

ABSTRACT
The genome organizer special AT‐rich sequence binding protein 1 (SATB1) regulates specific functions through chromatin remodeling in T helper cells. It was recently reported by our team that T cells from SATB1 conditional knockout (SATB1cKO) mice, in which the Satb1 gene is deleted from hematopoietic cells, impair phosphorylation of signaling molecules in response to T cell receptor (TCR) crosslinking. However, in vivo T cell responses upon antigen presentation in the absence of SATB1 remain unclear. In the current study, it was shown that SATB1 modulates T cell antigen responses during the induction and effector phases. Expression of SATB1 was upregulated in response to TCR stimulation, suggesting that SATB1 is important for this antigen response. The role of SATB1 in TCR responses and induced experimental autoimmune encephalomyelitis (EAE) was therefore examined using the myelin oligodendrocyte glycoprotein peptide 35‐55 (MOG35‐55) and pertussis toxin. SATB1cKO mice were found to be resistant to EAE and had defects in IL‐17‐ and IFN‐γ‐producing pathogenic T cells. Thus, SATB1 expression appears necessary for T cell function in the induction phase. To examine SATB1 function during the effector phase, a tamoxifen‐inducible SATB1 deletion system, SATB1cKO‐ER‐Cre mice, was used. Encephalitogenic T cells from MOG35‐55‐immunized SATB1cKO‐ER‐Cre mice were transferred into healthy mice. Mice that received tamoxifen before the onset of paralysis were resistant to EAE. Furthermore, no disease progression occurred in recipient mice treated with tamoxifen after the onset of EAE. Thus, SATB1 is essential for maintaining TCR responsiveness during the induction and effector phases and may provide a novel therapeutic target for T cell‐mediated autoimmune diseases.
Keywords: autoimmune disease, experimental autoimmune encephalomyelitis model, special AT‐rich sequence binding protein 1, T cell receptor response
Abbreviations
- CFA
complete Freund's adjuvant
- DLN
draining lymph node
- EAE
experimental autoimmune encephalomyelitis
- ER
estrogen receptor
- MAR
matrix attachment region
- MOG
myelin oligodendrocyte glycoprotein
- MOG35‐55
myelin oligodendrocyte glycoprotein peptide 35‐55
- OVA
ovalbumin
- SATB1
special AT‐rich sequence binding protein 1
- SATB1cKO mice
SATB1 conditional knockout mice
- SATB1cKOL
conditional knockout mice lacking SATB1 in T cells
- SATB1cKOV
conditional knockout mice lacking SATB1 in hematopoietic cells
- SRG3
SWI3‐related gene
- TCR
T cell receptor
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick‐end labeling
- WT
wild‐type
Chromatin structure modulates accessibility to target genes and is important for regulation of gene expression and cellular functions. Chromosomal organizers regulate chromatin topology, forming transcription‐regulatory compartments 1, 2. MARs or scaffold‐associated regions in DNA sequences are thought to mediate chromatin loop formation, which is important for compaction of genomic DNA and organization of chromatin into units of genomic function 3, 4. MARs frequently colocalize with enhancer regions and the findings of several studies have suggested that MARs mediate positive or negative regulation of gene expression 5, 6, 7. Special AT‐rich sequence binding protein 1 (SATB1), a MAR‐binding protein 8, is predominantly expressed in the thymus. SATB1 recruits adenosine triphosphate‐utilizing chromatin assembly and remodeling factor and nucleosome‐remodeling factor complex to specific DNA sites and functions as an epigenetic regulator that facilitates the functioning of several chromatin‐remodeling factors 9. A subsequent study demonstrated that SATB1 expression is highly associated with thymocyte differentiation 10. In addition, SATB1 regulates expression of multiple T helper cells genes and modulates effector T cell function by holding promoter regions of cytokine genes in close proximity 11. Thus, chromatin remodeling by SATB1 maintains the function of peripheral T cells.
Signals from TCRs are critical for activation of T cells and, together with those from cytokine receptors, activate diverse signaling pathways that control both the fate and functions of activated T cells 12. Naïve CD4 T cells begin a process of differentiation into effector T cells upon stimulation via cell surface receptors. Naïve CD4 T cells can also differentiate into distinct T helper subsets, such as Th1, Th2, Th17 and Tfh cells, as defined by the different cytokines present in the microenvironment 13. In addition to the appropriate cytokines, antigen recognition by TCRs is the most critical mediator of functional differentiation of Th subsets 14. These processes are transcriptionally regulated and involve induction of specific transcription factors 15. Chromatin remodeling, which controls the accessibility of transcription factors to their target genes, represents the next most critical regulatory mechanism in Th cell differentiation 16. We recently reported that T cells from SATB1‐deficient mice severely impaired proximal signaling following TCR engagement in vitro 17, suggesting that SATB1 supports T cell differentiation into Th subsets and/or maintenance of Th subsets. However, the contribution of SATB1 to supporting T cell function after antigen stimulation remains unknown.
In this study, we examined in vivo T cell responses in conditional knockout mice lacking SATB1 in hematopoietic cells (SATB1cKOV) or in T cells (SATB1cKOL). To this end, we used EAE. SATB1cKOV mice and SATB1cKOL mice are resistant to EAE induced with MOG35‐55. We demonstrated that T cells derived from both lines of SATB1cKO mice failed to proliferate and produce cytokines in response to protein antigens. In the transfer EAE model, induction of Satb1‐deletion after the onset of paralysis prevented disease progression. Our results suggest that SATB1 plays an important role in the priming and maintenance of T cell responses in vivo.
MATERIALS AND METHODS
Mice
SATB1‐floxed mice were generated as described previously 17. Vav‐Cre mice, ER‐Cre mice and OT‐II TCR transgenic mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Lck‐Cre mice were obtained from the laboratory animal resource bank at Nibiohn (Osaka, Japan). SATB1 conditional knockout mice were generated by crossing Satb1‐floxed with Vav‐Cre mice, Lck‐Cre mice and ER‐Cre mice to generate SATB1cKOV mice, SATB1cKOL mice and SATB1cKOe mice, respectively. The ER ligand‐binding domain was fused to the Cre recombinase (ER‐Cre) in Cre expressing mice to generate tamoxifen‐inducible conditional knockout mice. Given that administration of tamoxifen can induce nuclear translocation of ER‐Cre and subsequent inactivation of target genes containing the loxP sequence, SATB1cKOe mice were generated and used to induce deletion of Satb1 in CD4 T cells during the effector phase. OTII mice express transgenic TCRs specific for chicken OVA 323‐339 peptide. Given that these mice are useful for analyzing antigen (OVA) specific T cell responses, SATB1cKOe mice were generated and proliferation of T cells in the absence or presence of SATB1 assayed. C57BL/6 mice were purchased from Charles River Laboratories (Kanagawa, Japan). C57BL/6 CD45.1 mice and RAG2−/− mice were bred at the Toho University animal facility under specific pathogen‐free conditions in accordance with the institutional guidelines 18. All experiments using mice were approved by the Toho University Administrative Panel for Animal Care (17‐53‐311) and Recombinant DNA (17‐53‐303). The mice used were aged 8−12 weeks.
Real‐time PCR
Real‐time PCR was performed as described previously 19. Total RNA was isolated from cells using Isogen (Nippon Gene, Toyama, Japan). RNA (500 ng/reaction) was reverse transcribed using a High‐Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA, USA). For quantitative analysis, RT‐PCR was conducted using a TaqMan Gene Expression Assay kit (Applied Biosystems). Mm00487425_01 for Fos, Mm01268940_m1 for Satb1, Mm00519943_m1 for Il23r, Mm01168134_m1 for Ifng, Mm00475162_m1 for Foxp3, Mm01261022_m1 for Rorc and Mm02619580_g1 for actin were used as primers on an Applied Biosystems 7500 Fast system. β‐actin was used as an endogenous reference for normalization. Quantitative real‐time PCR experiments were repeated twice in triplicate.
EAE induction
Mice were immunized s.c. in the flank on Day 0 with 150 μg of MOG35‐55 peptide in CFA containing 5 mg/mL H37RA (Difco Laboratories, Detroit, MI, USA), as previously described 20. Pertussis toxin (200 ng; List Laboratories, Campbell, CA, USA) was injected intraperitoneally on Days 0 and 2. For passive transfer EAE, donor mice were immunized as describe above. Ten days later, DLN cells were cultured at 4 × 106 cells/mL with 10 mM MOG35‐55 peptide for 3 days in RPMI1640 culture medium with IL‐23, anti‐IL‐4 and anti‐IFN‐γ antibodies, as previously described 20. Next, 107 CD4 T cells were purified using negative selection kinetics on a MACS system (Miltenyi Biotec, Bergisch Gladbach, Germany) and transferred i.v. into naïve and 500‐rad X‐irradiated mice. Mice were graded for EAE on a clinical scale of 0–6 as follows: 0, no disease; 1, complete loss of tail tone; 2, hindlimb weakness; 3, hindlimb paralysis; 4, complete hind and partial forelimb paralysis; 5, hind and forelimb paralysis; and 6, death.
Recall responses
DLN cells were prepared from immunized mice and cultured for 72 hr with MOG35‐55 peptide or OVA. They were then pulsed for 6 hr with 3H‐thymidine (Amersham Biosciences, Little Chalfont, UK) and assayed for incorporation of 3H‐thymidine using Topcount (Perkin Elmer, Waltham, MA, USA), as previously described 21. Supernatants were collected at 24 hr and assayed for IL‐2, or at 72 hr for IL‐17 and IFN‐γ, with OptEIA ELISA kits (BD Biosciences, Franklin Lakes, NJ, USA). Cells were stained with anti‐mouse CD4 and CD8 antibodies, then washed with washing buffer (1% FCS, 0.1% sodium azide in PBS) and fixed with Cytofix/Cytoperm kit (BD Biosciences) for 20 min. Intracellular staining was performed using anti‐mouse IL‐17‐PE, IFN‐γ‐PE and Foxp3‐FITC antibodies for 30 min. In some experiments, isotype control antibodies used under similar conditions indicated the specific binding of test antibodies. Cells were acquired on a BD LSRFortessa X‐20 instrument using DIVA software. Data were analyzed using FlowJo software.
Tamoxifen treatment
Tamoxifen (Sigma–Aldrich, St. Louis, MO, USA) was resuspended in ethanol, diluted in corn oil to a final concentration of 10 mg/mL and heated at 37°C until dissolved. 4‐hydroxytamoxifen (Sigma) was dissolved in ethanol. To delete Satb1 in donor CD4 T cells, 100 μL of tamoxifen solution or oil were injected intraperitoneally into recipient mice adoptively transferred with CD4 T cells from SATB1cKOe mice once a day for three consecutive days, 3 days after cell transfer or 1 day after disease onset.
For the in vivo T cell proliferation assay, naïve CD4 T cells derived from SATB1cKOe‐OTII mice were labeled with the Cell Proliferation Dye eFluor 670 (eBioscience, San Diego, CA, USA). The labeled CD4 T cells were adoptively transferred into Rag2−/− mice i.v.. Recipient mice were treated with tamoxifen or oil for three consecutive days after cell transfer and injected i.v. with OVA on Day 1 after the transfer. Proliferation of donor T cells was quantitated by flow cytometry using a Cell Proliferation Dye. For in vitro deletion of Satb1, 0.2 μM of 4‐hydroxytamoxifen was administered into the culture medium to induce Cre‐mediated deletions.
Apoptosis assay
CD4 T cells prepared from WT or SATB1cKOL mice were stimulated with plate‐bound anti‐CD3 (1 μg/mL) and anti‐CD28 (1 μg/mL) antibodies for the indicated times to induce TCR‐mediated apoptosis. Apoptosis was determined using a MEBSTAIN apoptosis TUNEL kit (MBL, Nagoya, Japan).
Statistical analysis
Data are presented as means ± SEM. Statistical analysis was performed using GraphPad Prism 6.0 (GraphPad Software, San Diego, CA, USA). A P value < 0.05 was considered to denote significance. Statistical significance was assessed using the unpaired Student's t‐test or Mann–Whitney U test.
RESULTS
TCR stimuli induce SATB1 expression
T cells express numerous genes to gain effector function after antigen stimulation. If SATB1 plays an essential role in T cell function, SATB1 expression should be upregulated in response to TCR stimulation. We therefore assessed transcription of Satb1 in naïve CD4 T cells from WT mice. In response to TCR crosslinking with anti‐CD3 and anti‐CD28 antibodies, Satb1 mRNA expression gradually increased over 60 min, peaked at 120 min, and was substantially diminished at 12 hr (Fig. 1a). Given that transcription had been induced, we quantitated SATB1 protein by immunoblotting. As shown in Figure 1c, TCR crosslinking also led to increases in amounts of SATB1 and c‐Fos protein. Although the changes in SATB1 expression occurred slightly later than those in the immediate early gene c‐Fos (Fig. 1b), these results suggest that SATB1 plays a role in regulation of T cell function.
Figure 1.
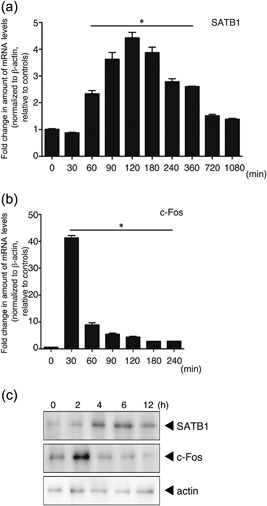
Expression of SATB1 in T cells. Naïve CD4 T cells prepared from WT mice were stimulated with anti‐CD3 and anti‐CD28 antibodies for the indicated times. mRNA expression of (a) SATB1 and (b) c‐Fos in these cells was analyzed using real‐time PCR. Transcription of these genes was quantitated relative to β‐actin. *values differ significantly from those in control cells (0 min) under the same conditions (P < 0.05). These results are representative of three independent experiments. (c) Protein expression for SATB1 and c‐Fos, determined by immunoblotting using anti‐SATB1 and anti‐c‐Fos antibodies. Results of one experiment, representative of three independent experiments, is shown.
SATB1 conditional knockout mice are resistant to EAE induction
We bred SATB1f/f mice with Vav‐Cre or Lck‐Cre mice to generate mice with SATB1cKOV or SATB1cKOL cKO of Satb1. To examine the role of SATB1 in T cell responses, we generated C57BL/6 WT, SATB1cKOV and SATB1cKOL T cell‐mediated autoimmune disease mouse models of EAE. These mice were immunized s.c. with MOG35‐55 peptide in CFA on Day 0 and i.v. injected with pertussis toxin on Days 0 and 2. As shown in Figure 2, all WT mice developed EAE that became evident on Day 12, whereas SATB1cKOV and SATB1cKOL mice failed to develop EAE during the 35 days following immunization. Histological examination of lumbar spinal cords from WT mice 20 days after immunization clearly demonstrated cellular infiltration in the spinal parenchyma (Fig. 2b). No such infiltration was observed in the spinal cords of SATB1cKOV mice, consistent with the observed lack of EAE development (Fig. 2b).
Figure 2.
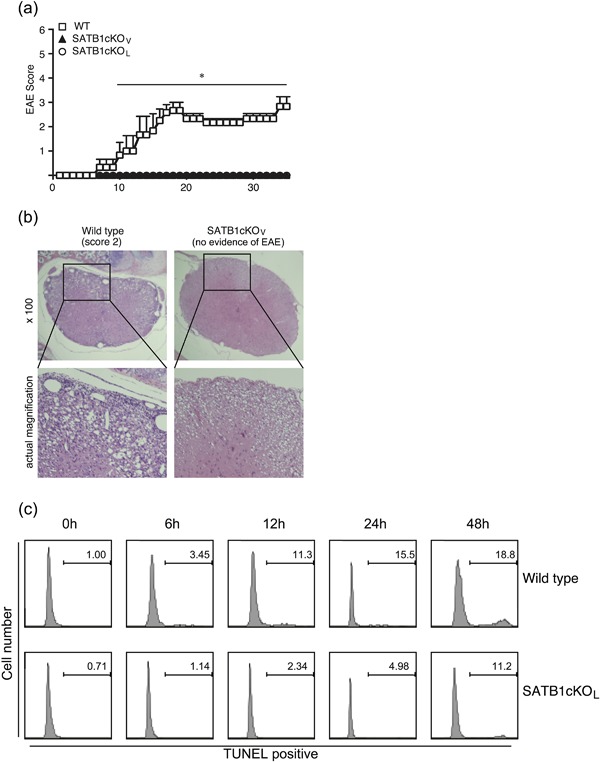
Failure of SATB1 conditional knockout mice to develop EAE. (a) Mice were immunized s.c. with MOG peptide in CFA, injected i.v. with pertussis toxin on Days 0 and 2, and assessed by clinical scores for 35 days after immunization. * P < 0.05. (b) Photomicrographs of spinal sections stained with hematoxylin and eosin (Day 20) after immunization × 250. Experiments were conducted three times, with essentially similar results. (c) Apoptotic T cell induction by TCR crosslinking was detected by TUNEL staining. Representative flow cytometry plots are shown. [Color figure can be viewed at http://wileyonlinelibrary.com]
We recently reported that thymic development is impaired in SATB1cKOV mice. We propose that peripheral T cells may have been activated due to homeostatic expansion, and were thus susceptible to apoptosis induced following TCR stimulation; which, in turn, may have led to EAE resistance. Moreover, SATB1‐deficient T cells may be susceptible to cell death induced following TCR activation. To examine this possibility, we stimulated CD4 T cells with plate‐bound anti‐CD3 (1 μg/mL) and anti‐CD28 (1 μg/mL) antibodies to induce TCR‐mediated apoptosis (activation‐induced cell death). We demonstrated that the percentage of CD4 T cells exhibiting TUNEL‐positive staining increased from 1% to 18.8% in a time‐dependent manner, indicating that WT T cells undergo activation‐induced cell death following TCR crosslinking (Fig. 2c). In contrast, SATB1‐deficient T cells from SATB1cKOL mice were resistant to TCR‐mediated cell death, suggesting that hyperactivation of T cells does not occur, even in the absence of SATB1.
SATB1 conditional knockout mice fail to generate IL‐17 and IFN‐γ‐producing T cells
To examine whether pathogenic T cells are generated in SATB1 conditional knockout mice, we compared the in vitro recall responses of DLN cells from WT, SATB1cKOV and SATB1cKOL mice. DLN cells were prepared 10 days after immunization, before there was evidence of EAE in WT mice. Although we observed proliferative recall responses to various doses of the MOG35‐55 peptide in DLN cells from WT mice, we did not detect cellular proliferation in DLN cells from either of the SATB1 conditional knockout mice (Fig. 3a). Next, we analyzed cytokine production in response to treatment with the MOG35‐55 peptide. There was much greater IL‐2 production in T cells from DLN cells in WT mice than in those from SATB1cKOV and SATB1cKOL mice, indicating that proliferation of SATB1‐deficient T cells was defective (Fig. 3a). We detected dose‐dependent production of IFN‐γ and IL‐17 in cultures of DLN cells from WT mice and SATB1 conditional knockout mice; however, production of each of these cytokines was severely diminished in SATB1cKOV and SATB1cKOL mice (Fig. 3a). Given that SATB1 regulates thymic development of T cells 17, our failure to induce a T cell response can be attributed to the fact that SATB1 knockout mice possess very few peripheral T cells. However, the number of CD4 T cells prepared from SATB1cKOL mice represented 18% to 20% of all lymph node cells (Fig. 3b). The CD4‐positive T cell population was slightly smaller in SATB1cKOL than WT mice (where the number of CD4 T cells represented approximately 25% of all lymph node cells), suggesting that the absence of a CD4 T cell population is not the conclusive underlying explanation for the failure to induce T cell responses and the presence of EAE resistance in SATB1 conditional knockout mice. The reduced in vitro IL‐17 and IFN‐γ production by DLN cells derived from SATB1cKOV and SATB1cKOL mice suggests a defect in Th17 and Th1 generations. To examine this possibility, we prepared DLN cells 10 days after immunization, incubated them with the MOG peptide, and assessed them for intracellular IL‐17 and IFN‐γ staining. As shown in Figure 3b, we found much lower percentages of CD4+ IL17+ Th17 cells in DLN cells from SATB1cKOL mice than in those from WT mice (0.26% vs. 2.1% of total cells and 1.3% vs. 8.3% of CD4 T cells, respectively). Moreover, there were far fewer CD4+ IFN‐γ+ Th1 cells in SATB1cKOL mice than in WT mice (0.17% vs. 2.2% of total cells and 0.09% vs. 9.1% of CD4 T cells, respectively).
Figure 3.
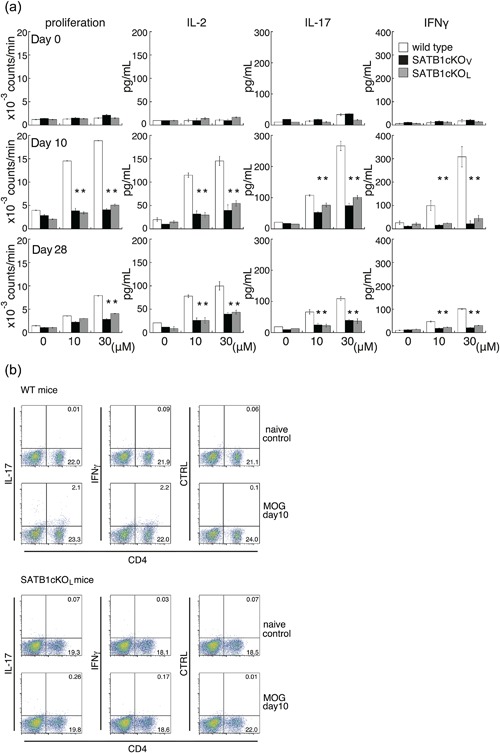
In vitro recall response to the MOG peptide for DLN cells from WT, SATB1cKOV, and SATB1cKOL mice. (a) Mice were immunized with MOG in CFA. DLN cells were prepared 10 or 28 days after immunization and incubated with MOG peptide at the indicated doses; proliferation was assessed on Day 3. Production of IL‐2 (Day 1), IL‐17 (Day 3), and IFN‐γ (Day 3) was determined by ELISA. (b) DLN cells were prepared from naïve WT and SATB1cKOL mice or from mice on Day 10 after immunization, as described in the legend of Figure 3(a). DLN cells were incubated with MOG peptide, after which intracellular expression of IL‐17 and IFN‐γ was assessed. The numbers in the light quadrants represent the percentages of all cells. * P < 0.05. One representative result of three independent experiments is shown. [Color figure can be viewed at http://wileyonlinelibrary.com]
To examine whether the abortive production of these cytokines by DLN cells from both lines of SATB1cKO mice might represent a specific response to the MOG peptide, we analyzed recall responses after immunization with OVA emulsified in CFA (Fig. 4). We prepared DLN cells 10 or 28 days after immunization, incubated them with OVA, and assessed proliferation of T cells and cytokine production. Similar to T cell responses against the MOG peptide, we observed a reduction in cytokine production, cell proliferation and generation of Th17/Th1 cells in cells from SATB1‐deficient mice (Fig. 4). These diminished recall responses in SATB1cKOV and SATB1cKOL mice were maintained for at least four weeks, suggesting that T cell priming requires SATB1 for induction of antigen responses and that SATB1 generally modulates T cell function in response to TCR signaling.
Figure 4.
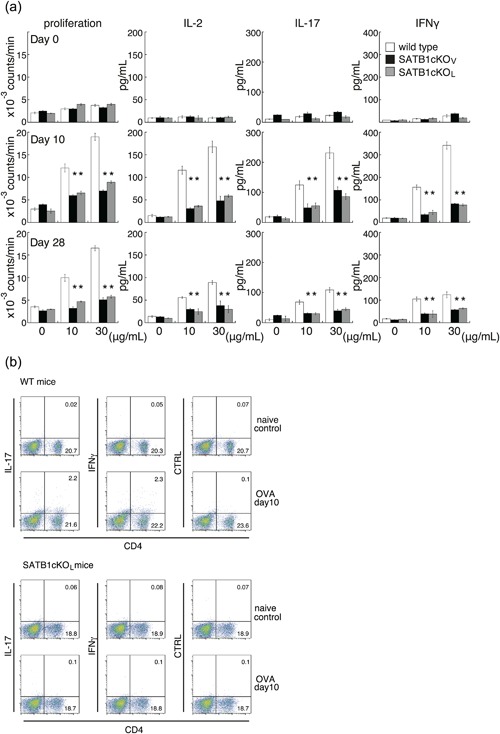
In vitro recall response to OVA for DLN cells from WT, SATB1cKOV and SATB1cKOL mice. (a) Mice were immunized with OVA in CFA. DLN cells were prepared 10 or 28 days after immunization and incubated with OVA at the indicated doses; proliferation was assessed on Day 3. Production of IL‐2 (Day 1), IL‐17 (Day 3), and IFN‐γ (Day 3) was determined by ELISA. (b) DLN cells were prepared from naïve WT and SATB1cKOL mice or from mice on Day 10 after immunization, as described in the legend of Figure 4(a). DLN cells were incubated with OVA, after which intracellular expression of IL‐17 and IFN‐γ was assessed. The numbers in the light quadrants represent the percentages of all cells. * P < 0.05. One representative result of three independent experiments is shown. [Color figure can be viewed at http://wileyonlinelibrary.com]
Mice lacking SATB1 displayed EAE resistance and defective induction of pathogenic T cells. These results led us to hypothesize that an immune regulatory microenvironment could be established in SATB1‐deficient mice. To evaluate this possibility, we generated EAE pathogenic Th17 cells from WT mice and transferred them into SATB1cKOV and SATB1cKOL mice. As shown in Figure 5, WT, SATB1cKOV, and SATB1cKOL mice receiving MOG‐reactive CD4 T cells developed EAE of similar severity. These data suggest that the attenuated EAE phenotype in SATB1‐deficient mice is T cell‐mediated. Taken together, these findings suggest that T cells derived from SATB1‐deficient mice fail to differentiate into pathogenic T cells during the priming or induction phases.
Figure 5.
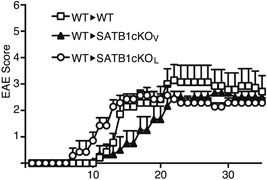
Transfer of encephalitogenic T cells induces EAE in SATB1‐deficient mice. DLN cells were prepared from WT mice 10 days after immunization and incubated with MOG peptide and IL‐23 for 3 days. CD4 T cells were transferred i.v. into naïve WT, SATB1cKOV or SATB1cKOL mice (n = 10). Representative results of three independent experiments are shown as mean EAE scores ± SD.
SATB1 maintains antigen responsiveness after T cell priming
Our findings suggest that pathogenic T cells are generated by a SATB1‐dependent mechanism. In addition to its regulatory function during the priming phase, SATB1 may also regulate T cell function during the effector phase of EAE. To investigate whether SATB1 is necessary for maintaining the self‐reactivity of T cells after establishment of pathogenic responses, we generated SATB1cKOe mice by crossing SATB1‐floxed mice with ER‐Cre mice to inducibly delete Satb1. Ten days after s.c. immunization, we incubated DLN cells from SATB1cKOe mice for 3 days with MOG35‐55 peptide and IL‐23; after which, we adoptively transferred CD4 T cells i.v. into WT mice. All recipient control mice developed EAE with similar clinical scores and time courses (Fig. 6a). These results suggest that SATB1cKOe mice normally generate neuroreactive T cells. Three days after the transfer, we injected recipient mice intraperitoneally with tamoxifen every other day for a total of three injections (Days 3, 5 and 7) to delete Satb1 in donor T cells. In contrast to control conditions, SATB1‐deficient T cells did not induce paralysis (Fig. 6a).
Figure 6.
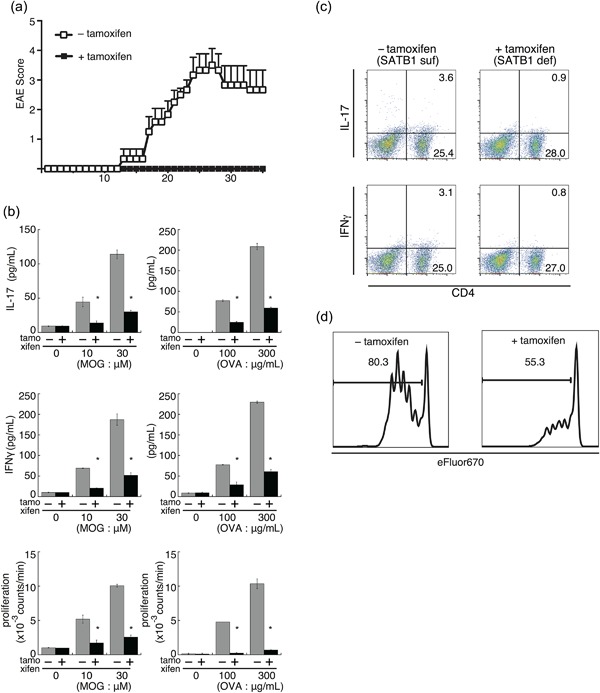
SATB1 is an essential factor for T cell responses during the effector phase of EAE. (a) Encephalitogenic Th17 cells were prepared from DLN cells derived from SATB1cKOe mice after immunization and stimulated with MOG peptide and IL‐23. CD4 T cells were transferred into naïve WT mice and Satb1 deleted by tamoxifen injection. Clinical status was monitored for 35 days. (b) SATB1cKOe mice were immunized with MOG peptide or OVA in CFA and administered tamoxifen on Days 11 and 13 to delete Satb1. CD4 T cells were prepared from these mice on Day 14 and restimulated with the MOG peptide or OVA presented by irradiated splenocytes from CD45.1 mice. Cytokine production and T cell proliferation were assessed. *, Values differ significantly from those in control cells (SATB1‐sufficient cells) under the same conditions (P < 0.05). (c) IL‐17+ and IFNγ+ CD4 T cells in (b) were analyzed by intracellular staining for cytokines and flow cytometry. (d) CD4 T cells from SATB1cKOe‐OTII mice were labeled with Cell Proliferation Dye eFluor 670 and transferred into RAG2−/− mice. Recipient mice were treated with tamoxifen and then injected with OVA. T cell proliferation was assessed via eFluor 670 dilution in treated (right) versus untreated (left) mice. Plot areas gated on OVA 323‐339 tetramer+ CD4+. [Color figure can be viewed at http://wileyonlinelibrary.com]
To examine the role of SATB1 in cytokine production by T cells, we immunized SATB1cKOe mice with MOG peptide or OVA in CFA and induced deletion of Satb1 in these mice in the presence or absence of tamoxifen on Days 11 and 13 after immunization. On Day 14, we prepared CD4 T cells from DLN cells from these mice and stimulated them with antigen‐loaded irradiated splenocytes from CD45.1 mice as antigen‐presenting cells. Recall cytokine production and intracellular staining assays showed dose‐dependent production of IFN‐γ and IL‐17 by cultures of SATB1‐sufficient CD4 T cells (Fig. 6b). In contrast, we barely detected cytokine production in response to MOG peptide or OVA in cultures of SATB1‐deficient CD4 T cells (Fig. 6b). We confirmed the defect in cytokine production by intracellular staining (Fig. 6c), which suggests that pathogenic CD4 T cells cannot produce cytokines in the absence of SATB1 expression. These results suggest that the lack of SATB1 in CD4 T cells after priming suppresses production of effector cytokines for EAE induction.
We also assessed whether proliferation of CD4 T cells depends on Satb1 expression. Similar to cytokine production, proliferative recall responses were diminished in cultures of SATB1‐deficient CD4 T cells (Fig. 6b). To investigate the role of SATB1 in antigen‐specific T cell responses in vivo, we used OTII mice and OVA 323‐339 tetramer. OTII mice express T cell receptors specific for OVA 323‐339 peptide and the OVA tetramer is able to detect antigen‐specific CD4 T cell responses. We transferred OVA‐specific CD4 T cells prepared from SATB1cKOe‐OTII mice into RAG2−/− mice. We then labeled donor CD4 T cells with the eFluor 670 cell proliferation‐specific dye and transferred them into recipient mice, after which we treated some of these mice with tamoxifen and administered OVA to all of them to stimulate donor OTII cells. Three days after immunization, we assessed dye‐division proliferation by flow cytometry. As shown in Figure 6d, most tetramer‐positive donor CD4 T cells (>80%) from mice that had not received tamoxifen showed a divided profile. In contrast, SATB1‐deficient donor CD4 T cells failed to proliferate in response to OVA. These results using SATB1cKOe mice suggest that: (i) SATB1 in CD4 T cells is an essential factor for maintaining antigen receptor responses; and (ii) SATB1 plays an important role in T cell responses that is independent of its roles in thymic development 17.
Encephalitogenic T cells lacking SATB1 partially retain the properties of Th17 cells
Th17 cells play an essential role in induction of EAE 22, 23. There were enough Th17 cells for induction of adoptive transfer of EAE under SATB1‐sufficient conditions (Fig. 6a). The same number of tamoxifen‐mediated SATB1‐deficient CD4 T cells did not induce EAE paralysis. Moreover, encephalitogenic activity in T cells was almost abolished following treatment with tamoxifen (Fig. 6). We suspect that deletion of Satb1 induces Th17 cells to change from being pathogenic to being suppressive. Furthermore, given that SATB1 negatively regulates expression of Foxp3 transcription factor 24, which regulates the generation and function of Treg cells, loss of SATB1 may induce maturation of Treg cells. To investigate this possibility, 10 days after s.c. immunization, we incubated DLN cells prepared from SATB1cKOe mice with MOG35‐55 peptide and IL‐23 (Th17 skew condition) for 3 days, after which we adoptively transferred CD4 T cells i.v. into CD45.1 WT mice. We treated some of the recipient mice with tamoxifen on Day 2 after the transfer. Next, we FACS‐sorted CD45.2 donor CD4 T cells and assessed expression of Th17 cell‐related genes. As shown in Figure 7a, there was significantly less Satb1 in the presence of tamoxifen. The Rorc and Il23r genes were similarly expressed in SATB1‐sufficient and SATB1‐deficient CD45.2 CD4 T cells, suggesting that CD4 T cells lacking SATB1 partially maintain encephalitogenic properties. In contrast, transcription of Il17a and Ifng pathogenic cytokines was reduced in SATB1‐deficient CD4 T cells. Flow cytometry also showed that SATB1‐deficient cells had smaller intracellular IL‐17 and IFN‐γ‐positive populations (Fig. 7b). We next assessed whether CD4 T cells upregulate Foxp3 expression in the absence of SATB1. In contrast to naïve CD4 T cells, SATB1‐sufficient Th17 cells showed weaker Foxp3 expression (Fig. 7a,b). Moreover, there was less rather than more Foxp3 mRNA and protein in SATB1‐deficient Th17 cells than in SATB1‐sufficient Th17 cells (Fig. 7a,b). These results suggest that, even in the absence of SATB1 expression, Th17 cells induced following treatment with MOG peptide and IL‐23 partially retain Th17 properties, without undergoing reprogramming to suppressive Th lineages. Combined with the results shown in Figure 5, these data indicate that SATB1 contributes to the antigen responses of CD4 T cells during the effector phase.
Figure 7.
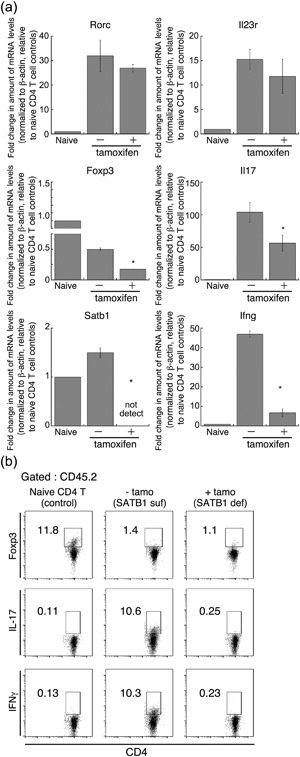
Encephalitogenic gene expression in Th17 cells not expressing SATB1. To prepare Th17 cells, DLN cells (CD45.2) from MOG immunized SATB1cKOe mice were skewed using MOG peptide and IL‐23 and then transferred into recipient mice (CD45.1). Some of these mice were treated with tamoxifen on Days 1, 3, and 5 after the transfer. On Day 7, CD4+ CD45.2+ transferred donor T cells were sorted from the spleen and lymph nodes of recipient mice using a FACS Aria III cell sorter. (a) Quantitative PCR was used to measure mRNA expression of Il17A, Ifng, Rorc, Il23r, Satb1 and Foxp3 in donor CD4 T cells. *Values differ significantly from those in control cells (SATB1‐sufficient Th17 cells) under the same conditions (P < 0.05). Experiments were conducted three times, with essentially similar results. (b) CD45.2+ CD4+ Th17 cells from SATB1‐deficient mice (with tamoxifen) or SATB1‐sufficient mice (without tamoxifen), or control CD4 T cells from naïve C57BL/6 mice were assessed for intracellular IL‐17 or Foxp3 expression. The numbers in the squares are the percentages of CD45.2+ CD4+ cells.
Induction of Satb1 deletion in CD4 T cells after the onset of EAE prevents disease progression
Finally, to examine whether controlling SATB1 function in T cells has any therapeutic potential for suppressing progression of EAE, we immunized SATB1cKOe mice with MOG35‐55 peptide in CFA. Ten days after immunization, we incubated DLN cells for 3 days with MOG peptide and IL‐23, after which we adoptively transferred CD4 T cells into WT mice to induce transfer EAE. After detecting the onset of the limp tail phenotype (score 1 for EAE), we treated diseased mice with tamoxifen to induce Satb1 deletion. The disease progressed and became more severe in control mice treated with tamoxifen‐free oil. In contrast, signs of EAE improved significantly in mice injected with tamoxifen and the disease did not progress or become more severe (Fig. 8). These results suggest that T cells lacking SATB1 attenuate antigen responsiveness during the effector phase and that controlling SATB1 activity in T cells allows the host to survive in the presence of autoimmune disease.
Figure 8.
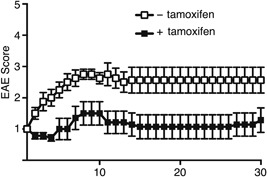
Satb1 deletion in Th17 cells after the onset of EAE alleviates clinical evidence of disease in EAE mice. Encephalitogenic Th17 cells were prepared from DLN cells derived from SATB1cKOe mice after immunization and stimulated with MOG peptide and IL‐23. CD4 T cells were transferred into naïve WT mice, and Satb1 deleted by tamoxifen injection after the onset of EAE. Clinical status was monitored for 30 days.
DISCUSSION
Expression of SATB1 in thymocytes is highest at the double‐positive stage and decreases as the maturation stage progresses 17. We first attempted to clarify expression of SATB1 in T cells after crosslinking of antigen receptors. A quantitative analysis in CD4 T cells indicated that SATB1 is clearly expressed in response to TCR stimulation. This increase in SATB1 expression is considered to induce changes in the structure of chromosomes, leading to increased expression of factors involved in the effector function of the T helper cell subset. These data suggest that SATB1 is induced by TCR signaling and that it plays important roles in peripheral naïve T cells in regulating generation of antigen‐specific T cells and maintaining the function of effector T cells. In general, CD4 T cells differentiate into T helper cells after stimulation of TCRs and cytokine receptors. SATB1 plays an important role in Th2 cells by controlling chromosomal structure 11. Differentiation of naïve T cells into Th2 cells (priming stage) is controlled by nuclear events such as epigenetic modifications and transcription. The transcription machinery prevents spontaneous differentiation of naïve T cells into Th2 cells. Upon TCR stimulation, naïve T cells are stimulated with a polarizing signal through the IL‐4 receptor pathway to upregulate expression of GATA‐3. GATA‐3 drives Th2 differentiation in a cell‐autonomous fashion. During the priming stage, the Th2‐related cytokine gene locus undergoes chromatin remodeling and epigenetic modifications, which contribute to Th2 cell‐related gene expression. SATB1 modulates formation of these chromosomal hubs 11. After differentiation, Th2 cells produce Th2‐related cytokines upon TCR stimulation and GATA‐3 upregulation. Similar to that in Th2 cells, chromatin remodeling may contribute to regulation of encephalitogenic T cells 25. SWI/SNF is an adenosine triphosphate‐dependent chromatin‐remodeling complex. SRG3 (a SWI3‐related gene) enhances the function of the SWI/SNF complex by stabilizing it 25, 26. SRG3‐SWI/SNF‐mediated chromatin remodeling in CD4 T cells facilitates EAE via induction of Th17 cells 25. In addition, epigenetic modification of histones following chromatin remodeling occurs during generation of Th17 cells from naïve T cells. A cis element, conserved non‐coding sequence 2, interacts with the promoters of Il17 and Il17f 27, 28. Conserved non‐coding sequence 2 is bound by p300 and JMJD3 (JmjC domain‐containing protein 3) histone remodeling enzymes, which modify the acetylation status of histone H3 and inhibit the suppressive histone marker H3K27me3, respectively, increasing accessibility of RORγt to the Il17 and Il17f gene promoters 28. These findings suggest that SATB1 activity manipulates chromatin structure and leads to development of Th17 cells. In this study, we demonstrated that SATB1‐deficient mice have severe impairment of production of EAE‐related cytokines such as IFN‐γ and IL‐17. This indicates that SATB1 is necessary for generation of encephalitogenic Th17 cells. However, we did not determine how modulation of chromatin structure in pathogenic T cells mediated by SATB1 regulates T cell responses. While it is still necessary to identify the molecular mechanism underlying the SATB1‐mediated differentiation of T cells, the results of this study suggest that SATB1 is an essential factor in formation of the chromatin hub during the priming stage of Th cells.
In this study, we showed that SATB1cKOV and SATB1cKOL mice are completely resistant to active induction of EAE, which suggests that SATB1 is required for generation of pathogenic Th cells. We also found that pathogenic T cells lacking SATB1 expression, which we induced by tamoxifen treatment on Day 2 after transfer into WT mice, failed to induce EAE. We therefore investigated the possibility that controlling SATB1 activity after the onset of symptoms leads to inhibition of disease progression. We prepared MOG‐reactive Th17 cells from SATB1cKOe mice and transferred them into healthy recipient mice. These Th17 cells induced paralysis and resulted in progressively severe disease; we used cells in which Satb1 deletion had not been induced as a control. In contrast, mice injected with tamoxifen to induce deletion of SATB1 did not exhibit palsied tails and legs or disease progression. These results suggest that SATB1 is necessary not only for induction of pathogenic gene expression during the priming phase, but also for retaining the pathogenic activity of T cells during the effector phase. We have previously reported that peripheral T cells from SATB1‐deficient mice show weak TCR signaling 17. Given that TCR signaling is a common essential factor during the priming and effector phases of EAE, SATB1 and its related molecules may regulate antigen responses. Our results suggest that donor encephalitogenic T cells barely respond to CNS‐antigens and are not sufficiently pathogenic to induce autoimmune disease. Although further studies are required to determine the molecular mechanism underlying SATB1‐mediated T cell responses, our findings have important implications for regulation of SATB1 activity in the treatment of EAE and/or Th17‐mediated diseases.
We and others investigators have reported that SATB1 is required for development of hematopoietic cells and T cells 17, 29. In the thymic stage, T cell progenitors develop into both conventional T cells and thymic Treg cells. SATB1 binds Treg cell‐specific super‐enhancers and upregulates expression of Treg‐related genes such as Foxp3 24. Thus, SATB1‐dependent Treg super‐enhancer activation is crucial for Treg cell development. In addition, SATB1 regulates Foxp3 expression in conventional T cells 24. Deletion of Satb1 in mature CD4 T cells derepresses Foxp3 expression; SATB1‐deficient conventional T cells express Foxp3. A proportion of these Foxp3‐positive T cells convert from conventional T cells on expressed CD25 on the cell surface and their activity is suppressed 24. Treatment of donor T cells with tamoxifen reduced SATB1 expression and EAE pathogenicity in our transfer EAE model. In contrast with the findings of a previous study, Foxp3 expression is decreased in SATB‐deficient T cells. On the basis of these results, we reasoned that the loss of pathogenicity in donor T cells in the absence of SATB1 expression is independently regulated via SATB1‐mediated derepression of Foxp3 expression.
In summary, this is the first report on the role of SATB1 expression in induction of autoimmune diseases during in vivo pathogenic T cell responses. Our work demonstrates that SATB1 regulates pathogenic Th17 cell generation and effector function. Finally, we found that deletion of Satb1 suppresses the self‐reactive T cell‐mediated autoimmune disease, EAE, suggesting that SATB1 may be a promising target for treatment of T cell‐mediated autoimmune diseases.
DISCLOSURE
The authors declare that they have no financial conflicts of interest.
ACKNOWLEDGMENTS
We would like to thank Editage (http://www.editage.jp) for English language editing. We also thank Kayo Tsuburaya, Tsukiko Sato and Maho Miyazaki for their excellent technical assistance. This work was supported by a Dr. Takeshi Yanase Grant from the Toho University School of Medicine to Y. A., a grant from the Takeda Science Foundation to T. K., a Japan Society for the Promotion of Science Grants‐in‐Aid for Scientific Research (25460600 and 17K08892 to T.K., 24390121 and 26670240 to M.K.), a Strategic Research Foundation Grant‐aided Project for Private Schools at Heisei 26th (S1411015) from the Ministry of Education, Culture, Sports, Science and Technology (to M.K.), a Research Promotion Grant from the Toho University Graduate School of Medicine (14‐02 to M.K.), the Public Foundation of the Vaccination Research Center (to M.K.), and a Grant‐in Aid for Private University Research Branding Project from MEXT (to M.K.).
REFERENCES
- 1. Phillips J.E., Corces, V.G. (2009) CTCF: Master weaver of the genome. Cell 137: 1194–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schneider R., Grosschedl R (2007) Dynamics and interplay of nuclear architecture, genome organization, and gene expression. Genes Dev 21: 3027–43. [DOI] [PubMed] [Google Scholar]
- 3. Cockerill P.N., Yuen M.H., Garrard W.T. (1987) The enhancer of the immunoglobulin heavy chain locus is flanked by presumptive chromosomal loop anchorage elements. J Biol Chem 262: 5394–97. [PubMed] [Google Scholar]
- 4. Zlatanova J.S., van Holde K.E. (1992) Chromatin loops and transcriptional regulation. Crit Rev Eukaryot Gene Exp 2: 211–24. [PubMed] [Google Scholar]
- 5. Banan M., Rojas I.C., Lee W.H., King H.L., Harriss J.V., Kobayashi R, Webb C.F., Gottlieb P.D. (1997) Interaction of the nuclear matrix‐associated region (MAR)‐binding proteins, SATB1 and CDP/Cux, with a MAR element (L2a) in an upstream regulatory region of the mouse CD8a gene. J Biol Chem 272: 18440–52. [DOI] [PubMed] [Google Scholar]
- 6. Wang Z., Goldstein A., Zong R.T., Lin D, Neufeld E.J., Scheuermann R.H., Tucker P.W. (1999) Cux/CDP homeoprotein is a component of NF‐muNR and represses the immunoglobulin heavy chain intronic enhancer by antagonizing the bright transcription activator. Mol Cell Biol 19: 284–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhong X.P., Carabana J., Krangel M.S. (1999) Flanking nuclear matrix attachment regions synergize with the T cell receptor delta enhancer to promote V(D)J recombination. Proc Natl Acad Sci USA 96: 11970–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dickinson L.A., Joh T., Kohwi Y., Kohwi‐Shigematsu T. (1992) A tissue‐specific MAR/SAR DNA‐binding protein with unusual binding site recognition. Cell 70: 631–45. [DOI] [PubMed] [Google Scholar]
- 9. Yasui D., Miyano M., Cai S., Varga‐Weisz P., Kohwi‐Shigematsu T. (2002) SATB1 targets chromatin remodelling to regulate genes over long distances. Nature 419: 641–5. [DOI] [PubMed] [Google Scholar]
- 10. Alvarez J.D., Yasui D.H., Niida H., Joh T., Loh D.Y., Kohwi‐Shigematsu T. (2000) The MAR‐binding protein SATB1 orchestrates temporal and spatial expression of multiple genes during T‐cell development. Genes Dev 14: 521–35. [PMC free article] [PubMed] [Google Scholar]
- 11. Cai S., Lee C.C., Kohwi‐Shigematsu T. (2006) SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat Genet 38: 1278–88. [DOI] [PubMed] [Google Scholar]
- 12. Sharpe A.H. (2009) Mechanisms of costimulation. Immunol Rev 229: 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dong C. (2008) TH17 cells in development: An updated view of their molecular identity and genetic programming. Nat Rev Immunol 8: 337–48. [DOI] [PubMed] [Google Scholar]
- 14. Nakayama T., Yamashita M. (2010) The TCR‐mediated signaling pathways that control the direction of helper T cell differentiation. Semin Immunol 22: 303–9. [DOI] [PubMed] [Google Scholar]
- 15. Li P., Spolski R., Liao W., Leonard W.J. (2014) Complex interactions of transcription factors in mediating cytokine biology in T cells. Immunol Rev 261: 141–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Falvo J.V., Jasenosky L.D., Kruidenier L., Goldfeld A.E. (2013) Epigenetic control of cytokine gene expression: Regulation of the TNF/LT locus and T helper cell differentiation. Adv Immunol 118: 37–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kondo M., Tanaka Y., Kuwabara T., Naito T., Kohwi‐Shigematsu T., Watanabe A. (2016) SATB1 plays a critical role in establishment of immune tolerance. J Immunol 196: 563–72. [DOI] [PubMed] [Google Scholar]
- 18. Kuwabara T., Tanaka Y., Ishikawa F., Kondo M., Sekiya H., Kakiuchi T. (2012) CCR7 ligands up‐regulate IL‐23 through PI3‐kinase and NF‐kappa B pathway in dendritic cells. J Leukoc Biol 92: 309–18. [DOI] [PubMed] [Google Scholar]
- 19. Kuwabara T., Kasai H., Kondo M. (2016) Acetylation modulates IL‐2 receptor signaling in T Cells. J Immunol 197: 4334–43. [DOI] [PubMed] [Google Scholar]
- 20. Kuwabara T., Ishikawa F., Yasuda T., Aritomi K., Nakano H., Tanaka Y., Okada Y., Lipp M., Kakiuchi T. (2009) CCR7 ligands are required for development of experimental autoimmune encephalomyelitis through generating IL‐23‐dependent Th17 cells. J Immunol 183: 2513–21. [DOI] [PubMed] [Google Scholar]
- 21. Aritomi K., Kuwabara T., Tanaka Y., Nakano H., Yasuda T., Ishikawa F., Kurosawa H., Kakiuchi T. (2010) Altered antibody production and helper T cell function in mice lacking chemokines CCL19 and CCL21‐Ser. Microbiol Immunol 54: 691–701. [DOI] [PubMed] [Google Scholar]
- 22. Gaffen S.L., Jain R., Garg A.V., Cua D.J. (2014) The IL‐23‐IL‐17 immune axis: From mechanisms to therapeutic testing. Nat Rev Immunol 14: 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuwabara T., Ishikawa F., Kondo M., Kakiuchi T. (2017) The role of IL‐17 and related cytokines in inflammatory autoimmune diseases. Mediators Inflamm 2017: 3908061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kitagawa Y., Ohkura N., Kidani Y., Vandenbon A., Hirota K., Kawakami R., Yasuda K., Motooka D., Nakamura S., Kondo M., Taniuchi I., Kohwi‐Shigematsu T., Sakaguchi S. (2017) Guidance of regulatory T cell development by Satb1‐dependent super‐enhancer establishment. Nat Immunol 18: 173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee S.W., Park H.J., Jeon S.H., Lee C., Seong R.H., Park S.H., Hong S. (2015) Ubiquitous over‐expression of chromatin remodeling factor SRG3 ameliorates the T cell‐mediated exacerbation of EAE by modulating the phenotypes of both dendritic cells and macrophages. PLoS ONE 10: e0132329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sohn D.H., Lee K.Y., Lee C., Oh J., Chung H., Jeon S.H., Seong R.H. (2007) SRG3 interacts directly with the major components of the SWI/SNF chromatin remodeling complex and protects them from proteasomal degradation. J Biol Chem 282: 10614–24. [DOI] [PubMed] [Google Scholar]
- 27. Akimzhanov A.M., Yang X.O., Dong C. (2007) Chromatin remodeling of interleukin‐17 (IL‐17)‐IL‐17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem 282: 5969–72. [DOI] [PubMed] [Google Scholar]
- 28. Wang X., Zhang Y., Yang X.O., Nurieva R.I., Chang S.H., Ojeda S.S., Kang H.S., Schluns K.S., Gui J., Jetten A.M., Dong C. (2012) Transcription of Il17 and Il17f is controlled by conserved noncoding sequence 2. Immunity 36: 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Satoh Y., Yokota T., Sudo T., Kondo M., Lai A., Kincade P.W., Kouro T., Iida R., Kokame K., Miyata T., Habuchi Y., Matsui K., Tanaka H., Matsumura I., Oritani K., Kohwi‐Shigematsu T., Kanakura Y. (2013) The Satb1 protein directs hematopoietic stem cell differentiation toward lymphoid lineages. Immunity 38: 1105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]