

ABSTRACT
Previous studies have examined various immune evasion strategies of human cytomegalovirus (HCMV) to gain understanding of its pathogenesis. Although the mechanism that underlies immunocyte destruction near HCMV‐infected lesions has yet to be established, it is here shown that substances produced by HCMV‐infected cells induce death in several types of immunocytes, but not in fibroblasts or astrocytomas. These substances contain HCMV proteins and were termed HCMV‐associated insoluble substance (HCMVAIS). The mechanism by which HCMVAIS induces cell death was characterized to improve understanding the death of immunocytes near HCMV‐infected lesions. HCMVAIS were found to trigger production of intracellular nicotinamide adenine dinucleotide phosphate oxidase‐derived reactive oxygen species (ROS), resulting in cell death, this effect being reversed following treatment with ROS inhibitors. Cell death was not induced in splenocytes from NOX‐2 knockout mice. It was hypothesized that DNA damage induced by oxidative stress initiates poly ADP‐ribose polymerase‐1 (PARP‐1)‐mediated cell death, or parthanatos. HCMVAIS‐induced cell death is accompanied by PARP‐1 activation in a caspase‐independent manner, nuclear translocation of apoptosis‐inducing factor (AIF), and DNA fragmentation, which are typical features of parthanatos. Treatment with an AIF inhibitor decreased the rate of HCMVAIS‐induced cell death, this being confirmed by hematoxylin and eosin staining; cell death in most HCMV‐positive foci in serial section samples of a large intestine with HCMV infection was TUNEL‐positive, cleaved caspase 3‐negative and CD45‐positive. Taken together, these data suggest that HCMV inhibits local immune responses via direct killing of immunocytes near HCMV‐infected cells through ROS‐induced parthanatos by HCMVAIS.
Keywords: human cytomegalovirus, parthanatos, reactive oxygen species
Abbreviations
- AIF
apoptosis‐inducing factor
- CMV
cytomegalovirus
- DMSO
dimethyl sulfoxide
- d.p.i.
days post‐infection
- DPI
diphenyleneiodonium chloride
- FasL
Fas ligand
- HCMV
human cytomegalovirus
- HCMVAIS
HCMV‐associated insoluble substances
- HRPE
human retinal pigment epithelium
- LCA
leukocyte common antigen
- MOCKAIS
mock‐associated insoluble substance
- NAC
N‐acetyl‐L‐cysteine
- NADPH
nicotinamide adenine dinucleotide phosphate
- NOX
NADPH oxidase
- NP
N‐phenylmaleimide
- PARP‐1
poly ADP‐ribose polymerase‐1
- PBMC
peripheral mononuclear cell
- PI
propidium iodide
- ROS
reactive oxygen species
- STS
staurosporine
- TRAIL
TNF‐related apoptosis‐inducing ligand
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Human cytomegalovirus causes congenital fetal infections, asymptomatic lifelong infections in healthy individuals and severe clinical complications in immunocompromised patients 1, 2, 3. Severe infections in immunocompetent patients have also been reported 4. Following primary infection, HCMV uses a variety of mechanisms to evade the host immune system and establish lifelong latency 5. These latent infections can be reactivated under certain conditions 6. However, despite the many immune evasion strategies used by HCMV, strong humoral and cellular immune responses are evident. These contradictory phenomena can be explained, in part, by the waxing and waning of each immune evasion strategy as the disease progresses and by differences in the microenvironments of virally infected sites.
Characteristic inclusions can be found in HCMV‐infected lesions 7; however, the mechanisms that drive their formation and resolution remain poorly understood. These lesions can form in spite of active host immune responses to HCMV, which should result in viral clearance during the immediate‐early to late phases of infection. HCMV‐infected cells exhibit additional immune evasion strategies in certain locations, such as the eye, including induction of active apoptosis of lymphocytes via up‐regulation of FasL and soluble FasL expression in HCMV‐infected HRPE cells 8. However, death of nearby lymphocytes due to HCMV‐infected HRPE cells cannot be generalized across all HCMV‐infected lesions because the retina occupies a unique immune privileged area, many cells not expressing FasL after HCMV infection.
Other viruses besides HCMV also employ strategies for killing immune cells. HIV infection increases expression of TRAIL and sensitizes T cells to TRAIL‐mediated apoptosis 9, whereas reovirus induces release of TRAIL and the TRAIL apoptotic pathway 10. In this study, we identified substances produced by HCMV‐infected cells that can induce the death of various hematopoietic cell lines and human PBMCs in the vicinity of HCMV‐infected cells. The substances were termed HCMV‐associated insoluble substance (HCMVAIS). Understanding HCMVAIS‐induced cell death is important in determining the mechanism(s) underlying immunocyte destruction near HCMV‐infected lesions. The nomenclature of cell death continues to evolve according to the morphological appearance, enzymological criteria, functional aspects and immunological characteristics; terms for cell death including apoptosis, necrosis, pyroptosis, autophagy, oncosis, ferroptosis and parthanatos 11, 12. We determined that the mechanism underlying the death of immunocytes near HCMV‐infected cells increases production of NOX‐derived ROS. ROS play an important role in caspase‐independent cell death in many cell types 13, 14, 15. DNA damage induced by ROS can initiate parthanatos 16, which is a form of programmed cell death that occurs through PARP‐1 12. Nuclear translocation of AIF and DNA fragmentation (in a caspase‐independent manner) are key steps in parthanatos 17. Herein we report the activation of PARP‐1, nuclear translocation of AIF and DNA fragmentation in Jurkat cells and PBMCs following exposure to HCMVAIS from HCMV‐infected cells. We have also confirmed that HCMVAIS‐induced death is independent of activation of caspases 3, 7 and 9. Together, these data suggest that HCMVAIS‐induced ROS initiate PARP‐1‐mediated parthanatos in Jurkat cells and human PBMCs. These findings provide evidence of a additional HCMV immune evasion strategy, thus aiding our understanding of the mechanism(s) underlying immunocyte destruction near HCMV‐infected lesions.
MATERIALS AND METHODS
Cells, tissues and viruses
Human embryo lung fibroblasts (HEL 299; ATCC CCL‐137), Jurkat (ATCC TIB‐152), THP‐1 (ATCC TIB‐202), K562 (ATCC CCL‐243), HL‐60 (ATCC CCL‐240), U937 (ATCC CRL‐1593.2), Raji (ATCC CCL‐86), V653 (ATCC CRL‐8375), U373MG (ATCC HTB‐17), Saos‐2 (ATCC HTB‐85), 22Rv1 (ATCC CRL‐2505) and human embryonic kidney cells 293 (HEK‐293; ATCC CRL‐1573) cells were grown in DMEM (Dulbecco's modified Eagle's medium; HyClone, Marietta, OH, USA) or RPMI‐1640 (HyClone) supplemented with 10% FBS (HyClone) and 1× penicillin‐streptomycin (Gibco Life Technologies, Carlsbad, CA, USA) at 37°C in a humidified incubator with a 5% CO2 atmosphere. HCMV Towne (ATCC VR‐977) was maintained as described previously 18. HEL 299 cells were infected with HCMV at a MOI of 4 unless otherwise stated.
Human peripheral blood was drawn from healthy donors after voluntary informed consent. PBMCs were collected at the Ficoll‐Paque interface (GE Healthcare, Little Chalfront, UK) after centrifugation at 700 g for 30 min. PBMCs were cultured in RPMI1640 containing 5% human serum (blood type AB) at 37°C in a 5% CO2 atmosphere. Human tissues infected with HCMV were selected from archives of the Department of Pathology in the Asan Medical Center. The presence of HCMV‐infected cells was confirmed by identification by pathologistsof the characteristic inclusion bodies in hematoxylin and eosin‐stained sections.
Animals
Twelve‐week‐old female, specific‐pathogen‐free C57BL/6 mice were purchased from Koatech (Seoul, Korea). C57BL/6 nox2‐/‐ mice were housed under specific pathogen‐free conditions at Ewha Womans University and provided by Ehwa Woman's University College of Medicine 19, 20. Animal studies were conducted under protocols approved by the Seoul National University Institutional Animal Care and Use Committee. Fragments of spleen were squeezed with forceps and splenocytes were prepared by lysis of red blood cells.
Antibodies and reagents
The mAb MCMVA 135 21 was used to neutralize HCMV infections. Monoclonal anti‐HCMV IE1 (6IE1) and IE2 (12IE2) (generously provided by E‐S Huang), anti‐HCMV UL44 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti‐HCMV pp65 (Santa Cruz) were used to detect HCMV antigens. Rabbit antibodies against caspases 3, 7 and 9, AIF, and PARP‐1 (Cell Signaling, Danvers, MA, USA) and monoclonal anti‐GAPDH (Merck, Darmstadt, Germany) were used as primary antibodies.
Staurosporine (1 μM) was used as a positive control for induction of apoptosis. Caspase inhibitors Z‐VAD‐FMK (pan‐caspase inhibitor), Z‐VDVAD‐FMK (caspase‐2 inhibitor), Z‐DEVD‐FMK (caspase‐3 inhibitor), Z‐IETD‐FMK (caspase‐8 inhibitor) and Z‐LEHD‐FMK (caspase‐9 inhibitor), were purchased from R&D Systems (Minneapolis, MN, USA); all caspase inhibitors were used at a final concentration of 20 μM. NP (50 μM) was used to inhibit AIF release. Neutralizing antibodies to FasL and TRAIL (R&D Systems) were used at a final concentration of 2 μg/mL. NAC (5 μM) and DPI (25 μM) were used to inhibit generation of ROS. All reagents were purchased from Sigma (St. Louis, MO, USA) unless otherwise stated.
Fractionation of cells and culture supernatants
Culture supernatants were collected from HCMV‐ or mock‐infected HEL 299 cells 4 d.p.i. and fractionated into supernatant in supernatant and insoluble pellet in supernatant by centrifugation at 800 g for 5 min. Following collection of culture supernatants, the remaining cells were scraped, washed twice with PBS and collected by centrifugation at 1600 g for 5 min. The final cell pellets were resuspended in 200 μL PBS, frozen in liquid nitrogen, thawed three times and then fractionated into supernatant in cell pellet and insoluble pellet in cell pellet by centrifugation at 16,000 g for 10 min.
DNA fragmentation assay
Jurkat cells were treated with substances isolated from HCMV‐ or mock‐infected HEL 299 cells for 24 hr at 37°C in a humidified incubator with 5% CO2; cells were also treated with STS as a positive control for DNA fragmentation, 50% DMSO and three freeze‐thaw cycles as negative controls. DNA was isolated from cells using a QIAamp DNA mini kit (Qiagen, Venlo, Limburg, the Netherlands) according to the manufacturer's protocol. Isolated DNA was separated on a 1.5% agarose gel containing ethidium bromide at 50 volts for 45 min, and visualized under a UV transilluminator using a standard 1 kb DNA ladder (Thermo Scientific, Marietta, OH, USA).
Analysis of cell death by flow cytometry
Cell death was quantified by flow cytometry using PI staining or TUNEL using an APO‐BRDUTM Kit (Merck), according to the manufacturer's instructions. Briefly, ∼1 × 106 cells were fixed with 70% ethanol and stored overnight at −20°C. The cells were then washed twice with washing buffer and incubated for at least 30 min in staining solution at 4°C. Staining solutions contained either 1 mg/mL RNase A and 50 μg/mL PI for PI staining methods, or TdT reaction reagents and antibody solution containing BrdU‐fluorescein isothiocyanate for TUNEL staining. DNA content or DNA fragmentation in cells was measured using a FACScantoII (Becton‐Dickinson, San Jose, CA, USA). Cell cycle status was analyzed by using FlowJo flow cytometry analysis software (Tree Star, Ashland, OR, USA). Cell death experiments were repeated three times; results are expressed as mean ± SEM.
Western blot analyses
Harvested cells were treated with lysis buffer containing 50 mM Tris‐HCl (pH 7.4), 0.25% sodium deoxycholate, 150 mM NaCl, 1% NP‐40, 1 mM EDTA, 1 mM NaF, 1 mM Na3V4 and a protease inhibition cocktail (Calbiochem, San Diego, CA, USA) for 30 min on ice. Cleared cell lysates were then separated on 4%–20% Tris‐Glycine gels (Invitrogen, Carlsbad, CA, USA), or 10%, 12% or 15% SDS‐polyacrylamide gel by electrophoresis, and electro‐transferred to polyvinylidene difluoride membrane (Merck) in Tris‐glycine transfer buffer containing 20% methanol (Merck). Membranes were then probed with specific primary antibodies, followed by HRP‐conjugated goat anti‐mouse IgG (Calbiochem) or HRP‐conjugated goat anti‐rabbit IgG (Calbiochem) as the secondary antibody. All membranes were developed using ECL reagents (Pierce Thermo), according to the manufacturer's instructions. Images were obtained using a LAS‐4000 imager (Fujifilm, Tokyo, Japan).
Measurement of intracellular ROS
Intracellular ROS generation was measured in live cells by using dichlorofluorescin diacetate and examining specimens under a confocal microscope. Approximately 2 × 104 cells were incubated in culture media containing 10 μM dichlorofluorescin diacetate for 30 min at 37°C, washed twice and then resuspended in culture media. Changes in fluorescent signals in live cells were obtained using a Fluoview FV1000 confocal laser scanning microscope (Olympus, Tokyo, Japan), and analyzed using Fluoview software (Olympus).
Immunohistochemical staining
Immunohistochemical stains for LCA and CMV were performed on formalin fixed paraffin embedded tissues. The tissue used was from a female patient with HCMV proctitis with microperforation and ulceration. All stains were carried out using an autoimmunostainer Benchmark XT with an Optiview DAB Detection Kit (Ventana Medical Systems, Tucson, AZ, USA) according to the manufacturer's instructions and using the reagents supplied with the kit. In brief, sections of 4 µm were mounted on silanized charged slides and allowed to dry for 10 min at room temperature, followed by 20 min in an incubator at 65°C. After deparaffinization, heat‐induced epitope retrieval using standard Cell Conditioning 1 was performed for 24 min. Subsequently, slides were incubated with mouse anti‐LCA (clone 2B11+PD7/26, dilution 1:400; DAKO, Glostrup, Denmark) and anti‐CMV (clone 8B1.2, 1G5.2&2D4.2, dilution 1:200; Cell Marque, Rocklin, CA, USA) for 16 min at 37°C. Anti‐mouse IgG HRP Multimer was then added and incubated for 8 min. Stains were then visualized by using DAB. Finally, the slides were counterstained with hematoxylin for 4 min.
Cleaved caspase 3 was stained with rabbit anti‐cleaved caspase 3 antibody (Cell Signaling) and a Bond polymer detection kit (Leica, Newcastle Upon Tyne, UK) using Leica Bond‐max autostainer according to manufacturer's protocol. In brief, after deparaffinization and dewaxing, slides were heat‐pretreated at 100°C for 20 min with epitope retrieval solution 2 (pH 9.0). Primary antibody was reacted for 30 min, followed by polymer for 8 min and DAB substrate for 10 min. Slides were counterstained with hematoxylin for 1 min.
A TUNEL assay was performed using an ApopTag Plus Peroxidase In Situ Apoptosis Kit (Chemicon, Temecula, CA, USA) according to the manufacturer's manual. In brief, after deparaffinization and dewaxing slides were pretreated with proteinase K (20 µg/mL) for 30 min at room temperature. Equilibration buffer was incubated for 5 min, after which TdT enzyme was incubated in a humidified chamber at 37°C for 1 hr 30 min, followed by peroxidase‐conjugated anti‐digoxigenin for 30 min. Color was developed with peroxidase substrate containing DAB for 5 min. Slides were counterstained in Mayer's hematoxylin for 10 s.
Study approval
All experimental protocols using human materials were reviewed and approved by the Institutional Review Board of Seoul National University Hospital (C‐1306‐021‐494) and Asan Medical Center (No. 2014‐0602). The animal study protocol was reviewed and approved by the Seoul National University Institutional Animal Care and Use Committee (SNU‐120723‐2).
RESULTS
HCMV‐infected fibroblasts induce death of nearby immunocytes in a dose‐dependent manner
Human CMV‐infected HRPE cells kill CD4+ T cells by up‐regulating FasL, enabling evasion of immune systems 8. However, HCMV cannot up‐regulate FasL on all types of infected cells because cells that permit HCMV replication, such as epithelial cells, endothelial cells, and fibroblasts, rarely express FasL. Jurkat and THP‐1 cells were co‐cultured with HCMV‐infected cell‐derived substances to investigate whether non‐FasL‐expressing cells can induce the death of neighboring cells with HCMV infection. Jurkat and THP‐1 cells died following treatment with HCMV‐infected cell‐derived substances, but not after treatment with mock‐infected cell‐derived substances (Fig. 1a,b). To better understand the toxicity of these substances, these treatments were administered to a wide range of cell types. The hematopoietic cell lines U937, Raji, K562, HL‐60, Jurkat and V653 were readily killed by HCMV‐infected cell‐derived substances, whereas the non‐hematopoietic cell lines HEL 299, HEK‐293, U373MG and 22Rv1 survived (Fig. S1). These data suggest that substances from HCMV‐infected cells, which rarely express FasL, induce the death of nearby immunocytes. Jurkat cells were chosen as a representative cell line for investigating the mechanism(s) of cell death induced by HCMV‐infected cell‐derived substances.
Figure 1.
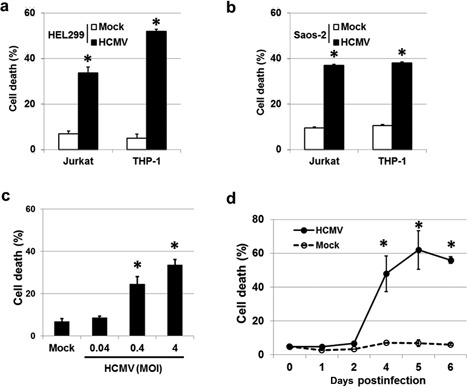
HCMV‐infected cells induce cell death in surrounding cells. Jurkat and THP‐1 cells were killed following exposure to substances released from HCMV‐infected (a) HEL 299 and (b) Saos‐2 cells. (c) Death rate of Jurkat cells following exposure to HCMV‐infected HEL 299 cells. HEL 299 cells were infected with HCMV at various MOI, and co‐cultured with Jurkat cells for 72 hr. The percentage of dead cells increased with increasing viral titer in a dose‐dependent manner. (d) Death of Jurkat cells following exposure to culture supernatants isolated from HCMV‐ or mock‐infected HEL 299 cells. Culture supernatants were harvested from HCMV‐ and mock‐infected HEL 299 cells for 6 days, and incubated in the presence of Jurkat cells for 24 hr. HCMV‐infected HEL 299 cells exhibited a significantly increased death rate 2 days post‐infection. Bars indicate the means ± SEM from three independent experiments. Statistical significance between experimental means (P value) was determined using Student's t‐test.
To examine whether HCMV infection is directly linked to this type of cell death, HEL 299 cells were infected with HCMV at a MOI of 0.04, 0.4 or 4 and incubated for 72 hr. Jurkat cells were then added to HCMV‐infected plates and co‐cultured for an additional 72 hr. The percentage of dead Jurkat cells ranged from 8.7% to 33.7%, with cell death increasing in a dose‐dependent manner (Fig. 1c), suggesting that induced death is dependent on HCMV infection. Next, the time frame was expanded. Culture supernatants were harvested from HCMV‐ and mock‐infected HEL 299 cells at 0, 1, 2, 4, 5 and 6 d.p.i. and incubated in the presence of Jurkat cells for 24 hr. Cell death was evident from between 3 and 4 d.p.i. and increased thereafter (Fig. 1d), suggesting that some death‐inducing substances are released from HCMV‐infected cells after 4 d.p.i.. Taken together, these findings suggest that substances produced by HCMV‐infected cells induce death of immunocytes in a dose‐dependent manner.
Jurkat cell death is induced by substances from HCMV‐infected fibroblasts
To better understand this mechanism of cell death, whether well‐known death‐inducing factors (FasL or TRIAL) or HCMV‐direct infection of Jurkat cells is involved in the death of Jurkat cells was examined. Cell death assays were conducted in Jurkat cells treated with culture supernatants from HCMV‐infected HEL 299 cells, along with neutralizing antibodies against FasL, TRAIL, or FasL plus TRAIL. The rate of cell death was unaffected by all neutralizing antibodies tested, with control, FasL−, TRAIL−, and FasL plus TRAIL‐treated cells exhibiting death rates of 75.3%, 73.5%, 73.7%, and 73.8%, respectively (Fig. 2a). These results demonstrate that neither FasL nor TRAIL is involved in the Jurkat cell death induced by these substances.
Figure 2.
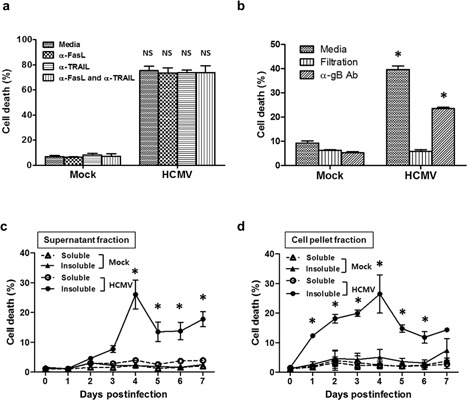
Insoluble substances isolated from HCMV‐infected HEL 299 culture supernatants and ruptured cells induce cell death in Jurkat cells independently of FasL, TRAIL and direct HCMV infection. Pretreatment of culture supernatants with neutralizing antibodies to (a) FasL and TRAIL or (b) HCMV did not affect death rates in Jurkat cells. Filtered culture supernatants from HCMV‐infected HEL 299 cells also failed to induce death in Jurkat cells. Bars in (a) and (b) indicate means ± SEM from three independent experiments. The kinetics of cell death in Jurkat cells following exposure to soluble and insoluble substances isolated from (c) culture supernatants or (d) ruptured cells. Cell death was readily induced by substances in the insoluble fractions of HCMV‐infected cells. All experiments were performed in triplicate. Figures 2c and d each depict the results of one representative experiment. Statistical significance between experimental means (P value) was determined using Student's t‐test.
The rate of cell death in Jurkat cells was also unaffected by neutralizing antibodies targeting HCMV gB (37.1% vs. 23.5% for control and treated samples, respectively), suggesting that direct infection of Jurkat cells by HCMV is not the cause of death of Jurkat cells. Furthermore, filtrates of the culture supernatant of HCMV‐infected HEL 299 cells did not induce cell death (Fig. 2b). Mock‐infected fibroblasts failed to induce cell death under any of the conditions tested.
Whereas Jurkat cells were unaffected by culture supernatant filtrates, the insoluble fractions of culture supernatants and ruptured cells from HCMV‐infected HEL 299 cells induced death at 3 d.p.i. and 1 d.p.i., respectively (Fig. 2c,d). Peak death rates for Jurkat cells were observed at 4 d.p.i. for both preparations; cell death was not detected following treatment with the soluble fractions of HCMV‐infected HEL 299 cells or any fraction derived from mock‐infected cells (Fig. 2c,d). These results demonstrate that the death‐inducing substances are present in the insoluble fractions and formed in HCMV‐infected fibroblasts from 1 d.p.i., being released into culture media in sufficient quantities by 3 d.p.i.
To characterize the substances from HCMV‐infected HEL 299 cells, western blot analyses were performed using antibodies targeting representative HCMV proteins. HCMV UL44 and pp65 were found at their respective molecular weights, along with additional smaller molecules, whereas HCMV IE1‐72 and IE2‐86 were weakly expressed (Fig. S2a). Boiling of substances from HCMV‐infected HEL 299 cells for 5 min did not affect the cell death rate (Fig. S2b), suggesting that the responsible substances from HCMV‐infected HEL 299 cells contain HCMV proteins and are heat‐stable; thus, these substances may not comprise a single compound. As mentioned above, these substances were termed HCMVAIS, whereas those from mock‐infected cells were termed MOCKAIS.
HCMVAIS induces ROS production via NOX, and contributes to cell death
Next, the mechanism of HCMVAIS‐induced cell death was investigated. Relative to MOCKAIS treatment, HCMVAIS treatment of Jurkat cells resulted in substantial intracellular ROS production. Pretreatment with either NAC or DPI prevented ROS production, amounts of ROS being maintained at or near baseline (Fig. 3a,b). This decrease in ROS production was directly correlated with cell survival; pretreatment with NAC reduced the percentage of dead cells from 59.4% to 33.5% in Jurkat cells and from 56.3% to 4.1% in PBMCs (Fig. 3c,d). Together, these data show that an increase in intracellular ROS in human mononuclear cells is critical for HCMVAIS‐induced cell death.
Figure 3.
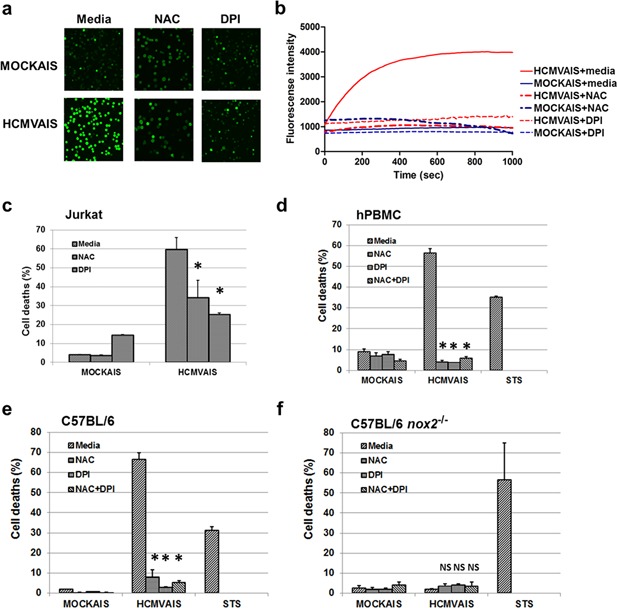
NOX‐mediated production of ROS plays a role in HCMVAIS‐associated cell death. (a) ROS production by HCMVAIS. Intracellular ROS was greater in Jurkat cells following exposure to HCMVAIS (bottom left) than in MOCKAIS (top left). Intracellular ROS in Jurkat cells following exposure to HCMVAIS was significantly attenuated by treatment with NAC (bottom middle, top middle) or DPI (bottom right, top right), indicating NOX‐mediated ROS production. Representative images from live confocal microscopy (Olympus, Tokyo, Japan) at 600 s are shown; original magnification ×400. (b) The kinetics of ROS generation in live Jurkat cells treated with HCMVAIS as visualized by confocal microscopy. HCMVAIS induced intracellular ROS in Jurkat cells; no effects were evident in MOCKAIS‐treated cells. (c and d) Intracellular ROS remained at baseline amounts after treatment with the ROS inhibitors NAC or DPI: (c) Jurkat cells, (d) human PBMCs exposed to HCMVAIS. (e and f) Intracellular ROS is involved in cell death. The effects of HCMVAIS on splenocytes derived from (e) C57BL/6 wild‐type and (f) nox‐2−/− mice were examined. These results suggest that HCMVAIS‐mediated ROS production is driven by NOX. Bars indicate the means ± SEM from three independent experiments. Cell death was measured by TUNEL assay. Statistical significance between experimental means (P value) was determined using Student's t‐test. [Color figure can be viewed at http://wileyonlinelibrary.com]
To determine the source of ROS generation, the effects of the NOX inhibitor DPI on HCMVAIS‐induced intracellular ROS generation was investigated. The Jurkat cell death rate decreased from 59.4% to 25.7% following treatment with DPI (Fig. 3c); additionally, pretreatment with DPI almost completely inhibited HCMVAIS‐induced death in human PBMCs (Fig. 3d). These results suggest that NOX‐derived intracellular ROS is the primary mechanism by which HCMVAIS causes cell death. To verify the involvement of NOX‐2 in cell death, splenocytes from nox‐2 −/− C57BL/6 mice were examined following treatment with HCMVAIS. The death rates of splenocytes from wild‐type C57BL/6 mice were found to be 66.8% and 1.9% following treatment with HCMVAIS and MOCKAIS, respectively (Fig. 3e), compared with 2.0% and 2.4%, respectively, in splenocytes from nox‐2 −/− C57BL/6 mice (Fig. 3f). These results clearly show that HCMVAIS induces cell death in Jurkat cells and murine splenocytes via NOX‐derived ROS.
PARP‐1, AIF and DNA fragmentation are involved in HCMVAIS‐induced cell death of Jurkat cells and human PBMCs
Increases in intracellular ROS can cause DNA damage in human cells 22. PARP‐1 participates in DNA damage repair by binding to DNA strand breaks and modifying nuclear proteins using nicotinamide adenine dinucleotide as the substrate 23. To determine whether PARP‐1 is involved in this induced cell death, PARP‐1 cleavage was assayed in Jurkat cells and human PBMCs after treatment with HCMVAIS; STS‐treated cells were used as a positive control. An 89 kDa fragment of PARP‐1 was detected in Jurkat cells and PBMCs treated with HCMVAIS (Fig. 4a,b), suggesting activation of PARP‐1 by increased intracelluar ROS. Parthanatos is mediated by PARP‐1 24. Because previous studies have shown that nuclear translocation of AIF is a characteristic feature of PARP‐1‐mediated parthanatos 17, 25, nuclear translocation of AIF was investigated by confocal microscopy. AIF was detected in the nuclei of HCMVAIS‐treated Jurkat cells and in the cytoplasm of MOCKAIS‐treated cells (Fig. 4c), suggesting translocation of AIF to the nucleus. To confirm the involvement of AIF in this cell death pathway, AIF release was inhibited by NP. Inhibition of AIF release decreased the rate of Jurkat cell death (Fig. 4d). Caspase‐independent DNA fragmentation is also a distinct feature of parthanatos 17. To confirm parthanatos, it was next determined whether DNA fragmentation is involved in HCMVAIS‐induced death. HCMVAIS, but not MOCKAIS, treatment was found to induce DNA fragmentation in Jurkat cells. STS was used as a positive inducer of DNA fragmentation, whereas repeated freeze‐thaw cycles and DMSO were used as negative controls (Fig. 4e). To determine the involvement of caspase activation in this process, various caspase inhibitors were added to Jurkat cells following treatment with HCMVAIS. Jurkat cell death rates were unaffected by HCMVAIS treatment, regardless of the caspase inhibitor used (Fig. S3a). To confirm the involvement of caspase activation, caspases 3, 7 and 9 were assayed in Jurkat cells and human PBMCs after treatment with HCMVAIS; STS‐treated cells were used as a positive control. No cleavage products were detected following treatment with any of the three caspases tested (Fig. S3b,c), or in cells treated with MOCKAIS. These results suggest that HCMVAIS‐induced ROS causes PARP‐1‐mediated parthanatos via nuclear translocation of AIF and DNA fragmentation in a caspase‐independent manner.
Figure 4.
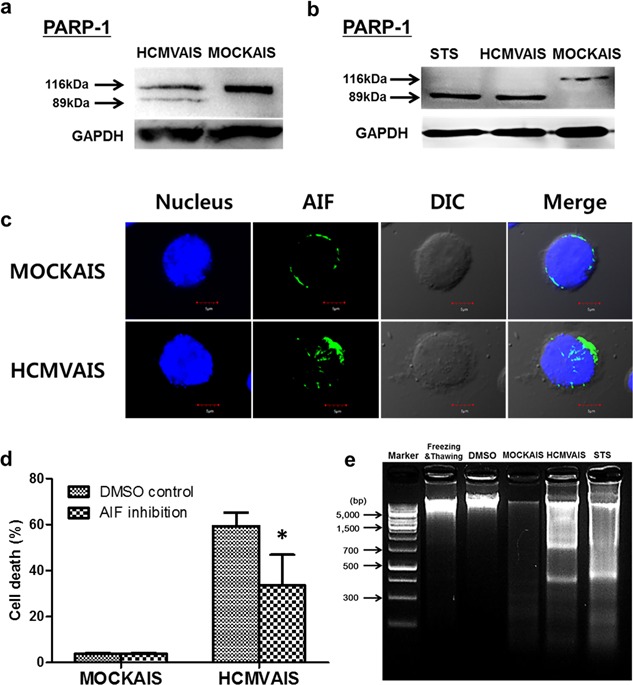
Activation of PARP‐1, nuclear translocation of AIF and DNA fragmentation are involved in the death of cells treated with HCMVAIS. Cleavage of PARP‐1 following treatment with HCMVAIS was detected by western blot in (a) Jurkat cells and (b) human PBMCs. STS‐treatment was used as a positive control for caspase cleavage. (c) Nuclear translocation of AIF in Jurkat cells with HCMVAIS treatment by confocal microscopy. Jurkat cells were adhered, fixed, permeabilized, and stained with AIF antibody and Alexa Flour 488‐labeled secondary antibody and the nuclei visualized with ToPro‐3. (d) Inhibition of AIF release with NP (50 mM) decreases the rate of Jurkat cell death. Bars indicate the means ± SEM from three independent experiments. Cell death was measured by TUNEL assay. (e) HCMVAIS induces DNA fragmentation in Jurkat cells; no fragmentation was seen in following treatment with MOCKAIS. STS was used as a positive control for DNA fragmentation; repeated freeze‐thaw cycles and 50% DMSO were used as negative controls. Images are representative of two independent experiments. Statistical significance between experimental means (P value) was determined using Student's t‐test. [Color figure can be viewed at http://wileyonlinelibrary.com]
Cell death in HCMV‐infected foci in the pathological tissue
To characterize the nature of cell death near HCMV‐infected cells in vivo, HCMV‐infected tissue was examined to detect the presence of HCMV antigens, leukocyte common antigen (CD45), cleaved caspase 3, and fragmented DNA. CD45(+) and TUNEL(+) cells were observed; however, cleaved caspase 3(+) cells were not detected in HCMV‐infected foci (Fig. 5, left rectangles). CD45, TUNEL and cleaved caspase 3 were variably stained in HCMV‐negative foci (Fig. 5, right rectangles), suggesting that leukocytes near HCMV‐infected cells are killed by the pathway without caspase 3 activation, supporting our in vitro results.
Figure 5.
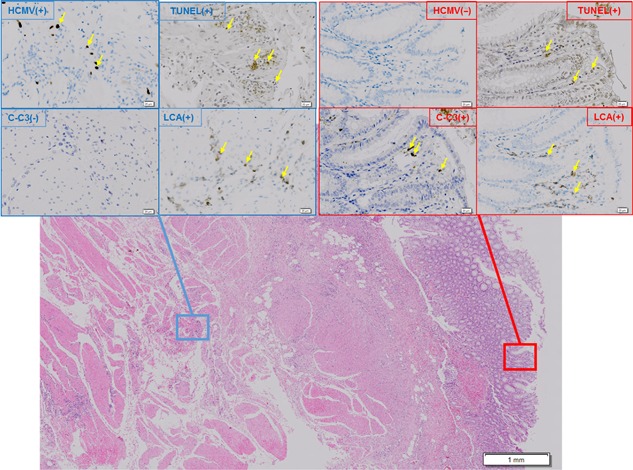
Immunohistochemical staining of HCMV‐infected foci in human large intestine. TUNEL(+), CD45(+), and cleaved caspase 3(−) cells were found in HCMV‐infected foci in sections of human large intestine. After serially sectioned tissues had been reacted with antibody to HCMV antigens, leukocyte common antigen (CD45), cleaved caspase 3, and TdT reagents and peroxidase‐conjugated respective antibody and DAB were successfully applied, and finally counterstained with hematoxylin as described in detail in Materials and Methods. Microphotographs demonstrate representative areas with HCMV(+) and HCMV(−). Scale bars indicate the magnification in each area. Yellow arrows indicate positive staining for HCMV, TUNEL, LCA or caspase3. [Color figure can be viewed at http://wileyonlinelibrary.com]
DISCUSSION
Here, we demonstrated the involvement of ROS, PARP‐1, AIF and DNA fragmentation in the parthanatos of hematopoietic cells following treatment with HCMVAIS, a collection of insoluble substances derived from HCMV‐infected fibroblasts.
The mechanism by which HCMV establishes latency outside the original site of infection remains unknown. We recently showed that HCMV causes latent infection in CD34+ human progenitor cells via epidermal growth factor receptor signaling 26. Previous studies have suggested a role for CD34+ human progenitor cells as a reservoir of latent infection 27, 28, 29; however, the mode of viral transmission to these precursor cells has not yet been established. The limited ability of free virions to infect cells via known cell surface receptor indicates that direct transmission of HCMV from infected cells to monocytic precursor cells is the more likely route. We originally attempted to model viral cell‐to‐cell transmission by adding THP‐1 monocytic precursor cells to HCMV‐infected fibroblasts and Saos‐2 osteosarcoma cells, but were unable to detect the presence of HCMV because of increased death of THP‐1 cells (Fig. 1a,b). We therefore chose to investigate the mechanism by which HCMV increases cell death in the present model.
We began by investigating representative apoptosis‐related molecules such as FasL and TRAIL, which play a major role in regulation of lymphocyte homeostasis 30. Inhibition of FasL and TRAIL, which are targeted by other viruses for apoptosis of lymphocytes and immune evasion 8, 9, 10, failed to prevent cell death induced by the substances produced by HCMV‐infected fibroblasts (Fig. 2a).
Evidence that the cytotoxic substances produced by HCMV‐infected fibroblasts (Fig. 1a) and Saos‐2 (Fig. 1b) are not associated with any previously known apoptosis‐related molecule led us to further investigate the nature and mechanism of action of these substances. We found that the cytotoxic substances derived from HCMV‐infected fibroblasts are insoluble complexes that appear to be formed in the cells as early as 1 d.p.i., but are not released from them until 3 d.p.i. (Fig. 2c,d). We termed these immunocytotoxic substances as HCMVAIS and the equivalent fraction isolated from mock‐infected fibroblasts MOCKAIS. Our data showed that HCMVAIS are produced from both of HCMV‐infected fibroblasts and Saos‐2 cells, suggesting HCMV could beis involved in the production of HCMVAIS in permissive cells such as fibroblasts and Saos‐2. To confirm death effect with other HCMV strains, we also examined that clinical strain TB40 and found that itE, which is a clinical strain, shows aed s similar death effect to as Jurkat cells (85%), suggesting HCMVAIS could beis produced from HCMV‐infected cells, regardless of HCMV strains. Although we expect that HCMVAIS is comprises heat‐stable complexes and may not representrather than a single compound (Fig. S2), direct analysis of the insolubility and complexity of the HCMVAIS hindered direct analysiswas limited due to the insolubility and complexity of the substances. Thus, further analyses are required to identify specific death‐inducing substances within these complexes.
We observed more intracellular ROS in Jurkat cells exposed to HCMVAIS than in MOCKAIS‐treated controls. Under oxidative stress, ROS, including free radicals such as superoxide, hydroxyl radicals and hydrogen peroxide, are generated in large amounts and induce widespread cellular damage and cell death 31. We here showed that the frequency of cell death in HCMVAIS‐treated Jurkat cells decreases following addition of NAC, a potent scavenger of free radicals and inhibitor of ROS generation. Intracellular ROS are generated by the mitochondria, peroxisomes, endoplasmic reticulum and NOX complex 32, 33. Among the compounds tested, we found that DPI, an inhibitor of NOX, significantly attenuates cell death in Jurkat cells and human PBMCs exposed to HCMVAIS (Fig. 3c,d), indicating that NOX is the source of HCMVAIS‐induced ROS. We further confirmed the involvement of this complex in cell death using splenocytes isolated from nox‐2−/− mice, in which NOX‐2 is disrupted enough to prevent HCMVAIS‐mediated cell death (Fig. 3e,f).
Increased intracellular ROS caused by HCMVAIS can cause DNA damage in Jurkat cells. DNA damaged by ROS activates PARP‐1 to initiate repair by binding to DNA strand breaks and modifying nuclear proteins using NAD as a substrate 23. Activation of PARP‐1 is also involved in parthanatos 16, 34. Parthanatos has unique features, including caspase‐independent PARP‐1 activation, AIF release into the nucleus and DNA fragmentation 25. We speculated that HCMVAIS‐induced ROS initiates parthanatos in Jurkat cells and showed here that HCMVAIS‐induced ROS initiates PARP‐1‐mediated cell death through nuclear translocation of AIF and DNA fragmentation via a caspase‐independent pathway (Figs. 4 and S3). The lack of a suitable animal model for HCMV infection makes it difficult to examine HCMV‐mediated immune cell death in vivo; however, investigation of increased cell death in the vicinity of HCMV‐infected cells may be possible in human subjects. In this respect, we observed cell death in HCMV‐infected foci in tissues from the large intestine (Fig. 5). The nature of cell death was TUNEL(+), CD45(+), and cleaved caspase 3(−), consistent with our in vitro experiments.
T cells appear to be particularly sensitive to ROS, which play an essential role in both T cell activation and the death of activated T cells 35. This increased sensitivity likely accounts for much of the differences in HCMVAIS sensitivity between cell types and may confer additional survival advantages to HCMV. HCMV in smooth muscle cells up‐regulates ROS and activates NF‐κB to express viral or cellular genes, which may increase the efficiency of HCMV infectivity and gene expression 36. If HCMVAIS increases the amount of intracellular ROS in nearby fibroblasts, it may provide an additional survival benefit by eliminating nearby immune cells, allowing HCMV to evade local immune surveillance.
Depending on the immune status of the individual and the site of infection, HCMV infection can cause a wide range of symptoms. A correlation between HCMV reactivation and a reduced relapse rate of a specific type of leukemia was recently reported 37. One possible explanation for this correlation is the destruction of newly formed leukemic cells by insoluble substances produced by HCMV‐infected cells, as described herein. Such a possibility is made more likely by the concurrence of HCMV reactivation and hematopoiesis, in addition to dysregulation of the cytokines that drive hematopoietic cells in HCMV‐infected cells 38, 39.
In summary, the data presented herein show that the increased death rate of nearby hematopoietic cells during HCMV infection is the result of increased ROS production caused by insoluble substances produced by HCMV‐infected cells (Fig. 6). This destruction of nearby immune cells through induction of intracellular ROS production manifests as PARP‐1 and AIF‐mediated parthanatos and may enable HCMV to evade immune surveillance.
Figure 6.
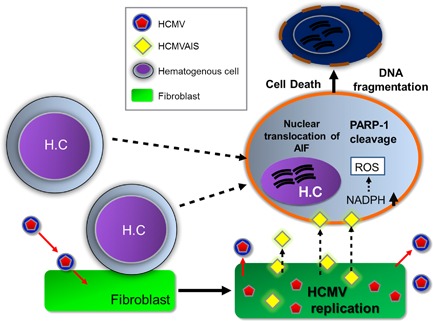
Proposed model of HCMVAIS‐mediated cell death. After HCMV infection, HCMVAIS is formed within cells and then released into the surrounding environment. HCMVAIS interacts with nearby hematopoietic cells, leading to increased production of intracellular ROS via NOX. Large amounts of ROS induce cleavage of PARP‐1, resulting in DNA fragmentation by nuclear translocation of AIF and cell death. H.C., Hematogenous cell. [Color figure can be viewed at http://wileyonlinelibrary.com]
DISCLOSURE
The authors declared they have no conflicts of interest.
The English in this document has been checked by at least two professional editors, both native speakers of English. For a certificate, please see:
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's website.
Fig. S1. Sensitivity of hematopoietic and non‐hematopoietic cell lines to substances produced by HCMV‐ or mock‐infected HEL 299 cells. Hematopoietic and non‐hematopoietic cell lines were treated with HCMV‐infected cells‐derived substances. The cell death rate was significantly higher in hematopoietic cells cultured in the presence of HCMV‐infected HEL 299 cells than in non‐hematopoietic and mock‐infected cells. Bars indicate the means ± SEM from three independent experiments. Statistical significance between experimental means (P value) was determined using Student's t‐test.
Fig. S2. Characteristics of the insoluble substances isolated from HCMV‐infected HEL 299 cells. (a) Insoluble substances isolated from HCMV‐infected HEL 299 cells contain HCMV proteins. Western blot analysis identified HCMV immediate early (IE1‐72 and IE2‐86), early (UL44) and late (pp65) proteins in the insoluble fraction of HCMV‐infected HEL 299 cells. Pip, insoluble pellet in the cell pellet; sip, supernatant in the cell pellet; pis, insoluble pellet in the supernatant; sis, supernatant in the supernatant. (b) Insoluble substances from HCMV‐infected HEL 299 cells are heat‐stable. Cell death rates are unaffected by heating of insoluble substances. Bars indicate the means ± SEM from three independent experiments. Statistical significance between experimental means (P value) was determined using Student's t‐test.
Fig. S3. HCMVAIS‐induced death occurs in a caspase‐independent manner. (a) HCMVAIS‐mediated cell death is independent of caspase activation. Inhibition of caspase activity with various caspase inhibitors failed to reduce the death rate of Jurkat cells following treatment with HCMVAIS. Cleavage of caspases 3, 7 and 9 in (b) Jurkat cells and (c) human PBMCs was detected by western blot following treatment with HCMVAIS. STS‐treatment was used as a positive control for caspase cleavage. This DNA fragmentation is independent of caspase activation. Bars indicate the means ± SEM from three independent experiments.
ACKNOWLEDGMENTS
We would like to thank Moa Sa and Byoung‐Jun Kim for their assistance with animal experiments, Professor Hyung‐ran Kim for the care of nox‐2−/−‐ mice and Dr. Hyun‐Je Kim for drawing peripheral blood from human subjects.
REFERENCES
- 1. Conboy T.J., Pass R.F., Stagno S., Alford C.A., Myers G.J., Britt W.J., Mccollister F.P., Summers M.N., Mcfarland C.E., Boll T.J. (1987) Early clinical manifestations and intellectual outcome in children with symptomatic congenital cytomegalovirus infection. J Pediatr 111: 343–8. [DOI] [PubMed] [Google Scholar]
- 2. Jacobson M.A., Mills J. (1988) Serious cytomegalovirus disease in the acquired immunodeficiency syndrome (AIDS). Clinical findings, diagnosis, and treatment. Ann Intern Med 108: 585–94. [DOI] [PubMed] [Google Scholar]
- 3. Light J.A., Burke D.S. (1979) Association of cytomegalovirus (CMV) infections with increased recipient mortality following transplantation. Transplant Proc 11: 79–82. [PubMed] [Google Scholar]
- 4. Rafailidis P.I., Mourtzoukou E.G., Varbobitis I.C., Falagas M.E. (2008) Severe cytomegalovirus infection in apparently immunocompetent patients: A systematic review. Virol J 5: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Castillo J.P., Kowalik T.F. (2002) Human cytomegalovirus immediate early proteins and cell growth control. Gene 290: 19–34. [DOI] [PubMed] [Google Scholar]
- 6. Jackson S.E., Mason G.M., Wills M.R. (2011) Human cytomegalovirus immunity and immune evasion. Virus Res 157: 151–60. [DOI] [PubMed] [Google Scholar]
- 7. Gajl‐Peczalska K. (1967) Cytomegalic inclusion disease. Arch Dis Child 42: 14–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chiou S.H., Liu J.H., Hsu W.M., Chen S.S., Chang S.Y., Juan L.J., Lin J.C., Yang Y.T., Wong W.W., Liu C.Y., Lin Y.S., Liu W.T., Wu C.W. (2001) Up‐regulation of Fas ligand expression by human cytomegalovirus immediate‐early gene product 2: A novel mechanism in cytomegalovirus‐induced apoptosis in human retina. J Immunol 167: 4098–103. [DOI] [PubMed] [Google Scholar]
- 9. Jeremias I., Herr I., Boehler T., Debatin K.M. (1998) TRAIL/Apo‐2‐ligand‐induced apoptosis in human T cells. Eur J Immunol 28: 143–52. [DOI] [PubMed] [Google Scholar]
- 10. Clarke P., Meintzer S.M., Gibson S., Widmann C., Garrington T.P., Johnson G.L., Tyler K.L. (2000) Reovirus‐induced apoptosis is mediated by TRAIL. J Virol 74: 8135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Galluzzi L., Vitale I., Abrams J.M., Alnemri E.S., Baehrecke E.H., Blagosklonny M.V., Dawson T.M., Dawson V.L., El‐Deiry W.S., Fulda S., Gottlieb E., Green D.R., Hengartner M.O., Kepp O., Knight R.A., Kumar S., Lipton S.A., Lu X., Madeo F., Malorni W., Mehlen P., Nunez G., Peter M.E., Piacentini M., Rubinsztein D.C., Shi Y., Simon H.U., Vandenabeele P., White E., Yuan J., Zhivotovsky B., Melino G., Kroemer G. (2012) Molecular definitions of cell death subroutines: Recommendations of the nomenclature committee on cell death 2012. Cell Death Differ 19: 107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fatokun A.A., Dawson V.L., Dawson T.M. (2014) Parthanatos: Mitochondrial‐linked mechanisms and therapeutic opportunities. Br J Pharmacol 171: 2000–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim H.S., Lee M.S. (2005) Essential role of STAT1 in caspase‐independent cell death of activated macrophages through the p38 mitogen‐activated protein kinase/STAT1/reactive oxygen species pathway. Mol Cell Biol 25: 6821–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin Y., Choksi S., Shen H.M., Yang Q.F., Hur G.M., Kim Y.S., Tran J.H., Nedospasov S.A., Liu Z.G. (2004) Tumor necrosis factor‐induced nonapoptotic cell death requires receptor‐interacting protein‐mediated cellular reactive oxygen species accumulation. J Biol Chem 279: 10822–8. [DOI] [PubMed] [Google Scholar]
- 15. Sade H., Sarin A. (2004) Reactive oxygen species regulate quiescent T‐cell apoptosis via the BH3‐only proapoptotic protein BIM. Cell Death Differ 11: 416–23. [DOI] [PubMed] [Google Scholar]
- 16. Chiu L.Y., Ho F.M., Shiah S.G., Chang Y., Lin W.W. (2011) Oxidative stress initiates DNA damager MNNG‐induced poly(ADP‐ribose)polymerase‐1‐dependent parthanatos cell death. Biochem Pharmacol 81: 459–70. [DOI] [PubMed] [Google Scholar]
- 17. Wang Y., Dawson V.L., Dawson T.M. (2009) Poly(ADP‐ribose) signals to mitochondrial AIF: A key event in parthanatos. Exp Neurol 218: 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang E.S., Chen S.T., Pagano J.S. (1973) Human cytomegalovirus. I. purification and characterization of viral DNA. J Virol 12: 1473–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee K., Won H.Y., Bae M.A., Hong J.H., Hwang E.S. (2011) Spontaneous and aging‐dependent development of arthritis in NADPH oxidase 2 deficiency through altered differentiation of CD11b+ and Th/Treg cells. Proc Natl Acad Sci USA 108: 9548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pollock J.D., Williams D.A., Gifford M.A., Li L.L., Du X., Fisherman J., Orkin S.H., Doerschuk C.M., Dinauer M.C. (1995) Mouse model of X‐linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 9: 202–9. [DOI] [PubMed] [Google Scholar]
- 21. Cha Cy H.E. (1988) Production and characterization of monoclonal antibodies reactive with human cytomegalovirus. J Kor Soc Microbiol 23: 495–504. [Google Scholar]
- 22. Ames B.N. (1989) Endogenous oxidative DNA damage, aging, and cancer. Free Radic Res Commun 7: 121–8. [DOI] [PubMed] [Google Scholar]
- 23. Berger N.A., Petzold S.J. (1985) Identification of minimal size requirements of DNA for activation of poly(ADP‐ribose) polymerase. Biochemistry 24: 4352–5. [DOI] [PubMed] [Google Scholar]
- 24. Harraz M.M., Dawson T.M., Dawson V.L. (2008) Advances in neuronal cell death 2007. Stroke 39: 286–8. [DOI] [PubMed] [Google Scholar]
- 25. Yu S.W., Wang H., Poitras M.F., Coombs C., Bowers W.J., Federoff H.J., Poirier G.G., Dawson T.M., Dawson V.L. (2002) Mediation of poly(ADP‐ribose) polymerase‐1‐dependent cell death by apoptosis‐inducing factor. Science 297: 259–63. [DOI] [PubMed] [Google Scholar]
- 26. Kim J.H., Collins‐Mcmillen D., Buehler J.C., Goodrum F.D., Yurochko A.D. (2017) HCMV requires EGFR signaling to enter and initiate the early steps in the establishment of latency in CD34+ human progenitor cells. J Virol 91: pii: e01206–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mendelson M., Monard S., Sissons P., Sinclair J. (1996) Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J Gen Virol 77: 3099–102. [DOI] [PubMed] [Google Scholar]
- 28. Albright E.R., Kalejta R.F. (2013) Myeloblastic cell lines mimic some but not all aspects of human cytomegalovirus experimental latency defined in primary CD34+ cell populations. J Virol 87: 9802–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Buehler J., Zeltzer S., Reitsma J., Petrucelli A., Umashankar M., Rak M., Zagallo P., Schroeder J., Terhune S., Goodrum F. (2016) Opposing regulation of the EGF receptor: A molecular switch controlling cytomegalovirus latency and replication. PLoS Pathog 12: e1005655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Holtzman M.J., Green J.M., Jayaraman S., Arch R.H. (2000) Regulation of T cell apoptosis. Apoptosis 5: 459–71. [DOI] [PubMed] [Google Scholar]
- 31. Benassi B., Fanciulli M., Fiorentino F., Porrello A., Chiorino G., Loda M., Zupi G., Biroccio A. (2006) c‐Myc phosphorylation is required for cellular response to oxidative stress. Mol Cell 21: 509–19. [DOI] [PubMed] [Google Scholar]
- 32. Muller F. (2000) The nature and mechanism of superoxide production by the electron transport chain: Its relevance to aging. J Am Aging Assoc 23: 227–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han D., Williams E., Cadenas E. (2001) Mitochondrial respiratory chain‐dependent generation of superoxide anion and its release into the intermembrane space. Biochem J 353: 411–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andrabi S.A., Dawson T.M., Dawson V.L. (2008) Mitochondrial and nuclear cross talk in cell death: Parthanatos. Ann N Y Acad Sci 1147: 233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hildeman D.A., Mitchell T., Kappler J., Marrack P. (2003) T cell apoptosis and reactive oxygen species. J Clin Invest 111: 575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Speir E., Yu Z.X., Ferrans V.J., Huang E.S., Epstein S.E. (1998) Aspirin attenuates cytomegalovirus infectivity and gene expression mediated by cyclooxygenase‐2 in coronary artery smooth muscle cells. Circ Res 83: 210–6. [DOI] [PubMed] [Google Scholar]
- 37. Green M.L., Leisenring W.M., Xie H., Walter R.B., Mielcarek M., Sandmaier B.M., Riddell S.R., Boeckh M. (2013) CMV reactivation after allogeneic HCT and relapse risk: Evidence for early protection in acute myeloid leukemia. Blood 122: 1316–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kwon Y.J., Kim D.J., Kim J.H., Park C.G., Cha C.Y., Hwang E.S. (2004) Human cytomegalovirus (HCMV) infection in osteosarcoma cell line suppresses GM‐CSF production by induction of TGF‐beta. Microbiol Immunol 48: 195–9. [DOI] [PubMed] [Google Scholar]
- 39. Reddehase M.J., Dreher‐Stumpp L., Angele P., Balthesen M., Susa M. (1992) Hematopoietic stem cell deficiency resulting from cytomegalovirus infection of bone marrow stroma. Ann Hematol 64: A125–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's website.
Fig. S1. Sensitivity of hematopoietic and non‐hematopoietic cell lines to substances produced by HCMV‐ or mock‐infected HEL 299 cells. Hematopoietic and non‐hematopoietic cell lines were treated with HCMV‐infected cells‐derived substances. The cell death rate was significantly higher in hematopoietic cells cultured in the presence of HCMV‐infected HEL 299 cells than in non‐hematopoietic and mock‐infected cells. Bars indicate the means ± SEM from three independent experiments. Statistical significance between experimental means (P value) was determined using Student's t‐test.
Fig. S2. Characteristics of the insoluble substances isolated from HCMV‐infected HEL 299 cells. (a) Insoluble substances isolated from HCMV‐infected HEL 299 cells contain HCMV proteins. Western blot analysis identified HCMV immediate early (IE1‐72 and IE2‐86), early (UL44) and late (pp65) proteins in the insoluble fraction of HCMV‐infected HEL 299 cells. Pip, insoluble pellet in the cell pellet; sip, supernatant in the cell pellet; pis, insoluble pellet in the supernatant; sis, supernatant in the supernatant. (b) Insoluble substances from HCMV‐infected HEL 299 cells are heat‐stable. Cell death rates are unaffected by heating of insoluble substances. Bars indicate the means ± SEM from three independent experiments. Statistical significance between experimental means (P value) was determined using Student's t‐test.
Fig. S3. HCMVAIS‐induced death occurs in a caspase‐independent manner. (a) HCMVAIS‐mediated cell death is independent of caspase activation. Inhibition of caspase activity with various caspase inhibitors failed to reduce the death rate of Jurkat cells following treatment with HCMVAIS. Cleavage of caspases 3, 7 and 9 in (b) Jurkat cells and (c) human PBMCs was detected by western blot following treatment with HCMVAIS. STS‐treatment was used as a positive control for caspase cleavage. This DNA fragmentation is independent of caspase activation. Bars indicate the means ± SEM from three independent experiments.