

Abstract
Overexpression of BCLX and BFL1/A1 has been reported in various human malignancies and is associated with poor prognosis and drug resistance, identifying these prosurvival BCL2 family members as putative drug targets. We have generated transgenic mice that express human BFL1 or human BCLX protein throughout the haematopoietic system under the control of the Vav gene promoter. Haematopoiesis is normal in both the Vav‐BFL1 and Vav‐BCLX transgenic (TG) mice and susceptibility to spontaneous haematopoietic malignancies is not increased. Lymphoid cells from Vav‐BCLX TG mice exhibit increased resistance to apoptosis in vitro while most blood cell types form Vav‐BFL1 TG mice were poorly protected. Both transgenes significantly accelerated lymphomagenesis in Eμ‐MYC TG mice and, surprisingly, the Vav‐BFL1 transgene was the more potent. Unexpectedly, expression of transgenic BFL1 RNA and protein is significantly elevated in B lymphoid cells of Vav‐BFL1/Eμ‐MYC double‐transgenic compared to Vav‐BFL1 mice, even during the preleukaemic phase, providing a rationale for the potent synergy. In contrast, Vav‐BCLX expression was not notably different. These mouse models of BFL1 and BCLX overexpression in lymphomas should be useful tools for the testing the efficacy of novel human BFL1‐ and BCLX‐specific inhibitors.
Keywords: apoptosis, BCLX, BFL1/A1, lymphomagenesis, MYC, Vav‐promoter
Abbreviations
- AV
Annexin V
- DT
double‐transgenic
- Eµ‐MYC
c‐MYC gene driven by the IgH enhancer
- Glc
glucocorticoids
- STS
staurosporine
- TG
transgene/transgenic
Introduction
The physiological roles of BFL1/A1, an antiapoptotic member of the BCL2 family, are still poorly understood. A1 was discovered in 1993 as an early response gene in GM‐CSF‐treated bone marrow‐derived macrophages 1, 2 and later shown to be induced by antigen‐mediated activation in T and B cells 3, 4. In mice, A1 is produced by three independent genes (Bcl2a1‐a, Bcl2a1‐b and Bcl2a1‐d) 5 and is mainly expressed in the haematopoietic system where it is dynamically regulated in response to antigens or inflammatory cues engaging NF‐kB 6, NF‐AT 7, and PU.1 8 transcription factors. Mice that lack all functional Bcl2a1 genes do not exhibit major impairments in the development and composition of their immune system 9 or T cell‐mediated immune responses 10. The human homologue, BFL1, which is highly homologous to A1 (72% amino acid identity) is encoded by a single gene 11. Elevated BFL1 expression has been associated with many malignancies, including acute lymphoblastic leukaemia, chronic lymphocytic leukaemia and melanoma skin cancer 12, 13. In mouse models, lentiviral transduction of bone marrow cells with Bcl2a1‐a led to the development of B cell lymphomas in recipient mice 14 and cotransduction with human BFL1 and c‐MYC caused acute myelogenous leukaemia 15. Importantly, BFL1 mutants that escape ubiquitin‐mediated proteasomal degradation are more stable and accelerate tumour formation in the presence of a dominant negative, truncated version of p53 DD 16, indicating the importance of BFL1 expression levels for facilitating tumour formation.
BCLX is essential during early development as Bclx‐deficient mice die at embryonic day 13 due to defective erythropoiesis and massive cell death in the central nervous system 17, 18. In the haematopoietic system, BCLX deficiency leads to a loss of pre‐B cells 19, impaired erythropoiesis 20 and decreased platelet life span 21. Interestingly, although highly expressed in CD4+CD8+ double‐positive (DP) thymocytes, Bclx deletion does not substantially influence T cell development but only reduces the life span of DP thymocytes ex vivo 22, 23.
The transcription factor c‐MYC is a key transcriptional regulator involved in many cellular processes including metabolism, cell cycle and apoptosis 24. Aberrant expression of MYC is associated with a significant number of human malignancies 25, including human Burkitt's lymphomas, which harbour chromosome translocations linking the MYC gene with Ig heavy (IGH) or Ig light chain (IGL) loci 26. Eμ‐MYC transgenic mice, which model Burkitt's lymphoma to a certain degree, develop monoclonal pro‐/pre‐B and immature B cell lymphomas between 4 and 7 months of age 27, 28. Of note, BCL2 29, 30, MCL1 31 or BCLX 32 transgenes significantly accelerate lymphomagenesis in Eμ‐MYC mice, indicating the importance of overcoming apoptosis for MYC‐driven lymphomagenesis.
Little is known about the lymphomagenic potential of BFL1/A1. Using an shRNA‐based model to knock down A1 protein expression in mice, we recently observed that MYC‐induced lymphomas select against low A1 levels and that diminished A1 renders premalignant cells more susceptible to apoptosis ex vivo 33. Studies using mice totally lacking A1 also suggest that A1 contributes to tumour cell survival in the context of MYC overexpression 34. Moreover, the recent report of a patient with DLBCL 35 having a BFL1/IgH translocation as well as a MYC/IgL translocation suggests that BFL1 overexpression can act as a second hit in MYC‐driven B cell lymphomagenesis.
To investigate the impact of pan‐haematopoietic overexpression of BFL1 and BCLX, we have generated Vav‐BFL1 TG and Vav‐BCLX TG mice. We found that both the Vav‐BFL1 and the Vav‐BCLX transgenes can accelerate Eμ‐MYC‐driven lymphomagenesis and observed an unexpected interrelationship between MYC and BFL1 TG expression levels.
Results
Enforced expression of BFL1 or BCLX does not perturb haematopoiesis in mice
The BFL1 TG and BCLX TG mice were generated by pronuclear injection of oocytes using a haematopoietic‐specific transgenic vector driven by the Vav gene promoter 36. For each transgene, independent colonies were established from three PCR‐positive founders and the two lines showing detectable exogenous protein expression were chosen for further characterization (Fig. 1A,B), alongside previously derived Vav‐Mcl1 TG 31 and Vav‐BCL2 TG mice 37. The Vav‐BFL1 TG and Vav‐BCLX TG mice were healthy, showed normal fertility and did not exhibit any premature deaths within the first year of age, unlike Vav‐Mcl1 or Vav‐BCL2 transgenic mice, which develop auto‐immune and/or malignant disease 31, 37, 38.
Figure 1.
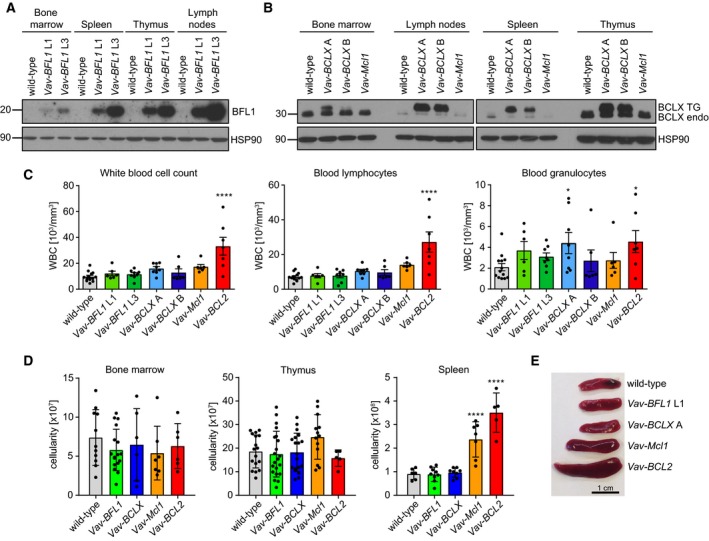
Characterization of transgene expression and composition of haematopoietic organs in Vav‐BFL1 and Vav‐BCLX TG mice. (A) Bone marrow, spleen, thymus and lymph nodes were isolated from 8–12‐week‐old wild‐type, Vav‐BFL1 (L1) and Vav‐BFL1 (L3) mice, respectively, and processed for western blotting using anti‐BFL1‐ and anti‐HSP90‐specific antibodies. (B) Bone marrow, lymph nodes, spleen and thymus were isolated from wild‐type, Vav‐BCLX (A), Vav‐BCLX (B) or Vav‐Mcl1 mice and processed for western analysis using anti‐BCLX‐ and anti‐HSP90‐specific antibodies. (C) Peripheral blood was sampled from mice of the indicated genotypes and white blood cell counts were determined by using a ScilVet abc blood counter (left bar graph). WBCs were further characterized as either lymphocytes (middle bar graph) or granulocytes (right bar graph). (D) Cell counts were determined from bone marrow (both femurs, left bar graph), thymus (middle bar graph) and spleen‐derived single‐cell suspensions (right bar graph). Data from Vav‐BFL1 TG line L1 and L3 and from Vav‐BCLX TG line A and line B were comparable and pooled for easier representation. (E) Representative spleen specimens from wild‐type, Vav‐BFL1 line L1, Vav‐BCLX line A, Vav‐Mcl1 and Vav‐BCL2 mice. Statistical analysis was performed using one‐way ANOVA with Dunnett's multiple comparison. *P < 0.05; ****P < 0.0001; n ≥ 4 ± SD.
To assess the impact of transgene expression on the haematopoietic compartment, adult mice were analysed between 8 and 12 weeks of age. First, we monitored the white blood cell (WBC) counts in the peripheral blood (PB). Different to Vav‐BCL2 TG mice neither Vav‐BCLX TG nor Vav‐BFL1 TG mice had significantly increased WBC numbers in the PB (Fig. 1C). Furthermore, neither Vav‐BFL1 nor Vav‐BCLX TG strains showed aberrant cellularity in bone marrow, thymus or spleen (Fig. 1D, TG lines were pooled to simplify data presentation), while Vav‐Mcl1 and Vav‐BCL2 TG mice showed splenomegaly (Fig. 1E), as reported before 31, 37.
Next, we examined the abundance of different lymphocyte subsets in primary and secondary lymphoid organs. Thymocyte development was normal in Vav‐BFL1 and Vav‐BCLX TG mice throughout all developmental stages (Fig. 2A), in contrast to Vav‐BCL2 TG mice which had decreased CD4+CD8+ DP thymocytes and increased CD4−CD8− double negative (DN) and CD4+ and CD8+ single‐positive (SP) cells, as reported previously 37. Furthermore, the composition of mature CD4+ and CD8+ T cells in the periphery was similar between all genotypes analysed (data not shown).
Figure 2.
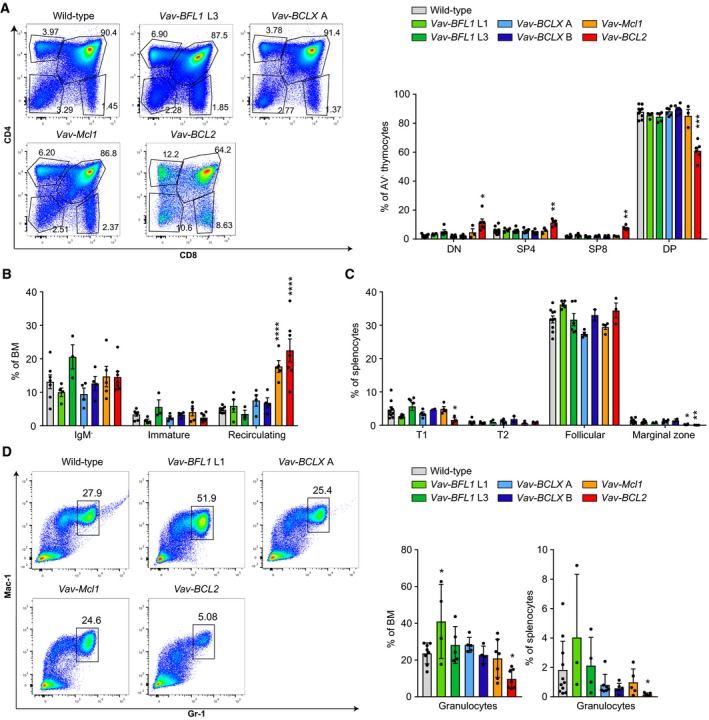
Leukocyte subset composition in Vav‐BFL1 and Vav‐BCLX TG mice. (A) Representative dot‐plots of thymocytes from wild‐type, Vav‐BFL1 TG line L3, Vav‐BCLX TG line A, Vav‐Mcl1 TG and Vav‐BCL2 TG mice stained with antibodies specific for CD8 or CD4; bar graph summarizing the results (n = 3–4/genotype). (B) Flow cytometry was used to assess the distribution of B220loIgM− pro‐/pre‐B cells, B220loIgM+IgD− immature B cells, and B220hiIgM+IgD+ recirculating B cells in the bone marrow (BM). (C) Splenocytes were analysed for the presence of B220+CD23−CD21loIgM+ transitional 1 (T1) B cells, B220+CD23+CD21hiIgMhi transitional 2 (T2) B cells, B220+CD23+CD21+IgM+ follicular B cells, and B220+CD23−CD21hiIgMhi marginal zone B cells. (D) Representative dot‐plots of bone marrow from wild‐type, Vav‐BFL1, Vav‐BCLX, Vav‐Mcl1 and Vav‐BCL2 TG mice showing CD11b/Mac‐1+Gr‐1hi granulocytes. Bar graphs: quantification of the granulocytes in the bone marrow (left graph) and spleen (right graph). Statistical analysis was performed by using one‐way ANOVA with Dunnett's multiple comparison. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; n ≥ 3 ± SD.
Regarding B cell development, Vav‐BFL1 and Vav‐BCLX TG mice did not show any major abnormalities in the bone marrow and spleen (Fig. 2B,C), unlike Vav‐BCL2 TG and Vav‐Mcl1 TG mice, which showed changes expected from previous studies 31, 37. One Vav‐BFL1 TG line (L3) displayed a trend towards an increase in immature B cells and a reduction in recirculating B cells (Fig. 2B), but this did not reach statistical significance nor was it seen in the second transgenic line. In the spleen, Vav‐BFL1 TG mice (L1) showed a tendency towards a loss of Transitional (T) 1 B cells, similar to what could be seen for Vav‐BCL2 mice (Fig. 2C), and a mild increase in follicular B cells (Fig. 2C).
We also examined the abundance of Mac‐1+Gr‐1hi granulocytes in the bone marrow and spleen. While the L1 Vav‐BFL1 TG line tended to have an increased percentage of granulocytes in the bone marrow, this was not observed in the L3 line. Granulocyte numbers were also normal in Vav‐BCLX and Vav‐Mcl1 TG mice (Fig. 2D), whereas Vav‐BCL2 TG mice had a significantly lower percentage of granulocytes in the bone marrow and spleen, as noted before 37.
Exogenous BFL1 is a weak antagonist of apoptosis
Next, we analysed the ability of the overexpressed proteins to protect cells from spontaneous and drug‐induced apoptosis. Equal numbers of mice from both lines of Vav‐BFL1 TG and Vav‐BCLX TG strains were analysed. No significant differences were detected and the data have therefore been pooled in Fig. 3. First, thymocytes were cultured for 4, 8, 12, 20, 30, 48 and 72 h, respectively, and analysed for Annexin V and 7AAD staining by flow cytometry. As shown previously 31, 37, Vav‐BCL2 and Vav‐Mcl1 TG thymocytes were strongly protected from spontaneous apoptosis (Fig. 3A). Thymocytes from both Vav‐BCLX TG lines also showed significantly delayed cell death, although not to the same extent, but cells from Vav‐BFL1 TG mice remained as sensitive as wild‐type controls (Fig. 3A). A similar picture was observed upon treatment with various apoptosis‐inducing stimuli, including irradiation, staurosporine (STS) or the glucocorticoid (Glc) corticosterone.
Figure 3.
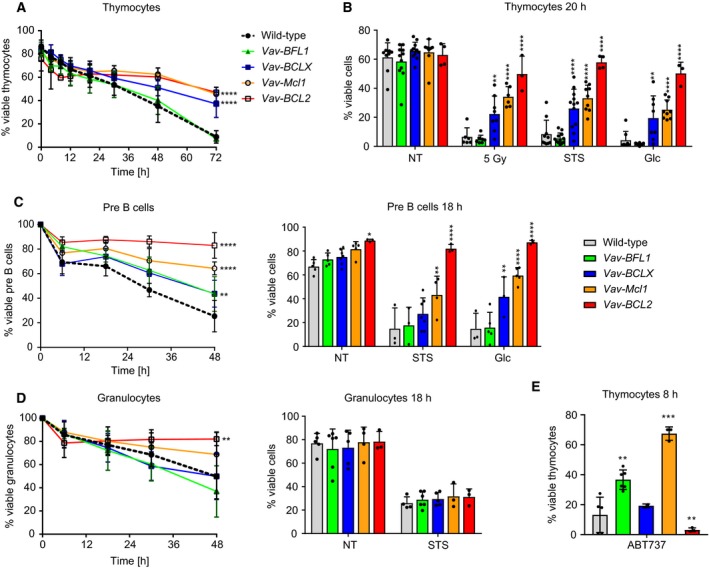
Transgenic BCLX protects lymphocytes more potently from apoptosis than BFL1. (A) Thymocytes from wild‐type, Vav‐BFL1 (both TG lines pooled), Vav‐BCLX (both TG lines pooled), Vav‐Mcl1 and Vav‐BCL2 TG mice were cultured in vitro for 72 h and apoptosis was assessed over time by flow cytometry. Cells negative for Annexin V and 7AAD were considered viable. (B) Thymocytes from wild‐type, Vav‐BFL1 (both lines pooled), Vav‐BCLX (both lines pooled), Vav‐Mcl1 and Vav‐BCL2 TG mice were either left untreated or exposed to 5 Gy γ‐irradiation, 100 nm staurosporine (STS) or 625 nm of the glucocorticoid (Glc), corticosterone respectively. Apoptosis was assessed after 20 h by flow cytometric analysis. (C) CD19+B220loIgM−CD25+ pre‐B cells from the same mice were sorted by FACS from bone marrow and cultured for 48 h. Spontaneous apoptosis was assessed over time (left graph). In addition, sorted pre‐B cells were either left untreated or treated with 100 nm staurosporine (STS) or 625 nm corticosterone (Glc), respectively and apoptosis was assessed after 18 h by flow cytometry (right graph). (D) Gr‐1+ Granulocytes from mice of the indicated genotypes were sorted from the bone marrow and cultured for 48 h. Spontaneous apoptosis was assessed over time (left graph). Additionally, sorted granulocytes were cultured for 18 h either in the absence or presence of STS and apoptosis was assessed by flow cytometry (right graph). (E) Thymocytes were cultured in vitro for 8 h in the presence of 5 μm ABT‐737 and thereafter analysed for Annexin V and 7AAD positivity by flow cytometry. Statistical analysis was performed by using a two‐way ANOVA with Dunnett's multiple comparison. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; n ≥ 3 ± SD.
Next we analysed pre‐B cells isolated by flow cytometry from bone marrow. Interestingly, Vav‐BFL1 and Vav‐BCLX TG pre‐B cells both showed significantly delayed spontaneous apoptosis when cultured in vitro (Fig. 3C), albeit not to the same degree as either Vav‐Mcl1 or Vav‐BCL2 TG pre‐B cells (Fig. 3C). However, although the Vav‐BCLX TG also protected pre‐B cells from STS and Glc‐induced apoptosis, Vav‐BFL1 expression did not (Fig. 3C, bar graph).
Neither the Vav‐BFL1 nor the Vav‐BCLX TG significantly enhanced the overall viability of bone marrow granulocytes in vitro, while Vav‐Mcl1 and especially Vav‐BCL2 TG expression did so effectively (Fig. 3D). Strikingly, none of the prosurvival proteins was able to protect granulocytes from STS‐induced cell death at the time‐point chosen for analysis (Fig. 3D bar graph).
Lastly, we treated thymocytes with ABT‐737, a BH3 mimetic that induces apoptosis by inhibiting BCL2, BCLX and BCLW. Confirming the functionality of the transgene, Vav‐BFL1 TG thymocytes were significantly protected from ABT‐737‐induced apoptosis, although the protection was not as pronounced as that seen in Vav‐Mcl1 TG thymocytes (Fig. 3E). As expected, thymocytes from Vav‐BCLX and Vav‐BCL2 mice were highly sensitive to ABT‐737.
Quantitative rather than qualitative differences in between different BCL2‐prosurvival proteins define the degree of protection from thymocyte apoptosis
We wished to clarify whether the degree of protection correlated with the level of transgene expression rather than with qualitative functional differences between the prosurvival proteins. To assist in the quantitation, we prepared transiently transfected 293T cells expressing cDNAs encoding for an HA and streptavidin (HS)‐tagged version of BCL2, BCLX, Mcl1 and BFL1 and loaded different amounts of these proteins next to 20 μg total protein lysates from the TG thymocytes (Fig. 4). Since the quantity of the HS‐tagged proteins were within a comparable range within the lysates from transfected 293T cells, as shown by the HA western blot, we were able to better judge the relative TG expression found in thymocytes using target‐specific antibodies. This comparison made it evident that the Vav‐BCL2 TG was expressed at much higher levels in the thymus when compared to the highest concentration of 293T lysate loaded. In contrast, the Vav‐BCLX and Vav‐Mcl1 TG thymocyte extracts showed comparable signals to those seen in the highest concentration of 293T cell lysate loaded. This suggests that the relative expression levels achieved in Vav‐BCLX and Vav‐Mcl1 TG thymocytes were comparable to each other but much lower than those of BCL2 in Vav‐BCL2 TG thymocytes. Strikingly, the BFL1 signal from the thymocyte lysates of Vav‐BFL1 TG mice was lower than the lowest signal generated from the HS‐BFL1 dilution series in 293T cell extracts. We conclude that thymocytes from Vav‐BCL2 TG mice express the highest amounts of transgenic protein, those from Vav‐BCLX and Vav‐Mcl1 TG express lower levels, albeit comparable to each other, and those from Vav‐BFL1 TG mice have the lowest expression. These results partly explain the differences observed in the relative resistance conveyed by the different transgenes to apoptosis‐inducing stimuli in culture.
Figure 4.
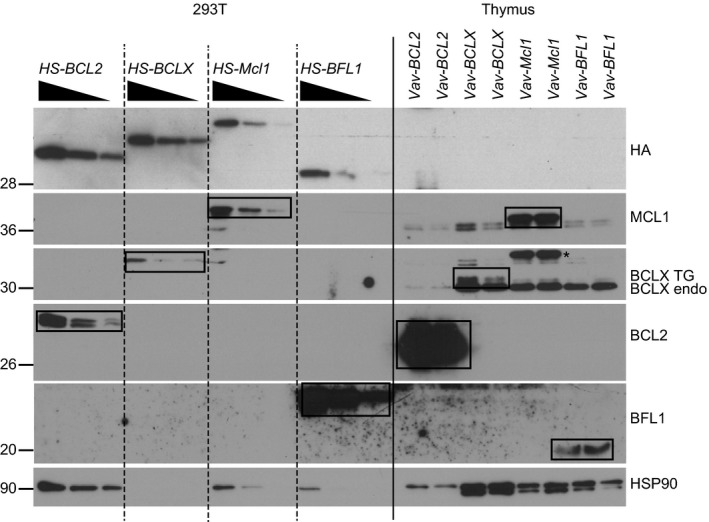
Comparative analysis reveals substantial differences in transgene expression levels across different Vav‐TG mice. 293T cells were transiently transfected with plasmids encoding HA/Streptavidin‐tagged BCL2, BCLX, MCL1 and BFL1 respectively. Comparable amounts of HA‐tagged proteins were loaded in different dilution next to 20 μg total thymocyte extract from Vav‐BCL2, Vav‐BCLX, Vav‐Mcl1 and Vav‐BFL1 TG mice respectively. Protein levels of HA‐tagged and transgenic BFL1, MCL1, BCLX, BCL2 and, for reference, HSP90 were assessed by western blotting.
BFL1‐MYC crosstalk accelerates B‐cell lymphomagenesis in mice
Since the Vav‐BCL2 and Vav‐Mcl1 transgenes accelerated Eμ‐MYC‐driven lymphomagenesis 30, 31, we aimed to test if this was also the case for Vav‐BCLX TG and Vav‐BFL1 TG mice. Therefore, Vav‐BCLX TG mice (line A) and Vav‐BFL1 TG lines 1 and 3 mice (data pooled) were intercrossed with Eμ‐MYC TG mice and monitored for acute signs of lymphomagenesis such as enlarged lymph nodes and shortness of breath. Of note, the Eμ‐MYC/Vav‐BFL1 double‐transgenic (DT) mice succumbed significantly faster to malignancy (median survival 53 days) than Eμ‐MYC/Vav‐BCLX DT mice (median survival 67 days) and Eμ‐MYC TG mice (median survival 139 days) (Fig. 5A). This result was unexpected since we had observed comparable protective capacity of the Vav‐BFL1 and Vav‐BCLX transgenes in cultured pre‐B cells (Fig. 3C) and significantly better protection by the Vav‐BCLX transgene against glucocorticoid treatment. Sick Eμ‐MYC/Vav‐BFL1 and Eμ‐MYC/Vav‐BCLX DT mice had similar WBC counts and these were in both cases significantly higher than that for sick Eμ‐MYC TG mice (Fig. 5B). Spleen weights were comparable between all three genotypes (Fig. 5C) while thymus weights were significantly higher in both Eμ‐MYC/Vav‐BFL1 and Eμ‐MYC/Vav‐BCLX DT mice than in Eμ‐MYC TG mice (Fig. 5D). Next, we analysed the phenotype of the tumours by flow cytometric analysis (Fig 5E, Tables S1–S3). The Eμ‐MYC mice developed mainly immature B220+CD19+IgM− tumours (Table S1). Most Eμ‐MYC/Vav‐BFL1 mice also developed pre‐B lymphomas (Fig. 5E, Table S2) but a significant number (7 of 11 mice analysed) additionally developed CD19−B220+CD4+ progenitor cell lymphomas that have been described before for Eμ‐MYC/Eμ‐BCL2 mice 29. In the Eμ‐MYC/Vav‐BFL1 mice, these progenitor tumours were largely restricted to the thymus (Fig. 5F, Table S2). However, the onset of disease was not significantly different between Eμ‐MYC/Vav‐BFL1 mice that harboured CD19−B220+CD4+ progenitor cell lymphomas in their thymus and those that did not (Fig. 5F, Kaplan–Meier plot). Interestingly, Eμ‐MYC/Vav‐BCLX DT tumours appeared more variable by showing IgM−, IgM+ and mixed phenotypes consisting of IgM+ and IgM− lymphoma cells respectively (Fig. 5E and Table S3). However, the different tumour types did not differ in the onset of disease (not shown).
Figure 5.
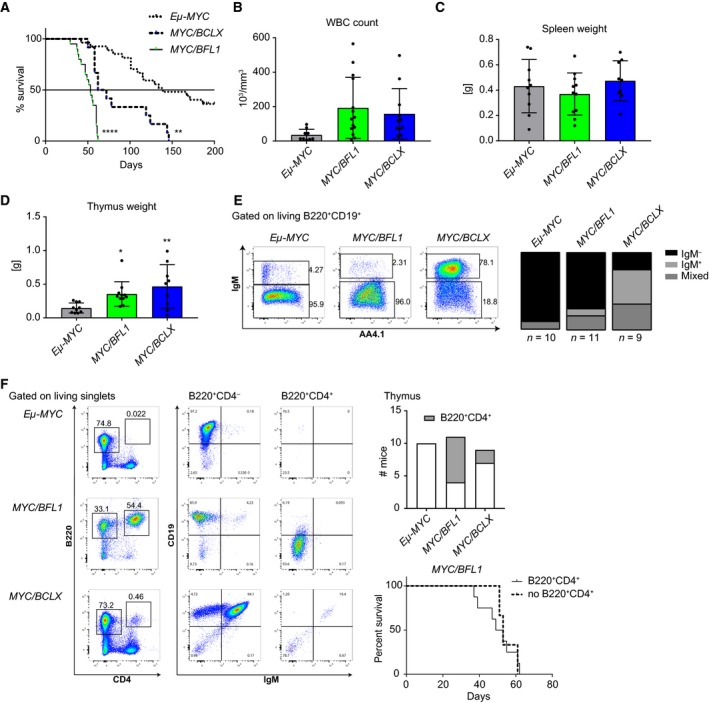
Vav‐BFL1 and Vav‐BCLX transgenes accelerate lymphomagenesis in Eμ‐Myc mice. (A) Kaplan–Meier plot of MYC‐induced lymphomagenesis in Eμ‐MYC TG (n = 27), Eμ‐MYC/Vav‐BFL1 (L3 and L1 pooled, n = 17) and Eμ‐MYC/Vav‐BCLX (A, n = 11) DT mice. (B) White blood cell counts (WBC) were analysed in tumour‐bearing mice. Thereafter, mice were sacrificed and (C) spleen and (D) thymus weights were assessed. (E) Representative dot‐plots of the tumour phenotype found in the spleen. Overall tumour phenotype quantification (see Tables S1–S3) is shown on the right. IgM− refers to CD19+B220+IgM− lymphoma cells, IgM+ refers to CD19+B220+IgM+ lymphoma cells, mixed refers to lymphomas containing both, CD19+B220+IgM− and CD19+B220+IgM+ tumour cell types (F) Representative dot‐plots of stem/progenitor cell lymphomas in the thymus. Other than B220+CD4− lymphoma cells B220+CD4+ lymphoma cells do not express CD19. Bar graph: Quantification of CD19−B220+CD4+ stem/progenitor cell lymphomas in the thymus. Kaplan–Meier plot: Survival curve of Eμ‐MYC/Vav‐BFL1 DT mice with or without B220+CD4+ stem/progenitor cell lymphomas in their thymus. Statistical analysis for tumour‐free survival was performed by using a log‐rank (Mantel–Cox) test. All other statistical analyses were performed by using a one‐way ANOVA with Holm‐Sidak's multiple comparison test compared to Eμ‐MYC TG. *P < 0.05; **P < 0.01; ****P < 0.0001; n ≥ 9 ± SD.
Next, we compared the premalignant phenotypes of Eμ‐MYC/Vav‐BFL1 and Eμ‐MYC/Vav‐BCLX DT mice. As Eμ‐MYC/Vav‐BFL1 DT mice can succumb to tumours as early as 29 days, we analysed the mice at 2 weeks of age in order to avoid transformed cells. Importantly, we analysed both Eμ‐MYC/Vav‐BFL1 lines to exclude potential side‐effects caused by random transgene insertion. First, we monitored white blood cell counts. Pups from both Eμ‐MYC/Vav‐BFL1 DT lines had five times higher WBC counts than Eμ‐MYC TG pups at that age (Fig. 6A). Interestingly, WBC counts from Eμ‐MYC/Vav‐BCLX DT pups although significantly higher than those of Eμ‐MYC TG mice were three times lower than in Eμ‐MYC/Vav‐BFL1 DT pups (Fig. 6A). Of note, the elevated WBC counts represent an early burst of pre‐B cells, caused by MYC overexpression 39, and the WBC counts subsided by 4 weeks of age (data not shown). Cell counts from the bone marrow and spleen of 2‐week‐old mice were comparable between all the genotypes (data not shown). However, the percentage of B220+ B lymphoid cells in the bone marrow was significantly higher in Eμ‐MYC/Vav‐BFL1 DT mice than in Eμ‐MYC TG and Eμ‐MYC/Vav‐BCLX DT mice (Fig. 6B left graph). Most B220+ cells were immature CD19+IgM− cells (data not shown), but a small proportion also represented CD19−CD4+ cells and was significantly higher in Eμ‐MYC/Vav‐BFL1 DT mice compared to the other genotypes (Fig. 6B right graph). However, this population was present in comparably low numbers (0.1% of all B220+ cells) in the blood (data not shown) and the spleen (Fig. 6D right graph) of all tested genotypes. Importantly, B220+ B lymphoid cells from Eμ‐MYC/Vav‐BFL1 and Eμ‐MYC/Vav‐BCLX DT bone marrows survived better in culture than those from Eμ‐MYC mice (Fig. 6C). B220+ B lymphoid cells were also increased proportionally in the spleen of both DT genotypes compared to Eμ‐MYC TG mice, although this reached significance only for Eμ‐MYC/Vav‐BFL1 DT mice (Fig. 6D left graph). Interestingly, Eμ‐MYC/Vav‐BFL1 DT mice had significantly more B220+CD19+IgM− B lymphoid cells in the spleen than Eμ‐MYC TG mice, and Eμ‐MYC/Vav‐BCLX DT mice showed significantly less (Fig. 6D middle graph) but no increase in B220+CD19−CD4+ cells (Fig. 6D right graph). Furthermore, only the Eμ‐MYC/Vav‐BFL1 splenic B lymphoid cells showed increased survival in culture (Fig. 6E).
Figure 6.
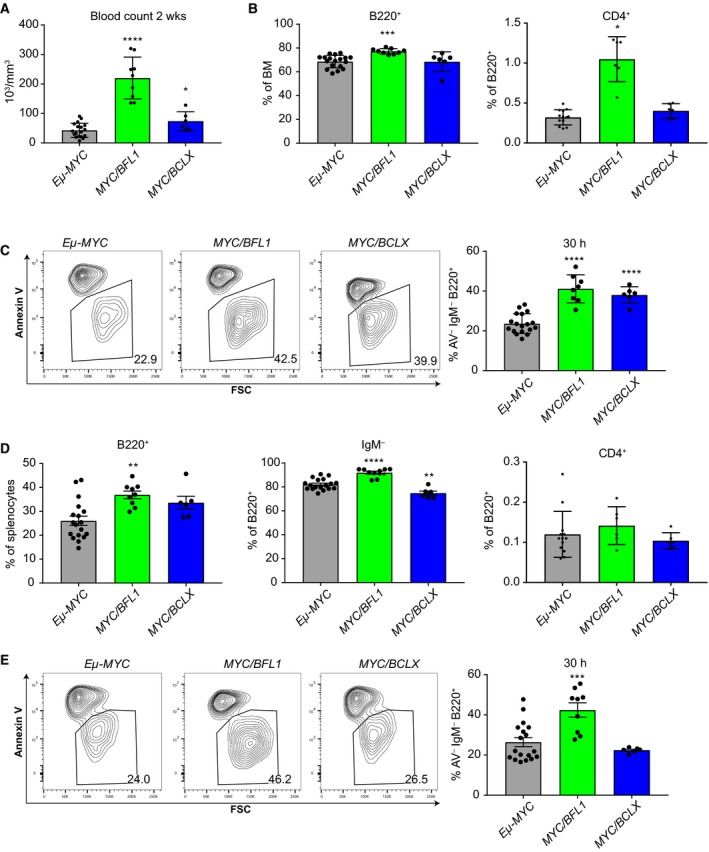
BFL1 overexpression protects premalignant immature B cells from MYC‐induced apoptosis. (A) White blood cell counts from 2–week‐old premalignant Eμ‐MYC, Eμ‐MYC/Vav‐BFL1 (L1 and L3 pooled), and Eμ‐MYC/Vav‐BCLX (line A) mice were assessed. (B) Percentage of B220+ B lymphoid cells in the bone marrow was analysed by flow cytometry (left bar). B220+ cells were further discriminated into CD19−CD4+ cells (right bar). (C) Total bone marrow was cultured for 30 h and the abundance of living (Annexin V−) B220+IgM− immature B lymphoid cells was assessed by flow cytometry. (D) Abundance of total B220+ B lymphoid cells in the spleen was analysed by flow cytometry (left graph). B220+ cells were further discriminated into CD19+IgM− (middle graph) and CD19−CD4+ cells (right graph). (E) Total splenocytes were cultured for 30 h and abundance of living (Annexin V−) B220+IgM− immature B lymphoid cells was assessed by flow cytometry. Statistical analysis was performed by using a one‐way ANOVA with Dunnett's multiple comparison test compared to Eμ‐MYC control. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; n ≥ 3 ± SD.
MYC overexpression increases transgenic BFL1 RNA and protein levels
We were surprised by the strong acceleration of disease in Eμ‐MYC/Vav‐BFL1 DT mice and the enhanced survival capacity of premalignant Eμ‐MYC/Vav‐BFL1 immature B cells as we had not observed major impact from expression of Vav‐BFL1 alone. We, therefore, wondered whether Vav‐BFL1 transgene expression had changed in the Eμ‐MYC TG background. Indeed, western blots revealed a striking difference in BFL1 levels in total splenocytes isolated from 2‐week‐old Eμ‐MYC/Vav‐BFL1 DT mice compared to those from age‐matched Vav‐BFL1 TG mice (Fig. 7A). Importantly, both transgenic lines showed highly enhanced BFL1 expression on the Eμ‐MYC TG background. This phenomenon did not appear to be a global event since other BCL2 family members were not affected. Furthermore, we showed that the enhanced transgene expression was not Vav promoter dependent since the endogenous VAV protein was not increased by MYC overexpression, but rather mildly reduced. Importantly, the increase in BFL1 expression by MYC overexpression happened to the same extent in both independently generated Vav‐BFL1 TG lines, minimizing potential transgene insertion effects. Interestingly, BFL1 mRNA was found increased by approximately ninefold in Vav‐BFL1/Eμ‐MYC DT splenocytes when compared to mRNA levels found in Vav‐BFL1 TG mice (Fig. 7A, bar graph). Endogenous A1 expression levels were not found substantially upregulated in total splenocytes by MYC overexpression, neither on mRNA nor on protein levels (Fig. 7B and data not shown). Together these findings argue for enhanced BFL1 mRNA stability or reduced protein turnover. MYC protein expression was also slightly higher in Eμ‐MYC/Vav‐BFL1 DT splenocytes than in Eμ‐MYC TG samples (Fig. 7A) although MYC mRNA levels were not significantly elevated (not shown), indicative of a feed‐forward loop where increased cell death resistance allows cells to tolerate increased MYC protein levels. In tumour samples, we found that BFL1 expression was further elevated when compared to splenocytes from 2‐week‐old premalignant Eμ‐MYC/Vav‐BFL1 DT mice (Fig. 7B). In order to determine BFL1 expression levels in the different tumour cell subsets, we FACS‐sorted B220+CD19+ tumour cells and B220−CD19− nontumour cells from the spleen and B220+CD19+ and B220+CD19−CD4+ tumour cells from the thymus of diseased Eμ‐MYC/Vav‐BFL1 DT mice. BFL1 protein levels were only detectable in B lymphoid tumour cells, while they were absent in non‐B220+ cells (Fig. 7C). Furthermore, we could not detect any quantitative differences in the BFL1 expression between B220+CD19+ and B220+CD19−CD4+ tumour populations.
Figure 7.
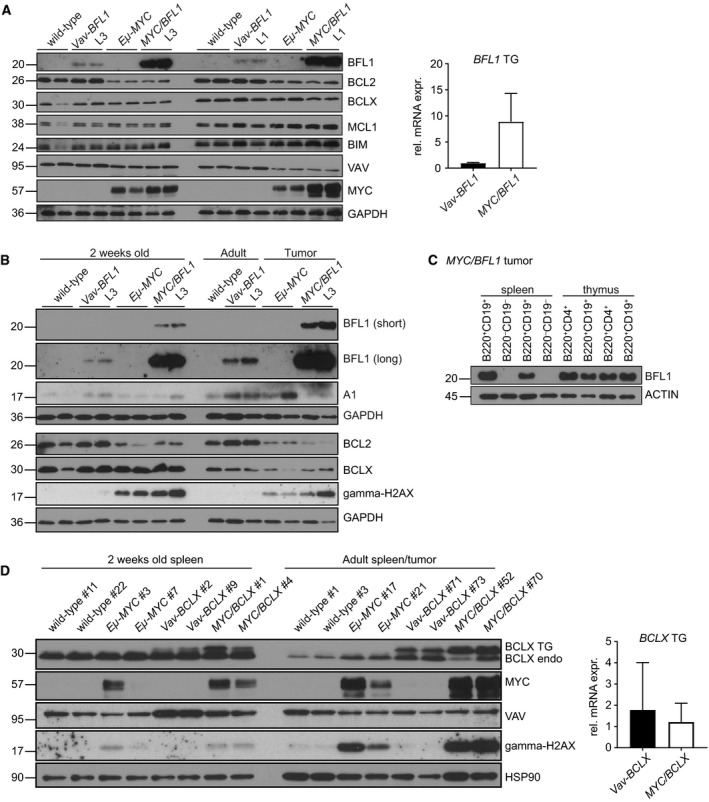
MYC elevates BFL1 mRNA and protein levels (A) Spleens were dissected from 2‐week‐old wild‐type, Vav‐BFL1 (L3) TG, Eμ‐MYC TG, Eμ‐MYC/Vav‐BFL1 (L3) DT, Vav‐BFL1 (L1) TG and Eμ‐MYC/Vav‐BFL1 (L1) DT mice and, following erythrocyte depletion, cells were lysed in NP‐40 containing buffer for western analysis using antibodies specific for the indicated proteins. mRNA was isolated from spleens from 2‐week‐old Vav‐BFL1 (L3) TG and Eμ‐MYC/Vav‐BFL1 (L3) DT mice, transcribed into cDNA and assessed for BFL1 expression levels. (B) Spleens from 2‐week‐old wild‐type, Vav‐BFL1 (L3) TG, Eμ‐MYC TG and Eμ‐MYC/Vav‐BFL1 (L3) DT mice as well as from adult wild‐type and Vav‐BFL1 (L3) TG and tumour‐bearing Eμ‐MYC TG and Eμ‐MYC/Vav‐BFL1 (L3) DT mice were isolated and erythrocyte‐depleted cell preparations were lysed in NP‐40 containing lysis buffer. Western blots were probed for indicated proteins. (C) B220+CD19+ B lymphoid and B220−CD19− non‐B lymphoid cells were isolated by FACS from spleens of tumour‐bearing Eμ‐MYC/Vav‐BFL1 DT mice. Furthermore, B220+CD19+ and B220+CD19−CD4+ cells, respectively, were isolated from thymocytes of tumour‐bearing Eμ‐MYC/Vav‐BFL1 DT mice. Western blot analysis was performed as described in (A). (D) Spleens from 2‐week‐old wild‐type, Vav‐BCLX (line A) TG, Eμ‐MYC TG and Eμ‐MYC/Vav‐BCLX (A) DT mice as well as from adult wild‐type and Vav‐BCLX (A) TG and tumour‐bearing Eμ‐MYC TG and Eμ‐MYC/Vav‐BCLX (line A) DT mice were isolated and erythrocyte‐depleted cells were lysed in NP‐40 containing lysis buffer. Western blots were probed for the indicated proteins using specific antibodies. mRNA was isolated from spleens from 2‐week‐old Vav‐BCLX (line A) TG, and Eμ‐MYC/Vav‐BCLX (line A) DT mice, transcribed into cDNA and assessed for FLAG‐BCLX expression levels.
Comparable increases in transgenic BCLX protein expression were not observed in splenocytes from premalignant Eμ‐MYC/Vav‐BCLX DT vs. premalignant Vav‐BCLX TG mice, although levels increased mildly in Eμ‐MYC/Vav‐BCLX tumours (Fig. 7D). Furthermore, BCLX TG mRNA was not influenced by exogenous MYC expression in 2‐week‐old spleen extracts (Fig. 7D, bar graph). We conclude that Eμ‐MYC overexpression positively influences expression of transgenic Vav‐BFL1, at both the protein and RNA level, but not the Vav‐BCLX transgene.
Discussion
The physiological importance of BFL1/A1 is still poorly understood, despite increased recent efforts. In a broad variety of immune cells, including neutrophils, granulocytes, B and T cells, A1 is rapidly inducible in response to diverse stimuli, including antigen receptor stimulation, GM‐CSF, BAFF receptor or CD40‐ligation 40, 41, predictive of crucial roles in inflammation and immunity. Nevertheless, complete deficiency of A1 does not impair the normal development or function of the immune system nor does it influence the normal behaviour and life span of mice 9, 10.
Here, we describe the generation and characterization of mice that overexpress human BFL1 or human BCLX under the control of a haematopoietic‐specific vector driven by the Vav gene promoter 42. In contrast to Vav‐BCL2 37 and Vav‐Mcl1 31 TG mice, the overall composition and cellularity of all major lymphoid organs was essentially normal in both Vav‐BCLX and Vav‐BFL1 mice (Figs 1 and 2). Interestingly, while the Vav‐BCLX TG partially protected both thymocytes and pre‐B cells from spontaneous and drug‐induced apoptosis, the Vav‐BFL1 TG provided only a minor survival benefit in pre‐B cells or when thymocytes were treated with ABT‐737 (Fig. 3). In general, the graded survival benefits observed in the different mouse models correlated directly with the level of transgenic protein expressed (Figs 3 and 4). It remains unclear why both Vav‐BFL1 TG lines express such low amounts of BFL1, especially when compared to the two Vav‐BCLX TG lines that were generated contemporaneously, but this likely reflects the short half‐life of BFL1 43, 44.
Surprisingly, when we crossed Vav‐BFL1 TG with Eμ‐MYC TG mice, expression of the Vav‐BFL1 TG was boosted (Fig. 7) and lymphomagenesis was dramatically accelerated (Fig. 5). Indeed, the median survival of Eμ‐MYC/Vav‐BFL1 DT lymphomas was only 53 days, comparable to that observed for Eμ‐MYC mice lacking the BH3‐only protein BIM 45, 46. While the Vav‐BCLX transgene also accelerated lymphomagenesis in Eμ‐MYC mice (median survival 67 days), previously described Eμ‐Bclx/Eμ‐MYC DT mice died at an age of only 6 weeks (42 days) 32. The difference is likely to reflect the relative level of BCLX protein achieved.
Intriguing differences were observed in the tumour phenotypes. C57BL/6 Eμ‐MYC TG mice can develop either pro‐/pre‐B or B lymphomas 34, 45, with the former dominating in our colony. Intriguingly, our Eμ‐MYC/Vav‐BCLX DT mice developed mainly IgM+ B cell lymphomas, like Eμ‐MYC mice lacking BIM 45 or BMF 46. It has been reported that Eμ‐MYC‐driven IgM− immature B lymphomas are more aggressive and develop faster than their IgM+ counterparts 30. This observation would be consistent with the prolonged tumour‐latency observed for Eμ‐MYC/Vav‐BCLX DT mice compared to Eμ‐MYC/Vav‐BFL1 DT mice that only develop IgM− tumours. However, within the group of Eμ‐MYC/Vav‐BCLX DT mice, no differences in latency were observed between the different immunophenotypes (not shown).
Interestingly, Eμ‐MYC/Vav‐BFL1 DT mice developed mainly immature (B220+ IgM−) lymphomas, with a significant proportion (64%) that additionally developed CD19−B220+CD4+ stem/progenitor cell lymphomas in the thymus. Stem/progenitor cell lymphomas also dominated in Eμ‐BCL2/Eμ‐MYC DT, Eμ‐Bclx/Eμ‐MYC DT and Vav‐Mcl1/Eμ‐MYC DT mice 29, 31, 32, while Vav‐BCL2/Eμ‐MYC DT mice developed IgM−CD19+CD43+ pro‐B cell tumours 30. Interestingly, WBC counts were massively increased in premalignant Eμ‐MYC/Vav‐BFL1 DT mice compared to Eμ‐MYC TG littermate controls or age‐matched Eμ‐MYC/Vav‐BCLX DT mice. Presumably the larger pool of precursor cells and their reduced susceptibility to apoptosis explains the accelerated lymphomagenesis.
Lastly, we are intrigued by the elevated expression of BFL1 protein and mRNA in premalignant Eμ‐MYC/Vav‐BFL1 TG splenocytes compared to Vav‐BFL1 TG samples (Fig. 7A,B). Intriguingly, BFL1 was upregulated to the same extent in both Eμ‐MYC/Vav‐BFL1 DT lines (Fig. 7A) and was already apparent at 2 weeks of age, arguing against clonal expansion of cells with stronger Vav‐BFL1 expression during transformation. Eμ‐MYC/Vav‐BCLX DT cells did not show elevated transgene expression compared to Vav‐BCLX TG littermates (Fig. 7D). Since MYC influences global gene expression 47, the elevation of transgenic BFL1 expression might reflect integration of the transgene within MYC accessible sites, in both independently generated Vav‐BFL1 TG lines. It might also be possible that c‐MYC stabilizes the transgenic BFL1 mRNA in the context of an artificial 3′ UTR as endogenous A1 was not found elevated (Fig. 7B). The molecular mechanism responsible for this intriguing phenomenon remains to be investigated further.
Together, our findings underline the major impact of elevated BFL1 on tumour development, an effect that might not be confined to MYC‐induced lymphomas. They also emphasize the potential usefulness of the development of BFL1‐specific inhibitors for cancer treatment and the mouse model described here might be perfectly suited for their preclinical testing.
Materials and methods
Mice
Vav‐BCL2 37, Vav‐Mcl1 31, and Eμ‐MYC 48 mice were described previously. Vav‐BFL1 and Vav‐BCLX TG mice were generated by pronuclear injection of the VavP vectors 36 encoding human FLAG‐BFL1 or human FLAG‐BCLX cDNA into C57BL/6N oocytes. The founder lines were designated as C57BL/6N‐Tg(Vav‐BFL1)676, 677, 686Biat and C57BL/6N‐Tg(Vav‐BCLX)670, 671, 672Biat, herein referred as Vav‐BFL1 TG line L1, L2, L3 and Vav‐BCLX TG line A, B, C. To generate Eμ‐MYC/Vav‐BFL1 or Eμ‐MYC/Vav‐BCLX DT offspring, Vav‐BFL1 or Vav‐BCLX females were mated with Eμ‐MYC TG males. Tumour onset was determined by regular palpation (three times a week) and by monitoring of short breath, activity and/or scruffy fur. All mice were maintained on a C57BL/6N genetic background. All experiments were performed in accordance with Austrian legislation (BMWF‐66.011/0008‐11/3b/2014). For the production of transgenic mice an experiment license was granted under BMWF‐68.205/0258‐II/3b/2011.
Haematopoietic cell analysis and flow cytometry
Peripheral blood was analysed with a scilVet abc blood counter (Viernheim, Germany) or by flow cytometric analysis after red blood cell lysis using 0.168 m ammonium chloride in PBS. Single‐cell suspensions were prepared from thymus, lymph nodes (axillary, brachial, inguinal and mesenteric), bone marrow (from femurs) and spleen, and viable cells were counted using a Neubauer counting chamber by trypan blue exclusion. Cell composition was determined by staining with cell surface marker‐specific antibodies and flow cytometric analysis using a LSR Fortessa (BD Biosciences, Franklin Lakes, NJ, USA). Stained single‐cell suspensions were sorted using a FACS ARIA III (BD Biosciences). The following labelled monoclonal antibodies were used: anti‐Gr‐1 (RB6‐8C5), anti‐CD11b (M1/70), anti‐B220 (RA3‐6B2), anti‐TCRβ (HS7‐597), anti‐CD4 (GK1.5), anti‐CD8 (53‐6.7), anti‐CD25 (PC61), anti‐CD44 (IM7), anti‐CD62L (MEL‐14), anti‐CD19 (6D5), anti‐CD93 (AA4.1), anti‐c‐kit (ACK2), anti‐IgM (eb121‐15F9), anti‐IgD (11‐26c.2a), anti‐CD21 (7G6), anti‐CD23 (B3B4). Cell viability was determined by using Annexin V‐FITC and 7AAD (both eBioscience, ThermoFisher Scientific, Inc., Grand Island, NY, USA). FCS files were analysed by using flowjo Version 10.4 for Windows.
Cell culture, apoptosis assays and chemical compounds
Total thymocytes, sorted pre‐B cells or sorted granulocytes were cultured in RPMI‐1640 medium (Sigma‐Aldrich, St. Louis, MO, USA) supplemented with 10% FCS (Sigma‐Aldrich, F7524), 250 μm l‐glutamine (PAA Laboratories, Vienna, Austria, M11‐004), 100 U·mL−1 penicillin, 100 μg·mL−1 streptomycin (PAA Laboratories, P11‐010), 100 μm nonessential amino acids (Gibco, ThermoFisher Scientific, Inc., 1091607), 1 mm sodium pyruvate (Gibco, 1046485) and 50 μm β‐mercapthoethanol (AppliChem, Darmstadt, Germany) at 37 °C in a humidified atmosphere containing 5% CO2. Apoptosis was induced either by γ‐irradiation with 5 Gy, 100 nm staurosporine (Sigma, S6942), 625 nm corticosterone (Sigma, C‐2503) or 5 μm ABT‐737 (ApexBio, Houston, TX, USA, A8194 Batch No.1).
Immunoblotting
For the comparative western blot analysis, 293T cells were transiently transfected with 1 μg/6‐well pTO‐HS‐BCL2, pTO‐HS‐BCLX, pTO‐HS‐Mcl1, and pTO‐HS‐BFL1 constructs by using polyethylenimine 49 and harvested after 24 h. Cell pellets were lysed using NP‐40 containing lysis buffer [50 mm Tris pH 8.0, 150 mm NaCl, 0.5% NP‐40, 50 mm NaF, 1 mm Na3VO4, 1 mm PMSF, one tablet protease inhibitors (EDTA free, Roche, Basel, Switzerland) per 10 mL and 30 μg·mL−1 DNaseI (Sigma‐Aldrich)] and protein concentration was quantified with Bradford reagent (500‐0006, Bio‐Rad, Munich, Germany). Twenty to thirty micrograms of total protein was loaded on 12% Bis‐Tris acrylamide gels and blotted on Amersham™ Hybond™—ECL nitrocellulose membranes (GE Healthcare, Little Chalfont, UK). The following antibodies were used for protein detection: rabbit anti‐BFL1 (kindly provided by Jannie Borst 50), rat anti‐mouse A1 (WEHI, 6D6, 2 μg·mL−1) 51, rabbit anti‐BIM/BOD (Enzo, Farmingdale, NY, USA, polyclonal, ADI‐AAP‐330‐E, 0.2 μg·mL−1), rabbit anti‐MCL1 (polyclonal, Rockland, Pottstown, PA, USA, Cat# 600‐401‐394, 2.2 μg·mL−1), rabbit anti‐BCLX (54H6, Cell Signaling, Danvers, MA, USA, Cat# CS2764, 1 : 1000), mouse anti‐BCL2 (7/Bcl‐2, BD Biosciences, 0.5 μg·mL−1), mouse anti‐HA (HA.11, Covance, Princeton, NJ, USA, 1 : 1000), rabbit anti‐MYC (Y69, Abcam, Cambridge, UK, ab32072), rabbit anti‐VAV1 (Cell Signaling #2502, 1 : 1000), rabbit anti‐gamma‐H2A.X (Ser139, Cell Signaling #2577, 1 : 1000), rabbit anti‐GAPDH (14C10, Cell Signaling #2118, 1 : 5000), rabbit anti‐beta‐Actin (polyclonal, Cell Signaling #4967, 1:1000) and mouse anti‐HSP90 (F‐8, Santa Cruz, Dallas, TX, USA, Cat# sc‐13119, 0.2 μg·mL−1). All primary antibodies were diluted in 5% BSA in PBST and blots were incubated overnight at 4 °C.
mRNA isolation and quantitative real‐time PCR analysis
RNA was isolated using TRIzol (ThermoFisher Scientific, Inc.) and quantified with a NanoDrop 1000 Spectrophotometer (ThermoFisher Scientific, Inc.). Two hundred nanograms of RNA was reverse transcribed into cDNA using iScript cDNA Synthesis Kit (Bio‐Rad) according to the manufacturer's instruction. Quantitative real‐time PCR was performed with a 100th part of the cDNA in a StepOnePlus System (ThermoFisher Scientific, Inc.) using Bimake SYBR Green (Bimake, Houston, TX, USA) according to the manufacturer's instructions and 100 nm of the following primers: Flag‐BFL1 forward primer ACA AAG ACG ATG ACG ATA AAA CAG A and reverse primer AGC ACT CTG GAC GTT TTG CT; Flag‐BCLX forward primer: CAA AGA CGA TGA CGA TAA AGG ATC T and reverse primer TCC AGC TGT ATC CTT TCT GGG A; Actin‐beta forward primer: ACT GGG ACG ACA TGG AGA AG and reverse primer GGG GTG TTG AAG GTC TCA AA. PCR conditions were 95 °C for 10 min, 40 cycles of (95 °C for 15 s and 60 °C for 60 s), 95 °C for 15 s, 60 °C for 60 s followed by a melting curve with 0.3 °C increment steps up to 95 °C for 15 s. Results were normalized to Actin‐beta expression to be compared with the ΔΔC t relative quantification method.
Statistical analysis
Statistical analysis was performed using graphpad prism Version 7.03. for Windows, GraphPad Software, La Jolla, CA, USA, http://www.graphpad.com. Used tests are indicated in the figure legends.
Conflict of interest
The authors declare no financial conflict of interest.
Author contributions
ST performed experiments, analysed data, wrote paper and prepared figures; MDH and AMM performed experiments and analysed data; VL provided reagents, performed experiments and discussed data; TR generated transgenic mice, SC provided transgenic mice, discussed data, revised paper; AV conceived and planned study, and wrote paper.
Supporting information
Table S1. Phenotypic characterization of lymphomas arising in Eμ‐MYC TG mice.
Table S2. Phenotypic characterization of lymphomas arising in Eμ‐MYC/Vav‐BFL1 DT mice.
Table S3. Phenotypic characterization of lymphomas arising in Eμ‐MYC/Vav‐BCLX DT mice.
Acknowledgements
We thank all members of the Villunger laboratory for their support and advice. Special thanks to K. Rossi, C. Soratroi and I. Gaggl for animal care and excellent technical assistance, S. Herzog for reagents and L. Fava for helpful discussion. We also thank M. Herold (WEHI) for the anti‐A1 antibody and J. Borst (NKI) for the anti‐BFL1 antiserum. This work was supported by grants from the Austrian Science Fund (FWF), Grant I 3271 (FOR‐2036), the MCBO Doctoral College ‘Molecular Cell Biology and Oncology’ (W1101) and the ‘Österreichische Krebshilfe Tirol’. MDH and ST are supported by a DOC‐fellowship from the Austrian Academy of Science (ÖAW).
Contributor Information
Selma Tuzlak, Email: selma.tuzlak@i-med.ac.at.
Andreas Villunger, Email: andreas.villunger@i-med.ac.at.
References
- 1. Lin EY, Orlofsky A, Berger MS & Prystowsky MB (1993) Characterization of A1, a novel hemopoietic‐specific early‐response gene with sequence similarity to bcl‐2. J Immunol 151, 1979–1988. [PubMed] [Google Scholar]
- 2. Orlofsky A, Somogyi RD, Weiss LM & Prystowsky MB (1999) The murine antiapoptotic protein A1 is induced in inflammatory macrophages and constitutively expressed in neutrophils. J Immunol 163, 412–419. [PubMed] [Google Scholar]
- 3. Verschelde C, Walzer T, Galia P, Biemont MC, Quemeneur L, Revillard JP, Marvel J & Bonnefoy‐Berard N (2003) A1/Bfl‐1 expression is restricted to TCR engagement in T lymphocytes. Cell Death Differ 10, 1059–1067. [DOI] [PubMed] [Google Scholar]
- 4. Grumont RJ, Rourke IJ & Gerondakis S (1999) Rel‐dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation‐induced apoptosis. Genes Dev 13, 400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hatakeyama S, Hamasaki A, Negishi I, Loh DY, Sendo F & Nakayama K (1998) Multiple gene duplication and expression of mouse bcl‐2‐related genes, A1. Int Immunol 10, 631–637. [DOI] [PubMed] [Google Scholar]
- 6. Zong W‐X, Edelstein LC, Chen C, Bash J & Gélinas C (1999) The prosurvival Bcl‐2 homolog Bfl‐1/A1 is a direct transcriptional target of NF‐κB that blocks TNFα‐induced apoptosis. Genes Dev 13, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ulleras E, Karlberg M, Moller Westerberg C, Alfredsson J, Gerondakis S, Strasser A & Nilsson G (2008) NFAT but not NF‐kappaB is critical for transcriptional induction of the prosurvival gene A1 after IgE receptor activation in mast cells. Blood 111, 3081–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jenal M, Batliner J, Reddy VA, Haferlach T, Tobler A, Fey MF, Torbett BE & Tschan MP (2010) The anti‐apoptotic gene BCL2A1 is a novel transcriptional target of PU.1. Leukemia 24, 1073–1076. [DOI] [PubMed] [Google Scholar]
- 9. Schenk RL, Tuzlak S, Carrington EM, Zhan Y, Heinzel S, Teh CE, Gray DH, Tai L, Lew AM, Villunger A et al (2017) Characterisation of mice lacking all functional isoforms of the pro‐survival BCL‐2 family member A1 reveals minor defects in the haematopoietic compartment. Cell Death Differ 24, 534–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tuzlak S, Schenk RL, Vasanthakumar A, Preston SP, Haschka MD, Zotos D, Kallies A, Strasser A, Villunger A & Herold MJ (2017) The BCL‐2 pro‐survival protein A1 is dispensable for T cell homeostasis on viral infection. Cell Death Differ 24, 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Herman MD, Nyman T, Welin M, Lehtio L, Flodin S, Tresaugues L, Kotenyova T, Flores A & Nordlund P (2008) Completing the family portrait of the anti‐apoptotic Bcl‐2 proteins: crystal structure of human Bfl‐1 in complex with Bim. FEBS Lett 582, 3590–3594. [DOI] [PubMed] [Google Scholar]
- 12. Vogler M, Butterworth M, Majid A, Walewska RJ, Sun XM, Dyer MJ & Cohen GM (2009) Concurrent up‐regulation of BCL‐XL and BCL2A1 induces approximately 1000‐fold resistance to ABT‐737 in chronic lymphocytic leukemia. Blood 113, 4403–4413. [DOI] [PubMed] [Google Scholar]
- 13. Hind CK, Carter MJ, Harris CL, Chan HT, James S & Cragg MS (2015) Role of the pro‐survival molecule Bfl‐1 in melanoma. Int J Biochem Cell Biol 59, 94–102. [DOI] [PubMed] [Google Scholar]
- 14. Metais JY, Winkler T, Geyer JT, Calado RT, Aplan PD, Eckhaus MA & Dunbar CE (2012) BCL2A1a over‐expression in murine hematopoietic stem and progenitor cells decreases apoptosis and results in hematopoietic transformation. PLoS One 7, e48267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beverly LJ & Varmus HE (2009) MYC‐induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene 28, 1274–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fan G, Simmons MJ, Ge S, Dutta‐Simmons J, Kucharczak J, Ron Y, Weissmann D, Chen CC, Mukherjee C, White E et al (2011) Defective ubiquitin‐mediated degradation of antiapoptotic Bfl‐1 predisposes to lymphoma. Blood 115, 3559–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Motoyama N, Wang FP, Roth KA, Sawa H, Nakayama K, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S et al (1995) Massive cell death of immature hematopoietic cells and neurons in Bcl‐x deficient mice. Science 267, 1506–1510. [DOI] [PubMed] [Google Scholar]
- 18. Sochalska M, Tuzlak S, Egle A & Villunger A (2015) Lessons from gain‐ and loss‐of‐function models of pro‐survival Bcl2 family proteins: implications for targeted therapy. FEBS J 282, 834–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Malin S, McManus S, Cobaleda C, Novatchkova M, Delogu A, Bouillet P, Strasser A & Busslinger M (2010) Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro‐B cell development. Nat Immunol 11, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wagner KU, Claudio E, Rucker EB 3rd, Riedlinger G, Broussard C, Schwartzberg PL, Siebenlist U & Hennighausen L (2000) Conditional deletion of the Bcl‐x gene from erythroid cells results in hemolytic anemia and profound splenomegaly. Development 127, 4949–4958. [DOI] [PubMed] [Google Scholar]
- 21. Debrincat MA, Josefsson EC, James C, Henley KJ, Ellis S, Lebois M, Betterman KL, Lane RM, Rogers KL, White MJ et al (2012) Mcl‐1 and Bcl‐x(L) coordinately regulate megakaryocyte survival. Blood 119, 5850–5858. [DOI] [PubMed] [Google Scholar]
- 22. Ma A, Pena JC, Chang B, Margosian E, Davidson L, Alt FW & Thompson CB (1995) Bclx regulates the survival of double‐positive thymocytes. Proc Natl Acad Sci USA 92, 4763–4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dzhagalov I, Dunkle A & He YW (2008) The anti‐apoptotic Bcl‐2 family member Mcl‐1 promotes T lymphocyte survival at multiple stages. J Immunol 181, 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eilers M & Eisenman RN (2008) Myc's broad reach. Genes Dev 22, 2755–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gabay M, Li Y & Felsher DW (2014) MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med 4, a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perry RP (1983) Consequences of myc invasion of immunoglobulin loci: facts and speculation. Cell 33, 647–649. [DOI] [PubMed] [Google Scholar]
- 27. Adams JM (1985) Oncogene activation by fusion of chromosomes. Nature 315, 242–243. [DOI] [PubMed] [Google Scholar]
- 28. Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL & Adams JM (1988) The Eμ‐myc transgenic mouse: a model for high‐incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med 167, 353–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Strasser A, Harris AW, Bath ML & Cory S (1990) Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl‐2. Nature 348, 331–333. [DOI] [PubMed] [Google Scholar]
- 30. Schuster C, Berger A, Hoelzl MA, Putz EM, Frenzel A, Simma O, Moritz N, Hoelbl A, Kovacic B, Freissmuth M et al (2011) The cooperating mutation or “second hit” determines the immunologic visibility toward MYC‐induced murine lymphomas. Blood 118, 4635–4645. [DOI] [PubMed] [Google Scholar]
- 31. Campbell KJ, Bath ML, Turner ML, Vandenberg CJ, Bouillet P, Metcalf D, Scott CL & Cory S (2010) Elevated Mcl‐1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells and enhances drug‐resistance. Blood 116, 3197–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Swanson PJ, Kuslak SL, Fang W, Tze L, Gaffney P, Selby S, Hippen KL, Nunez G, Sidman CL & Behrens TW (2004) Fatal acute lymphoblastic leukemia in mice transgenic for B cell‐restricted bcl‐xL and c‐myc. J Immunol 172, 6684–6691. [DOI] [PubMed] [Google Scholar]
- 33. Sochalska M, Schuler F, Weiss JG, Prchal‐Murphy M, Sexl V & Villunger A (2017) MYC selects against reduced BCL2A1/A1 protein expression during B cell lymphomagenesis. Oncogene 36, 2066–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mensink M, Anstee NS, Robati M, Schenk RL, Herold MJ, Cory S & Vandenberg CJ (2018) Anti‐apoptotic A1 is not essential for lymphoma development in Eμ‐Myc mice but helps sustain transplanted Eμ‐Myc tumour cells. Cell Death Differ. https://doi.org/10.1038/s41418-017-0045-8 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Akasaka T, Kishimori C, Fukutsuka K, Nakagawa M, Takeoka K, Hayashida M, Honjo G & Ohno H (2017) The novel double‐hit, t(8;22)(q24;q11)/MYC‐IGL and t(14;15)(q32;q24)/IGH‐BCL2A1, in diffuse large B‐cell lymphoma. Cancer Genet 214–215, 26–31. [DOI] [PubMed] [Google Scholar]
- 36. Ogilvy S, Metcalf D, Gibson L, Bath ML, Harris AW & Adams JM (1999) Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood 94, 1855–1863. [PubMed] [Google Scholar]
- 37. Ogilvy S, Metcalf D, Print CG, Bath ML, Harris AW & Adams JM (1999) Constitutive bcl‐2 expression throughout the hematopoietic compartment affects multiple lineages and enhances progenitor cell survival. Proc Natl Acad Sci USA 96, 14943–14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Egle A, Harris AW, Bath ML, O'Reilly L & Cory S (2004) VavP‐Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood 103, 2276–2283. [DOI] [PubMed] [Google Scholar]
- 39. Croxford JL, Tang ML, Pan MF, Huang CW, Kamran N, Phua CM, Chng WJ, Ng SB, Raulet DH & Gasser S (2013) ATM‐dependent spontaneous regression of early Emu‐myc‐induced murine B‐cell leukemia depends on natural killer and T cells. Blood 121, 2512–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ottina E, Tischner D, Herold MJ & Villunger A (2012) A1/Bfl‐1 in leukocyte development and cell death. Exp Cell Res 318, 1291–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vogler M (2011) BCL2A1: the underdog in the BCL2 family. Cell Death Differ 19, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ogilvy S, Elefanty AG, Visvader J, Bath ML, Harris AW & Adams JM (1998) Transcriptional regulation of vav, a gene expressed throughout the hematopoietic compartment. Blood 91, 419–430. [PubMed] [Google Scholar]
- 43. Kucharczak JF, Simmons MJ, Duckett CS & Gelinas C (2005) Constitutive proteasome‐mediated turnover of Bfl‐1/A1 and its processing in response to TNF receptor activation in FL5.12 pro‐B cells convert it into a prodeath factor. Cell Death Differ 12, 1225–1239. [DOI] [PubMed] [Google Scholar]
- 44. Herold MJ, Zeitz J, Pelzer C, Kraus C, Peters A, Wohlleben G & Berberich I (2006) The stability and anti‐apoptotic function of A1 are controlled by its C terminus. J Biol Chem 281, 13663–13671. [DOI] [PubMed] [Google Scholar]
- 45. Egle A, Harris AW, Bouillet P & Cory S (2004) Bim is a suppressor of Myc‐induced mouse B cell leukemia. Proc Natl Acad Sci USA. 101, 6164–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Frenzel A, Labi V, Chmelewskij W, Ploner C, Geley S, Fiegl H, Tzankov A & Villunger A (2010) Suppression of B‐cell lymphomagenesis by the BH3‐only proteins Bmf and Bad. Blood 115, 995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kress TR, Sabo A & Amati B (2015) MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 15, 593–607. [DOI] [PubMed] [Google Scholar]
- 48. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD & Brinster RL (1985) The c‐myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318, 533–538. [DOI] [PubMed] [Google Scholar]
- 49. Longo PA, Kavran JM, Kim MS & Leahy DJ (2014) Single cell cloning of a stable mammalian cell line. Methods Enzymol 536, 165‐172. [DOI] [PubMed] [Google Scholar]
- 50. Werner AB, de Vries E, Tait SW, Bontjer I & Borst J (2002) Bcl‐2 family member Bfl‐1/A1 sequesters truncated bid to inhibit is collaboration with pro‐apoptotic Bak or Bax. J Biol Chem 277, 22781–22788. [DOI] [PubMed] [Google Scholar]
- 51. Lang MJ, Brennan MS, O'Reilly LA, Ottina E, Czabotar PE, Whitlock E, Fairlie WD, Tai L, Strasser A & Herold MJ (2014) Characterisation of a novel A1‐specific monoclonal antibody. Cell Death Dis 5, e1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Phenotypic characterization of lymphomas arising in Eμ‐MYC TG mice.
Table S2. Phenotypic characterization of lymphomas arising in Eμ‐MYC/Vav‐BFL1 DT mice.
Table S3. Phenotypic characterization of lymphomas arising in Eμ‐MYC/Vav‐BCLX DT mice.