

Abstract
The Loeys–Dietz syndrome (LDS) is a connective tissue disorder affecting the cardiovascular, skeletal, and ocular system. Most typically, LDS patients present with aortic aneurysms and arterial tortuosity, hypertelorism, and bifid/broad uvula or cleft palate. Initially, mutations in transforming growth factor‐β (TGF‐β) receptors (TGFBR1 and TGFBR2) were described to cause LDS, hereby leading to impaired TGF‐β signaling. More recently, TGF‐β ligands, TGFB2 and TGFB3, as well as intracellular downstream effectors of the TGF‐β pathway, SMAD2 and SMAD3, were shown to be involved in LDS. This emphasizes the role of disturbed TGF‐β signaling in LDS pathogenesis. Since most literature so far has focused on TGFBR1/2, we provide a comprehensive review on the known and some novel TGFB2/3 and SMAD2/3 mutations. For TGFB2 and SMAD3, the clinical manifestations, both of the patients previously described in the literature and our newly reported patients, are summarized in detail. This clearly indicates that LDS concerns a disorder with a broad phenotypical spectrum that is still emerging as more patients will be identified. All mutations described here are present in the corresponding Leiden Open Variant Database.
Keywords: aneurysm, Loeys–Dietz syndrome, SMAD2, SMAD3, TGFB2, TGFB3
1. BACKGROUND
The Loeys–Dietz syndrome (LDS, MIM# 609192, 610168, 613795, 614816, 615582) is an autosomal dominant connective tissue disorder with widespread systemic involvement. In 2005, Loeys and Dietz were the first to describe this disorder, which is—in its most typical presentation—characterized by vascular tortuosity and aneurysm in association with craniofacial and skeletal manifestations (Loeys et al., 2005; Loeys et al., 2006). On the one hand, LDS shows significant clinical overlap with Marfan syndrome (MFS, MIM# 154700), with vascular and skeletal features including aortic root aneurysm, arachnodactyly, scoliosis, and pectus deformity. On the other hand, a clear distinction between LDS and MFS can be made based on typical LDS findings such as widespread aortic/arterial aneurysm and tortuosity, club foot, craniosynostosis, hypertelorism, and bifid/broad uvula or cleft palate. Other LDS‐specific features can include cervical spine malformation and/or instability, translucent skin with easy bruising and dystrophic scars, severe allergic tendency, and bowel inflammation including eosinophilic esophagitis/gastritis and/or inflammatory bowel disease. Soon after the initial description, the phenotypic spectrum was expanded to less syndromic presentations, including those that overlap with vascular Ehlers–Danlos syndrome (Loeys et al., 2005; Loeys et al., 2006). Mutations in the genes encoding the transforming growth factor β (TGF‐β) receptor I (TGFBR1) and TGF‐β receptor II (TGFBR2) subunits were the first reported genetic causes of LDS. More recently, mutations in four additional genes, namely the mothers against decapentaplegic homolog 2 and 3 (SMAD2/3) and the TGF‐β 2 and 3 ligand (TGFB2/3), have been shown to cause an LDS‐like phenotype (Bertoli‐Avella et al., 2015; Boileau et al., 2012; Lindsay et al., 2012; Micha et al., 2015; van de Laar et al., 2011).
SMAD2 and SMAD3, located on the long arm of chromosome 18 and 15, span about 130 kb and consist of 11 and 9 exons, respectively. Both the SMAD2 and SMAD3 proteins belong to the receptor‐activated (R)‐SMAD family, intracellular effectors of the canonical TGF‐β signaling pathway. The activating ligands of this pathway include TGF‐β2 and TGF‐β3, which are encoded by the TGFB2 and TGFB3 genes. These genes include 8 and 7 exons and are positioned on the long arm of chromosome 1 and 14, respectively. Including TGFBR1/2, all genes related to LDS spectrum disorders are part of the TGF‐β signaling pathway. Additionally, fibrillin‐1, which is encoded by the FBN1 gene and deficient in MFS, binds to the large latent TGF‐β complex and contributes to TGF‐β bioavailability and activation (Chaudhry et al., 2007; Isogai et al., 2003; Neptune et al., 2003). This clearly highlights the key role of the TGF‐β pathway in the pathogenesis of aortic aneurysm development in LDS and LDS‐like disorders. However, the exact pathogenic mechanisms remain controversial.
Initially, it was suggested that two types of LDS could be distinguished: patients with LDS type 1 (LDSI) displayed typical craniofacial features, where LDS type 2 (LDSII) patients had more pronounced cutaneous features. Recently, this classification was revised and these LDS types are now thought to be part of a phenotypic spectrum of disease. Therefore, LDS type 1 (LDS1) and LDS type 2 (LDS2) now designate to the disease‐responsible genes; TGFBR1 and TGFBR2, respectively. Mutations in SMAD3 were initially described as the genetic cause of aneurysms‐osteoarthritis syndrome, but because of the many overlapping clinical features with LDS, including hypertelorism, bifid uvula, arterial tortuosity, and widespread and aggressive aneurysms, it is now also classified as LDS type 3 (LDS3) (MacCarrick et al., 2014). Similarly, patients with mutations in TGFB2 share clinical manifestations with LDS, and are for this reason now diagnosed with LDS type 4 (LDS4). More recently, patients with mutations in SMAD2 and TGFB3 (LDS5) were also shown to present with LDS‐like features (Bertoli‐Avella et al., 2015; Micha et al., 2015). It is thus suggested that a mutation in any of these six genes in combination with the presence of arterial aneurysm or dissection should be sufficient for the diagnosis of LDS (MacCarrick et al., 2014). Since most literature so far has focused on LDS1 and LDS2, this review will highlight known and novel SMAD2/3 and TGFB2/3 mutations and will summarize and compare the clinical features of affected individuals with those reported in the literature.
2. VARIANTS
Each published mutation was checked for accuracy and compared to the respective wild‐type reference sequence. When a different reference sequence was used, nucleotide and codon numbers were converted so their annotation matched with reference transcripts: NM_001135599.2 for TGFB2, NM_003239.3 for TGFB3, NM_005901.5 for SMAD2 and NM_005902.3 for SMAD3. The A nucleotide of the translation initiation codon (ATG) was designated as position +1. All variants are submitted to the corresponding Leiden Open Variant Database (LOVD) (Fokkema et al., 2011) http://143.169.238.105/LOVD/genes/TGFB2; http://143.169.238.105/LOVD/genes/TGFB3; http://143.169.238.105/LOVD/genes/SMAD2; http://143.169.238.105/LOVD/genes/SMAD3).
We report 7, 67, 30, and 15 different variants for SMAD2, SMAD3, TGFB2, and TGFB3, respectively (Tables 1, 2, and 3 and Figures 1 and 2). These variants were either published as full article cited in PubMed or newly identified in this study. We did not include variants from abstracts (without full article) or the ClinVar database since little or no clinical information is available on these individuals. Except for the five variants listed in Table 4, these mutations are not found in the ExAC database (http://exac.broadinstitute.org/; accessed April 2017). To obtain an indication on the pathogenicity, all variants are classified according to the ACMG guidelines (Suppl. Table S1) (Richards et al., 2015).
Table 1.
Previously described and novel SMAD3 gene mutations
| Exon | c‐Notation | p‐Notation | Domain | Reference | Times reported | Effect |
|---|---|---|---|---|---|---|
| 1 | c.2T > C | p.Met1Thr | MH1 | Haller et al. (2015) | 1 | Pathogenic |
| 1 | c.3G > A | p.Met1Ile | MH1 | Fitzgerald et al. (2014) | 1 | Pathogenic |
| 2 | c.221G > A | p.Arg74Gln | MH1 | Current study | 2 | Likely pathogenic |
| 2 | c.266G > A | p.Cys89Tyr | MH1 | Zhang et al. (2015) | 1 | Pathogenic |
| 2 | c.269G > A | p.Arg90His | MH1 | Current study | 1 | Likely pathogenic |
| 2 | c.281G > T | p.Trp94Leu | MH1 | Blinc et al. (2015) | 1 | VUS |
| 2 | c.290T > G | p.Leu97Arg | MH1 | Current study | 1 | VUS |
| 2 | c.300_301insAGGGCCGGCAGGC | p.His101Argfs*14 | MH1 | Current study | 1 | Pathogenic |
| 2 | c.313delG | p.Ala105Profs*11 | MH1 | van de Laar et al. (2012) | 1 | Pathogenic |
| 2 | c.335C > T | p.Ala112Val | MH1 | (Regalado et al., 2011) | 1 | Likely pathogenic |
| 2 | c.370C > A | p.Pro124Thr | MH1 | (Garcia‐Bermudez et al., 2017) | 1 | VUS |
| 2 | c.374A > G | p.Tyr125Cys | MH1 | Current study | 1 | Likely pathogenic |
| 2 | c. 374A > C | p.Tyr125Ser | MH1 | Current study | 1 | Likely pathogenic |
| 3 | c.401‐6G > A | p.Val134Aspfs*33 | MH1 | ( Campens et al., 2015 ), Current study | 2 | Pathogenic |
| 3 | c.401_405dup | p.Pro136Phefs*52 | MH1 | (Berthet et al., 2015) | 1 | Pathogenic |
| 3 | c.455delC | p.Pro152Hisfs*34 | Linker | ( Courtois et al., 2017 ) | 1 | Pathogenic |
| 3 | c.511G > T | p.Glu171* | Linker | Current study | 1 | Pathogenic |
| 3 | c.532+1G > C | Linker | Current study | 1 | Pathogenic | |
| 4 | c.539_540insC | p.Pro180Thrfs*7 | Linker | (van de Laar et al., 2012), Current study | 2 | Pathogenic |
| 4 | c.546delT | p.Gly183Alafs*3 | Linker | (Campens et al., 2015) | 1 | Pathogenic |
| 4 | c.584_585insTC | p.Gln195Hisfs*3 | Linker | (Campens et al., 2015) | 1 | Pathogenic |
| 5 | c.652delA | p.Asn218Thrfs*23 | Linker | (Regalado et al., 2011) | 1 | Pathogenic |
| 6 | c.668delC | p.Pro223Glnfs*18 | Linker | (Aubart et al., 2014) | 1 | Pathogenic |
| 6 | c.715G > A | p.Glu239Lys | MH2 | (Regalado et al., 2011), (Campens et al., 2015) | 4 | Pathogeic |
| 6 | c.728G > C | p.Arg243Pro | MH2 | Current study | 1 | Likely pathogenic |
| 6 | c.733G > A | p.Gly245Arg | MH2 | (Aubart et al., 2014) | 1 | Likely pathogenic |
| 6 | c.741_742delAT | p.Thr247Profs*61 | MH2 | (van de Laar et al., 2011) | 1 | Pathogenic |
| 6 | c.742T > C | p.Phe248Leu | MH2 | (Aubart et al., 2014) | 1 | Likely pathogenic |
| 6 | c.748G > A | p.Ala250Thr | MH2 | Current study | 1 | Likely pathogenic |
| 6 | c.762delC | p.Met255* | MH2 | Current study | 1 | Pathogenic |
| 6 | c.772G > C | p.Asp258His | MH2 | Current study | 1 | Likely pathogenic |
| 6 | c.782C > T | p.Thr261Ile | MH2 | (van de Laar et al., 2011) | 1 | Pathogenic |
| 6 | c.788C > T | p.Pro263Leu | MH2 | (van de Laar et al., 2012) | 1 | VUS |
| 6 | c.797C > A | p.Ser266* | MH2 | (Haller et al., 2015) | 1 | Pathogenic |
| 6 | c.803G > T | p.Arg268Leu | MH2 | ( Schubert et al., 2016 ), Current study | 2 | Likely pathogenic |
| 6 | c.836G > A | p.Arg279Lys | MH2 | (Regalado et al., 2011) | 2 | Likely pathogenic |
| 6 | c.859C > T | p.Arg287Trp | MH2 | (van de Laar et al., 2011), (Campens et al., 2015) | 2 | Likely pathogenic |
| 6 | c.860G > A | p.Arg287Gln | MH2 | ( Aubart et al., 2014 ), Current study | 2 | Likely pathogenic |
| 6 | c.861delG | p.Arg288Aspfs*53 | MH2 | Current study | 1 | Pathogenic |
| 7 | c.862_871+1dupAGACACATCGG | p.Arg292Aspfs*53 | MH2 | (Aubart et al., 2014) | 1 | Pathogenic |
| 7 | c.887T > C | p.Leu296Pro | MH2 | (Campens et al., 2015) | 1 | Likely pathogenic |
| 7 | c.988A > G | p.Thr330Ala | MH2 | (Ye et al., 2013) | 1 | Likely pathogenic |
| 7 | c.1009+1G > A | MH2 | Current study | 1 | Pathogenic | |
| 7 | c.1009+2T > A | MH2 | (Nevidomskyte et al., 2017) | 1 | Pathogenic | |
| 8 | c.1045G > C | p.Ala349Pro | MH2 | (van de Laar et al., 2012) | 1 | Likely pathogenic |
| 8 | c.1080dupT | p.Glu361* | MH2 | Current study | 1 | Pathogenic |
| 8 | c.1081G > T | p.Glu361* | MH2 | (van de Laar et al., 2012) | 1 | Pathogenic |
| 8 | c.1091A > G | p.Tyr364Cys | MH2 | Current study | 1 | Likely pathogenic |
| 8 | c.1102C > T | p.Arg368* | MH2 | ( Aubart et al., 2014 ), Current study | 2 | Pathogenic |
| 8 | c.1141G > C | p.Gly381Arg | MH2 | Current study | 1 | Likely pathogenic |
| 9 | c.1155‐2A > G | MH2 | (Campens et al., 2015) | 1 | Pathogenic | |
| 9 | c.1170_1179del | p.Ser391Alafs*7 | MH2 | (Wischmeijer et al., 2013) | 1 | Pathogenic |
| 9 | c.1179_1180dupC | p.Cys394Leufs*4 | MH2 | (Aubart et al., 2014) | 1 | Pathogenic |
| 9 | c.1185G > C | p.Trp395Cys | MH2 | Current study | 1 | VUS |
| 9 | c.1199T > C | p.Leu400Pro | MH2 | Current study | 1 | VUS |
| 9 | c.1208C > T | p.Pro403Leu | MH2 | (Martens et al., 2013) | 1 | Likely pathogenic |
| 9 | c.1222G > C | p.Asp408His | MH2 | Current study | 1 | Likely pathogenic |
| 9 | c.1224C > A | p.Asp408Glu | MH2 | Current study | 1 | Likely pathogenic |
| 9 | c.1243G > C | p.Gly415Arg | MH2 | Current study | 1 | Likely pathogenic |
| 9 | c.1247C > T | p.Ser416Phe | MH2 | Current study | 1 | Likely pathogenic |
| 9 | c.1259G > A | p.Arg420His | MH2 | (Proost et al., 2015) | 1 | VUS |
| 9 | c.1265C > T | p.Ser422Phe | MH2 | Current study | 1 | Likely pathogenic |
| 9 | c.1267A > G | p.Ser423Gly | MH2 | (Aubart et al., 2014) | 1 | Likely pathogenic |
| 9 | c.1268G > A | p.Ser423Asn | MH2 | Current study | 1 | Likely pathogenic |
| 9 | c.1274C > T | p.Ser425Phe | MH2 | Current study | 1 | VUS |
| Deletion from exon 2 onwards: Chr2.hg19: g.(67,408,242)_(67,603,013)del | (Hilhorst‐Hofstee et al., 2013) | 1 | Pathogenic | |||
| Deletion 15q22.33 | Current study | 1 | Pathogenic | |||
Mutations identified in this study are indicated in bold.
Table 2.
Previously described and novel TGFB2 gene mutations
| Exon | c‐Notation | p‐Notation | Domain | Reference | Times reported | Effect |
|---|---|---|---|---|---|---|
| 1 | c.236A > T | p.Gln79Leu | LAP | Current study | 1 | Likely pathogenic |
| 1 | c.244G > T | p.Ala82Ser | LAP | Current study | 1 | VUS |
| 1 | c.274G > T | p.Glu92* | LAP | Current study | 1 | Pathogenic |
| 1 | c.294_308delCTACGCCAAGGAGGT | p.Ala100_Tyr104del | LAP | (Lindsay et al., 2012) | 1 | Likely pathogenic |
| 1 | c.297 > A | p.Tyr99* | LAP | (Lindsay et al., 2012) | 1 | Pathogenic |
| 1 | c.304G > T | p.Glu102* | LAP | (Boileau et al., 2012) | 1 | Pathogenic |
| 1 | c.305_307delAGG | p.Glu102del | LAP | Current study | 1 | Likely pathogenic |
| 3 | c.475C > T | p.Arg159* | LAP | ( Renard et al., 2013 ), Current study | 3 | Pathogenic |
| 3 | c.518_519 insT | p.Lys174Glufs*18 | LAP | Current study | 1 | Pathogenic |
| 3 | c.577C > T | p.Arg193Trp | LAP | ( Campens et al., 2015 ), Current study | 2 | Likely pathogenic |
| 4 | c.673G > T | p.Glu225* | LAP | Current study | 1 | Pathogenic |
| 5 | c.687C > A | p.Cys229* | LAP | (Boileau et al., 2012) | 1 | Pathogenic |
| 5 | c.839‐1G > A | p.Gly280Aspfs*41 | LAP | (Ritelli et al., 2014) | 1 | Pathogenic |
| 6 | c.873_888dup | p.Asn297* | LAP | (Boileau et al., 2012) | 1 | Pathogenic |
| 6 | c.979C > T | p.Arg327Trp | RKKR motif | (Lindsay et al., 2012), (Renard et al., 2013), (Schubert et al., 2016) | 3 | Pathogenic |
| 6 | c.980G > A | p.Arg327Gln | RKKR motif | ( Renard et al., 2013 ), ( Campens et al., 2015 ), Current study | 5 | Pathogenic |
| 6 | c.988C > T | p.Arg330Cys | RKKR motif | ( Lindsay et al., 2012 ), ( Campens et al., 2015 ), Current study | 4 | Pathogenic |
| 6 | c.1000delG | p.Ala334Arg*fs25 | Cytokine | Current study | 1 | Pathogenic |
| 7 | c.1021_1025delTACAA | p.Tyr341Cysfs*25 | Cytokine | (Boileau et al., 2012) | 1 | Pathogenic |
| 7 | c.1042C > T | p.Arg348Cys | Cytokine | ( Gago‐Diaz et al., 2014 ), Current study | 2 | Likely pathogenic |
| 7 | c.1097C > A | p.Pro366His | Cytokine | (Lindsay et al., 2012) | 1 | Likely pathogenic |
| 7 | c.1106_1110delACAAT | p.Tyr369Cysfs*26 | Cytokine | (Lindsay et al., 2012) | 1 | Pathogenic |
| 7 | c.1125delT | p.Gly376Glufs*17 | Cytokine | (Renard et al., 2013) | 1 | Pathogenic |
| 7 | c.1165dupA | p.Ser389Lysfs*8 | Cytokine | (Leutermann et al., 2014) | 1 | Pathogenic |
| 7 | c.1170+1G > A | Cytokine | Current study | 1 | Pathogenic | |
| 8 | c.1234G > C | p.Asp412His | Cytokine | Current study | 1 | VUS |
| Entire gene Chr1.hg19: g.(215,588,712)_(222,145,072)del | (Lindsay et al., 2012) | 1 | Pathogenic | |||
| Entire gene Chr1.hg19:g.(216,672,181)_(220,202,575)del | (Lindsay et al., 2012) | 1 | Pathogenic | |||
| Entire gene Chr1.hg19: g.(214,271,966)_(219,506,825)del | (Fontana et al., 2014) | 1 | Pathogenic | |||
| Entire gene Chr1.hg19:g.(215,713,272)_(218,899,968)del | Current study | 1 | Pathogenic | |||
Mutations identified in this study are indicated in bold.
Table 3.
Previously described and novel TGFB3 and SMAD2 gene mutations
| Gene | Exon | c‐notation | p‐notation | Domain | Reference | Times reported | Effect |
|---|---|---|---|---|---|---|---|
| TGFB3 | 1 | c.106A > T | p.Lys36* | LAP | Current study | 1 | Pathogenic |
| TGFB3 | 2 | c.437delT | p.Leu146Hisfs*68 | LAP | Current study | 1 | Pathogenic |
| TGFB3 | 4 | c.704delA | p.Asn235Metfs*11 | LAP | (Bertoli‐Avella et al., 2015) | 1 | Pathogenic |
| TGFB3 | 4 | c.754+2T > C | p.Glu216_Lys251del | LAP | (Bertoli‐Avella et al., 2015) | 1 | Pathogenic |
| TGFB3 | 5 | c.787G > C | p.Asp263His | LAP | ( Bertoli‐Avella et al., 2015 ), Current study | 3 | Likely pathogenic |
| TGFB3 | 5 | c.796C > T | p.Arg266Cys | LAP | Current study | 1 | VUS |
| TGFB3 | 5 | c.898C > T | p.Arg300Trp | RKKR motif | ( Bertoli‐Avella et al., 2015 ), Current study | 5 | Pathogenic |
| TGFB3 | 5 | c.899G > A | p.Arg300Gln | RKKR motif | ( Matyas et al., 2014 ), Current study | 2 | Pathogenic |
| TGFB3 | 5 | c.898C > G | p.Arg300Gly | RKKR motif | (Kuechler et al., 2015) | 1 | Likely pathogenic |
| TGFB3 | 6 | c.965T > C | p.Ile322Thr | Cytokine | (Bertoli‐Avella et al., 2015) | 1 | Likely pathogenic |
| TGFB3 | 6 | c.979G > T | p.Asp327Tyr | Cytokine | Current study | 1 | Likely pathogenic |
| TGFB3 | 6 | c.1095C > A | p.Tyr365* | Cytokine | (Bertoli‐Avella et al., 2015) | 1 | Pathogenic |
| TGFB3 | 7 | c.1157delT | p.Leu386Argfs*21 | Cytokine | (Bertoli‐Avella et al., 2015) | 1 | Pathogenic |
| TGFB3 | 7 | c.1202T > C | p.Leu401Pro | Cytokine | ( Bertoli‐Avella et al., 2015 ), Current study | 2 | Likely pathogenic |
| TGFB3 | 7 | c.1226G > A | p.Cys409Tyr | Cytokine | (Rienhoff et al., 2013) | 1 | Pathogenic |
| SMAD2 | 8 | c.954T > A | p.Asn318Lys | MH2 | Current study | 1 | Likely pathogenic |
| SMAD2 | 9 | c.1082A > C | p.Asn361Thr | MH2 | Current study | 1 | Likely pathogenic |
| SMAD2 | 10 | c.1163A > G | p.Gln388Arg | MH2 | (Micha et al., 2015) | 1 | Likely pathogenic |
| SMAD2 | 10 | c.1190C > A | p.Ser397Tyr | MH2 | Current study | 1 | Likely pathogenic |
| SMAD2 | 11 | c.1346T > C | p.Leu449Ser | MH2 | (Micha et al., 2015) | 1 | Likely pathogenic |
| SMAD2 | 11 | c.1369G > A | p.Gly457Arg | MH2 | (Micha et al., 2015) | 1 | Likely pathogenic |
| SMAD2 | 11 | c.1400C > T | p.Ser467Leu | MH2 | Current study | 1 | VUS |
Mutations identified in this study are indicated in bold.
Figure 1.
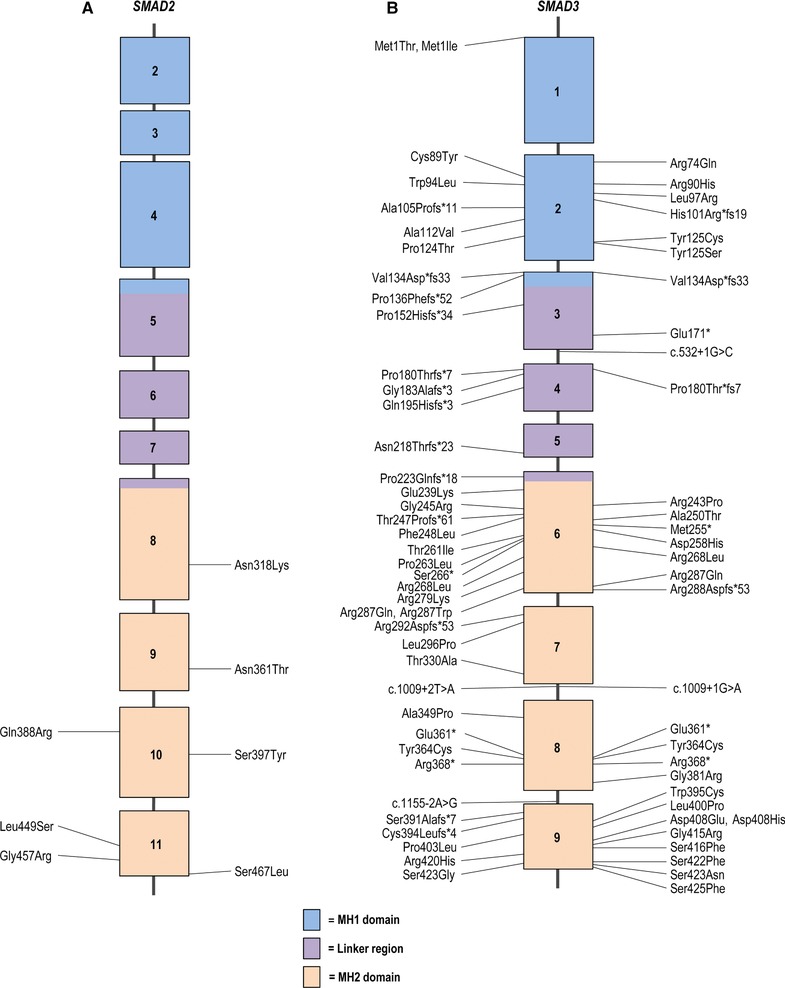
Schematic representation of the SMAD2 and SMAD3 gene with their protein coding domains. Boxes represent exons 1–11 and 1–9, respectively. On the left side of the schematic are the previously reported mutations, whereas on the right side mutations identified in this study are described. For SMAD2, the first depicted exon is exon 2 because exon 1 is 5′UTR. Mutations are annotated at the protein level, with exception of splice site mutations (reference transcript: NM_005901.5 and NM_005902.3 for SMAD3)
Figure 2.
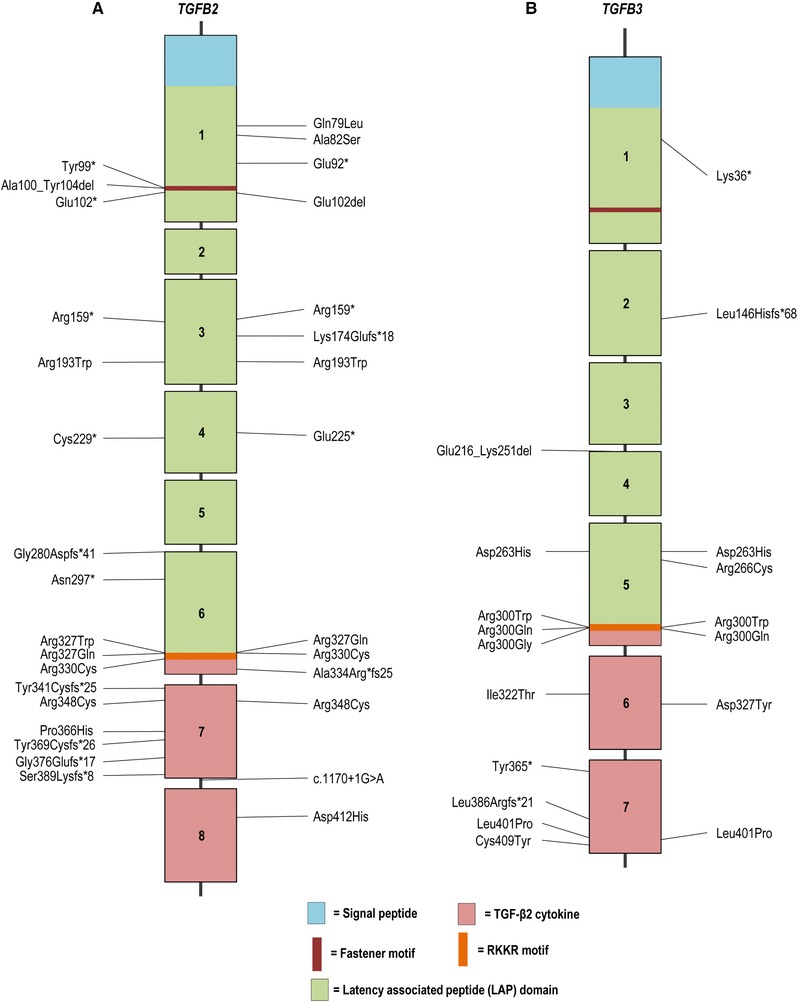
Schematic representation of the TGFB2 and TGFB3 gene with their protein coding domains. Boxes represent exons 1–8 and 1–7, respectively. On the left side of the schematic are the previously reported mutations, whereas on the right side mutations identified in this study are described. Mutations are annotated at the protein level (reference transcript: NM_001135599.2 for TGFB2 and NM_003239.3 for TGFB3)
Table 4.
Pathogenic variants in SMAD2, SMAD3, TGFB2, and TGFB3 present in ExAC
| Gene | Variant (p‐notation) | Times in ExAC |
|---|---|---|
| SMAD2 | p.Gln388Arg | 1/121108 |
| SMAD3 | p.Arg420His | 1/120914 |
| TGFB2 | p.Arg193Trp | 1/121376 |
| TGFB3 | p.Ile322Thr | 1/121368 |
| p.Asp327Tyr | 1/121378 |
For SMAD3, 40 variants were previously reported (Aubart et al., 2014; Berthet, Hanna, Giraud, & Soubrier, 2015; Blinc et al., 2015; Burke, Shalhub, & Starnes, 2015; Campens et al., 2015; Courtois et al., 2017; Fitzgerald, Bhat, Conard, Hyland, & Pizarro, 2014; Garcia‐Bermudez, Moustafa, Barros‐Membrilla, & Tizon‐Marcos, 2017; Haller et al., 2015; Hilhorst‐Hofstee et al., 2013; Martens et al., 2013; Nevidomskyte et al., 2017; Proost et al., 2015; Regalado et al., 2011; Schubert, Landis, Shikany, Hinton, & Ware, 2016; van de Laar et al., 2011; van de Laar et al., 2012; van der Linde et al., 2012; Wischmeijer et al., 2013; Yao et al., 2003; Ye et al., 2013; Zhang et al., 2015), whereas 38 probands were identified in this study, with 27 mutations that have never been reported (Table 1 and Figure 1). These SMAD3 mutations include 61% missense mutations, 23% frameshift mutations, 7% nonsense mutations, 6% splice site mutations, and 3% whole or partial gene deletions (Figure 3). The 30 TGFB2 mutations include 20 previously published pathogenic variants (Boileau et al., 2012; Campens et al., 2015; Fontana et al., 2014; Gago‐Diaz et al., 2014; Leutermann et al., 2014; Lindsay et al., 2012; Renard et al., 2013; Ritelli et al., 2014; Schubert et al., 2016) and 10 newly identified mutations (Table 2 and Figure 2). In five new probands, previously reported TGFB2 mutations were identified. New mutations include 30% missense mutations, 23% frameshift mutations, 23% nonsense mutations, 14% whole or partial gene deletions, 7% intragenic in‐frame deletions, and 3% splice site mutations (Figure 3). For SMAD2 and TGFB3, far fewer mutations have been reported (Table 3) (Bertoli‐Avella et al., 2015; Kuechler et al., 2015; Matyas, Naef, Tollens, & Oexle, 2014; Micha et al., 2015; Rienhoff et al., 2013). In addition to the three previously reported SMAD2 mutations, we identified four new missense mutations, bringing the total to seven missense mutations identified so far (Micha et al., 2015). For TGFB3, we identified four additional probands with a mutation that was previously published and four new mutations. The majority of the TGFB3 mutations consist of missense mutations (60%), whereas 20% are frameshift mutations, 13% are nonsense mutations, and 7% of mutations affect a splice site.
Figure 3.
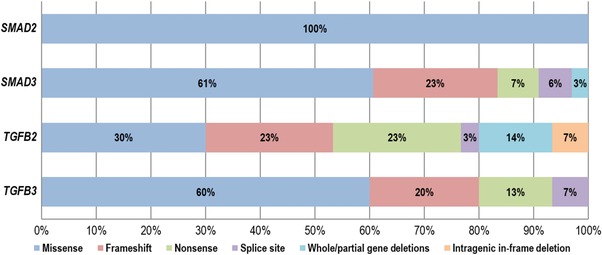
Frequencies of the different types of SMAD2, SMAD3, TGFB2, and TGFB3 mutations identified so far
No mutational hotspot is observed for SMAD2 and SMAD3 mutations, but the majority (100% and 63%) reside in the MH2 domain, a well conserved region responsible for the oligomerization of SMAD2 or SMAD3 with SMAD4 and subsequent Smad‐dependent activation of downstream transcription (Figure 1). For TGFB2, 23% of mutations are located in the exons coding for the active TGF‐β2 cytokine and 28% reside in the RKKR‐motif, a proteolytic cleavage site responsible for releasing mature TGF‐β2 from the latency‐associated peptide (LAP). Of the 17 mutations so far identified in the LAP domain of TGFB2, 76% are nonsense or frameshift mutations, whereas 24% are missense mutations. For TGFB3, 35% of mutations affect the RKKR‐motif. Remarkably, of the 23 currently known TGFB3 mutations, three substitute histidine for aspartic acid at amino acid position 263. This arginine belongs to the RGD‐motif that mediates binding to integrins and subsequent integrin‐mediated activation (release) of TGFβ3 from its latent complex (Shi et al., 2011). Since all described patients with this mutation originate from the same county, this might represent a founder mutation (a hypothesis currently under investigation).
3. BIOLOGICAL RELEVANCE
For a long time, it was thought that MFS, which is caused by mutations in the fibrillin‐1 gene, and related connective tissue disorders, was caused by a pure structural deficiency of the extracellular matrix (ECM), leading to tissue weakening of the aortic wall and resulting in an increased risk of aneurysm progression and tear. Although some manifestations of MFS, such as lens dislocation and aortic aneurysm could be explained in this way, loss of structural tissue integrity could not easily reconcile the skeletal overgrowth phenotype observed in MFS patients or the low muscle mass and fat stores characteristic of this condition. Mouse models, like Fbn1+/C1039G, have significantly changed our understanding of disease pathogenesis. It has been shown that altered TGF‐β signaling plays an important role in the lung, valve, skeletal muscle, and aortic pathology of fibrillin‐1‐deficient mice (Cohn et al., 2007; Habashi et al., 2006; Neptune et al., 2003; Ng et al., 2004), indicating that fibrillin‐1 is not only a structural component of the ECM but also a key regulator of TGF‐β signaling (Dallas, Miyazono, Skerry, Mundy, & Bonewald, 1995; Isogai et al., 2003). This new hypothesis was confirmed by a key experiment in which the mutant phenotype in fibrillin‐1‐deficient mice could be attenuated by the administration of TGF‐β‐ neutralizing antibodies (Cohn et al., 2007; Habashi et al., 2006; Neptune et al., 2003; Ng et al., 2004). Shortly after this, the central role of TGF‐β dysregulation was proven by the identification of mutations in the TGFBR1 and TGFBR2 genes as the cause of LDS. The effect of these mutations on the TGF‐β pathway is, however, complex as heterozygous loss‐of‐function mutations associate with paradoxical activation of TGF‐β signaling, which was demonstrated by increased phosphorylation of Smad proteins and increased output of TGF‐β‐driven genes.
3.1. TGF‐β pathway
TGF‐β is the prototype of a family of secreted polypeptide growth factors essential in development, differentiation, cell growth, migration, apoptosis, and ECM production (Derynck & Akhurst, 2007; Massague, Blain, & Lo, 2000). In humans, three TGF‐β ligand isoforms exist, TGF‐β1, TGF‐β2, and TGF‐β3, encoded by the TGFB1, TGFB2, and TGFB3 genes, respectively. TGF‐β is secreted from cells as part of a large latent complex, that consists of the mature TGF‐β cytokine, a dimer of its processed amino terminal propeptide (LAP) and one of three latent TGFβ binding protein‐isoforms (LTBP1, 3, or 4). The latter binds to ECM components such as fibronectin or microfibrils composed of fibrillin‐1. Upon release from this complex, TGF‐β binds to the type II TGF‐β receptor subunit (TGFβR2), which alters its conformation and phosphorylates the type I TGF‐β receptor subunit (TGFβR1), also known as activin receptor‐like kinase 5 (ALK5). In the canonical pathway, TGFβRI then phosphorylates SMAD2 and SMAD3 proteins that transmit the TGFβ signal to the nucleus upon association with SMAD4. SMADs are transcription factors that partner with other factors that drive TGFβ‐mediated gene transcription (Derynck & Zhang, 1996; Feng & Derynck, 2005; Massague et al., 2005; Zhang, 2009). Alternatively, ligand‐activated TGFβ receptors can also initiate non‐canonical signaling cascades, including RhoA and the mitogen‐activated protein kinases ERK, JNK, and p38 (Derynck & Zhang, 2003; Lee et al., 2007; Yamashita et al., 2008).
One of the most unambiguous indications that TGF‐β signaling plays a major role in aneurysm pathogenesis was provided by the finding that TGFBR1/2 mutations result in LDS. Aortic wall tissue of TGFBR1 or 2 mutant patients showed increased nuclear accumulation of phosphorylated SMAD2 (pSMAD2) and enhanced expression of TGF‐β driven gene products such as connective tissue growth factor (CTGF), illustrating enhanced TGF‐β signaling. The latter is paradoxical as most of the TGFBR1 or 2 mutations are predicted to lead to a decrease of the serine‐threonine kinase activity of the receptor (Loeys et al., 2005). An analogous observation was made in medial vascular smooth muscle cells (VSMC) of patients with a SMAD3 mutation (van de Laar et al., 2011). Finally, in accordance with the findings for SMAD3, increased pSMAD2 and CTGF expression was observed in the aortic media of patients with either loss‐of‐function TGFB2 or TGFB3 mutations, further confirming the paradoxical observation of increased TGF‐β signaling in vivo (Bertoli‐Avella et al., 2015; Lindsay et al., 2012).
In addition to increased expression of pSMAD2 and TGF‐β‐driven gene products, alternative expression of TGFβ‐ligands is observed in LDS patients as well. In human TGFB2 or TGFB3‐deficient aortas, an increase in TGFB1 expression was observed (Bertoli‐Avella et al., 2015; Lindsay et al., 2012). An analogous observation was made in aneurysmal aortic media of SMAD3 mutation positive patients (van de Laar et al., 2011). Aortic wall tissue of these patients also revealed an increase in cytoplasmic and nuclear total SMAD3 immunostaining, indicating that other compensatory mechanisms lead to further dysregulation of the pathway (van de Laar et al., 2011). TGFB2 mutation positive patients showed, besides an increase in TGFB1, also an enhanced expression of TGFB2 in their aortic wall tissue (Boileau et al., 2012). Although TGFB2 mutations are predicted to result in haploinsufficiency, the exact mechanisms on how loss‐of‐function mutations lead to a paradoxical activation of TGF‐β signaling remain elusive.
Different hypotheses have arisen to address this issue but further experimental validation is mandatory. The first hypothesis states that a potential cell‐autonomous mechanism results in upregulation of TGF‐β signaling (Lindsay & Dietz, 2011). TGF‐β ligand can activate both the canonical and non‐canonical pathway. By a negative feedback loop, the canonical pathway can regulate TGF‐β pathway activity. Mutations in LDS genes (TGFBR1/2, SMAD3, TGFB2/3) cause a decrease in canonical signaling hereby downregulating the feedback inhibition in order to restore the canonical signaling pathway. This will result in increased ligand expression and excessive activation of the non‐canonical signaling pathway. Further supporting evidence for the contribution of non‐canonical signaling came from the observation that treatment with a specific ERK inhibitor, RDEA119, abrogates pathological aortic root growth in an MFS mouse model hereby rescuing the aortic aneurysm phenotype (Holm et al., 2011; Habashi et al., 2011). Secondly, as shown by increased expression of TGFB1, both in TGFB2‐deficient patients and mice, a shift in the use of specific TGF‐β cytokine isoforms might contribute to the upregulation of the TGF‐β cascade (Lindsay et al., 2012). Thirdly, a non‐cell autonomous effect (paracrine) offers an alternative explanation for the observed increased TGFB1 expression and concomitant enhanced TGF‐β signaling. The VSMC of the thoracic aorta have different embryonic origins: the cells of the ascending aorta are derived from the second heart field and cardiac neural crest, whereas the cells in the descending aorta originate from the somatic mesoderm (proximal descending aorta) or splanchnic mesoderm (distal descending aorta) (Majesky, 2007). Aneurysms can occur throughout the aorta, implicating that there is not a common origin for all VSMCs at sites prone to aneurysm formation. However, aneurysm formation always occurs at locations where cells of divergent origins can interact (at the transition regions) (Gallo et al., 2014; Lindsay & Dietz, 2011). Because cells from distinct origins respond in different ways to TGF‐β stimulation, it is likely that cells of one lineage are more prone to perturbed TGF‐β signaling compared to cells of another origin (Topouzis & Majesky, 1996). Cell types that are more sensitive towards heterozygous LDS mutations might attempt to compensate for the initial loss in TGF‐β signaling by secreting excessive amounts of TGF‐β ligand in their direct environment, leading to overdrive of TGF‐β signaling in neighboring cells which are intrinsically less vulnerable to LDS mutations (Gallo et al., 2014). Finally, since increased expression of pSMAD2 is observed in the aortic media of LDS patients, this might indicate that other TGF‐β related pathways such as angiotensin II or activin signaling cascades are involved as well (Bernard, 2004; Rodriguez‐Vita et al., 2005).
3.2. Mouse models
For SMAD3, TGFB2, and TGFB3, multiple mouse models have been developed through the years confirming the hypotheses about the molecular and cellular mechanisms underlying LDS pathogenesis. Mouse models for SMAD2 have also been developed but Smad2 knockout mice embryos die before E8,5, due to defective egg cylinder elongation and germ layer formation, which entangles aortic size examination (Waldrip, Bikoff, Hoodless, Wrana, & Robertson, 1998; Weinstein et al., 1998).
Different Smad3 mouse models initially confirmed the importance of SMAD3 and the TGF‐β pathway for cartilage integrity. Homozygous mutant mice in which Smad3 exon 8 is targeted (Smad3 ex8/ex8) develop progressive degenerative cartilage resembling human osteoarthritis (Yang et al., 1999). Additionally, Smad3 knock‐out mice (Smad3 −/−) show phenotypes similar to human osteoarthritis (Li et al., 2009). However, initially, these mouse models were not reported to develop a vascular phenotype (Bonniaud et al., 2004; Li et al., 2009). After identification of SMAD3 as an aortic aneurysm disease causing gene, the vascular phenotype of the Smad3 knock‐out mice was studied in more detail (Ye et al., 2013). Necropsy of the Smad3−/− mice revealed that the majority died from a ruptured aneurysm. Additionally, ultrasound imaging of these mutant mice revealed progressive aortic root and ascending aortic dilation as early as 2 months.
Sanford et al reported that Tgfb2‐null mice died shortly after birth and had small, thin walled ascending aortas in addition to other developmental defects involving different organ systems (Sanford et al., 1997). In a later study, Bartram et al. (2001) also observed that Tgfb2‐null mice die during gestation due to congenital heart disease and display aortic anomalies. Therefore, Lindsay et al. studied the Tgfb2 heterozygous knockout mouse model to study aneurysm formation in more detail. Tgfb2+/− mice showed dilatations of the aortic annulus and aortic root, a pattern similar to that of LDS and MFS patients (Loeys et al., 2005; Mc Kusick, 1955), confirming that loss‐of‐function of one Tgfb2 allele is sufficient to cause aortic root aneurysm. As Western blot of Tgfb2+/− mouse aortas showed a similar increase in phosphorylation of Smad2/3 and Erk1/2, recapitulating the observations in the human aorta, it can be concluded that increased activation of TGF‐β signaling underlies the observed phenotype (Lindsay et al., 2012). Besides Tgfb2+/‐ mice, the aortas of double‐heterozygous knock‐out mice, Tgfb2+/−; Fbn1C+/C1039G, were examined as well. The aortic root dimensions of Tgfb2+/−; Fbn1C+/C1039G were significantly increased compared with the wild‐type mice at 2 and 4 months of age. Also elastic fiber fragmentation, higher collagen deposition and increased nuclear accumulation of pSmad2 in the aortic media were observed. Tgfb1 mRNA levels in the proximal aorta of Tgfb2+/−; Fbn1+/C1039G mice at 2 months of age were elevated, which is similar to the observations in human. This is again underlining the paradoxical TGF‐β pathway activation and stressing the complex pathogenesis that goes together with TGF‐β dysregulation.
In 1995, both Proetzel et al. (1995) and Kaartinen et al. (1995) generated, independently, a Tgfb3 null mutant mouse model. Homozygous Tgfb3 knockout mice died within 20–24 hr of birth displaying defective palatogenesis but no other concomitant craniofacial or growth abnormalities. Additionally, abnormal pulmonary development was observed. Azhar et al. (2003) described irregularities in the curvature and position of the aortic arches in Tgfb3 knockout mice, but no major cardiac defects have been reported. More recently, Doetschman et al. (2012) developed Tgfb3 conditional knockout mice that died at birth from cleft palate defects, similar to Tgfb3−/− mice, but there was no record of cardiovascular abnormalities in these mice. Further evaluation of the aortic sizes of Tgfb3 conditional knockout or haploinsufficient mice definitely offer some interesting and necessary research prospects.
4. CLINICAL AND DIAGNOSTIC RELEVANCE
The most important clinical finding in LDS patients is dilatation of the aortic root at the level of the sinuses of Valsalva, a feature that nearly all LDS patients will develop ultimately. Aneurysms of the ascending or descending aorta are less frequently observed. Dissection and rupture of these aortic aneurysms tend to occur at a younger age and at smaller diameters in LDS patients compared to MFS patients (Loeys et al., 2006). The aortic phenotype in patients with SMAD3 mutations is very similar to TGFBR1/2 patients, whereas TGFB2 and TGFB3 cardiovascular features tend to be milder, although severe aortic presentation at young age has also been observed. Non‐penetrance seems more common in TGFB2/3 families. Using CT or MRI, 3D reconstruction of images from the head to pelvis is needed to identify arterial tortuosity, present in most individuals with a TGFBR1/2, TGFB2, or SMAD3 mutation, and aneurysms in the rest of the arterial tree. This is essential because aneurysms distant from the aortic root can be easily overlooked using echocardiography.
Skeletal features in LDS include joint hyperlaxity, scoliosis, arachnodactyly, pectus deformity, and club foot, showing some overlap with those of MFS. Additionally, osteoarthritis and hernia (mostly inguinal) have been frequently observed in all LDS types (Table 5). Although a significant overlap exists between the phenotypic characteristics of SMAD2/3, TGFB2/3 patients and prior TGFBR1/2 reported features, some differences seem to emerge (Table 5). For example cervical spine instability has not yet been seen in TGFB2/3 mutations patients, and craniosynostosis has only been reported once in a SMAD3 patient. For the first time, we report on cleft palate in patients with TGFB2 mutations. The number of patients with SMAD2 and TGFB3 mutations is probably too low to draw any significant conclusions so far. For TGFB2 and SMAD3, a detailed summary of the clinical manifestations, both of the patients previously described in the literature and our newly reported patients, can be found in Suppl. Tables S2 and S3. For TGFB3 and SMAD2, detailed clinical features of all patients reported so far are listed in Suppl. Tables S4 and S5.
Table 5.
Comparison of the clinical manifestations of patients with SMAD2, SMAD3, TGFB2, and TGFB3 mutations
| TGFB2 | TGFB3 | SMAD3 | SMAD2 | |
|---|---|---|---|---|
| Hypertelorism | + | + | + | + |
| Bifid uvula/cleft palate | + | + | + | − |
| Exotropia | + | + | + | ? |
| Craniosynostosis | − | − | + | ? |
| Cervical spine instability | − | + | + | ? |
| Retrognathia | + | + | + | − |
| Scoliosis | + | + | + | + |
| Club foot | + | + | + | − |
| Osteo‐arthritis | + | + | + | + |
| Dural ectasia | + | ? | + | + |
| Pneumothorax | + | − | + | + |
| Hernia | + | + | + | + |
| Dissection at young age | + | ? | + | − |
| Arterial tortuosity | + | − | + | + |
+ indicates presence of the clinical feature, − indicates absence of the clinical features and a question mark illustrates presence of the clinical feature is unknown.
Although no formal diagnostic criteria have been developed, genetic testing of the LDS genes should be considered in the following scenarios: (1) patients with the typical clinical trial of hypertelorism, cleft palate/bifid uvula and arterial tortuosity/aneurysm; (2) early onset aortic aneurysm with variable combination of other features including arachnodactyly, camptodactyly, club feet, craniosynostosis (all types), blue sclerae, thin skin with atrophic scars, easy bruising, joint hypermobility, bicuspid aortic valve, patent ductus arteriosus, atrial and ventricular septum defects; (3) sporadic young probands with aortic root dilatation/dissection; (4) families with autosomal dominant thoracic aortic aneurysms, especially those families with early onset aortic/arterial dissection, aortic disease beyond the aortic root (including cerebral arteries); (5) patients with a MS‐like phenotype, especially those without ectopia lentis, but with aortic and skeletal features not fulfilling the MS diagnostic criteria; (6) patients with clinical features reminiscent of vascular Ehlers–Danlos syndrome (thin skin with atrophic scars, easy bruising, joint hypermobility) and normal type III collagen biochemistry and/or normal COL3A1 genetic testing.
The diagnosis of LDS is established in a proband (by definition a person without a known family history of LDS), who has a heterozygous pathogenic variant in SMAD2, SMAD3, TGFB2, TGFB3, TGFBR1, or TGFBR2 and either of the following (MacCarrick et al., 2014): (1) aortic root enlargement (defined as an aortic root z‐score greater than or equal to 2.0) or type A dissection or (2) compatible systemic features including characteristic craniofacial, skeletal, cutaneous and/or vascular manifestations found in combination. Special emphasis is given to arterial tortuosity, prominently including the head and neck vessels, and to aneurysms or dissections involving medium‐to‐large muscular arteries throughout the arterial tree. In the presence of a family history of documented LDS, the diagnosis can be made in at‐risk relatives on the basis of molecular genetic testing, even if vascular involvement or other features are not yet apparent.
5. FUTURE PROSPECTS
The recent identification of SMAD2,3 and TGFB2,3 as disease causing genes responsible for LDS phenotypes further pinpoints altered TGF‐β signaling as the culprit in aortic aneurysm pathology. As we anticipate that more genes will cause similar clinical LDS phenotypes, there is a clear rationale for the development of gene panels. This makes it possible to screen multiple candidate genes at one go, taking advantage of the next‐generation sequencing techniques, which are more time and cost efficient compared with the elaborate Sanger sequencing technique. As shown by recent studies, this will facilitate the assessment of an accurate genetic diagnosis for LDS patients hereby surely benefiting patient management (Campens et al., 2015; Proost et al., 2015).
Cardiovascular manifestations of LDS can be managed in a medical and/or surgical treatment strategy. Since vascular disease is more aggressive in LDS compared with MFS, prophylactic aortic surgery is already being recommended at smaller aortic root dimensions of 4.0‐4.5 cm (MacCarrick et al., 2014). In order to reduce aortic wall‐shear stress and eventually aortic dilatation, beta‐blockers, such as atenolol, have been the first‐line treatment (Shores, Berger, Murphy, & Pyeritz, 1994). However, since aortic‐root tissue of LDS patients shows excessive TGF‐β activation and signaling, therapies that reduce TGF‐β pathway activation offer attractive therapeutic targets. Indeed, angiotensin II type 1 receptor blockers, such as losartan, reduce the rate of aortic root growth by lowering the expression of TGFβ ligands, receptors, and activators (Everett, Tufro‐McReddie, Fisher, & Gomez, 1994; Fukuda et al., 2000; Habashi et al., 2006; Naito et al., 2004). Although a randomized clinical trial comparing losartan with atenolol did not show a significant difference in the rate of aortic‐root dilatation between these two treatment groups, the observation that aortic‐root z‐scores decrease especially in younger patients advocates to start therapy earlier in the disease course (Lacro et al., 2014). Alternatively, the use of ERK inhibitors (RDEA119) and angiotensin II type 2 receptor agonists offer interesting alternative treatment options (Loeys, 2015).
The full spectrum of phenotypes associated with some genes of the TGF‐β related vasculopathies, such as FBN1 and TGFBR1/2, has been described extensively (Arslan‐Kirchner et al., 2011; Cook, Carta, Galatioto, & Ramirez, 2015; Lerner‐Ellis et al., 2014; Ramachandra et al., 2015; Romaniello et al., 2014). For the more recently identified LDS genes (TGFB2,3 and SMAD2,3), the phenotypic spectrum has not been fully studied so far. By summarizing the clinical manifestations of TGFB2,3 and SMAD2,3 mutation patients, this manuscript extends the spectrum of phenotypes associated with these LDS genes. However, the screening of larger cohorts of patients with a LDS‐like phenotype, whether or not using gene panels, will be required to reveal the full phenotypic spectrum of LDS.
Supporting information
Supporting Information Table S1
Supporting Information Table S2
Supporting Information Table S3
Supporting Information Table S4
Supporting Information Table S5
ETHICAL COMPLIANCE
All study participants or their legal guardians gave informed consent at the respective sample‐contributing centers. The study was approved by the ethical committee of the Antwerp University Hospital.
DISCLOSURE STATEMENT
The authors declare no conflict of interest.
Schepers D, Tortora G, Morisaki H, et al. A mutation update on the LDS‐associated genes TGFB2/3 and SMAD2/3 . Human Mutation. 2018;39:621–634. https://doi.org/10.1002/humu.23407
Contract grant sponsors: University of Antwerp (Lanceringsproject); the Fund for Scientific Research, Flanders (FWO, Belgium, G.0356.17); The Loeys‐Dietz Foundation; The Dutch Heart Foundation (2013T093); The Fondation Leducq (MIBAVA – Leducq 12CVD03)).
Communicated by Stylianos E. Antonarakis
REFERENCES
- Arslan‐Kirchner, M. , Epplen, J. T. , Faivre, L. , Jondeau, G. , Schmidtke, J. , De Paepe, A. , & Loeys, B. (2011). Clinical utility gene card for: Loeys‐Dietz syndrome (TGFBR1/2) and related phenotypes. European Journal of Human Genetics, 19(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubart, M. , Gobert, D. , Aubart‐Cohen, F. , Detaint, D. , Hanna, N. , d'Indya, H. , … Jondeau, G. (2014). Early‐onset osteoarthritis, Charcot‐Marie‐Tooth like neuropathy, autoimmune features, multiple arterial aneurysms and dissections: An unrecognized and life threatening condition. PLoS One, 9(5), e96387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azhar, M. , Schultz Jel, J. , Grupp, I. , Dorn, G. W., II , Meneton, P. , Molin, D. G. , … Doetschman, T. (2003). Transforming growth factor beta in cardiovascular development and function. Cytokine and Growth Factor Reviews, 14(5), 391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartram, U. , Molin, D. G. , Wisse, L. J. , Mohamad, A. , Sanford, L. P. , Doetschman, T. , … Gittenberger‐de Groot, A. C. (2001). Double‐outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF‐beta(2)‐knockout mice. Circulation, 103(22), 2745–2752. [DOI] [PubMed] [Google Scholar]
- Bernard, D. J. (2004). Both SMAD2 and SMAD3 mediate activin‐stimulated expression of the follicle‐stimulating hormone beta subunit in mouse gonadotrope cells. Molecular Endocrinology, 18(3), 606–623. [DOI] [PubMed] [Google Scholar]
- Berthet, E. , Hanna, N. , Giraud, C. , & Soubrier, M. (2015). A case of rheumatoid arthritis associated with SMAD3 gene mutation: A new clinical entity? Journal of Rheumatology, 42(3), 556. [DOI] [PubMed] [Google Scholar]
- Bertoli‐Avella, A. M. , Gillis, E. , Morisaki, H. , Verhagen, J. M. , de Graaf, B. M. , van de Beek, G. , … Loeys, B. L. (2015). Mutations in a TGF‐beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. Journal of American College of Cardiology, 65(13), 1324–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinc, A. , Maver, A. , Rudolf, G. , Tasic, J. , Pretnar Oblak, J. , Berden, P. , & Peterlin, B. (2015). clinical exome sequencing as a novel tool for diagnosing Loeys‐Dietz syndrome type 3. European Journal of Vascular and Endovascular Surgery, 50, 816–821. [DOI] [PubMed] [Google Scholar]
- Boileau, C. , Guo, D. C. , Hanna, N. , Regalado, E. S. , Detaint, D. , Gong, L. , … Milewicz, D. M. (2012). TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nature Genetics, 44(8), 916–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonniaud, P. , Kolb, M. , Galt, T. , Robertson, J. , Robbins, C. , Stampfli, M. , … Gauldie, J. (2004). Smad3 null mice develop airspace enlargement and are resistant to TGF‐beta‐mediated pulmonary fibrosis. Journal of Immunology, 173(3), 2099–2108. [DOI] [PubMed] [Google Scholar]
- Burke, C. , Shalhub, S. , & Starnes, B. W. (2015). Endovascular repair of an internal mammary artery aneurysm in a patient with SMAD‐3 mutation. Journal of Vascular Surgery, 62(2), 486–488. [DOI] [PubMed] [Google Scholar]
- Campens, L. , Callewaert, B. , Muino Mosquera, L. , Renard, M. , Symoens, S. , De Paepe, A. , … De Backer, J. (2015). Gene panel sequencing in heritable thoracic aortic disorders and related entities ‐ results of comprehensive testing in a cohort of 264 patients. Orphanet Journal of Rare Diseases, 10(1), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry, S. S. , Cain, S. A. , Morgan, A. , Dallas, S. L. , Shuttleworth, C. A. , & Kielty, C. M. (2007). Fibrillin‐1 regulates the bioavailability of TGFbeta1. Journal of Cell Biology, 176(3), 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn, R. D. , van Erp, C. , Habashi, J. P. , Soleimani, A. A. , Klein, E. C. , Lisi, M. T. , … Dietz, H. C. (2007). Angiotensin II type 1 receptor blockade attenuates TGF‐beta‐induced failure of muscle regeneration in multiple myopathic states. Nature Medicine, 13(2), 204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook, J. R. , Carta, L. , Galatioto, J. , & Ramirez, F. (2015). Cardiovascular manifestations in Marfan syndrome and related diseases; multiple genes causing similar phenotypes. Clinical Genetics, 87(1), 11–20. [DOI] [PubMed] [Google Scholar]
- Courtois, A. , Coppieters, W. , Bours, V. , Defraigne, J. O. , Colige, A. , & Sakalihasan, N. (2017). A novel SMAD3 mutation caused multiple aneurysms in a patient without osteoarthritis symptoms. European Journal of Medical Genetics, 60(4), 228–231. [DOI] [PubMed] [Google Scholar]
- Dallas, S. L. , Miyazono, K. , Skerry, T. M. , Mundy, G. R. , & Bonewald, L. F. (1995). Dual role for the latent transforming growth factor‐beta binding protein in storage of latent TGF‐beta in the extracellular matrix and as a structural matrix protein. Journal of Cell Biology, 131(2), 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck, R. , & Akhurst, R. J. (2007). Differentiation plasticity regulated by TGF‐beta family proteins in development and disease. Nature Cell Biology, 9(9), 1000–1004. [DOI] [PubMed] [Google Scholar]
- Derynck, R. , & Zhang, Y. (1996). Intracellular signalling: The mad way to do it. Current Biology, 6(10), 1226–1229. [DOI] [PubMed] [Google Scholar]
- Derynck, R. , & Zhang, Y. E. (2003). Smad‐dependent and Smad‐independent pathways in TGF‐beta family signalling. Nature, 425(6958), 577–584. [DOI] [PubMed] [Google Scholar]
- Doetschman, T. , Georgieva, T. , Li, H. , Reed, T. D. , Grisham, C. , Friel, J. , … Azhar, M. (2012). Generation of mice with a conditional allele for the transforming growth factor beta3 gene. Genesis, 50(1), 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett, A. D. , Tufro‐McReddie, A. , Fisher, A. , & Gomez, R. A. (1994). Angiotensin receptor regulates cardiac hypertrophy and transforming growth factor‐beta 1 expression. Hypertension, 23(5), 587–592. [DOI] [PubMed] [Google Scholar]
- Feng, X. H. , & Derynck, R. (2005). Specificity and versatility in tgf‐beta signaling through Smads. Annual Review of Cell and Developmental Biology, 21, 659–693. [DOI] [PubMed] [Google Scholar]
- Fitzgerald, K. K. , Bhat, A. M. , Conard, K. , Hyland, J. , & Pizarro, C. (2014). Novel SMAD3 mutation in a patient with hypoplastic left heart syndrome with significant aortic aneurysm. Case Reports in Genetic, 2014, 591516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokkema, I. F. , Taschner, P. E. , Schaafsma, G. C. , Celli, J. , Laros, J. F. , & den Dunnen, J. T. (2011). LOVD v.2.0: The next generation in gene variant databases. Human Mutation, 32(5), 557–563. [DOI] [PubMed] [Google Scholar]
- Fontana, P. , Genesio, R. , Casertano, A. , Cappuccio, G. , Mormile, A. , Nitsch, L. , … Melis, D. (2014). Loeys‐Dietz syndrome type 4, caused by chromothripsis, involving the TGFB2 gene. Gene, 538(1), 69–73. [DOI] [PubMed] [Google Scholar]
- Fukuda, N. , Hu, W. Y. , Kubo, A. , Kishioka, H. , Satoh, C. , Soma, M. , … Kanmatsuse, K. (2000). Angiotensin II upregulates transforming growth factor‐beta type I receptor on rat vascular smooth muscle cells. American Journal of Hypertension, 13(2), 191–198. [DOI] [PubMed] [Google Scholar]
- Gago‐Diaz, M. , Blanco‐Verea, A. , Teixido‐Tura, G. , Valenzuela, I. , Del Campo, M. , Borregan, M. , … Brion, M. (2014). Whole exome sequencing for the identification of a new mutation in TGFB2 involved in a familial case of non‐syndromic aortic disease. Clinica Chimica Acta, 437, 88–92. [DOI] [PubMed] [Google Scholar]
- Gallo, E. M. , Loch, D. C. , Habashi, J. P. , Calderon, J. F. , Chen, Y. , Bedja, D. , … Dietz, H. C. (2014). Angiotensin II‐dependent TGF‐beta signaling contributes to Loeys‐Dietz syndrome vascular pathogenesis. Journal of Clinical Investigation, 124(1), 448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Bermudez, M. , Moustafa, A. H. , Barros‐Membrilla, A. , & Tizon‐Marcos, H. (2017). Repeated loss of consciousness in a young woman: A suspicious SMAD3 mutation underlying spontaneous coronary artery dissection. Canadian Journal of Cardiology, 33(2), 292 e1–292 e3. [DOI] [PubMed] [Google Scholar]
- Habashi, J. P. , Judge, D. P. , Holm, T. M. , Cohn, R. D. , Loeys, B. L. , Cooper, T. K. , … Dietz, H. C. (2006). Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science, 312(5770), 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller, G. , Alvarado, D. M. , Willing, M. C. , Braverman, A. C. , Bridwell, K. H. , Kelly, M. , … Dobbs, M. B. (2015). Genetic risk for aortic aneurysm in adolescent idiopathic scoliosis. Journal of Bone and Joint Surgery. American Volume, 97(17), 1411–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilhorst‐Hofstee, Y. , Scholte, A. J. , Rijlaarsdam, M. E. , van Haeringen, A. , Kroft, L. J. , Reijnierse, M. , … Breuning, M. H. (2013). An unanticipated copy number variant of chromosome 15 disrupting SMAD3 reveals a three‐generation family at serious risk for aortic dissection. Clinical Genetics, 83(4), 337–344. [DOI] [PubMed] [Google Scholar]
- Holm, T. M. , Habashi, J. P. , Doyle, J. J. , Bedja, D. , Chen, Y. , van Erp, C. , … Cohn, R. D. (2011). Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 332(6027), 358–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai, Z. , Ono, R. N. , Ushiro, S. , Keene, D. R. , Chen, Y. , Mazzieri, R. , … Sakai, L. Y. (2003). Latent transforming growth factor beta‐binding protein 1 interacts with fibrillin and is a microfibril‐associated protein. Journal of Biological Chemistry, 278(4), 2750–2757. [DOI] [PubMed] [Google Scholar]
- Kaartinen, V. , Voncken, J. W. , Shuler, C. , Warburton, D. , Bu, D. , Heisterkamp, N. , & Groffen, J. (1995). Abnormal lung development and cleft palate in mice lacking TGF‐beta 3 indicates defects of epithelial‐mesenchymal interaction. Nature Genetics, 11(4), 415–421. [DOI] [PubMed] [Google Scholar]
- Kuechler, A. , Altmuller, J. , Nurnberg, P. , Kotthoff, S. , Kubisch, C. , & Borck, G. (2015). Exome sequencing identifies a novel heterozygous TGFB3 mutation in a disorder overlapping with Marfan and Loeys‐Dietz syndrome. Molecular and Cellular Probes, 29, 330–334. [DOI] [PubMed] [Google Scholar]
- Lacro, R. V. , Dietz, H. C. , Sleeper, L. A. , Yetman, A. T. , Bradley, T. J. , Colan, S. D. , … Loeys, B. L. (2014). Atenolol versus losartan in children and young adults with Marfan's syndrome. New England Journal of Medicine, 371(22), 2061–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. K. , Pardoux, C. , Hall, M. C. , Lee, P. S. , Warburton, D. , Qing, J. , … Derynck, R. (2007). TGF‐beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO Journal, 26(17), 3957–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner‐Ellis, J. P. , Aldubayan, S. H. , Hernandez, A. L. , Kelly, M. A. , Stuenkel, A. J. , Walsh, J. , & Joshi, V. A. (2014). The spectrum of FBN1, TGFbetaR1, TGFbetaR2 and ACTA2 variants in 594 individuals with suspected Marfan syndrome, Loeys‐Dietz syndrome or thoracic aortic aneurysms and dissections (TAAD). Molecular Genetics and Metabolism, 112(2), 171–176. [DOI] [PubMed] [Google Scholar]
- Leutermann, R. , Sheikhzadeh, S. , Brockstadt, L. , Rybczynski, M. , van Rahden, V. , Kutsche, K. , … Rosenberger, G. (2014). A 1‐bp duplication in TGFB2 in three family members with a syndromic form of thoracic aortic aneurysm. European Journal of Human Genetics, 22(7), 944–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C. G. , Liang, Q. Q. , Zhou, Q. , Menga, E. , Cui, X. J. , Shu, B. , … Wang, Y. J. (2009). A continuous observation of the degenerative process in the intervertebral disc of Smad3 gene knock‐out mice. Spine, 34(13), 1363–1369. [DOI] [PubMed] [Google Scholar]
- Lindsay, M. E. , & Dietz, H. C. (2011). Lessons on the pathogenesis of aneurysm from heritable conditions. Nature, 473(7347), 308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay, M. E. , Schepers, D. , Bolar, N. A. , Doyle, J. J. , Gallo, E. , Fert‐Bober, J. , … Loeys, B. L. (2012). Loss‐of‐function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nature Genetics, 44(8), 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeys, B. L. (2015). Angiotensin receptor blockers: A panacea for Marfan syndrome and related disorders? Drug Discovery Today, 20(2), 262–266. [DOI] [PubMed] [Google Scholar]
- Loeys, B. L. , Chen, J. , Neptune, E. R. , Judge, D. P. , Podowski, M. , Holm, T. , … Dietz, H. C. (2005). A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nature Genetics, 37(3), 275–281. [DOI] [PubMed] [Google Scholar]
- Loeys, B. L. , Schwarze, U. , Holm, T. , Callewaert, B. L. , Thomas, G. H. , Pannu, H. , … De Paepe, A. M. (2006). Aneurysm syndromes caused by mutations in the TGF‐beta receptor. New England Journal of Medicine, 355(8), 788–798. [DOI] [PubMed] [Google Scholar]
- MacCarrick, G. , Black, J. H., III , Bowdin, S. , El‐Hamamsy, I. , Frischmeyer‐Guerrerio, P. A. , Guerrerio, A. L. , … Dietz, H. C., III (2014). Loeys‐Dietz syndrome: A primer for diagnosis and management. Genetics in Medicine, 16(8), 576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majesky, M. W. (2007). Developmental basis of vascular smooth muscle diversity. Arteriosclerosis, Thrombosis and Vascular Biology, 27(6), 1248–1258. [DOI] [PubMed] [Google Scholar]
- Martens, T. , Van Herzeele, I. , De Ryck F, Renard, M. , De Paepe, A. , Francois K, … De Backer, J. , (2013). Multiple aneurysms in a patient with aneurysms‐osteoarthritis syndrome. Annals of Thoracic Surgery, 95(1), 332–335. [DOI] [PubMed] [Google Scholar]
- Massague, J. , Blain, S. W. , & Lo, R. S. (2000). TGFbeta signaling in growth control, cancer, and heritable disorders. Cell, 103(2), 295–309. [DOI] [PubMed] [Google Scholar]
- Massague, J. , Seoane, J. , & Wotton, D. (2005). Smad transcription factors. Genes Development, 19(23), 2783–2810. [DOI] [PubMed] [Google Scholar]
- Matyas, G. , Naef, P. , Tollens, M. , & Oexle, K. (2014). De novo mutation of the latency‐associated peptide domain of TGFB3 in a patient with overgrowth and Loeys‐Dietz syndrome features. American Journal of Medical Genetics A, 164A(8), 2141–2143. [DOI] [PubMed] [Google Scholar]
- Mc Kusick, V. (1955). The cardiovascular aspects of Marfan's syndrome: A heritable disorder of connective tissue. Circulation, 11(3), 321–342. [DOI] [PubMed] [Google Scholar]
- Micha, D. , Guo, D. C. , Hilhorst‐Hofstee, Y. , van Kooten, F. , Atmaja, D. , Overwater, E. , … van Dijk, F. S. (2015). SMAD2 mutations are associated with arterial aneurysms and dissections. Human Mutation, 36, 1145–1149. [DOI] [PubMed] [Google Scholar]
- Naito, T. , Masaki, T. , Nikolic‐Paterson, D. J. , Tanji, C. , Yorioka, N. , & Kohno, N. (2004). Angiotensin II induces thrombospondin‐1 production in human mesangial cells via p38 MAPK and JNK: A mechanism for activation of latent TGF‐beta1. American Journal of Physiology‐Renal Physiology, 286(2), F278–F287. [DOI] [PubMed] [Google Scholar]
- Neptune, E. R. , Frischmeyer, P. A. , Arking, D. E. , Myers, L. , Bunton, T. E. , Gayraud, B. , … Dietz, H. C. (2003). Dysregulation of TGF‐beta activation contributes to pathogenesis in Marfan syndrome. Nature Genetics, 33(3), 407–411. [DOI] [PubMed] [Google Scholar]
- Nevidomskyte, D. , Shalhub, S. , Aldea, G. S. , Byers, P. H. , Schwarze, U. , Murray, M. L. , & Starnes, B. (2017). Endovascular repair of internal mammary artery aneurysms in 2 sisters with SMAD3 mutation. Annals of Vascular Surgery, 41, 283.e5–283.e9. [DOI] [PubMed] [Google Scholar]
- Ng, C. M. , Cheng, A. , Myers, L. A. , Martinez‐Murillo, F. , Jie, C. , Bedja, D. , … Dietz, H. C. (2004). TGF‐beta‐dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. Journal of Clinical Investigation, 114(11), 1586–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proetzel, G. , Pawlowski, S. A. , Wiles, M. V. , Yin, M. , Boivin, G. P. , Howles, P. N. , … Doetschman, T. (1995). Transforming growth factor‐beta 3 is required for secondary palate fusion. Nature Genetics, 11(4), 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proost, D. , Vandeweyer, G. , Meester, J. A. , Salemink, S. , Kempers, M. , Ingram, C. , … Van Laer, L. (2015). Performant mutation identification using targeted next generation sequencing of fourteen thoracic aortic aneurysm genes. Human Mutation, 36, 808–814. [DOI] [PubMed] [Google Scholar]
- Ramachandra, C. J. , Mehta, A. , Guo, K. W. , Wong, P. , Tan, J. L. , & Shim, W. (2015). Molecular pathogenesis of Marfan syndrome. International Journal of Cardiology, 187, 585–591. [DOI] [PubMed] [Google Scholar]
- Regalado, E. S. , Guo, D. C. , Villamizar, C. , Avidan, N. , Gilchrist, D. , McGillivray, B. , … Milewicz, D. M. (2011). Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circulation Research, 109(6), 680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard, M. , Callewaert, B. , Malfait, F. , Campens, L. , Sharif, S. , del Campo, M. , … De Backer, J. (2013). Thoracic aortic‐aneurysm and dissection in association with significant mitral valve disease caused by mutations in TGFB2. International Journal of Cardiology, 165(3), 584–587. [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , & Gastier‐Foster, J. (2015). ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rienhoff, H. Y., Jr. , Yeo, C. Y. , Morissette, R. , Khrebtukova, I. , Melnick, J. , Luo, S. , … Whitman, M. (2013). A mutation in TGFB3 associated with a syndrome of low muscle mass, growth retardation, distal arthrogryposis and clinical features overlapping with Marfan and Loeys‐Dietz syndrome. American Journal of Medical Genetics A, 161A(8), 2040–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritelli, M. , Chiarelli, N. , Dordoni, C. , Quinzani, S. , Venturini, M. , Maroldi, R. , … Colombi, M. (2014). Further delineation of Loeys‐Dietz syndrome type 4 in a family with mild vascular involvement and a TGFB2 splicing mutation. BMC Medical Genetics, 15, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Vita, J. , Sanchez‐Lopez, E. , Esteban, V. , Ruperez, M. , Egido, J. , & Ruiz‐Ortega, M. (2005). Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor‐beta‐independent mechanism. Circulation, 111(19), 2509–2517. [DOI] [PubMed] [Google Scholar]
- Romaniello, F. , Mazzaglia, D. , Pellegrino, A. , Grego, S. , Fiorito, R. , Ferlosio, A. , … Orlandi, A. (2014). Aortopathy in Marfan syndrome: An update. Cardiovascular Pathology, 23(5), 261–266. [DOI] [PubMed] [Google Scholar]
- Sanford, L. P. , Ormsby, I. , Gittenberger‐de Groot, A. C. , Sariola, H. , Friedman, R. , Boivin, G. P. , … Doetschman, T. (1997). TGFbeta2 knockout mice have multiple developmental defects that are non‐overlapping with other TGFbeta knockout phenotypes. Development, 124(13), 2659–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert, J. A. , Landis, B. J. , Shikany, A. R. , Hinton, R. B. , & Ware, S. M. (2016). Clinically relevant variants identified in thoracic aortic aneurysm patients by research exome sequencing. American Journal of Medical Genetics A, 170A(5), 1288–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, M. , Zhu, J. , Wang, R. , Chen, X. , Mi, L. , Walz, T. , & Springer, T. A. (2011). Latent TGF‐beta structure and activation. Nature, 474(7351), 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shores, J. , Berger, K. R. , Murphy, E. A. , & Pyeritz, R. E. (1994). Progression of aortic dilatation and the benefit of long‐term beta‐adrenergic blockade in Marfan's syndrome. New England Journal of Medicine, 330(19), 1335–1341. [DOI] [PubMed] [Google Scholar]
- Topouzis, S. , & Majesky, M. W. (1996). Smooth muscle lineage diversity in the chick embryo. Two types of aortic smooth muscle cell differ in growth and receptor‐mediated transcriptional responses to transforming growth factor‐beta. Developmental Biology, 178(2), 430–445. [DOI] [PubMed] [Google Scholar]
- van de Laar, I. M. , Oldenburg, R. A. , Pals, G. , Roos‐Hesselink, J. W. , de Graaf, B. M. , Verhagen, J. M. , … Bertoli‐Avella, A. M. (2011). Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early‐onset osteoarthritis. Nature Genetics, 43(2), 121–126. [DOI] [PubMed] [Google Scholar]
- van de Laar, I. M. , van der Linde, D. , Oei, E. H. , Bos, P. K. , Bessems, J. H. , Bierma‐Zeinstra, S. M. , … Wessels, M. W. (2012). Phenotypic spectrum of the SMAD3‐related aneurysms‐osteoarthritis syndrome. Journal of Medical Genetics, 49(1), 47–57. [DOI] [PubMed] [Google Scholar]
- van der Linde, D. , van de Laar, I. M. , Bertoli‐Avella, A. M. , Oldenburg, R. A. , Bekkers, J. A. , Mattace‐Raso, F. U. , … Roos‐Hesselink, J. W. (2012). Aggressive cardiovascular phenotype of aneurysms‐osteoarthritis syndrome caused by pathogenic SMAD3 variants. Journal of American College of Cardiology, 60(5), 397–403. [DOI] [PubMed] [Google Scholar]
- Waldrip, W. R. , Bikoff, E. K. , Hoodless, P. A. , Wrana, J. L. , & Robertson, E. J. (1998). Smad2 signaling in extraembryonic tissues determines anterior‐posterior polarity of the early mouse embryo. Cell, 92(6), 797–808. [DOI] [PubMed] [Google Scholar]
- Weinstein, M. , Yang, X. , Li, C. , Xu, X. , Gotay, J. , & Deng, C. X. (1998). Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proceedings of the National Academy of Sciences of the United States of America, 95(16), 9378–9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischmeijer, A. , Van Laer, L. , Tortora, G. , Bolar, N. A. , Van Camp, G. , Fransen, E. , … Loeys, B. L. (2013). Thoracic aortic aneurysm in infancy in aneurysms‐osteoarthritis syndrome due to a novel SMAD3 mutation: Further delineation of the phenotype. American Journal of Medical Genetics A, 161A(5), 1028–1035. [DOI] [PubMed] [Google Scholar]
- Yamashita, M. , Fatyol, K. , Jin, C. , Wang, X. , Liu, Z. , & Zhang, Y. E. (2008). TRAF6 mediates Smad‐independent activation of JNK and p38 by TGF‐beta. Molecular Cell, 31(6), 918–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. , Letterio, J. J. , Lechleider, R. J. , Chen, L. , Hayman, R. , Gu, H. , … Deng, C. (1999). Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF‐beta. EMBO Journal, 18(5), 1280–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, J. Y. , Wang, Y. , An, J. , Mao, C. M. , Hou, N. , Lv, Y. X. , … Yang, X. (2003). Mutation analysis of the Smad3 gene in human osteoarthritis. European Journal of Human Genetics, 11(9), 714–717. [DOI] [PubMed] [Google Scholar]
- Ye, P. , Chen, W. , Wu, J. , Huang, X. , Li, J. , Wang, S. , … Xia, J. (2013). GM‐CSF contributes to aortic aneurysms resulting from SMAD3 deficiency. Journal of Clinical Investigation, 123(5), 2317–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Zhou, M. , Liu, C. , Liu, C. , Qiao, T. , Huang, D. , … Liu, Z. (2015). A novel mutation of SMAD3 identified in a Chinese family with aneurysms‐osteoarthritis syndrome. Biomed Research International, 2015, 968135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. E. (2009). Non‐Smad pathways in TGF‐beta signaling. Cell Research, 19(1), 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Table S1
Supporting Information Table S2
Supporting Information Table S3
Supporting Information Table S4
Supporting Information Table S5