analysis of wild barley accessions (faststructure single population ancestry component > 95%). Only first two PCs are shown. A percentage of the total variation explained by the PCs is shown in parentheses.
Principal component analysis of wild barley genotypes identified nine clusters, which were significantly different between each other (pairwise P‐values < 1e–55) and corresponded to the wild barley populations defined by faststructure (Fig. 3d; Table S7). The patterns of genetic diversity revealed by PCA mirrored the patterns of geographic distribution of the wild barley populations within the Fertile Crescent.
A putative wild ancestor could be assigned for 1232 loci separately in each domesticated genotype (Fig. 4a). In all, 60% of the loci were monophyletic, i.e. descended from the same wild barley population in all domesticated genotypes, whereas ancestry of 40% of the loci could be traced back to several wild populations across the domesticated genotypes. For further analyses, we separated the dataset into two parts: the candidate domestication loci, to identify geographical origin of the domestication sweep events even without knowing direct targets of selection; and the rest of the genome, which we tentatively termed neutral, to search for the genome‐wide signatures of independent founder lineages.
Figure 4.
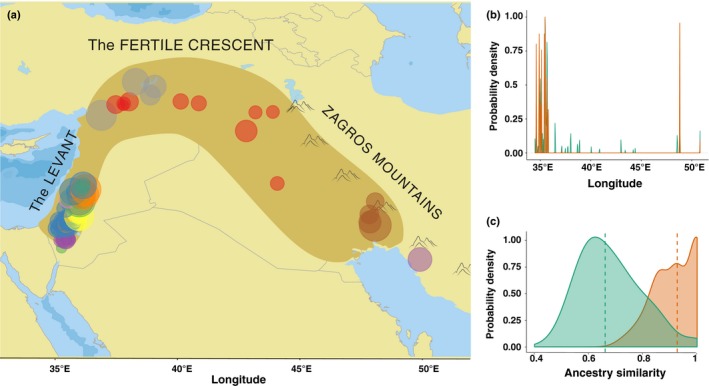
Origin and ancestral composition of the domesticated barley genomes. (a) Geographic distribution of wild barley accessions, which carry ancestral haplotypes of the domesticated barley (Hordeum vulgare ssp. vulgare) loci, within the Fertile Crescent. The colors correspond to the nine wild barley (H. vulgare ssp. spontaneum) populations: Carmel & Galilee (pink); Golan Heights (orange); Hula Valley & Galilee (green); Judean Desert & Jordan Valley (yellow); Lower Mesopotamia (brown); Negev Mountains (magenta); North Levant (gray); Sharon, Coastal Plain & Judean Lowlands (blue); and Upper Mesopotamia (red). (b) Longitudinal distribution of the ancestral wild haplotypes for the neutral (green) and domestication sweep loci (orange) shown as kernel density plots. (c) Distribution of the pairwise ancestry similarity coefficients estimated for the neutral (green) and domestication sweep loci (orange) shown as kernel density plots. The vertical dashed lines indicate the median similarity values for the corresponding subgroups.
The geographical distribution of the ancestral haplotypes of the neutral loci suggested that wild barley populations from every sampled part of the Fertile Crescent contributed to the domesticated genomes (Fig. 4a). In contrast to the neutral loci, the longitudinal distribution of the haplotypes ancestral to the domestication sweep loci was significantly different (Kolmogorov–Smirnov test, P‐value < 1e–12) (Fig. 4b). The domestication sweep loci descended from two discernible clusters of wild barley genotypes in the eastern and western parts of the Fertile Crescent (Fig. 4b). Intriguingly, the proportions of individual contributions of the ancestral wild populations did not differ noticeably between the domesticated genotypes, hinting at a single, highly admixed progenitor lineage at the root of domesticated barley (Fig. S13).
To gather further evidence on this hypothesis, we quantified the similarity of the ancestry patterns in the genomes of the domesticates by pairwise correlation of the sorted ancestry palettes of individual accessions (Fig. S14). The ancestry palettes of the neutral loci were only moderately similar (median 0.64), which means that, for multiple loci, the patterns of ancestry were not consistent across the domesticated genotypes (Fig. 4c). Apparently this dissimilarity of the ancestral patterns at multiple loci resulted from a gene flow, which randomly shuffled alleles descending from different ancestral wild populations between the domesticated genotypes. By contrast, the ancestry palettes of the domestication sweep loci were remarkably similar (median 0.96) across the domesticated genotypes compared with the neutral genome, indicating that the randomizing effect of gene flow was considerably weaker at the genomic regions maintaining the domestication syndrome (Fig. 4c). It is noteworthy that this difference between the similarity of the ancestry palettes in the neutral and domestication sweep loci did not arise from the unbalanced number of loci in the subgroups (Fig. S15). In both subsets, the heatmaps of the ancestry similarity did not reveal any clear‐cut patterns, e.g. presence of several discernible clusters, which could be interpreted as a signal of distinct founder lineages (Fig. S16).
Several domestication models may explain the discovered ancestry patterns. One of the candidate scenarios implicates independent protodomesticated lineages, which originated from several founder events in the eastern and western parts of the Fertile Crescent. These lineages could have been combined by means of gene flow into a single admixed progenitor lineage, which was at the root of the domesticated barley genotypes (Fig. S17). An alternative hypothesis suggests that a single founder lineage of domestication may have experienced gene flow from the wild populations and that the selective sweeps occurred sequentially in the background of the ancestral fragments of heterogeneous origins (Fig. S18). In both cases, continuing gene flow between wild and domesticated subspecies further randomized the ancestry patterns of the modern domesticated genotypes, particularly at the selectively neutral loci.
Discussion
Selection scans reveal multiple domestication sweeps in barley
Our understanding of the genes and traits that constitute the barley domestication syndrome is extremely limited. Here, the selection scans identified a domestication sweep at the Btr1/2 locus, which modulates spike brittleness – the only studied example of a crucial domestication trait (sensu Abbo et al., 2014). We also discovered multiple novel candidate domestication genes, which are implicated in the regulation of light signaling, circadian clock, and carbohydrate metabolism pathways. It is noteworthy that the domestication loci detected in this study may be only a representative sample of the truly selected loci. Some loci might have experienced selection regimes leaving signatures that escape detection, confounded with the effects of demography, or that are missed because of the gaps in certain regions of the genetic and physical maps (Teshima et al., 2006; Mascher et al., 2013b, 2017; Beier et al., 2017).
Intriguingly, we found examples of genes carrying selection signatures in both barley and other crops, suggesting convergence of domestication‐related selection on homologous developmental pathways and protein complexes in different crop species. The most prominent examples were a circadian clock gene of the EID1 family, the homolog of which is a domestication gene in tomato (Müller et al., 2016), and genes SPA and CUL4 encoding components of the E3 ubiquitin‐ligase COP1‐CUL4‐SPA, which was targeted by domestication of common bean. The COP1‐CUL4‐SPA complex is a critical part of the far‐red light signaling, photoperiod and circadian clock pathways (Zhu et al., 2015).
This finding adds to the growing evidence that components of the circadian clock, light signaling and shade‐avoidance pathways were targets of adaptive selection during domestication or further adaptation to new agronomic environments in various crop species (Faure et al., 2012; Zakhrabekova et al., 2012; Müller et al., 2016; Shor & Green, 2016). However, the evolutionary role played by such modifications in the domestication syndrome has not been understood. Müller et al. (2016) suggested that modification of the circadian clock was a human‐mediated adaptation of cultivated tomato, which was domesticated in the equatorial regions, to long photoperiods of the northern latitudes. In our study, many of the domesticated barley genotypes originated from the same latitude where barley domestication ensued, which makes, in the case of barley, the scenario of adaptation to a latitudinal cline less plausible. An alternative hypothesis might be linked to the fact that many crop plants are cultivated in dense stands that result in dramatic changes in the light environment and, as a consequence, alter plant architecture compared with their wild ancestor species. Therefore, we propose that such common patterns in the crop adaptation to agricultural practices might be the key to understanding the involvement of the modulators of light signaling, circadian clock and shade‐avoidance pathways in domestication.
Ancestry of the candidate domestication loci relates the mosaic model and the process of domestication
Identification of the candidate domestication genes enables the phylogeorgaphic origin of the domestication sweep events to be predicted, which, together with the surveys using neutral markers revealing the closest wild ancestor of the domesticated populations at the genome‐wide level, represent two complementary approaches to untangling domestication histories (Badr et al., 2000; Matsuoka et al., 2002; Morrell & Clegg, 2007; Huang et al., 2012; Civáň et al., 2015; Poets et al., 2015; Pourkheirandish et al., 2015). Here, the heterogeneous ancestry of the candidate domestication loci provided compelling evidence that multiple domestication sweeps occurred in the background of various founder populations of wild barley. The ancestral populations of the domestication sweep loci were confined to the eastern and western parts of the Fertile Crescent.
The dominant narrative of the barley domestication history has long since revolved around the idea of the two independent domesticated lineages originating in the Levantine (west) and Zagros (east) horns of the Fertile Crescent (hereafter east–west model). It stems from the finding suggesting the existence of the Occidental and Oriental types of domesticated barley corresponding to the btr1 and btr2 types, respectively (Takahashi, 1955). Later, the east–west model was supported by molecular analyses of the barley population structure, as well as by archaeological studies (Azhaguvel & Komatsuda, 2007; Morrell & Clegg, 2007; Riehl et al., 2012, 2013; Tanno & Willcox, 2012; Fang et al., 2014; Morrell et al., 2014). However, the patterns of geographical distribution of the functional btr1 and btr2 mutations challenged the simplicity of the east–west model – in addition to differentiation of the btr1 and btr2 mutations along the east–west gradient in the Fertile Crescent, another latitudinal cline in the btr1 and btr2 allele frequencies became apparent (Pourkheirandish et al., 2015).
Our findings based on the origin of the domestication sweep loci expand the east–west model by suggesting that not only two but multiple protodomesticated lineages may have existed in the past in the Levantine and Zagros clusters of the origin of agriculture. The presence of a third mutation conferring the nonbrittle rachis phenotype of domesticated barley supports this hypothesis (Civáň & Brown, 2017).
Recently, based on the genome‐wide SNP genotyping data, Poets et al. (2015) presented the mosaic model of barley domestication, which suggests that the genomes of modern barley landraces consist of a mixture of ancestral blocks originating in the five wild barley populations from different parts of the Fertile Crescent (Allaby, 2015; Poets et al., 2015). We found that, in contrast to the domestication sweep regions, the neutral partition of the domesticated barley genomes comprised ancestral blocks that descended from all nine wild barley populations corroborating the mosaic model. Moreover, the ancestral patterns at the neutral loci were not very similar across the genotypes. This indicates that, after domestication, the gene flow between the wild and domesticated subspecies and the domesticates themselves continued reshuffling the ancestral blocks in the modern domesticated genotypes, thus erasing the genome‐wide signatures of independent protodomesticated lineages. A simulation study demonstrated that, in the case of neutral markers, the gene flow between the independent domestication lineages indeed hinders identification of the founder events (Allaby et al., 2008). By contrast, the domestication sweep loci had nearly uniform ancestry patterns across the genotypes. This shows the importance of retaining specific ancestral alleles of the domestication genes, which are critical for maintaining the domestication syndrome traits.
Involvement of gene flow in domestication has been documented in other crop species (Huang et al., 2012; Civáň et al., 2013; Hufford et al., 2013). The model of rice domestication is arguably the most vivid example. In rice, two different, possibly extinct wild Oryza rufipogon populations were ancestors of the indica and japonica domesticated subspecies; however, the domestication sweep loci originated once in japonica and were later introgressed into the indica lineage (Fuller, 2011; Huang et al., 2012; Huang & Han, 2015; Choi et al., 2017; but see Civáň et al., 2015). We suggest that, in contrast to the rice model, in the barley domestication history, gene flow was not an isolated event but probably a continuous process, which ensued in the early domestication era and was apparently facilitated by modern breeding. Indeed, the genome of a 6000‐yr‐old barley landrace carried signatures of the wild barley introgressions, thus confirming instances of the gene flow in the early domesticates (Mascher et al., 2016).
We have yet to understand the exact nature and sequence of demographic events that formed the complex mosaic ancestry patterns during the apparently protracted process of barley domestication. Involvement of several wild populations and abundant continuous gene flow in the process of barley domestication greatly complicates explicit modeling of a realistic demographic history of its domestication. What is clear, however, is that it was not constrained to a single center of domestication and involved intensive exchange of the early domesticates between the Neolithic farming communities. Recent evidence predicting migration between the early agriculturalist settlements of the eastern and western parts of the Fertile Crescent hints at the likelihood of such a scenario (Lazaridis et al., 2016).
To further unravel barley domestication history, characterization of direct targets of selection within the domestication sweep regions is of the utmost importance. Our catalog of the candidate domestication loci will facilitate future efforts to characterize novel domestication genes, which modulate as yet unstudied aspects of the barley domestication syndrome.
Author contributions
A.P. and M.v.K. planned and designed the research. J.A. and C.B conducted the enrichment sequencing. A.P. analyzed the data. A.P. and M.v.K. wrote and revised the manuscript.
Supporting information
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Selection of target genes.
Fig. S2 The data analysis pipeline – read filtering, mapping, SNP calling and genotyping.
Fig. S3 Characteristics of coverage.
Fig. S4 Distribution of SNP markers over the barley chromosomes.
Fig. S5 Correlation of the ancestry coefficients estimated using faststructure and instruct.
Fig. S6 Genomic signatures of domestication selective sweeps (Mascher_2013 map).
Fig. S7 Procedure for estimating the ancestry of domesticated barley haplotypes.
Fig. S8 Population structure of wild barley (K = 9) determined by faststructure and instruct.
Fig. S9 The maximum likelihood (ML) unrooted phylogeny of 230 nonadmixed barley accessions.
Fig. S10 The Neighbor‐Net phylogenetic network of 359 barley accessions.
Fig. S11 Distribution of the wild barley populations within the Fertile Crescent.
Fig. S12 Distribution of the wild barley populations in Israel, the West Bank and Gaza.
Fig. S13 Unsorted ancestral palettes of the candidate domestication loci.
Fig. S14 Sorted ancestry palettes of the domesticated barley genotypes.
Fig. S15 Estimation of the median ancestry coefficients in the unbalanced subgroups of loci.
Fig. S16 Heatmaps of the pairwise ancestry similarity coefficients.
Fig. S17 A simplified candidate demographic model implying multiple domestication lineages.
Fig. S18 A simplified candidate demographic model implying a single domestication lineage.
Table S1 Characteristics of wild and cultivated barley accessions
Table S2 Sequences selected for targeted enrichment design
Table S3 Gene ontology (GO) terms of selected Arabidopsis genes
Table S4 Composition of the mapping reference
Table S5 Characteristics of the enrichment assay and SNP calling
Table S6 Characteristics of outlier loci identified by the selection scans
Table S7 Statistical assessment of wild barley population clustering by ‘smartpca’
Methods S1 Selection of genes for targeted enrichment assay.
Methods S2 Mapping reference design.
Methods S3 Quality check, mapping and SNP calling pipeline.
Notes S1 Characteristics of the enrichment assay.
Notes S2 Wild barley population structure – a note of caution.
Acknowledgements
We cordially thank Kerstin Luxa, Teresa Bisdorf, Caren Dawidson, Elisabeth Kirst and Andrea Lossow for excellent technical assistance; Eyal Fridman, Hakan Özkan and Benjamin Kilian for barley seeds. This article benefited greatly from the critical comments of Angela Hancock and two anonymous referees. This work was supported by the Max Planck Society and by Deutsche Forschungsgemeinschaft grants (DFG SPP1530 ‘Flowering time control: from natural variation to crop improvement’) and the Excellence Cluster (EXC1028). A.P. was supported by an IMPRS fellowship from the Max Planck Society.
Contributor Information
Artem Pankin, Email: pankin@mpipz.mpg.de.
Maria von Korff, Email: korff@mpipz.mpg.de.
References
- Abbo S, Lev‐Yadun S, Gopher A. 2010. Agricultural origins: centers and noncenters; a Near Eastern reappraisal. Critical Reviews in Plant Science 29: 317–328. [Google Scholar]
- Abbo S, van‐Oss RP, Gopher A, Saranga Y, Ofner I, Peleg Z. 2014. Plant domestication versus crop evolution: a conceptual framework for cereals and grain legumes. Trends in Plant Sciences 19: 351–360. [DOI] [PubMed] [Google Scholar]
- Allaby RG. 2015. Barley domestication: the end of a central dogma? Genome Biology 16: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaby RG, Fuller DQ, Brown TA. 2008. The genetic expectations of a protracted model for the origins of domesticated crops. Proceedings of the National Academy of Sciences, USA 105: 13982–13986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azhaguvel P, Komatsuda T. 2007. A phylogenetic analysis based on nucleotide sequence of a marker linked to the brittle rachis locus indicates a diphyletic origin of barley. Annals of Botany 100: 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badr A, Sch R, Rabey HE, Effgen S, Ibrahim HH, Pozzi C, Rohde W, Salamini F. 2000. On the origin and domestication history of barley (Hordeum vulgare). Molecular Biology and Evolution 17: 499–510. [DOI] [PubMed] [Google Scholar]
- Beier S, Himmelbach A, Colmsee C, Zhang XQ, Barrero RA, Zhang Q, Li L, Bayer M, Bolser D, Taudien S et al 2017. Construction of a map‐based reference genome sequence for barley, Hordeum vulgare L.. Science Data 4: 170044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. 2015. Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JY, Platts AE, Fuller DQ, Wing RA, Purugganan MD. 2017. The rice paradox: multiple origins but single domestication in Asian rice. Molecular Biology and Evolution 34: 969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civáň P, Brown TA. 2017. A novel mutation conferring the nonbrittle phenotype of cultivated barley. New Phytologist 214: 468–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civáň P, Craig H, Cox CJ, Brown TA. 2015. Three geographically separate domestications of Asian rice. Nature Plants 1: 15164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civáň P, Ivaničová Z, Brown TA. 2013. Reticulated origin of domesticated emmer wheat supports a dynamic model for the emergence of agriculture in the Fertile Crescent. PLoS ONE 8: e81955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cu S, Roumeliotis S, Eglinton J. 2013. Alpha‐amylase allelic variation in domesticated and wild barley In: Guoping Z, Chengdao L, Xu L, eds. Advance in barley sciences. Dordrecht, the Netherlands: Springer, 57–68. [Google Scholar]
- Dai F, Nevo E, Wu D, Comadran J, Zhou M, Qiu L, Chen Z, Beiles A, Chen G, Zhang G. 2012. Tibet is one of the centers of domestication of cultivated barley. Proceedings of the National Academy of Sciences, USA 109: 16969–16973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST. 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson IK, Russell J, Powell W, Steffenson B, Thomas WTB, Waugh R. 2015. Barley: a translational model for adaptation to climate change. New Phytologist 206: 913–931. [DOI] [PubMed] [Google Scholar]
- Fang Z, Gonzales AM, Clegg MT, Smith KP, Muehlbauer GJ, Steffenson BJ, Morrell PL. 2014. Two genomic regions contribute disproportionately to geographic differentiation in wild barley. G3 (Bethesda) 4: 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure S, Turner AS, Gruszka D, Christodoulou V, Davis SJ, von Korff M, Laurie DA. 2012. Mutation at the circadian clock gene EARLY MATURITY 8 adapts domesticated barley (Hordeum vulgare) to short growing seasons. Proceedings of the National Academy of Sciences, USA 109: 8328–8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DQ. 2011. Pathways to Asian civilizations: tracing the origins and spread of rice and rice cultures. Rice 4: 78–92. [Google Scholar]
- Fuller DQ, Asouti E, Purugganan MD. 2012. Cultivation as slow evolutionary entanglement: comparative data on rate and sequence of domestication. Vegetation History and Archaeobotany 21: 131–145. [Google Scholar]
- Fuller DQ, Willcox G, Allaby RG. 2011. Early agricultural pathways: moving outside the ‘core area’ hypothesis in Southwest Asia. Journal of Experimental Botany 63: 617–633. [DOI] [PubMed] [Google Scholar]
- Gao H, Williamson S, Bustamante CD. 2007. A markov chain monte carlo approach for joint inference of population structure and inbreeding rates from multilocus genotype data. Genetics 176: 1635–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer K. 1984. Das domestikationssyndrom. Kult 32: 11–34. [Google Scholar]
- Harlan JR, Zohary D. 1966. Distribution of wild wheats and barley. Science 153: 1074–1080. [DOI] [PubMed] [Google Scholar]
- Heun M, Abbo S, Lev‐Yadun S, Gopher A. 2012. A critical review of the protracted domestication model for Near‐Eastern founder crops: linear regression, long‐distance gene flow, archaeological, and archaeobotanical evidence. Journal of Experimental Botany 63: 4333–4341. [DOI] [PubMed] [Google Scholar]
- Higgins JA, Bailey PC, Laurie DA. 2010. Comparative genomics of flowering time pathways using Brachypodium distachyon as a model for the temperate grasses. PLoS ONE 5: e10065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Han B. 2015. Rice domestication occurred through single origin and multiple introgressions. Nature Plants 2: 15207. [DOI] [PubMed] [Google Scholar]
- Huang X, Kurata N, Wei X, Wang Z‐X, Wang A, Zhao Q, Zhao Y, Liu K, Lu H, Li W et al 2012. A map of rice genome variation reveals the origin of cultivated rice. Nature 490: 497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. 2002. Generating samples under a Wright‐Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Hufford MB, Lubinksy P, Pyhäjärvi T, Devengenzo MT, Ellstrand NC, Ross‐Ibarra J. 2013. The genomic signature of crop‐wild introgression in maize. PLoS Genetics 9: e1003477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufford MB, Xu X, van Heerwaarden J, Pyhäjärvi T, Chia J‐M, Cartwright RA, Elshire RJ, Glaubitz JC, Guill KE, Kaeppler SM et al 2012. Comparative population genomics of maize domestication and improvement. Nature Genetics 44: 808–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution 23: 254–267. [DOI] [PubMed] [Google Scholar]
- Huson DH, Scornavacca C. 2012. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Systematic Biology 61: 1061–1067. [DOI] [PubMed] [Google Scholar]
- Innan H, Kim Y. 2004. Pattern of polymorphism after strong artificial selection in a domestication event. Proceedings of the National Academy of Sciences, USA 101: 10667–10672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Barley Genome Sequencing Consortium (IBGSC) . 2012. A physical, genetic and functional sequence assembly of the barley genome. Nature 491: 711–716. [DOI] [PubMed] [Google Scholar]
- Kilian B, Özkan H, Kohl J, von Haeseler A, Barale F, Deusch O, Brandolini A, Yucel C, Martin W, Salamini F. 2006. Haplotype structure at seven barley genes: relevance to gene pool bottlenecks, phylogeny of ear type and site of barley domestication. Molecular Genetics and Genomics 276: 230–241. [DOI] [PubMed] [Google Scholar]
- Kim Y, Stephan W. 2002. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160: 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koops P, Pelser S, Ignatz M, Klose C, Marrocco‐Selden K, Kretsch T. 2011. EDL3 is an F‐box protein involved in the regulation of abscisic acid signalling in Arabidopsis thaliana . Journal of Experimental Botany 62: 5547–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I. 2015. Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Molecular Ecology Resources 15: 1179–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis I, Nadel D, Rollefson G, Merrett DC, Rohland N, Mallick S, Fernandes D, Novak M, Gamarra B, Sirak K et al 2016. Genomic insights into the origin of farming in the ancient Near East. Nature 536: 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev‐Yadun S, Gopher A, Abbo S. 2000. The cradle of agriculture. Science 288: 1602–1603. [DOI] [PubMed] [Google Scholar]
- Lin T, Zhu G, Zhang J, Xu X, Yu Q, Zheng Z, Zhang Z, Lun Y, Li S, Wang X et al 2014. Genomic analyses provide insights into the history of tomato breeding. Nature Genetics 46: 1220–1226. [DOI] [PubMed] [Google Scholar]
- Liu Q, Zhou Y, Morrell PL, Gaut BS. 2017. Deleterious variants in Asian rice and the potential cost of domestication. Molecular Biology and Evolution 34: 908–924. [DOI] [PubMed] [Google Scholar]
- Mascher M, Gundlach H, Himmelbach A, Beier S, Twardziok SO, Wicker T, Radchuk V, Dockter C, Hedley PE, Russell J et al 2017. A chromosome conformation capture ordered sequence of the barley genome. Nature 544: 427–433. [DOI] [PubMed] [Google Scholar]
- Mascher M, Richmond TA, Gerhardt DJ, Himmelbach A, Clissold L, Sampath D, Ayling S, Steuernagel B, Pfeifer M, D'Ascenzo M et al 2013a. Barley whole exome capture: a tool for genomic research in the genus Hordeum and beyond. Plant Journal 76: 494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher M, Muehlbauer GJ, Rokhsar DS, Chapman J, Schmutz J, Barry K, Muñoz‐Amatriaín M, Close TJ, Wise RP, Schulman AH et al 2013b. Anchoring and ordering NGS contig assemblies by population sequencing (POPSEQ). Plant Journal 76: 718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher M, Schuenemann VJ, Davidovich U, Marom N, Himmelbach A, Hübner S, Korol A, David M, Reiter E, Riehl S et al 2016. Genomic analysis of 6,000‐year‐old cultivated grain illuminates the domestication history of barley. Nature Genetics 48: 1089–1093. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Vigouroux Y, Goodman MM, Sanchez J, Buckler E, Doebley J. 2002. A single domestication for maize shown by multilocus microsatellite genotyping. Proceedings of the National Academy of Sciences, USA 99: 6080–6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer RS, Purugganan MD. 2013. Evolution of crop species: genetics of domestication and diversification. Nature Reviews Genetics 14: 840–852. [DOI] [PubMed] [Google Scholar]
- Molina‐Cano JL, Moralejo M, Igartua E, Romagosa I. 1999. Further evidence supporting Morocco as a centre of origin of barley. Theoretical and Applied Genetics 98: 913–918. [Google Scholar]
- Morrell PL, Clegg MT. 2007. Genetic evidence for a second domestication of barley (Hordeum vulgare) east of the Fertile Crescent. Proceedings of the National Academy of Sciences, USA 104: 3289–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrell PL, Gonzales AM, Meyer KKT, Clegg MT. 2014. Resequencing data indicate a modest effect of domestication on diversity in barley: a cultigen with multiple origins. Journal of Heredity 105: 253–264. [DOI] [PubMed] [Google Scholar]
- Müller NA, Wijnen CL, Srinivasan A, Ryngajllo M, Ofner I, Lin T, Ranjan A, West D, Maloof JN, Sinha NR. 2016. Domestication selected for deceleration of the circadian clock in cultivated tomato. Nature Genetics 48: 89–93. [DOI] [PubMed] [Google Scholar]
- Negassa M. 1985. Patterns of phenotypic diversity in an Ethiopian barley collection, and the Arussi‐Bale Highland as a center of origin of barley. Hereditas 102: 139–150. [Google Scholar]
- Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, Bustamante C. 2005. Genomic scans for selective sweeps using SNP data. Genome Research 15: 1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankin A, von Korff M. 2017. Co‐evolution of methods and thoughts in cereal domestication studies: a tale of barley (Hordeum vulgare). Current Opinion in Plant Biology 36: 15–21. [DOI] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D. 2006. Population structure and eigenanalysis. PLoS Genetics 2: e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Živković D, Stamatakis A, Alachiotis N. 2013. SweeD: likelihood‐based detection of selective sweeps in thousands of genomes. Molecular Biology and Evolution 30: 2224–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poets AM, Fang Z, Clegg MT, Morrell PL. 2015. Barley landraces are characterized by geographically heterogeneous genomic origins. Genome Biology 16: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourkheirandish M, Hensel G, Kilian B, Senthil N, Chen G, Sameri M, Azhaguvel P, Sakuma S, Dhanagond S, Sharma R et al 2015. Evolution of the grain dispersal system in barley. Cell 162: 527–539. [DOI] [PubMed] [Google Scholar]
- Purugganan MD, Fuller DQ. 2011. Archaeological data reveal slow rates of evolution during plant domestication. Evolution 65: 171–183. [DOI] [PubMed] [Google Scholar]
- Quinlan AR. 2014. BEDTools: the Swiss‐army tool for genome feature analysis. Current Protocols in Bioinformatics 47: 11.12.1–11.12.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Stephens M, Pritchard JK. 2014. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197: 573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehl S, Benz M, Conard NJ, Darabi H, Deckers K, Nashli HF, Zeidi‐Kulehparcheh M. 2012. Plant use in three Pre‐Pottery Neolithic sites of the northern and eastern Fertile Crescent: a preliminary report. Vegetation History and Archaeobotany 21: 95–106. [Google Scholar]
- Riehl S, Zeidi M, Conard NJ. 2013. Emergence of agriculture in the foothills of the Zagros Mountains of Iran. Science 341: 65–67. [DOI] [PubMed] [Google Scholar]
- Ross‐Ibarra J, Morrell PL, Gaut BS. 2007. Plant domestication, a unique opportunity to identify the genetic basis of adaptation. Proceedings of the National Academy of Sciences, USA 104: 8641–8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell J, Mascher M, Dawson IK, Kyriakidis S, Calixto C, Freund F, Bayer M, Milne I, Marshall‐Griffiths T, Heinen S et al 2016. Exome sequencing of geographically diverse barley landraces and wild relatives gives insights into environmental adaptation. Nature Genetics 48: 1024–1030. [DOI] [PubMed] [Google Scholar]
- Saisho D, Purugganan MD. 2007. Molecular phylogeography of domesticated barley traces expansion of agriculture in the Old World. Genetics 177: 1765–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz J, McClean PE, Mamidi S, Wu GA, Cannon SB, Grimwood J, Jenkins J, Shu S, Song Q, Chavarro C et al 2014. A reference genome for common bean and genome‐wide analysis of dual domestications. Nature Genetics 46: 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Lai J. 2015. Patterns of genomic changes with crop domestication and breeding. Current Opinion in Plant Biology 24: 47–53. [DOI] [PubMed] [Google Scholar]
- Shor E, Green RM. 2016. The impact of domestication on the circadian clock. Trends in Plant Science 21: 281–283. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi R. 1955. The origin and evolution of cultivated barley. Advance Genetics 7: 227–266. [Google Scholar]
- Taketa S, Kikuchi S, Awayama T, Yamamoto S, Ichii M, Kawasaki S. 2004. Monophyletic origin of naked barley inferred from molecular analyses of a marker closely linked to the naked caryopsis gene (nud). Theoretical and Applied Genetics 108: 1236–1242. [DOI] [PubMed] [Google Scholar]
- Tanno K, Willcox G. 2012. Distinguishing wild and domestic wheat and barley spikelets from early Holocene sites in the Near East. Vegetation History and Archaeobotany 21: 107–115. [Google Scholar]
- Teshima KM, Coop G, Przeworski M. 2006. How reliable are empirical genomic scans for selective sweeps? Genome Research 16: 702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco D, Hough J, Aradhya M, Ross‐Ibarra J. 2016. Evolutionary genomics of peach and almond domestication. G3 (Bethesda) 6: 3985–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Yu Y, Haberer G, Marri PR, Fan C, Goicoechea JL, Zuccolo A, Song X, Kudrna D, Ammiraju JS et al 2014. The genome sequence of African rice (Oryza glaberrima) and evidence for independent domestication. Nature Genetics 46: 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelan SJ, Church DM, Ostell JM. 2001. Spidey: a tool for mRNA‐to‐genomic alignments. Genome Research 11: 1952–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Johnson CH. 2001. A clock‐and light‐regulated gene that links the circadian oscillator to LHCB gene expression. Plant Cell 13: 1411–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakhrabekova S, Gough SP, Braumann I, Müller AH, Lundqvist J, Ahmann K, Dockter C, Matyszczak I, Kurowska M, Druka A et al 2012. Induced mutations in circadian clock regulator Mat‐a facilitated short‐season adaptation and range extension in cultivated barley. Proceedings of the National Academy of Sciences, USA 109: 4326–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng K, Fu YX, Shi S, Wu CI. 2006. Statistical tests for detecting positive selection by utilizing high‐frequency variants. Genetics 174: 1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Yang Q, Yan X, Yu C, Su L, Zhang X, Zhu Y. 2017. Signatures of soft sweeps across the Dt1 locus underlying determinate growth habit in soybean [Glycine max (L.) Merr.]. Molecular Ecology 26: 4686–4699. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Jiang Y, Wang Z, Gou Z, Lyu J, Li W, Yu Y, Shu L, Zhao Y, Ma Y et al 2015. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nature Biotechnology 33: 408–414. [DOI] [PubMed] [Google Scholar]
- Zhu L, Bu Q, Xu X, Paik I, Huang X, Hoecker U, Deng XW, Huq E. 2015. CUL4 forms an E3 ligase with COP1 and SPA to promote light‐induced degradation of PIF1. Nature Communications 6: 7245. [DOI] [PubMed] [Google Scholar]
- Zhu D, Maier A, Lee J‐H, Laubinger S, Saijo Y, Wang H, Qu L‐J, Hoecker U, Deng XW. 2008. Biochemical characterization of Arabidopsis complexes containing CONSTITUTIVELY PHOTOMORPHOGENIC1 and SUPPRESSOR OF PHYA proteins in light control of plant development. Plant Cell 20: 2307–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zohary D, Hopf M, Weiss E. 2012. Domestication of plants in the Old World: the origin and spread of domesticated plants in Southwest Asia, Europe, and the Mediterranean Basin. Oxford, UK: Oxford University Press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Selection of target genes.
Fig. S2 The data analysis pipeline – read filtering, mapping, SNP calling and genotyping.
Fig. S3 Characteristics of coverage.
Fig. S4 Distribution of SNP markers over the barley chromosomes.
Fig. S5 Correlation of the ancestry coefficients estimated using faststructure and instruct.
Fig. S6 Genomic signatures of domestication selective sweeps (Mascher_2013 map).
Fig. S7 Procedure for estimating the ancestry of domesticated barley haplotypes.
Fig. S8 Population structure of wild barley (K = 9) determined by faststructure and instruct.
Fig. S9 The maximum likelihood (ML) unrooted phylogeny of 230 nonadmixed barley accessions.
Fig. S10 The Neighbor‐Net phylogenetic network of 359 barley accessions.
Fig. S11 Distribution of the wild barley populations within the Fertile Crescent.
Fig. S12 Distribution of the wild barley populations in Israel, the West Bank and Gaza.
Fig. S13 Unsorted ancestral palettes of the candidate domestication loci.
Fig. S14 Sorted ancestry palettes of the domesticated barley genotypes.
Fig. S15 Estimation of the median ancestry coefficients in the unbalanced subgroups of loci.
Fig. S16 Heatmaps of the pairwise ancestry similarity coefficients.
Fig. S17 A simplified candidate demographic model implying multiple domestication lineages.
Fig. S18 A simplified candidate demographic model implying a single domestication lineage.
Table S1 Characteristics of wild and cultivated barley accessions
Table S2 Sequences selected for targeted enrichment design
Table S3 Gene ontology (GO) terms of selected Arabidopsis genes
Table S4 Composition of the mapping reference
Table S5 Characteristics of the enrichment assay and SNP calling
Table S6 Characteristics of outlier loci identified by the selection scans
Table S7 Statistical assessment of wild barley population clustering by ‘smartpca’
Methods S1 Selection of genes for targeted enrichment assay.
Methods S2 Mapping reference design.
Methods S3 Quality check, mapping and SNP calling pipeline.
Notes S1 Characteristics of the enrichment assay.
Notes S2 Wild barley population structure – a note of caution.