

Abstract
Neuropilin‐1 (NRP1) is a transmembrane co‐receptor involved in binding interactions with variety of ligands and receptors, including receptor tyrosine kinases. Expression of NRP1 in several cancers correlates with cancer stages and poor prognosis. Thus, NRP1 has been considered a therapeutic target and is the focus of multiple drug discovery initiatives. Vascular endothelial growth factor (VEGF) binds to the b1 domain of NRP1 through interactions between the C‐terminal arginine of VEGF and residues in the NRP1‐binding site including Tyr297, Tyr353, Asp320, Ser346 and Thr349. We obtained several complexes of the synthetic ligands and the NRP1‐b1 domain and used X‐ray crystallography and computational methods to analyse atomic details and hydration profile of this binding site. We observed side chain flexibility for Tyr297 and Asp320 in the six new high‐resolution crystal structures of arginine analogues bound to NRP1. In addition, we identified conserved water molecules in binding site regions which can be targeted for drug design. The computational prediction of the VEGF ligand‐binding site hydration map of NRP1 was in agreement with the experimentally derived, conserved hydration structure. Displacement of certain conserved water molecules by a ligand's functional groups may contribute to binding affinity, whilst other water molecules perform as protein–ligand bridges. Our report provides a comprehensive description of the binding site for the peptidic ligands’ C‐terminal arginines in the b1 domain of NRP1, highlights the importance of conserved structural waters in drug design and validates the utility of the computational hydration map prediction method in the context of neuropilin.
Database
The structures were deposited to the PDB with accession numbers PDB ID: 5IJR, 5IYY, 5JHK, 5J1X, 5JGQ, 5JGI.
Keywords: ligand‐binding protein, neuropilin, SPR, vascular endothelial growth factor (VEGF), X‐ray crystallography
Abbreviations
- NRP1
Neuropilin‐1
- NRP1‐b1
B1 domain of Neuropilin‐1
- NRPs
Neuropilin‐1 and Neuropilin‐2
- PDB
protein data bank
- VEGF‐A165
vascular endothelial growth factor
Introduction
Neuropilins (NRPs) are transmembrane receptors that take part in protein–protein interactions, and are essential for formation of the mammalian nervous and vascular systems 1, 2. NRPs were initially discovered as neuronal receptors for class‐3 semaphorins and mediators of axonal guidance in the developing nervous system 3. NRPs also bind a number of growth factors, including vascular endothelial growth factors (VEGFs) 4, 5, transforming growth factor (TGF‐β) 6, 7 and hepatocyte growth factor (HGF) 8. Furthermore, NRPs are involved in the Hedgehog signal transduction pathway 9, and tumour immune regulation through regulatory T cells 10. Two homologous proteins, neuropilin‐1 (NRP1) and neuropilin‐2 (NRP2), share 44% sequence identity and a similar multi‐domain structure. The interaction of NRP1 with VEGF‐A165 has been extensively studied, and is implicated in the processes of angiogenesis 11, cell migration and metastasis 12, and evasion of an immune response in cancer 13, 14. VEGF‐A165 binds to a well‐defined binding site on the b1 domain of NRP1 (NRP1‐b1) through its C‐terminal arginine residue. It is possible that other ligands containing a C‐terminal arginine will also bind in a similar fashion 15. Although protein–protein interactions have been considered challenging targets in drug discovery 16, a number of small molecules and peptides have been identified as inhibitors of the VEGF‐A165–NRP1 interaction 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29. In some cases, it has been demonstrated that the inhibitors act through direct binding to the b1 domain of NRP1. These inhibitors, exemplified by EG00229, also incorporate an arginine residue and bind at the same site as the C‐terminus of VEGF‐A165 19.
We have conducted an extensive study of the C‐terminal arginine‐binding site in NRP1‐b1, focusing on protein side chain flexibility and conserved water molecules. Hydration of proteins is an important parameter to consider in drug design. Structural waters may play a key role in the function, shape and conformation of proteins, as well as in protein–ligand interactions 30, 31. The incorporation of structural waters in computational drug design may significantly improve the outcome of molecular docking results 32, 33, 34. Water molecules can act as bridges between the protein‐binding site and a ligand or cofactor via hydrogen bonds 32, 33, 34, 35. Disruption of these water networks by small molecule compounds may have an effect on the binding affinity of such molecules. Replacement of a tightly bound water molecule does not, however, necessarily improve binding affinity or ligand efficiency, and it may influence the pharmacodynamic profile of the molecule 32, 33, 34, 35. It is therefore important to consider structural or conserved waters in the binding site, and to explore their role in ligand‐binding efficiency either by replacing them with functional groups or by incorporating them as hydrogen‐bond bridges between the ligand and the protein. Unfortunately, due to poor structural understanding or due to lack of availability of this information from protein crystal structures (e.g. in low‐resolution structures), water molecules are often not included in structure‐based drug design. As an alternative, computational models may be used to predict the location of water molecules in protein structures 34, 36.
We selected a number of arginine analogues with simple structural variations to perform this study, and obtained high‐resolution structures of six compounds bound to NRP1‐b1. Surface plasmon resonance (SPR) was used to determine the dissociation constant (K D) of the arginine analogues to immobilised NRP1‐b1. The binding data, together with computational methods and crystallographic information, was used to rationalise the side chain flexibility and hydration of the binding site in NRP1‐b1. The new crystal structures were analysed alongside publicly available NRP1‐b1 structures in the protein data bank (PDB). This comprehensive analysis and knowledge of the detailed architecture of the VEGF‐A165 binding site in NRP1‐b1 will facilitate future drug design efforts.
Results and Discussion
Prediction of binding site hydration structure
We initiated our study into the binding site hydration structure of NRP1‐b1 domain by predicting the position of water molecules using computational models. We used a solvent analysis application within the software package MOE (Chemical Computing Group), which assesses the role of solvent in protein structures, using a three‐dimensional reference interaction site model (3D‐RISM). This method allows for the calculation of the solvation structure represented by the probability density function of finding interaction sites for the solvent molecules (in this case, water molecules) at the specific point around the solute molecule (in this case, our protein target NRP1‐b1) 34, 35, 36, 37. At the time this study was designed, there were 11 structures of NRP1 deposited in the PDB, 7 of which are of human origin (PDB codes: 4RN5, 4DEQ, 3I97, 2QQI, 2QQM, 2QQN, 1KEX), 2 mouse (PDB codes: 4GZ9, 4GZA) and 2 rat (PDB codes: 2ORX, 2ORZ). These structures corresponded either to the isolated b1 domain or tandem b1b2 and a2b1b2 domains of NRP1. The structure of EG00229‐bound NRP1‐b1 (PDB code: 3I97) was selected for initial computational studies (Fig. 1). Although this was not the structure of highest resolution available at the time, it was the only one with a synthetic ligand present at the binding site, and thus deemed appropriate for analysing the effect of ligand‐binding on the solvation structure of the receptor protein. The calculations were performed on the protein–ligand complex, the protein alone with the ligand excluded from the calculations and on the ligand alone. To visualise these calculations, the predicted distribution of water density was plotted on the binding site of NRP1‐b1 (Fig. 2). There is a clear overlap of the distribution of the predicted water density between the ligand‐bound and the ligand‐free protein state. However, the comparison of the predicted solvation distribution in the presence and absence of ligand shows that guanidine and carboxylic acid groups of EG00229 occupy two of the distinct patches of the solvation space predicted to exist in the absence of the ligand (regions G and C in Fig. 2). The predictions thus suggest that ligand binding is accompanied by solvent‐displacement. In addition to the areas of the molecular surface occupied by the guanidine and the carboxylate groups, four other regions, labelled 1–4, show water density in the binding site identified from the calculations performed with either the protein alone or NRP1‐b1‐EG00229 complex (Fig. 2).
Figure 1.
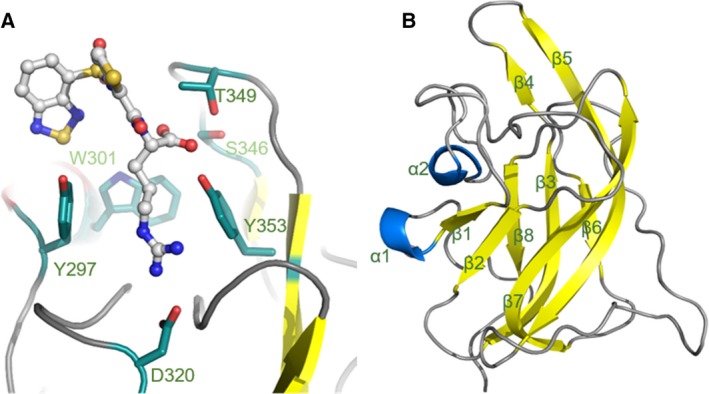
Structure of the binding site of NRP1‐b1 domain. (A) Ball and stick representation of EG00229 (carbon atoms are coloured grey, nitrogen blue, sulphur yellow and oxygen red) bound to NRP‐b1 (PDB entry 3I97). NRP1‐b1 domain residues involved in the non‐covalent interactions with the ligand are shown in sticks representation. (B) Ribbon diagram of NRP1‐b1 fold: β‐sheets are represented in yellow and α‐helixes in blue.
Figure 2.
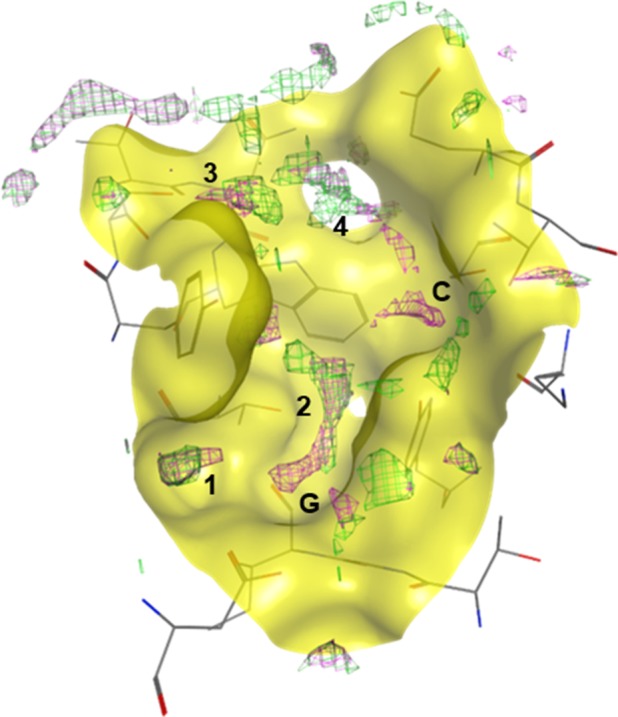
Prediction model of NRP1‐b1 binding site hydration. Computational analysis performed on PDB entry 3I97, where the green mesh represents the solvation prediction with EG00229 bound to NRP1‐b1, and the pink mesh represents the solvation prediction of NRP1‐b1 in the apo form. Labels 1–4 indicate the regions predicted to be occupied by water molecules in the binding site; G and C indicate the regions occupied by the arginine guanidine and carboxylate groups, respectively, if EG000229 is bound. In the apo form of NRP1‐b1, these regions are also predicted to be occupied by water molecules.
Selection of ligands for the study
EG00229 is a small molecule formed of an arginine residue with Nα‐substitution. Key interactions of EG00229 with NRP1‐b1 are established through hydrogen bonds between the guanidine and carboxylic acid groups of the ligand to the receptor. To gain more comprehensive understanding of the ligand‐binding site structure which would assist us in the design of NRP1 antagonists, we investigated interaction between NRP1‐b1 domain and multiple commercially available arginine analogues. The ligands selected for this study retain the arginine moiety as seen in EG00229, while providing structural diversity in the Nα‐site of the molecule (Fig. 3). l‐arginine (R1) and seven commercially available Nα‐substituted‐l‐arginine analogues R2–R8 were selected to explore their binding affinities and the solvation profile of the NRP1‐b1 ligand‐binding site experimentally. In addition, underivatised l‐homoarginine (R9) was also included in the study (Fig. 3). As we were only interested in probing the binding site and the crystallographic behaviour of the ligands, metabolic stability of the compounds was not considered. Thus, we were able to probe the binding site with a relatively simple, readily available and cheap set of analogues.
Figure 3.
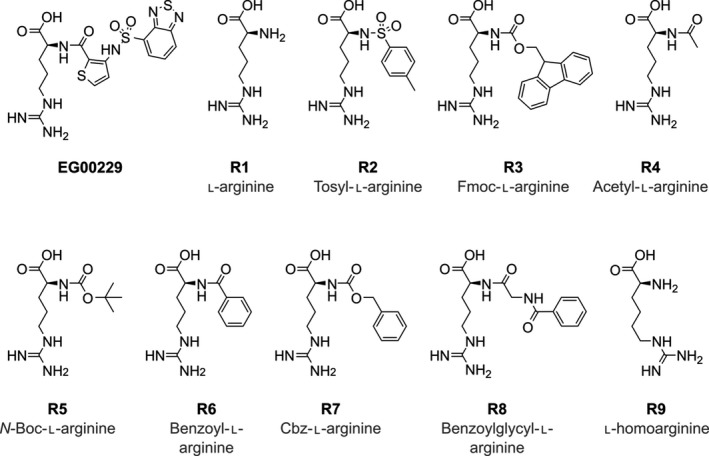
Chemical structures of selected arginine analogues R1–R9.
A surface plasmon resonance (SPR) method was used to determine the binding affinities of the ligands to NRP1‐b1, and crystallisation trials were performed with the preformed complexes between NRP1‐b1 and the individual ligands.
Ligand‐binding affinities
All arginine analogues showed very fast rates of association and dissociation to immobilised NRP1‐b1, as determined by surface plasmon resonance experiments (Fig. 4). Binding responses were consistent with a 1 : 1 binding interaction model and equilibrium binding affinities were determined using steady‐state binding levels to afford dissociation constants (Fig. 4). l‐Arginine R1 and l‐homoarginine R9 showed weak binding to NRP1‐b1, with dissociation constants (K Ds) of 325 ± 5 and 637 ± 70 μm, respectively. These are the weakest binding ligands tested, with all other Nα‐substituted analogues exhibiting higher affinities and lower corresponding dissociation constants. The data suggest that the free amine is not favourable for binding, and that the longer alkane chain on l‐homoarginine might de‐stabilise key interactions with the receptor. Interestingly, a large Nα substituent, as seen in FMOC‐l‐arginine (R3) also resulted in a weakly binding ligand, with a K D of 201 ± 106 μm; however, this molecule's poor solubility might have affected the results. Tosyl Nα‐substituted l‐arginine (R2), acetyl‐l‐arginine (R4), benzoyl‐l‐arginine (R6) and benzoylglycyl‐l‐arginine (R8) exhibited a medium range of affinities with K Ds between 70 and 22 μm (Fig. 4) without a clear structure/activity relationship. The two other analogues containing carbamate groups, N‐boc‐l‐arginine (R5) and Cbz‐l‐arginine (R7) exhibited highest affinities for NRP1‐b1 in the series, with K Ds of 3 ± 1 and 17 ± 1 μm, respectively.
Figure 4.
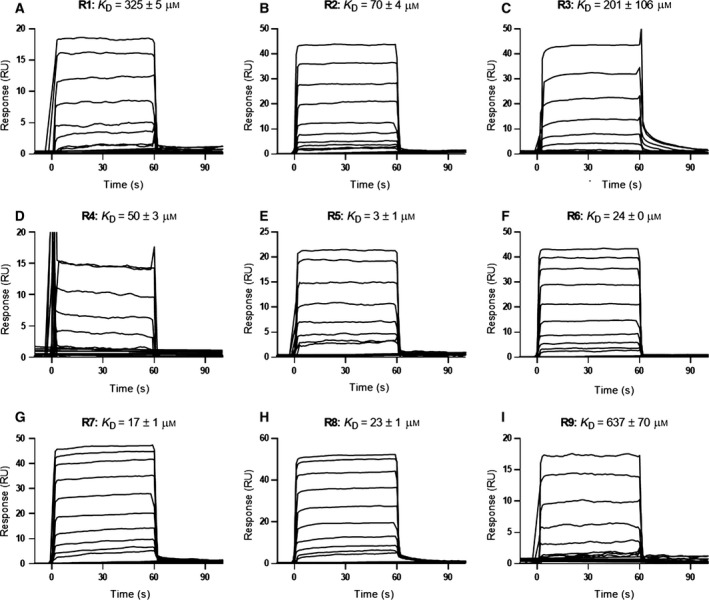
SPR measurements of arginine analogues R1–R9 binding to immobilised NRP1‐b1. Equilibrium dissociation constants (KD) were calculated using steady‐state binding levels and assuming a 1 : 1 binding model of the arginine analogues R1–R9 to immobilised NRP1‐b1. All sensorgrams are double‐referenced, using a blank surface and sample. Concentration ranges are as follows: R1 and R9 were tested at 12–1500 μm, R5 was tested at 0.16–20 μm, all other analogues were tested at 0.6–300 μm (n = 2).
The analogues were also tested in a ligand displacement essay assessing their potency to compete with the VEGF‐A165 for binding to NRP1‐b1. R5, the Arg analogue with the highest affinity in the SPR assay, exhibited clear sigmoidal curve in a ligand displacement essay with an IC50 of 2.6 ± 0.38 μm, which correlates extremely well with the SPR data (Fig. 5). However, while R6, R7 and R8 exhibited weak activity, we were unable to reproducibly determine their IC50 values as the binding curves did not display sigmoidal features.
Figure 5.
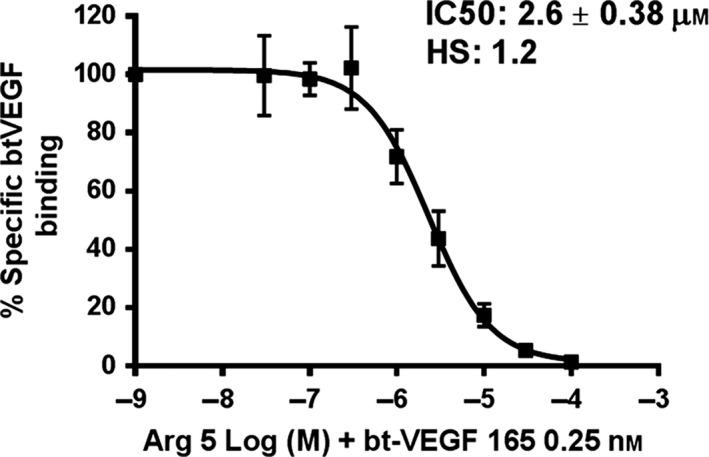
Inhibition of bt‐VEGF‐A165 binding to NRP1 by R5 arginine analogue. The various concentrations of R5 compound were added to the 96‐well plates precoated with NRP1‐b1 protein, followed by addition of 0.25 nm bt‐VEGF‐A165. Non‐specific binding of bt‐VEGF‐A165 to the plates was determined in the absence of NRP1‐b1. R5 analogue competed with bt‐VEGF‐A165 for binding to plates coated with NRP1 with an IC50 of 2.6 μm. Values presented are the means ± SEM obtained from three independent experiments each performed in duplicates.
Crystal structures of the NRP1‐b1 domain complexed with arginine analogues
Co‐crystallisation studies were attempted with all ligands selected and six complexes produced good‐quality crystals. All six complexes crystallised in the monoclinc (P21) space group with a dimer in the asymmetric unit for all complexes except for that of R5 analogue which contained a tetramer in the asymmetric unit. The structures were refined using data at the resolutions higher than 2.1 Å (for R5‐bound NRP1 b1); full data and refinement statistics are available in Table 1.
Table 1.
Data collection and refinement statistics. The values in parentheses are for the last resolution shell
| Ligand | R4 | R5 | R6 | R7 | R8 | R9 |
|---|---|---|---|---|---|---|
| PDB ID | 5JGI | 5J1X | 5JGQ | 5IYY | 5JHK | 5IJR |
| Data collection | ||||||
| Space group | P 21 | P 21 | P 21 | P 21 | P 21 | P 21 |
| Unit cell parameters (Å) | 40.65 | 41.59 | 40.79 | 41.52 | 40.38 | 40.14 |
| 89.20 | 90.48 | 89.57 | 88.94 | 90.78 | 88.94 | |
| 41.65 | 81.89 | 41.38 | 41.70 | 40.97 | 40.96 | |
| β (°) | 98.71 | 99.64 | 98.57 | 91.61 | 98.27 | 96.73 |
| Molecules per asymmetric unit | 2 | 4 | 2 | 2 | 2 | 2 |
| Resolution range (Å)a | 1.38 (1.40–1.38) | 2.10 (2.16–2.10) | 1.60 (1.63–1.60) | 1.60 (1.63–1.60) | 1.80 (1.84–1.80) | 1.52 (1.55–1.52) |
| Rmerge | 0.035 (0.136) | 0.103 (0.476) | 0.047 (0.442) | 0.117 (0.436) | 0.058 (0.552) | 0.048 (0.510) |
| CC(1/2) | 0.999 (0.987) | 0.995 (0.820) | 0.998 (0.844) | 0.994 (0.871) | 0.999 (0.872) | 1.00 (0.866) |
| <I/σ (I)> | 28.4 (8.6) | 9.1 (2.6) | 10.5 (2.0) | 9.9 (3.5) | 22.6 (3.2) | 22.8 (2.8) |
| Completeness (%) | 95.7 (70.0) | 99.8 (99.8) | 97.0 (75.3) | 99.7 (99.0) | 99.9 (99.9) | 95.4 (67.8) |
| Number of total reflections | 377 819 (10823) | 130 425 (10307) | 115 306 (2763) | 270 340 (12410) | 181 428 (10898) | 276 749 (7499) |
| Number of unique reflections | 57 512 (2073) | 34 913 (2843) | 37 578 (1441) | 39 753 (1908) | 27 093 (1632) | 41 807 (1464) |
| Multiplicity | 6.6 (5.2) | 3.7 (3.6) | 3.1 (1.9) | 6.8 (6.5) | 6.7 (6.7) | 6.6 (5.1) |
| Refinement statistics | ||||||
| Rwork/Rfree (%) | 15.10/16.91 | 18.16/22.32 | 16.05/19.30 | 14.8/18.7 | 16.74/21.43 | 18.11/21.29 |
| Mean B factor (Å2) | 13.705 | 22.39 | 19.59 | 13.42 | 24.74 | 20.08 |
| R.m.s deviations from ideal | ||||||
| Bond lengths (Å) | 0.020 | 0.013 | 0.020 | 0.023 | 0.016 | 0.018 |
| Bond angles (°) | 1.978 | 1.517 | 1.864 | 2.117 | 1.732 | 1.883 |
| Ramachadran plot (%) | ||||||
| Residues in favoured region | 94.28 | 92.88 | 93.33 | 93.36 | 92.51 | 95.05 |
| Residues in allowed region | 4.38 | 5.79 | 5.33 | 5.32 | 6.19 | 3.63 |
| Residues in disallowed region | 1.35 | 1.32 | 1.33 | 1.33 | 1.30 | 1.32 |
The values in parentheses correspond to the highest resolution shell.
The NRP1‐b1 domain belongs to the discoidin structural domain family that also includes homologous FV/VIII C domains. The fold in this type of domain is characterised by an eight‐stranded distorted jellyroll β‐barrel where a five‐stranded antiparallel β‐sheet packs against a three‐stranded antiparallel β‐sheet. The VEGF‐A165 binding site is located at the top of the β‐barrel core delineated by six juxtaposed loops connecting the β‐strands (Fig. 1B). All arginine analogues were found bound at the same site and are positioned in a similar way as the arginine moiety of EG00229 and the terminal arginine of VEGF‐A165 within the binding site of NRP1‐b1 (Figs 1 and 6). While in the crystal structures of the complexes containing ligands R4, R6, R8 and R9, clearly interpretable electron density was found in only one copy of the b1 domain within the dimeric asymmetric unit, in the structures of the complexes of the higher affinity ligands (R5 and R7), we were able to identify the arginine analogue bound to each of the b1 molecules within the asymmetric unit of the respective unit cells. In the structure of the complexes with the ligands R4–R8, the side chain guanidine group forms bidentate hydrogen bonds with Asp320. The aliphatic part of arginine is located in the groove between Tyr297 and Tyr353, with a conformation favouring a π–π stacking between the arginine side chain and the phenyl rings. The carboxylate group anchors the small molecules specifically through hydrogen‐bond interactions such that one of the oxygen atoms binds to the hydroxyl of Ser346, while the other carboxylate oxygen forms hydrogen bonds with the hydroxyl oxygen atoms from Thr349, and Tyr353. These interactions are analogous to that observed for the C‐terminus of the peptide ligands 38, 39 and are also conserved in a structure of acetate bound to NRP1‐b1 (PDB code: 4RN5).
Figure 6.
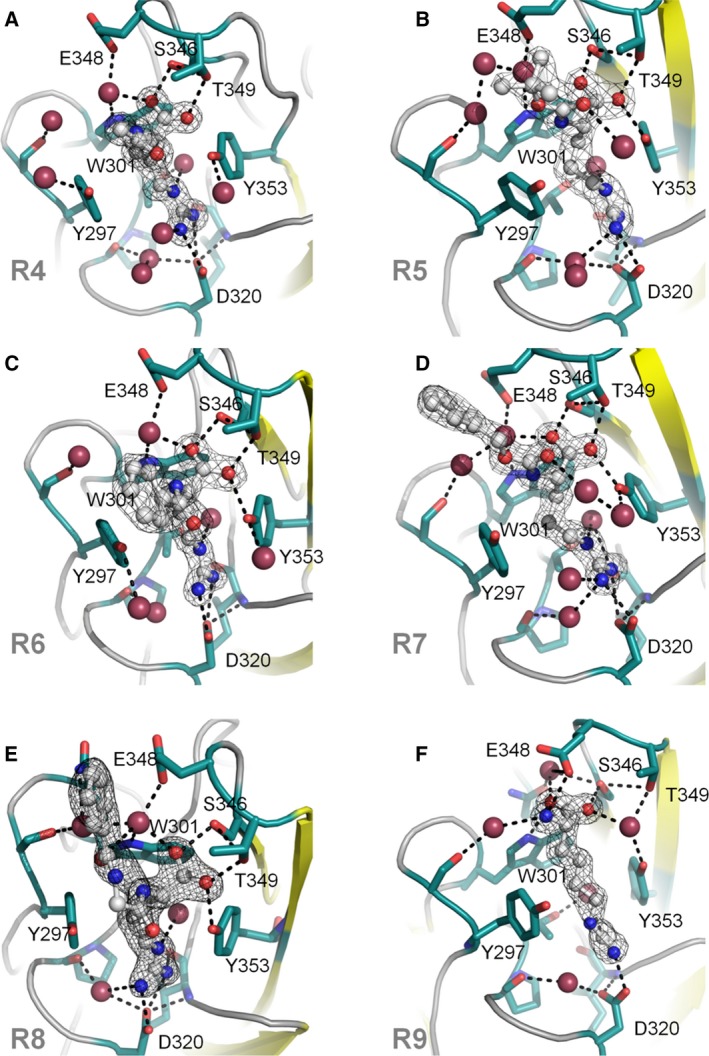
X‐ray crystal structures of arginine analogues R4–R9 bound to NRP1‐b1. Side chains of key residues in the binding site are shown in green, ligands are shown in grey, nitrogen atoms are shown in blue and oxygen atoms are shown in red. Oxygen atoms corresponding to water molecules are larger and shown in raspberry colour. The figures were generated in PyMOL. The electron density of the 2Fo‐Fc map is shown as a wire mesh and contoured to 1 sigma level; hydrogen bonds are represented as black dashes. (A) R4 (PDB entry 5JGI); (B) R5 (PDB entry 5J1X); (C) R6 (PDB entry 5JGQ); (D) R7 (PDB entry 5IYY); (E) R8 (PDB entry 5JHK); (F) R9 (PDB entry 5IJR).
Among the obtained crystal structures of the complexes, R9 is the only analogue without the Nα‐substituent, and with an extra methylene group in the side chain when compared to the natural amino acid arginine. Longer aliphatic chain affected a shift in the positioning of the carboxyl group such that it binds differently to all other ligands in the ligand‐bound structures presented here. Compared to other ligands, the carboxylate of R9 is moved by more than 2 Å towards the protein core such that one of the oxygen atoms interacts directly with the hydroxyl from Ser346, whereas the other oxygen atom forms direct hydrogen bond with the nitrogen atom from the side chain of the residue Trp301. Furthermore, the guanidine group now forms single hydrogen bond with Asp320 compared to bidentate interaction observed in the structures of the b1 complexes with the other arginine analogues. Although the free amino group of R9 engages in a hydrogen bond with Glu348, the total number of direct hydrogen bonds to the protein chain is four compared to five observed with the other analogues. This shift in binding mode is accompanied by a significant decrease in binding affinity, as shown by SPR.
Overall, the protein components of the six arginine analogue‐bound structures show high structural similarity, with only discernible conformational differences evident in an area of the b1 domain engaged in forming the intermolecular interaction within the dimer/tetramer of the asymmetric unit in the bound structures and the side chains of residues Tyr297 and Asp320 in the ligand‐binding site. The two side‐chain differences reflect the structural differences of the bound arginine analogues. The differences are best depicted by the changes in the torsion angle of the Y297 side chain as shown in Fig. 7 and Table 2.
Figure 7.
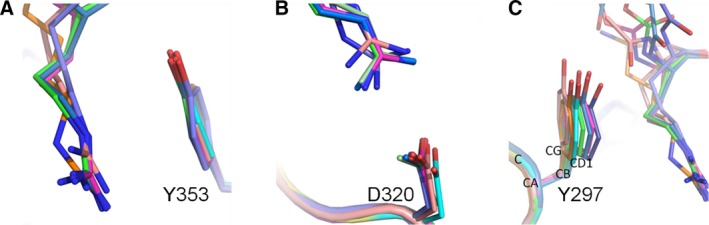
Side chain flexibility of Asp320 and Tyr297 in the NRP1‐b1 binding site. Effect of ligand binding on the side chain conformation of Y297, Y353 and D320 amino acids. The complexed protein structures (pdb IDs: 5JGI – dark pink, 5J1X – dark blue, 5JGQ – lilac, 5IYY – light pink, 5JHK – green, 5IJR – orange) were superimposed over the apo‐structure of NRP1‐b1 domain (pdb code: 1KEX, turquoise colour) (A) π–π stacking between the guanidino‐group of the bound small molecules and phenyl ring of Y353 residue. (B) A stick representation of the side chain rotamers of D320 residue of NRP1‐b1 protein as it has been observed in the X‐ray structures of the complexes. The interacting portion of the arginine analogues is shown as well. (C) A demonstration of the variability of the rotamer conformation of Y297 amino acid upon compound binding. As it is shown in Table 2, the differences between the values of torsion angles Chi1 and Chi2 of the ligand‐bound structures and those in the apo structure are significant and vary depending on the compound.
Table 2.
Side chain flexibility of D320 and Y297 in the NRP1‐b1‐binding site
| PDB ID | Ligand | Y297Chi1 | Y297Chi2 | D320Chi1 | D320Chi2 |
|---|---|---|---|---|---|
| 1KEX (apo) | −167.7 | 67.6 | −69.1 | −37.9 | |
| 5IJR, chain A | R4 | −178.3 | −88.1 | −58.3 | −55.1 |
| 5J1X, chain A | R5 | −173.8 | −80.6 | −63.5 | −44.0 |
| 5J1X, chain B | R5 | −172.6 | 83.0 | −62.2 | −41.9 |
| 5J1X, chain C | R5 | −173.7 | 84.1 | −61.4 | −41.8 |
| 5J1X, chain D | R5 | −175.1 | 83.6 | −61.4 | −41.5 |
| 5JGQ, chain B | R6 | 179.3 | 74.4 | −61.6 | −54.9 |
| 5IYY, chain A | R7 | −175.7 | 93.6 | −59.5 | −55.9 |
| 5IYY, chain B | R7 | −178.3 | 86.6 | −61.2 | −51.0 |
| 6JHK, chain A | R8 | −171.9 | −88.3 | −62.9 | −51.3 |
| 5JGI, chain B | R9 | −167.9 | 79.7 | −59.7 | −47.4 |
Analysis of ordered water molecules
The high‐resolution X‐ray crystal structures showed additional density consistent with water molecules in the binding site. To conduct an analysis of the conserved water molecules in the binding site of NRP1‐b1, we considered all new structures reported here together with the already publicly available structures. Only structures of human NRP1 with a resolution higher than 2.1 Å were selected for analysis. Table 3 summarises the selected X‐ray structures.
Table 3.
Selected X‐ray crystal structures for binding site hydration studies
| PDB code | Resolution (Å) | Protein domain | Ligand in binding site |
|---|---|---|---|
| 2QQI | 1.80 | b1b2 | (apo) |
| 4RN5 | 1.70 | b1 | Acetate ion |
| 1KEX | 1.90 | b1 | (apo) |
| 5JGI | 1.38 | b1 | R4 |
| 5J1X | 2.10 | b1 | R5 |
| 5JGQ | 1.60 | b1 | R6 |
| 5IYY | 1.60 | b1 | R7 |
| 6JHK | 1.80 | b1 | R8 |
| 5IJR | 1.52 | b1 | R9 |
The polypeptide chains were aligned to allow the visualisation of the crystallised water molecules in the binding site of NRP1‐b1. A water molecule was considered ‘conserved’ when observed in at least four out of ten superimposed molecules from the crystal structures (see methods for details). In total, five localised areas, labelled 1–5, on the molecular surface of the NRP1‐b1 ligand‐binding site were identified as containing the conserved water molecules (Fig. 8A). It was very encouraging to see that the computational hydration prediction using the MOE implementation of 3D‐RISM theory performed well with a structure of a lower resolution and this correlated with the experimentally derived solvent positions based on the X‐ray diffraction data. The 3D‐RISM predicted the location of four out of five experimentally determined conserved water molecules. The water molecule at site 5, near the surface of the NRP1‐b1 domain, was the only water molecule whose position was not predicted by this hydration analysis (Fig. 8). Free‐energy mapping suggests which water molecules are more stable and better targets for displacements in drug design (Table 4). In the apo NRP1‐b1 structure (1KEX), in addition to the water molecules corresponding to sites 1–4, crystallised waters are found at the sites commonly occupied by the carboxyl and guanidine groups of the arginine‐based compounds, as is also true for the prediction model. The calculated free energies for these two areas (−1.4 kcal·mol−1 and 0.12 kcal·mol−1 for the carboxylic acid site and guanidine site, respectively) indicate that these are stable waters, and that their replacement would be favourable.
Figure 8.
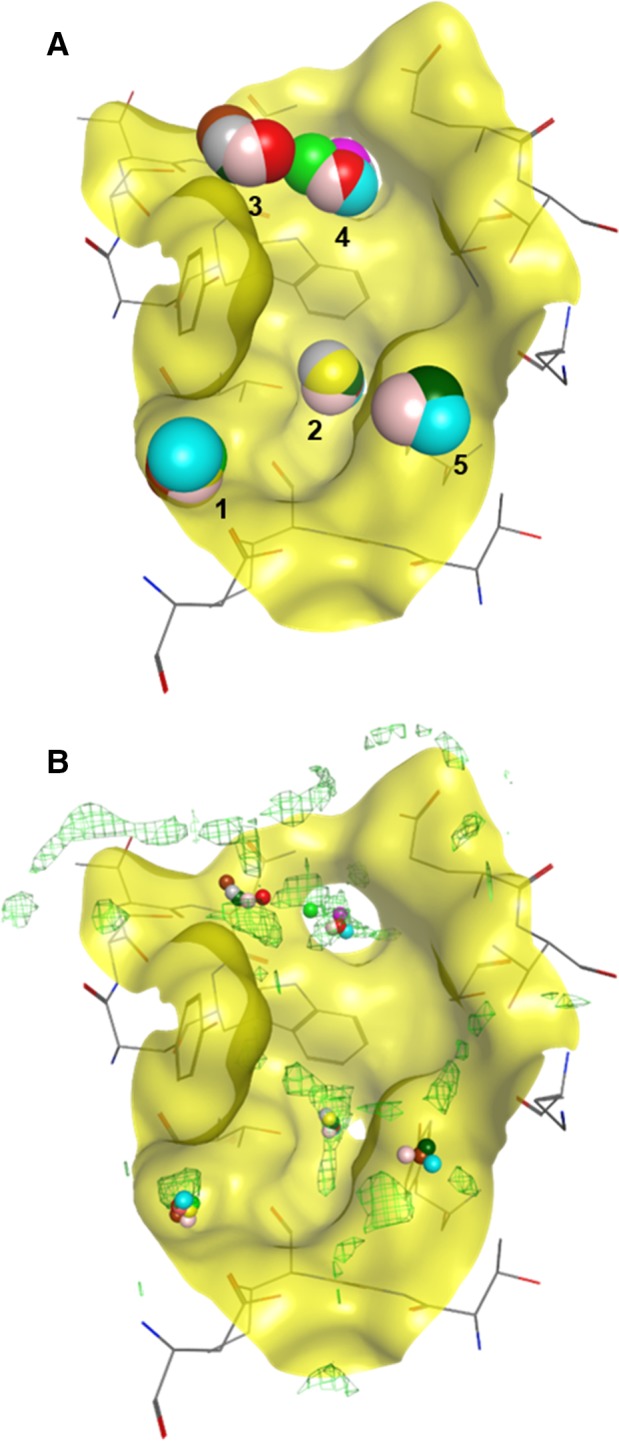
Analysis of hydration sites in the binding site of NRP1‐b1. (A) Superposition of nine high‐resolution X‐ray crystal structures (10 chains) (PDB entries: 4RN5, 2QQI, 1KEX, 5JGI, 5J1X, 5JGQ, 5IYY, 6JHK, 5IJR). 5 sites were identified and 4 or more structures show a water molecule conserved in that position. (B) Overlay of superposed structures showing conserved water molecules with the protein hydration prediction map. The computational model correctly predicted four sites where conserved water molecules were found.
Table 4.
Characterisation of water clusters in NRP1‐b1
| Cluster number | Free energy (kcal·mol−1) |
|---|---|
| 1 | 1.30 |
| 2 | 1.15 |
| 3 | −0.80 |
| 4 | 2.16 |
| 5 | Nd |
| COO‐site (seen only in apo structure) | −1.40 |
| Guanidine site (seen only in apo structure) | 0.12 |
Sites 1 and 2 are occupied by water molecules in 9 out of 10 analysed structures. These are deep areas in the binding site, which could be classified as sub‐pockets, and could be targeted for replacement in drug design with the aim of enhancing affinity. The water molecule in site 3 is present in five structures. This molecule establishes a hydrogen bond with the side chain of Ser298 and is present in both apo‐ and ligand‐bound structures. Calculated free energy of site 3 (−0.8 kcal·mol−1) suggests that this cluster could also be target for displacement by a ligand's functional group. In fact, in the crystal structure of EG0229 (PDB code: 3I97), this site is occupied by a sulphonamide group. The conserved water molecule in site 4 is engaged in a hydrogen‐bond interaction with the nitrogen atom on the Trp301. This is observed in six structures. Additionally, this molecule is also seen engaging in hydrogen bonds with E348. In the structures with ligands R4–R8, the water molecule in site 4 additionally forms a hydrogen bond with the oxygen atom of the arginine analogue carboxyl group. However, in the structure of R9‐bound NRP1, the conserved water molecule in site 4 has been replaced by an oxygen atom of the R9 carboxylate. As mentioned above, R9 shows very weak binding affinity to the NRP1‐b1 domain, which can be interpreted by the loss of favourable direct hydrogen bonds of R9 to the polypeptide chain together with a disruption of a water network caused by the displacement of the water molecule in site 4. This carboxyl/oxygen–water–Trp301/nitrogen interaction might be a key network in the binding of ligands to NRP1‐b1. We suggest this water molecule, which corresponds to the highest free‐energy site (2.16 kcal·mol−1), should be included in docking studies for drug design, and used as a bridge between the ligand and the protein instead of attempting to displace it or replace it with functional groups. The last conserved water molecule observed was in site 5, found in only four structures, and located near the protein surface, with a possible hydrogen bond to Tyr353. Since it is superficial water, we would not consider it significant for drug design.
Implications for structure‐based drug design
We studied a known binding site in the NRP1‐b1 domain. To disrupt ligand binding to this domain, a protein–protein interaction, with a potent small molecule, it was important to perform an in‐depth study of the architecture and the solvation map of the target site. NRP1 shows a well‐defined arginine‐binding site where the guanidine part can establish a bidentate hydrogen‐bond interaction with the side chain of Asp320, and the C‐terminal carboxyl group can establish hydrogen bonds with the side chain oxygen atoms from Thr349 and Ser346. Although these interactions are conserved when small molecules containing a Nα‐substituted arginine are bound to NRP1‐b1, the arginine analogues examined in this study exhibited varied affinities for the receptor. Both molecules with the highest affinity (R5 and R7) contained carbamate group.
We obtained the crystal structures of six arginine analogues bound to NRP1‐b1 and analysed these alongside other publicly available structures in the PDB. The binding site of NRP1‐b1 shows little main chain and/or side chain flexibility, with only Tyr297 and Asp320 showing significant variations, which are dependent on the ligand bound. Tyr297 has been previously shown as required for VEGF binding, with NRP1Y297A/Y297A homozygous knock‐in mice exhibiting reduced VEGF‐induced angiogenesis and tumourigenesis 40. The availability of a closely related set of protein–ligand structures at high resolution provided a rare opportunity to test the water prediction capabilities of the software. Our computational prediction of the NRP1‐b1 binding site hydration showed good correlation with the location of the observed crystallographic waters. Especially important are the insights gained into the stability of the individual waters in the binding sites and whether they are likely to be displaced upon ligand (inhibitor) binding. Structural water molecules were found reproducibly within five buried subpockets in the immediate vicinity of the NRP1‐b1 ligand‐binding site; four of these sites were highly conserved with the fifth, less conserved one, positioned near the protein surface. We propose that displacement of these waters by small molecules may add to their binding affinity, with the exception of the conserved water molecule in site 4 which should in turn be retained in the binding site during docking studies. Displacement of this water molecule resulted in a much weaker ligand, as demonstrated by R9 analogue. Our analysis highlights the importance of considering structural water molecules in structure‐based drug design. When high‐resolution crystal structures are not available to determine conserved water molecules, simple computational methods may be used as an alternative.
Experimental procedures
Materials
Arginine analogues were obtained from commercial sources: R1, R2, R8 (Sigma, Haverhill, UK), R3, R7, R9 (AK Scientific), R4, R5, R6 (Alfa Aesar, Heysham, UK), and used without further purification. All experiments were conducted using commercial reagents and standard procedures, unless stated otherwise.
Computational prediction of conserved water molecules and molecular modelling
Molecular modelling was performed using the software package MOE (Molecular Operating Environment, 2014.09; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2015).
Preparation of the input file
The structure used for the prediction of binding site hydration was PDB code: 3I97. Water and co‐crystallised ligands were removed from the structure. An arginine residue was modelled from the coordinates of EG00229 (ligand id: 8dr, PDB code: 3I97) and used to define the boundaries of the binding site. Hydrogen atoms were added to the structure by selecting the function ‘add hydrogen’ in MOE before the calculations were performed. The binding site for solvent prediction was defined by all atoms included in a 10 Å proximity from the ligand, extended by residues of those atoms.
Solvent analysis
Binding site hydration prediction was carried out using the ‘Solvent Analysis’ function in MOE. The application uses the three‐dimensional reference interaction site model (3D‐RISM) method 37, 41, 42 to analyse the role of solvent in proteins. It computes a time‐averaged distribution of water H and O densities, and free‐energy maps for analysing solvent stability and solvation contributions to binding free energy. The parameters used are as follows: the dimension of the grid spacing was 0.35 Å; a distance of 7 Å was set for the boundary box where atoms are extended; the convergence or precision of 3D‐RISM was setup to ‘tight’; the NDIIS (the Number of copies of Direct Inversion in the Iterative Subspace) was set to 5. This value balances the memory used and the convergence rate. The number of copies (N) of the various 3D grids retained in memory for accelerating convergence by extrapolation. Smaller values use less memory but slow down the convergence rate. A detailed description of these parameters can be found in the references 43.
Analysis of ordered water molecules
The SAS tool (Sequence annotated by Structure) 44 from the EMBL‐EBI website was used to retrieve all the related PDBs using 3I97 (NRP1‐b1) as a template. Only PDBs with a resolution better than 2.1 Å and sequence identity higher than 90% were retained for analysis. This resulted in three PDBs, which are 4RN5, 2QQI and 1KEX. In cases where the X‐ray crystal structure had more than one chain, both chains were analysed. Combined with the six structures obtained in‐house, a total of 9 PDBs or 10 chains were analysed. Among these structures, 2QQI and 1KEX are in the apo form.
Definition of binding site and conserved water molecule
The protein binding site was defined by a 5 Å proximity of all the ligand atoms present. Only the water molecules observed in the binding site were analysed. A water molecule is considered ‘conserved’ when observed in the same position of at least four superimposed crystal structures.
Surface plasmon resonance
Surface plasmon resonance experiments were performed using a Biacore 4000 instrument at a constant temperature of 25 °C. Sensor chips, buffer stock solutions and immobilisation reagents were purchased from GE Healthcare.
Chip preparation
PBS containing 0.05% surfactant P20 was used as the running buffer during immobilisation. NRP1‐b1 was immobilised onto a CM5 chip using random amine coupling. The four flow cells were treated in the same way to optimise throughput. In summary, immobilisation spots 1 and 2 were activated with the coupling reagents, 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide and N‐hydroxysuccinimide for 10 min. NRP1‐b1 at a concentration of 20 μg·mL−1 in 10 mm sodium acetate pH 5 was injected onto the surface for 10 and 5 min in spots 1 and 2, respectively, to generate surfaces with high and low density. The immobilisation levels ranged from 3487 to 3652 resonance units (RU) on spot 1 and from 1275 to 1756 RU on spot 2. Spot 3 was left unmodified and used as a reference.
Equilibrium affinity measurements
PBS containing 0.05% surfactant P20 was used as the running buffer and sample dilution buffer throughout these experiments. Dose‐responses were obtained using a two‐fold sample dilution, generating eight data points for the concentration range, and using an injection time of 60 s. Surface regeneration between injections was not necessary, but a wash step with 1M NaCl was included after injection of the highest concentration sample for each compound. All compounds were initially tested at a wide dilution range from 0.6 to 300 μm. Dilution series were then optimised for three compounds to obtain a more accurate measurements and dissociation constants. R1 and R9 were tested at 12–1500 μm, and R5 was tested at 0.16–20 μm. Binding responses to high‐ and low‐density surfaces were processed independently and the average ± SD is presented. K Ds reported are derived from steady‐state binding responses assuming a 1 : 1 interaction and therefore correspond to the equilibrium binding affinity of the compounds.
Protein purification and crystallisation
NRP1‐b1 was expressed in E. coli strain Rosetta‐gami2‐(DE3)pLysS (Novagen) and purified as previously described 19. The protein was concentrated to 9–10 mg·mL−1 and 10 μL was mixed with 1 μL ligand at 10–100 mm in water. The complex was crystallised in a 1 : 1 volume mixture with 10–30% w/v PEG 3350, and 0.2 m ammonium chloride at 16 °C using hanging drop vapour diffusion method and micro‐seeding with 1/10 dilution of seeds of apo‐NRP1‐b1 crystals. Seeds were prepared according to Hampton Research's seeding kit. Crystals appeared within 2–3 days with needle morphology. A single crystal was transferred to a solution containing the crystallisation condition plus 20 % v/v ethylene glycol and was afterwards flash‐frozen in liquid nitrogen.
X‐ray Crystallography
X‐ray diffraction data were collected at 100 K on beamline I04 at the Diamond Light Source, Didcot UK. All data sets were processed with xia2‐3d automated software system 45, 46, 47, 48, 49. Molecular replacement solutions were obtained by Phaser 50 using an apo NRP1‐b1 domain (PDB code 1KEX) as the search model. The refinement was carried out by Refmac5 51. The dictionary files for the fragments were generated using the acedgr programme from ccp4 software suite 52. Iterative rounds of building and refinement were carried out in COOT 53 and Refmac5. TLS (Translation/Libration/Screw) groups were generated automatically by Refmac5. Data collection and refinement statistics for all structures can be found in Table 1.
Cell‐Free bt‐VEGF‐A165‐binding assay
The assay was run as previously described 19. The 96‐well plates were precoated with NRP1‐b1 protein at 3 μg·mL−1 overnight at 4 °C. On the following day, the plates were treated with blocking buffer (PBS containing 1% BSA) and washed three times with wash buffer (PBS containing 0.1% Tween‐20). The various concentrations of compounds diluted in PBS containing 1% DMSO were added, followed by addition of 0.25 nm bt‐VEGF‐A165. After 2 h of incubation at room temperature, the plates were washed three times with wash buffer. The bt‐VEGF‐A165 bound to NRP1‐b1 was detected by streptavidin‐horseradish peroxidase conjugates and the enzyme substrate and measured using a Tecan Genios plate reader at 450 nm absorbance with a reference wavelength at 595 nm. Non‐specific binding was determined in the absence of NRP1‐b1‐coated wells of the plates and subtracted from the readings obtained in the presence of NRP1‐b1. GraphPad Prism was used to carry out a non‐linear regression analysis and to generate inhibition curve and IC50 value. IC50 values for the particular ligand were obtained based on the three separate experiments.
Conflict of interest
DS, IZ, PF and SD were consultants for Magnus Life Science.
Author contributions
CF, RR and TY carried out the X‐ray crystallography; AT assisted with protein preparation. EC did the computational work. JH and DS selected the compounds for analysis. AOL carried out VEGF displacement assays. FM performed the SPR experiments, analysed the results and wrote the paper. DS, IZ, PF and SD supervised the research team and edited the paper.
Acknowledgements
Funding from Magnus Life Science and BHF Programme Grant RG/11/11/29050 to IZ are gratefully acknowledged.
[The spelling of the name of the third author was amended on 20 March after original online publication.]
References
- 1. Pellet‐Many C, Frankel P, Jia H & Zachary I (2008) Neuropilins: structure, function and role in disease. Biochem J 411, 211–226. [DOI] [PubMed] [Google Scholar]
- 2. Prud'homme GJ & Glinka Y (2012) Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget 3, 921–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. He Z & Tessier‐Lavigne M (1997) Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell 90, 739–751. [DOI] [PubMed] [Google Scholar]
- 4. Soker S, Takashima S, Miao HQ, Neufeld G & Klagsbrun M (1998) Neuropilin‐1 is expressed by endothelial and tumor cells as an isoform‐specific receptor for vascular endothelial growth factor. Cell 92, 735–745. [DOI] [PubMed] [Google Scholar]
- 5. Migdal M, Huppertz B, Tessler S, Comforti A, Shibuya M, Reich R, Baumann H & Neufeld G (1998) Neuropilin‐1 is a placenta growth factor‐2 receptor. J Biol Chem 273, 22272–22278. [DOI] [PubMed] [Google Scholar]
- 6. Cao S, Yaqoob U, Das A, Shergill U, Jagavelu K, Huebert RC, Routray C, Abdelmoneim S, Vasdev M & Leof E (2010) Neuropilin‐1 promotes cirrhosis of the rodent and human liver by enhancing PDGF/TGF‐β signaling in hepatic stellate cells. J Clin Invest 120, 2379–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Glinka Y & Prud'homme GJ (2008) Neuropilin‐1 is a receptor for transforming growth factor β‐1, activates its latent form, and promotes regulatory T cell activity. J Leukoc Biol 84, 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sulpice E, Plouet J, Bergé M, Allanic D, Tobelem G & Merkulova‐Rainon T (2008) Neuropilin‐1 and neuropilin‐2 act as coreceptors, potentiating proangiogenic activity. Blood 111, 2036–2045. [DOI] [PubMed] [Google Scholar]
- 9. Hillman RT, Feng BY, Ni J, Woo W‐M, Milenkovic L, Gephart MGH, Teruel MN, Oro AE, Chen JK & Scott MP (2011) Neuropilins are positive regulators of Hedgehog signal transduction. Genes Dev 25, 2333–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Delgoffe GM, Woo S‐R, Turnis ME, Gravano DM, Guy C, Overacre AE, Bettini ML, Vogel P, Finkelstein D & Bonnevier J (2013) Stability and function of regulatory T cells is maintained by a neuropilin‐1‐semaphorin‐4a axis. Nature 501, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miao H‐Q, Lee P, Lin H, Soker S & Klagsbrun M (2000) Neuropilin‐1 expression by tumor cells promotes tumor angiogenesis and progression. FASEB J 14, 2532–2539. [DOI] [PubMed] [Google Scholar]
- 12. Jia H, Cheng L, Tickner M, Bagherzadeh A, Selwood D & Zachary I (2010) Neuropilin‐1 antagonism in human carcinoma cells inhibits migration and enhances chemosensitivity. Br J Cancer 102, 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hansen W (2013) Neuropilin 1 guides regulatory T cells into VEGF‐producing melanoma. Oncoimmunology 2, e23039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miyauchi J, Chen D, Choi M, Nissen J, Shroyer K, Djordjevic S, Zachary I, Selwood D & Tsirka S (2016) Ablation of neuropilin 1 from glioma‐associated microglia and macrophages slows tumor progression. Oncotarget 7, 9801–9814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Teesalu T, Sugahara KN, Kotamraju VR & Ruoslahti E (2009) C‐end rule peptides mediate neuropilin‐1‐dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci USA 106, 16157–16162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arkin MR & Wells JA (2004) Small‐molecule inhibitors of protein–protein interactions: progressing towards the dream. Nat Rev Drug Discov 3, 301–317. [DOI] [PubMed] [Google Scholar]
- 17. Jia H, Aqil R, Cheng L, Chapman C, Shaikh S, Jarvis A, Chan AW, Hartzoulakis B, Evans IM, Frolov A et al (2014) N‐terminal modification of VEGF‐A C terminus‐derived peptides delineates structural features involved in neuropilin‐1 binding and functional activity. ChemBioChem 15, 1161–1170. [DOI] [PubMed] [Google Scholar]
- 18. Novoa A, Pellegrini‐Moïse N, Bechet D, Barberi‐Heyob M & Chapleur Y (2010) Sugar‐based peptidomimetics as potential inhibitors of the vascular endothelium growth factor binding to neuropilin‐1. Bioorg Med Chem 18, 3285–3298. [DOI] [PubMed] [Google Scholar]
- 19. Jarvis A, Allerston CK, Jia H, Herzog B, Garza‐Garcia A, Winfield N, Ellard K, Aqil R, Lynch R, Chapman C et al (2010) Small molecule inhibitors of the neuropilin‐1 vascular endothelial growth factor A (VEGF‐A) interaction. J Med Chem 53, 2215–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jia H, Bagherzadeh A, Hartzoulakis B, Jarvis A, Löhr M, Shaikh S, Aqil R, Cheng L, Tickner M, Esposito D et al (2006) Characterization of a bicyclic peptide neuropilin‐1 (NP‐1) antagonist (EG3287) reveals importance of vascular endothelial growth factor exon 8 for NP‐1 binding and role of NP‐1 in KDR signaling. J Biol Chem 281, 13493–13502. [DOI] [PubMed] [Google Scholar]
- 21. Getz JA, Cheneval O, Craik DJ & Daugherty PS (2013) Design of a cyclotide antagonist of neuropilin‐1 and‐2 that potently inhibits endothelial cell migration. ACS Chem Biol 8, 1147–1154. [DOI] [PubMed] [Google Scholar]
- 22. Starzec A, Miteva MA, Ladam P, Villoutreix BO & Perret GY (2014) Discovery of novel inhibitors of vascular endothelial growth factor‐A–Neuropilin‐1 interaction by structure‐based virtual screening. Bioorg Med Chem 22, 4042–4048. [DOI] [PubMed] [Google Scholar]
- 23. Borriello L, Montès M, Lepelletier Y, Leforban B, Liu W‐Q, Demange L, Delhomme B, Pavoni S, Jarray R & Boucher JL (2014) Structure‐based discovery of a small non‐peptidic Neuropilins antagonist exerting in vitro and in vivo anti‐tumor activity on breast cancer model. Cancer Lett 349, 120–127. [DOI] [PubMed] [Google Scholar]
- 24. Kim Y‐J, Bae J, Shin T‐H, Kang SH, Jeong M, Han Y, Park J‐H, Kim S‐K & Kim Y‐S (2015) Immunoglobulin Fc‐fused, neuropilin‐1‐specific peptide shows efficient tumor tissue penetration and inhibits tumor growth via anti‐angiogenesis. J Control Release 216, 56–68. [DOI] [PubMed] [Google Scholar]
- 25. Fedorczyk B, Lipiński PFJ, Tymecka D, Puszko AK, Wilenska B, Perret GY & Misicka A (2017) Conformational latitude ‐ activity relationship of KPPR tetrapeptide analogues toward their ability to inhibit binding of vascular endothelial growth factor 165 to neuropilin‐1. J Pept Sci 23, 445–454. [DOI] [PubMed] [Google Scholar]
- 26. Grabowska K, Puszko AK, Lipinski PFJ, Laskowska AK, Wilenska B, Witkowska E, Perret GY & Misicka A (2017) Structure‐activity relationship study of a small cyclic peptide H‐c[Lys‐Pro‐Glu]‐Arg‐OH: a potent inhibitor of Vascular Endothelial Growth Factor interaction with Neuropilin‐1. Bioorg Med Chem 25, 597–602. [DOI] [PubMed] [Google Scholar]
- 27. Tymecka D, Lipinski PFJ, Fedorczyk B, Puszko A, Wileska B, Perret GY & Misicka A (2017) Structure‐activity relationship study of tetrapeptide inhibitors of the Vascular Endothelial Growth Factor A binding to Neuropilin‐1. Peptide 94, 25–32. [DOI] [PubMed] [Google Scholar]
- 28. Richard M, Chateau A, Jelsch C, Didierjean C, Manival X, Charron C, Maigret B, Barberi‐Heyob M, Chapleur Y, Boura C et al (2016) Carbohydrate‐based peptidomimetics targeting neuropilin‐1: synthesis, molecular docking study and in vitro biological activities. Bioorg Med Chem 24, 5315–5325. [DOI] [PubMed] [Google Scholar]
- 29. Liu W‐Q, Lepelletier Y, Montes M, Borriello L, Jarray R, Grepin R, Leforban B, Loukaci A, Benhida R, Hermine O et al (2018) NRPa‐308, a new neuropilin‐1 antagonist, exerts in vitro anti‐angiogenic and anti‐proliferative effects and in vivo anti‐cancer effects in a mouse xenograft model. Cancer Lett 414, 88–98. [DOI] [PubMed] [Google Scholar]
- 30. García‐Sosa AT (2013) Hydration properties of ligands and drugs in protein binding sites: tightly‐bound, bridging water molecules and their effects and consequences on molecular design strategies. J Chem Inf Model 53, 1388–1405. [DOI] [PubMed] [Google Scholar]
- 31. Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM & Shichida Y (2002) Functional role of internal water molecules in rhodopsin revealed by X‐ray crystallography. Proc Natl Acad Sci USA 99, 5982–5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu Y, Wang R, Yang C‐Y & Wang S (2007) Analysis of ligand‐bound water molecules in high‐resolution crystal structures of protein‐ligand complexes. J Chem Inf Model 47, 668–675. [DOI] [PubMed] [Google Scholar]
- 33. Michael J, Tirado‐Rives J & Jorgenson WL (2009) Energetics of displacing water molecules from protein binding sites: consequences for ligand optimization. J Am Chem Soc 131, 15403–15411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ross GA, Morris GM & Biggin PC (2012) Rapid and accurate prediction and scoring of water molecules in protein binding sites. PLoS ONE 7, e32036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu Y, Yang C‐Y & Wang S (2006) Binding free energy contributions of interfacial waters in HIV‐1 protease/inhibitor complexes. J Am Chem Soc 128, 11830–11839. [DOI] [PubMed] [Google Scholar]
- 36. Young T, Abel R, Kim B, Berne BJ & Friesner RA (2007) Motifs for molecular recognition exploiting hydrophobic enclosure in protein–ligand binding. Proc Natl Acad Sci USA 104, 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beglov D & Roux B (1997) An integral equation to describe the solvation of polar molecules in liquid water. J Phys Chem B 101, 7821–7826. [Google Scholar]
- 38. Vander Kooi CW, Jusino MA, Perman B, Neau DB, Bellamy HD & Leahy DJ (2007) Structural basis for ligand and heparin binding to neuropilin B domains. Proc Natl Acad Sci USA 104, 6152–6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Parker MW, Xu P, Li X & Vander Kooi CW (2012) Structural basis for selective vascular endothelial growth factor‐A (VEGF‐A) binding to neuropilin‐1. J Biol Chem 287, 11082–11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fantin A, Herzog B, Mahmoud M, Yamaji M, Plein A, Denti L, Ruhrberg C & Zachary I (2014) Neuropilin 1 (NRP1) hypomorphism combined with defective VEGF‐A binding reveals novel roles for NRP1 in developmental and pathological angiogenesis. Development 141, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kovalenko A & Hirata F (1999) Self‐consistent description of a metal–water interface by the Kohn–Sham density functional theory and the three‐dimensional reference interaction site model. J Chem Phys 110, 10095–10112. [Google Scholar]
- 42. Kovalenko A & Hirata F (2000) Potentials of mean force of simple ions in ambient aqueous solution. I. Three‐dimensional reference interaction site model approach. J Chem Phys 112, 10391–10402. [Google Scholar]
- 43. Stumpe MC, Blinov N, Wishart D, Kovalenko A & Pande VS (2011) Calculation of local water densities in biological systems: a comparison of molecular dynamics simulations and the 3D‐RISM‐KH molecular theory of solvation. J Phys Chem B 115, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Milburn D, Laskowski RA & Thornton JM (1998) Sequences annotated by structure: a tool to facilitate the use of structural information in sequence analysis. Protein Eng 11, 855–859. [DOI] [PubMed] [Google Scholar]
- 45. Winter G (2010) xia2: an expert system for macromolecular crystallography data reduction. J Appl Crystallogr 43, 186–190. [Google Scholar]
- 46. Evans P (2006) Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62, 72–82. [DOI] [PubMed] [Google Scholar]
- 47. Kabsch W (2010) Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sauter NK, Grosse‐Kunstleve RW & Adams PD (2004) Robust indexing for automatic data collection. J Appl Crystallogr 37, 399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang Z, Sauter NK, van den Bedem H, Snell G & Deacon AM (2006) Automated diffraction image analysis and spot searching for high‐throughput crystal screening. J Appl Crystallogr 39, 112–119. [Google Scholar]
- 50. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC & Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F & Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG & McCoy A (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Emsley P, Lohkamp B, Scott WG & Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]