

Abstract
Several families of protein kinases have been shown to play a critical role in the regulation of cell cycle progression, particularly progression through mitosis. These kinase families include the Aurora kinases, the Mps1 gene product and the Polo Like family of protein kinases (PLKs). The PLK family consists of five members and of these, the role of PLK1 in human cancer is well documented. PLK2 (SNK), which is highly homologous to PLK1, has been shown to play a critical role in centriole duplication and is also believed to play a regulatory role in the survival pathway by physically stabilizing the TSC1/2 complex in tumor cells under hypoxic conditions. As a part of our research program, we have developed a library of novel ATP mimetic chemotypes that are cytotoxic against a panel of cancer cell lines. We show that one of these chemotypes, the 6-arylsulfonyl pyridopyrimidinones, induces apoptosis of human tumor cell lines in nanomolar concentrations. The most potent of these compounds, 7ao, was found to be a highly specific inhibitor of PLK2 when profiled against a panel of 288 wild type, 55 mutant and 12 lipid kinases. Here, we describe the synthesis, structure activity relationship, in vitro kinase specificity and biological activity of the lead compound, 7ao.
Keywords: PLK, Kinase inhibitor, Apoptosis, Cytotoxicity
1. Introduction
In 2006, Weinstein and Joe1 proposed that cancer cells contain multiple genetic and epigenetic abnormalities and that despite this complexity, their growth and survival can often be impaired by the inactivation of a single oncogene. They proposed that this phenomenon, called ‘oncogene addiction,’ provides a rationale for molecular targeted therapy. The success of imatinib provides strong experimental support for the ‘Oncogene Addiction’ theory2 and has also created a great impetus for the discovery of putative ‘addictive kinases’ in various tumor model systems.
Since growth of most human tumors is dictated by the activation of kinase cascades, we designed an approach in which we combined the identification of ‘addictive kinases’ with drug discovery. To enable us to identify possible oncogenic kinases to which a tumor cell might be addicted, we synthesized a large number (approximately 300) of novel ATP mimetics that can act as kinase inhibitors and screened this compound library for compounds that can preferentially induce the death of human tumor cells without affecting normal cell viability. Our approach was to first identify a compound that is effective in tumor cell killing and then determine the identities of the kinases that are inhibited by this compound. This approach allowed two important aspects of drug discovery to occur simultaneously: the development of novel pharmaceuticals and the identification of new therapeutic targets. Here we report the synthesis of 7ao, which blocks tumor cell cycle progression in mitosis, causing apoptotic cell death and also inhibits PLK2 selectively.
There are five mammalian Polo kinases, termed Polo-like kinase (Plk) 1–5 which share two 30 amino acid conserved motifs, termed polo boxes, as well as additional residues in the kinase domain.3 PLKs play a critical role in cell cycle progression and are known to mediate various stages of mitosis including kinetochore division, centriole duplication and cytokinesis.
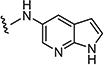
Within the PLK family, PLK1 is the best-characterized member and is the most closely related to yeast and Drosophila polo kinases.4 PLK1 is relevant to cancer biologists due to its over-expression in a variety of human tumor types and is linked to poor prognosis.5,6 PLK2, or Snk, was identified as an immediate-early gene product and a p53 target gene.7 PLK2 plays an important role in cell cycle control through the specific phosphorylation of centrosome-associated substrates. Specifically, PLK-2 regulates centriole duplication that occurs at the G1/S border and is coordinately regulated by CDK2/cyclin E and CDK2/cyclin A complexes, as well as PLK4.8,9 Studies have shown that inhibiting the expression of this gene by siRNA leads to mitotic catastrophe in paclitaxel-treated cells.10
While several pan-PLK inhibitors have been described in literature, no PLK2-specific small molecule inhibitor has been described. Here we report that 7ao (ON1231320) is a selective inhibitor of PLK2 and blocks tumor cell cycle progression in mitosis, causing apoptotic cell death. 7ao synergizes with paclitaxel, and is efficacious in inhibiting tumor growth in vivo suggesting that PLK2 is drug-gable kinase and a new selective target for cancer therapy.
2. Chemistry
The compounds presented in this report were prepared following the general approach as illustrated in Scheme 1, that has been described previously.11 Pyrimidines 2a–g were prepared from commercially available 4-chloro-2-methylthio-5-pyrimidinecarboxylic acid ethyl ester (1) by replacement of chlorine with ammonia or methylamine or ethylamine or propylamine or cyclopropylamine or cyclopentyl amine or cyclohexyl amine. The ester functional group was converted to an aldehyde via a two-step reduction–oxidation sequence employing lithium aluminum hydride followed by manganese(IV) oxide to give aldehydes 4a–g. Aldehydes 4a–g were converted to pyrido[2,3-d]-pyrimidin-7-ones 5a–z using Knoevenagel condensation. Thus, each aldehyde was treated with active methylene compounds (arylsulfonylacetic acid or arylmethanesulfonylacetic acid or N-arylmalonamic acid or N-arylsulfamoylacetic acid) in the presence of benzylamine to generate their corresponding intermediates 5a–z. The methyl sulfides 5a–z were oxidized to methyl sulfoxides 6a–z using m-chloroperbenzoic acid (m-CPBA). The methyl sulfoxide was then replaced with different aryl/heteroaryl amines, to obtain the target pyridopyrimidine compounds 7 as described in Scheme 1.
Scheme 1.

Synthesis of pyrido[2,3-d]pyrimidines. Reagents and conditions: (i) X-NH2, Et3N, THF; rt, 1–3 h, 80–95%; (ii) LiAlH4, THF, −10 °C–rt, 1 h, 80–86%; (iii) MnO2, CHCl3, rt, 24 h, 70–90%; (iv) active methylene compounds, BnNH2, AcOH, 100 °C, 6 h, 32–67%; (v) m-CPBA, CH2Cl2, rt, 3 h, 54–86%; (vi) Z, DMSO or toluene, 100 °C, 3–8 h, 45–65%.
The compounds 6aa–6ac were prepared by oxidation of 5aa–5ac with m-CPBA, which in turn was prepared by alkylation of 5u with corresponding alkyl iodides in the presence of Potassium carbonate in N,N-Dimethylformamide (DMF) at 50 °C as shown in Scheme 2.
Scheme 2.

Alkylation of 6-((2,4-difluorophenyl)sulfonyl)-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5u). Reagents and conditions: (i) X-I, NaH, DMF, 50 °C, 1 h, 55–60%; (ii) m-CPBA, CH2Cl2, rt, 3 h, 50–60%.
As shown in Scheme 3, the biotin conjugated compound (7u) was prepared by treating the compound 7t with biotin in the presence of EDC and DMAP at room temperature.
Scheme 3.

Synthesis of 4-(2-(1H-indol-5-ylamino)-7,8-dihydro-8-methyl-7-oxopyrido[2,3-d]pyrimidin-6-ylsulfonyl)phenyl-5-(hexahydro-2-oxo-1H-thieno[3,4-d] imidazol-6-yl)pentanoate (7u). Reagents and conditions: (i) EDC, DMAP, rt, 45%.
The active methylene compounds, arylsulfonylacetic acids (10a–j), arylmethanesulfonylacetic acids (13a and 13b), N-arylmalonamic acids (16a and 16b) and N-arylsulfamoylacetic acids (18a and 18b) were prepared as shown in Schemes 4–7 using the procedures reported earlier.12–15 As shown in Scheme 4, Arylthiols (8a–j) were treated with chloroacetic acid in the presence of NaOH and subsequent oxidation with 30% H2O2 to produce arylsulfanyl acetic acids (9a–j) and arylsulfonyl acetic acids (10a–j) respectively.
Scheme 4.

Synthesis of arylsulfonyl acetic acids. Reagents and conditions: (i) ClCH2CO2H, NaOH, MeOH, HCl, rt, 3 h, 55–66%; (ii) 30% H2O2, AcOH, rt, 24 h, 70–90%.
Scheme 7.

Synthesis of N-arylsulfamoylacetic acid. Reagents and conditions: (i) Et3N, DCM, rt, 3 h, 70–72%; (ii) 10% NaOH, rt, 1 h, 76–80%.
Arylmethyl bromides (11a and 11b) were treated with thioglycolic acid in the presence of sodium hydroxide to produce corresponding arylmethylsulfanyl acetic acids (12a and 12b). Oxidation of 12a and 12b with 30% H2O2 in acetic acid yielded the corresponding arylmethanesulfonyl acetic acids (13a and 13b) as reported in Scheme 5.
Scheme 5.

Synthesis of arylmethanesulfonyl acetic acids. Reagents and conditions: (i) NaOH, MeOH, rt, 3 h, 90–96%; (ii) 30% H2O2, AcOH, 24 h, 90–93%.
The reaction of aromatic amines (14a and 14b) with methyl-3-chloro-3-oxopropionate in the presence of triethylamine generated 3-anilino-3-oxopropionic acid methyl esters (15a and 15b), which, when subjected to hydrolysis, resulted in the formation of 3-anilino-3-oxopropionic acids (16a and 16b) as illustrated in Scheme 6.
Scheme 6.

Synthesis of N-arylmalonamic acids. Reagents and conditions: (i) Et3N, DCM, rt, 3 h, 76–77%; (ii) NaOH, rt, 1 h, 73–75%.
As depicted in Scheme 7, chlorosulfonylacetic acid ethyl ester was treated with aromatic amines (14a and 14b) in the presence of triethylamine in DCM to obtain the arylsulfamoylacetic acid ethyl esters (17a and 17b) in good yields. Hydrolysis of 17a and 17b with 10% NaOH in water generated the corresponding arylsulfamoylacetic acids (18a and 18b) in high yields.
Methyl arylsulfonyl esters were prepared as shown in Scheme 8. Aromatic nucleophilic substitution of fluorine in 3,4-difluoronitrobenzene (19) and 2,5-difluoronitrobenzene (20) by methyl 2-mercaptoacetate in the presence of DIPEA produced methyl 2-(2-fluoro-4-nitrophenylthio)acetate (21) and methyl (2-(4-fluoro-2-nitrophenylthio)acetate (22) respectively.
Scheme 8.

Synthesis of methyl arylsulfonyl esters. Reagents and conditions: (i) HSCH2CO2Me, DIPEA, THF, rt, 15 h, 85–96%; (ii) 30% H2O2, AcOH, 60 °C, 6 h, 90–99%; (iii) 10% Pd/C, H2, MeOH, rt, 4 h, 86–93%.
The thio compounds 21 and 22 were subjected to oxidation with 30% H2O2 in acetic acid to generate corresponding sulfonyl compounds, methyl 2-(2-fluoro-4-nitrophenylsulfonyl)acetate (23) and methyl (2-(4-fluoro-2-nitrophenylsulfonyl)acetate (24). The resulting compounds 23 and 24 were dissolved in methanol and reduced to corresponding amines 25 and 26 using 10% Pd/C in the presence of hydrogen gas.
3. Results and discussion
3.1. Structure–activity relationships (SARs)
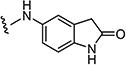
The cytotoxic data from the Tables 1–4 shows that the structure activity relationship (SAR) of these pyridopyrimidines (7a–7aar) is tightly regulated. Slight variation of X (N-8 position), Y (C-6 position) and Z (C-2 position) substitutions in 7a–7aar drastically affected cytotoxic activity of the molecules against tumor cell lines. Initially, when we started the synthesis of these pyridopyrimidines, we kept the methyl group constant at N-8 position of 7 and varied the C-6 position (Y) with different arylsulfonyl groups and then introduced different aromatic and heteroaryl amines at the Z (C-2 position) position of the pyridopyrimidine ring. Cytotoxicity data from Table 1 shows that keeping the arylsulfonyl groups constant at the C-6 position and introducing aromatic amines (7a–c) and heteroaryl amines (7d–7ap) at the C-2 position yielded heteroaryl amino compounds which were more active than the aryl amino substituted derivatives. Among the heterocyclic derivatives of 7d–7ap, bicyclic heterocyclic compounds (7d–7ak and 7an–ap) were found to be more active than monocyclic heterocyclic compounds (7al and 7am). Of the various bicyclic-heterocycles (7d–7ak and 7an–ap) tested, amino indoles (7d, 7e, 7h, 7i, 7l–o, 7t–7x, 7ac–ak, and 7an–ap) have shown better cytotoxicity compared to other benzo-fused five or six membered heterocyclic compounds (7f, 7g, 7j, 7k, 7p–s and 7y–7ab). Among the amino indoles tested for their cytotoxicity, 5-aminoindoles (7d, 7i, 7l, 7n, 7t, 7u, 7w, 7ad–ak and 7ao) showed higher activity compared to 4-amino indoles (7h, 7m, 7v, 7ac and 7an) and 6-amino indoles (7e, 7o, 7x and 7ap). These results suggest that 5-amino indole group is essential for the cytotoxic activity of the molecule and replacement of this moiety either by similar analogs (7f, 7j, 7p–s, 7y and 7z) or with corresponding 4th and 6th positional isomers (7e, 7h, 7m, 7o, 7v, 7x, 7ac, 7an and 7ap) results in substantial loss of activity of these compounds. Since 5-amino indole compounds showed best cytotoxic activity compared to the other analogs, this group was fixed at Z-position for subsequent SAR studies.
Table 1.
In vitro cytotoxicity of pyrido[2,3-d]pyrimidine (7a–ap) with variables at C2 and C6 positions
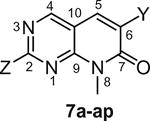
| ||||
|---|---|---|---|---|
|
| ||||
| Compd | Y | Z | IC50 (µM) | |
|
|
||||
| K562 | DU145 | |||
| 7a |
|
|
15 | 15 |
| 7b |
|
|
15 | 30 |
| 7c |
|
|
75 | 100 |
| 7d |
|
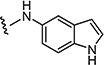
|
1 | 5 |
| 7e |
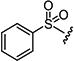
|
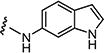
|
10 | 10 |
| 7f |
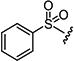
|
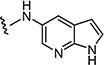
|
5 | 1 |
| 7g |
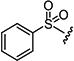
|
|
100 | 50 |
| 7h |
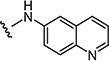
|
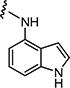
|
5 | 5 |
| 7i |
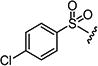
|
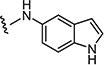
|
0.1 | 0.25 |
| 7j |
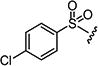
|
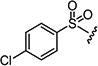
|
5 | 5 |
| 7k |
|
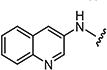
|
15 | 15 |
| 7l |
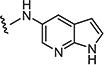
|
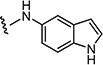
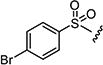
|
5 | 5 |
| 7m |
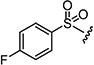
|
|
0.75 | 0.75 |
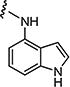
| 7n |
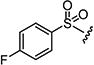
|
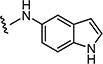
|
0.2 | 0.3 |
| 7o |
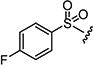
|
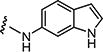
|
10 | 10 |
| 7p |
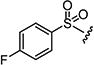
|
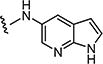
|
0.75 | 0.5 |
| 7q |
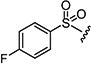
|
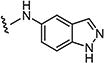
|
10 | 5 |
| 7r |
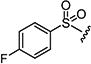
|
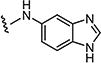
|
10 | 10 |
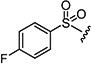
| 7s |
|
|
5 | 5 |
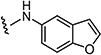
| 7t |
|
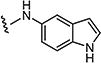
|
0.1 | 0.25 |
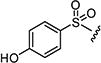
| 7u |
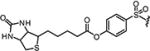
|
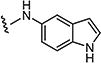
|
0.6 | 0.15 |
| 7v |
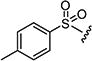
|
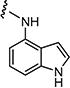
|
5 | 10 |
| 7w |
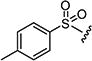
|
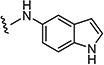
|
0.75 | 0.75 |
| 7x |
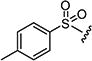
|
|
15 | 15 |
| 7y |
|
|
15 | 25 |
| 7z |
|
|
15 | 15 |
| 7aa |
|
|
10 | 10 |
| 7ab |
|
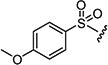
|
10 | 10 |
| 7ac |
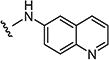
|
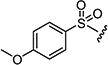
|
5 | 5 |
| 7ad |
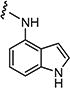
|
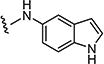
|
0.3 | 0.5 |
| 7ae |
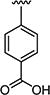
|
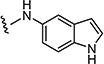
|
5 | 10 |
| 7af |
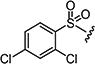
|
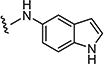
|
1 | 1 |
| 7ag |
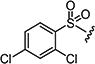
|
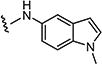
|
0.75 | 0.75 |
| 7ah |
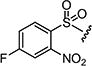
|
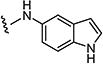
|
0.9 | 1.5 |
| 7ai |
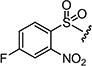
|
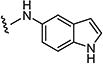
|
2 | 3 |
| 7aj |
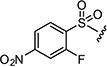
|
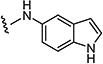
|
0.2 | 0.4 |
| 7ak |
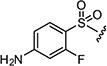
|
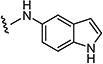
|
1.5 | 8 |
| 7al |
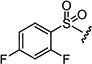
|
|
35 | 75 |
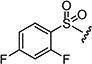
| 7am |
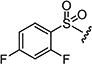
|
|
30 | 50 |
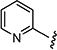
| 7an |
|
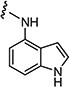
|
1.5 | 0.75 |
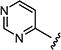
| 7ao |
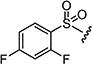
|
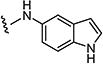
|
0.075 | 0.075 |
| 7ap |
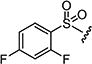
|
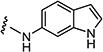
|
5 | 5 |
Table 4.
In vitro cytotoxicity and kinase profile of pyrido[2,3-d]pyrimidine (7aaj–aar) with variables at N8 position
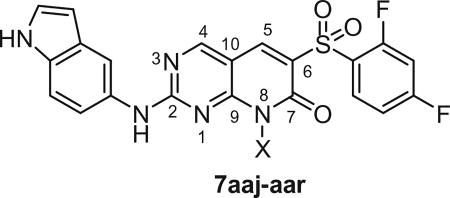
| |||
|---|---|---|---|
|
| |||
| Compd | X | IC50 (µM) | |
|
|
|||
| K562 | DU145 | ||
| 7aaj | H | 15 | 15 |
| 7aak |
|
0.75 | 0.75 |
| 7aal |
|
1 | 1 |
| 7aam |
|
5 | 10 |
| 7aan |
|
0.5 | 0.5 |
| 7aao |
|
5 | 5 |
| 7aap |
|
10 | 10 |
| 7aaq |
|
1 | 1 |
| 7aar |
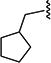
|
7 | 10 |
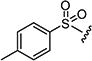
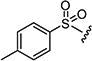
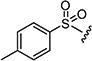
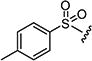
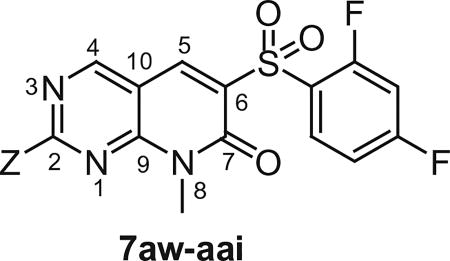
After optimizing the position of Z in 7 with 5-amino indole, we then focused our efforts on varying the Y position with different substitutions on the aromatic ring of the arylsulfonyl group. The cytotoxicity data (Table 1) indicated that arylsulfonyl ring with different substitutions exhibit moderate to excellent activity. Monosubstituted arylsulfonyl compounds (7i, 7l, 7n, 7t, 7u, 7w and 7ad) and di-substituted arylsulfonyl compounds (7af–ak and 7ao) were found to be more active than un-substituted arylsulfonyl compound (7d). Among mono-substituted compounds, bromo (7l) and carboxy (7ae) substituted compounds showed substantially less cytotoxic activity compared to chloro (7i), fluoro (7n), hydroxyl (7t), biotiniloxy (7u), methyl (7w) and methoxy (7ad) substituted compounds. Of the various di-substituted arylsulfonyl compounds, 2,4-dichloro (7ag), 2-fluoro-4-nitro (7aj) and 2,4-difluoro (7ao) compounds showed excellent activity compared to di-substituted 4-fluoro-2-nitro (7ah), 2-amino-4-fluoro (7ai) and 4-amino-2-fluoro (7ak) compounds. Surprisingly reduction of the nitro compounds (7ah and 7aj) to an amino group (7ai and 7ak) resulted in 10 fold less cytotoxic activity towards cancer cells. A comparison of the activities of all of the arylsulfonyl compounds listed in Table 1 showed that the molecule (7ao) with 2,4-difluoro arylsulfonyl at Y and 5-amino indole at Z position exhibits the best cytotoxic activity. In our attempts to achieve even more potent molecule than 7ao, we replaced arylsulfonyl group of the pyridopyrimidine ring with benzylsulfonyl (7aq and 7ar), arylcarbamoyl (7as and 7at) and arylsulfamyl (7au and 7av) (Table 2) groups. All these changes resulted in a drastic loss of biological activity indicating that 2,4-difluoro arylsulfonyl group is indispensable at that position for the cytotoxic activity of these compounds. Similar attempts were made to produce molecules that were more active than 7ao by introducing other bicyclic-heterocycles (7aw–7aai) (Table 3) at Z position of the pyridopyrimidine ring. However, these compounds were found to be substantially less active than 7ao. Only a few compounds (7ax, 7az, 7aaa and 7aai) showed a moderate cytotoxic activity indicating that 5-amino indole is critical for the activity at the Z position and is irreplaceable. Additional efforts were made to produce a more potent molecule than 7ao by varying the substitutions on the nitrogen at N-8 position of the pyridopyrimidine ring (7aaj–aar) (Table 4). The cytotoxicity data from Table 4 shows that any modifications at N-8 position of the ring other than methyl group results in a severe loss of activity. Only N-ethyl substitution (7aak) at N-8 position showed a moderate cell killing activity, which however, was 10 fold less than 7ao.
Table 2.
In vitro cytotoxicity and kinase profile of pyrido[2,3-d]pyrimidine (7aq–av) with variables at C6 positions
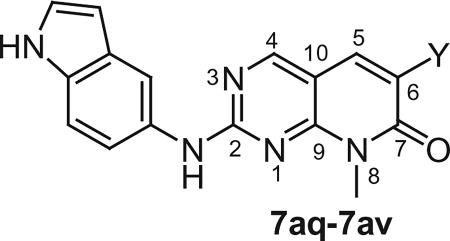
| |||
|---|---|---|---|
|
| |||
| Compd | Y | IC50 (µM) | |
|
|
|||
| K562 | DU145 | ||
| 7aq |
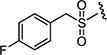
|
30 | 30 |
| 7ar |
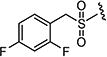
|
1 | 1 |
| 7as |
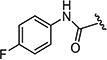
|
5 | 5 |
| 7at |
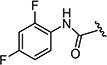
|
5 | 5 |
| 7au |
|
5 | 5 |
| 7av |
|
5 | 5 |
Table 3.
In vitro Cytotoxicity and kinase profile of pyrido[2,3-d]pyrimidine (7aw–aai) with variable at C2 position
| |||
|---|---|---|---|
|
| |||
| Compd | Z | IC50 (µM) | |
|
|
|||
| K562 | DU145 | ||
| 7aw |
|
50 | 30 |
| 7ax |
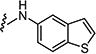
|
0.5 | 0.5 |
| 7ay |
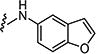
|
15 | 15 |
| 7az |
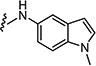
|
0.75 | 0.75 |
| 7aaa |
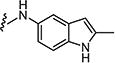
|
0.75 | 0.75 |
| 7aab |
|
75 | 100 |
| 7aac |
|
5 | 5 |
| 7aad |
|
30 | 75 |
| 7aae |
|
5 | 5 |
| 7aaf |
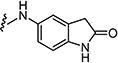
|
15 | 100 |
| 7aag |
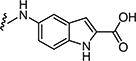
|
10 | 10 |
| 7aah |
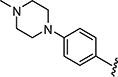
|
5 | 5 |
| 7aai |
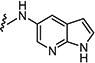
|
0.75 | 0.75 |
Results from the cytotoxicity data (Tables 1–4) suggested that 7ao was the best molecule in this class of compounds and hence we performed all subsequent in vitro and in vivo biological studies using this compound.
3.2. Biological results
3.2.1. In vitro anti-tumor activity of 7n, 7t, 7u and 7ao
We next tested the cytotoxicity of the most active compounds in tissue culture growth inhibitory (GI50) assays against a panel of 16 cancer cell lines (Table 5). These cell lines were chosen based upon the tissue of origin and aggressiveness of tumors propagated in xenograft models. This screen revealed four structurally related compounds (7n, 7t, 7u and 7ao) that inhibited the growth of all the 16 tumor cell lines tested, at concentrations between 25 and 2500 nM (Table 5). As can be seen from the data, compound 7ao was the most potent inhibitor of cancer cell growth followed by compounds 7n, 7t and 7u. We then tested 7ao, the most active of the four compounds, in an extended panel of tumor cell lines (Table 6) and observed growth inhibition of multiple tumor cell types, suggesting that 7ao inhibits cell proliferation by blocking one or more key component(s) of growth signaling pathways.
Table 5.
Evaluation of 7n, 7t, 7u and 7ao against a panel of human tumor cell lines
| Cell line | GI50 (µM) | |||
|---|---|---|---|---|
|
|
||||
| 7n | 7t | 7u | 7ao | |
| DU145 | 0.3 | 0.75 | 0.75 | 0.075 |
| MCF-7 | 0.25 | 0.5 | 0.5 | 0.075 |
| BT474 | 0.2 | 0.2 | 0.25 | 0.1 |
| SK-OV-3 | 0.2 | 0.75 | 0.4 | 0.075 |
| MIA-PaCa-2 | 0.075 | 0.1 | 0.1 | 0.075 |
| SK-MEL-28 | 0.3 | 0.5 | 0.5 | 0.2 |
| A549 | 0.2 | 0.75 | 2.5 | 0.075 |
| U87 | 0.4 | 1 | 1 | 0.02 |
| COLO-205 | 0.2 | 0.25 | 0.5 | 0.075 |
| HELA | 0.075 | 0.75 | 0.5 | 0.05 |
| H1975 | 0.1 | 0.75 | 0.75 | 0.075 |
| RAJI | 0.1 | 0.5 | 0.4 | 0.05 |
| U205 | 0.055 | 0.35 | 0.35 | 0.035 |
| K562 | 0.1 | 0.5 | 0.5 | 0.075 |
| GRANTA-519 | 0.06 | 0.25 | 0.5 | 0.04 |
Table 6.
Evaluation of 7ao against a panel of human tumor cell lines
| Cell line | Tumor type | 7ao GI50 (µM) |
|---|---|---|
| K562 | Chronic myelogenous leukemia | 0.075 |
| DU145 | Prostate | 0.075 |
| RAJI | Burkitt’s lymphoma (B-cell) | 0.05 |
| SK-MEL-28 | Skin melanoma | 0.2 |
| SK-OV-3 | Ovarian | 0.075 |
| MCF-7 | Breast | 0.075 |
| MIA-PaCa-2 | Pancreatic | 0.075 |
| A549 | Non small cell lung carcinoma | 0.075 |
| U87 | Glioblastoma | 0.2 |
| COLO-205 | Colo-rectal | 0.075 |
| BT-474 | Breast | 0.1 |
| HELA | Cervical | 0.075 |
| GRANTA-519 | Mantle cell lymphoma | 0.04 |
| JURKAT | acute t cell leukemia | 0.025 |
| U2OS | Osteosarcoma | 0.05 |
| A431 | Epidermoid | 0.03 |
| WI-38 | Normal fibroblast-lung | >5.0 |
| HFL-1 | Normal fibroblast-lung | >2.0 |
| H460 | Lung (large cell carcinoma) | 0.1 |
| BxPC-3 | Pancreatic | 0.075 |
| SU.86.86 | Pancreatic | 0.4 |
| BT-549 | Lung | 0.25 |
| H292 | Non-small cell lung carcinoma | 0.15 |
| H1650 | Non-small cell lung carcinoma | 0.2 |
| UM-UC-3 | Bladder | 0.3 |
| 5637 | Bladder | 0.125 |
| T24 | Bladder | 0.15 |
| SW780 | Bladder | 0.15 |
| U-937 | Histiocytic lymphoma (monocytic) | 0.125 |
| DAUDI | Burkitt’s lymphoma (B-cell) | 0.075 |
| HL-60 | Acute myeloid leukemia | 0.125 |
| TCC-SUP | Bladder | 0.25 |
| RT-4 | Bladder | 0.5 |
| PC-3 | Prostate | 0.15 |
| HCT-116 | Colo-rectal | 0.15 |
| CEM | T-ALL | 0.05 |
| Z-138 | Mantle cell lymphoma | 0.2 |
| PANC 10.05 | Pancreatic | 0.4 |
| PANC 03.25 | Pancreatic | 0.2 |
| ASPC-1 | Pancreatic | 0.3 |
| HPAF-II | Pancreatic | 0.25 |
| Capan-1 | Pancreatic | 0.075 |
| LNCaP | Prostate | 0.04 |
| BEL-7402 | Hepatocellular carcinoma | 0.05 |
| SNU-449 | Hepatocellular carcinoma | 0.4 |
| SNU-398 | Hepatocellular carcinoma | 0.2 |
| PLC/PRF/5 | Hepatocellular carcinoma | 0.75 |
| SNU-428 | Hepatocellular carcinoma | 0.75 |
| SNU-398 | Hepatocellular carcinoma | 0.15 |
| SNU-475 | Hepatocellular carcinoma | 7.5 |
| Hep-G2 | Hepatocellular carcinoma | 0.5 |
| SK-HEP-1 | Liver adenocarcinoma (metastatic) | 0.1 |
| H359 | Non-small cell lung carcinoma | 0.15 |
| H23 | Non-small cell lung carcinoma | 0.2 |
| H1734 | Non-small cell lung carcinoma | 0.15 |
| H827 | Non-small cell lung carcinoma | 0.5 |
| H1975 | Non-small cell lung carcinoma | 0.2 |
| MDA-MB-436 | Breast | 0.15 |
| MDA-MB-453 | Breast | 0.075 |
| HCC1806 | Breast | 0.1 |
3.2.2. Kinase inhibition profile of 7n and 7ao
To understand the molecular basis for the high potency exhibited by compounds 7n and 7ao, we tested in vitro kinase inhibitory activities of these two compounds against a panel of 355 functional protein kinases (Reaction Biology Corp) (see Supporting information Table 1 and Fig. 1 for 7ao). Interestingly, this kinase profiling study revealed that both compounds are highly specific inhibitors of Polo like kinase 2 (PLK2).10 We next examined the ability of all of the compounds described in Tables 1–4 to inhibit PLK2 in in vitro kinase assays using recombinant PLK2 kinase. The results of these experiments are shown in Table 7. The kinase assays performed with the three structurally related compounds 7n, 7t and 7u are shown in Figure 1. These studies suggest that inhibition of PLK2 is detrimental for tumor cell proliferation and/or survival. It is also interesting to note that 7ao, which is the most potent inhibitor of this kinase amongst the compounds tested, also exhibits highest cytotoxicity against tumor cells, suggesting a functional relationship between PLK2 inhibitory activity and cytotoxicity. Further in vitro kinase and differential scanning fluorimetric (DSF)16,17 testing revealed that 7ao is a selective inhibitor of PLK2 with no inhibitory activity against PLK1, PLK3 and PLK4 (Fig. 2A and B).
Figure 1.

In vitro PLK2 kinase inhibitory activity of 7n, 7t, 7u and 7ao. Ten nano grams of recombinant PLK2 was mixed with the indicated concentrations of compounds 7n, 7t, 7u and 7ao. Kinase assays were performed as described in Section 5.1 using dephosphorylated α-casein as substrate.
Table 7.
PLK2 inhibitory activity of 7d, 7m, 7n, 7t, 7ad and 7ao
| Compound | PLK2 IC50 (µM) |
|---|---|
| 7d | 1.23 |
| 7m | 6.08 |
| 7n | 1.07 |
| 7t | 0.47 |
| 7ad | 0.35 |
| 7ao | 0.31 |
Figure 2.

(A) Binding of 7ao (ON1231320) is specific towards PLK2 and not PLK1-recombinant proteins (PLK2 or PLK1 kinase domain) were incubated with different small molecules and assayed for binding by differential scanning fluorimetry (DSF). 7ao shows specific binding to PLK2 and not to PLK1. Controls using BI2536, show both proteins binding to the PLK pan inhibitor. (B) Recombinant human PLKs were incubated with the indicated concentrations of the compounds. The kinase activity was assayed by using recombinant casein or synuclein as substrate in the presence of γ32P-ATP. IC50 values were calculated from non-sigmoidal regression plots with variable slope using GraphPad Prism software.
3.2.3. Prediction of 7ao binding to the PLK2 ATP binding pocket
To gain a better understanding of the possible mode of 7ao binding to the PLK2 kinase domain which permits specificity, molecular docking and energy minimization studies were conducted using the PLK2-1C8 (PDB 4I6H) X-ray co-crystal structure. Prediction of the binding of the inhibitor to the PLK2 kinase domain shows that multiple interactions stabilize the binding of 7ao to PLK2 (Fig. 3A and B). Positioning of the difluoro phenyl moiety by van der Waals interactions with the side chains of residues Asn210, Phe212, Asp223 and the α-nitrogen atom within the main chain of Asp223 appear to be of high importance. In particular several of these interactions are observed with fluorine at position 2, noting the contribution of this atom for the observed binding. Phe212 is predicted to also contribute to the stabilization of pyridopyrimidine ring. In addition, Nitrogen atom at positions 1 and methyl group at positions 8 of the pyridopyrimidine ring are predicted to interact with Leu88. The carbonyl oxygen atom position 7 may be stabilized by interactions with the side chain of Cys96. The sulfone moiety of 7ao is predicted to be within a distance that permits van der Waal interactions with the side chains of Lys111 and the PLK2 gatekeeper residue, Leu159. Another main interaction in the form of a hydrogen bond is predicted to form between the nitrogen atom at position 1 of the pyridopyrimidine ring and the nitrogen atom in the main chain of Cys162. A second hydrogen bond is also likely to form between the amino group located between the pyridopyrimidine and the indole ring with the oxygen atom in the main chain of Cys162. Positioning of the amino indole group is mainly governed by interactions with side chains of Leu88, Tyr161 and Arg165. Overall, these predicted interactions result in the optimal positioning of 7ao to the ATP binding pocket of the PLK2 kinase domain.
Figure 3.

Model of 7ao binding to PLK2. 7ao binding to the kinase domain of PLK2 was predicted by docking and energy minimization using the X-ray crystal structure of PLK2-1C8 (4I6H) as a reference. Representations of the predicted lowest energy binding (PLK2/7ao) were prepared using PyMOL. (A) Ribbon representation of PLK2 kinase domain (green) bound to 7ao (magenta). 7ao is shown as stick. (B) Close up view showing proximal residues of PLK2 to 7ao predicted to be important for binding. (C) Superimposition of PLK1 crystal structure (2RKU) onto PLK2. PLK1 and PLK2 are shown as cyan and green ribbons, respectively.
Positioning of 7ao in the ATP binding pocket of the PLK2 kinase domain is dictated by multiple and distinct interactions that permit high affinity and specificity for this member of the PLK family (Fig. 3A and B). In particular, PLK2 Tyr161, which corresponds to PLK1 Leu132, seems to be a major determinant for selectivity within the PLK family. The predicted proximity of Tyr161 to the 5-amino indole moiety of 7ao will most likely facilitate an edge-to-face interaction, as has been described for another PLK2 ATP-mimetic inhibitor.18 This type of interaction will not occur with PLK1 (Fig. 3C). Another determinant for selectivity could be due to the higher flexibility of the PLK2 P-loop as compared to that which is seen in PLK-1 co-crystal structures. The position of the amino group at position 5 of the indole moiety in 7ao also appears to be critical for binding and specificity, since substitution at a different position (i.e., at position 4) may cause a steric clash with the hinge loop in PLK2. At the center of the molecule, the presence of carbonyl oxygen atom and a methyl group at positions 7 and 8 of the pyridopyrimidine ring are predicted to aid in the stabilization of the molecule in the PLK2 ATP binding site. The 2,4-difluoro benzene moiety at the other end of the small molecule fits deep within the binding site and is adequately accommodated between the side chains of three amino acid residues (Asn210, Phe212, Asp223). Other spatial orientations of this moiety do not appear to be favorable. Furthermore, given the predicted interactions of the difluoro benzene, substitution or removal of the highly electronegative fluorides will have a detrimental effect in the binding and activity of the molecule, which was seen with compounds 7d, 7l, 7ae. Finally, in addition to directly interacting with PLK2, the sulfone moiety might participate in electron delocalization from the adjacent difluoro benzene ring. The importance of the functionality of these groups at specific positions of the molecule is denoted by a loss of SAR activity as observed in the kinase and cytotoxicity assays and is supported by the modeling studies.
3.3. 7n and 7ao activate programmed cell death in human tumor cells
The cytotoxicity exhibited by these compounds against cancer cells prompted us to investigate the mechanism by which these compounds induced cell death. We employed a fluorescent/luminescent assay combination (Cell Titer Blue/Caspase Glo-3/7, Promega Corp) that measures both cell viability and Caspase 3/7 activity (a measure of apoptosis) to determine the effects of compounds 7n and 7ao on U2OS human osteosarcoma cells following a 24 h exposure (Fig. 4). The results show that compounds 7n and 7ao increase the activity of Caspases 3/7 in a dose-dependent manner, suggesting that both compounds induce apoptosis.
Figure 4.

Induction of caspase activity by 7n and 7ao. U2OS cells were treated with the indicated concentrations of each compound for 24 h. Cell viability (blue) and the induction of Caspase 3/7 activity (red) were measured as described in Section 5.1.
3.4. 7ao does not inhibit tubulin polymerization
To test the possibility that these compounds exert their growth inhibitory effect by directly inhibiting tubulin polymerization, we examined their effect on tubulin polymerization in a cell free system. MAP-rich tubulin (Cytoskeleton Inc., Denver, CO) was reconstituted in polymerization buffer and its polymerization kinetics assayed by UV/VIS spectrophotometry at 305 nm. The results of this study (Fig. 5) show that while compounds 7n and 7t inhibit tubulin polymerization in a vincristine-like manner (albeit at a 10-fold higher concentration), compound 7u only shows partial inhibitory activity. In contrast, compound 7ao did not appreciably inhibit tubulin polymerization, suggesting that this compound is likely to be less toxic to normal cells which tend to be sensitive to the toxic effects of tubulin depolymerizing agents.
Figure 5.

7ao does not affect in vitro tubulin polymerization. MAP-rich Tubulin was polymerized in the presence of DMSO, 1 µM Vincristine (positive control for inhibition of polymerization) or 10 µM of compounds 7n, 7t, 7u and 7ao as described in Section 5.1.
3.5. In vivo antitumor efficacy of 7ao
To determine the efficacy of compound 7ao in vivo using a tumor xenograft model, MDAMB-231 triple negative breast cancer cells were injected subcutaneously into nude mice. Once the tumors reached an average volume of 70 mm3, 7ao was administered on alternate days (Q2D) via intraperitoneal (IP) injection at a dose of 75 mg/kg body weight. This treatment with 7ao resulted in significant inhibition of tumor growth (86.5%) as compared to the control group (Fig. 6A), and no overt signs of toxicity were observed in the 7ao treated group (body weights shown in Fig. 6B). The potent in vivo anticancer efficacy of compound 7ao and its low toxicity profile in mice portend well for its development for potential clinical use.
Figure 6.

In vivo efficacy of 7ao in subcutaneous MDAMB-231 xenografts. Subcutaneous MDAMB-231 triple negative breast cancer xenografts were established in nude mice (n = 10 per group). Treatment was started when the average tumor volume reached 75 mm3. Compound 7ao at 75 mg/kg or vehicle was administered ip every other day (Q2D) and the tumor volumes (A) and body weights (B) measured over the course of the experiment.
3.6. Conclusion
To identify possible oncogenic kinases to which tumor cells might be addicted, we screened our compound library of ATP-mimetic kinase inhibitors for candidate compounds that can preferentially induce the death of human tumor cells without affecting normal cell viability. This approach led to the identification of a novel kinase inhibitor, 7ao, an arylsulfonyl pyrido-pyrimidinone, that specifically inhibits PLK2 isoform (SNK) and 7ao also causes mitotic catastrophe and apoptosis of cultured tumor cell lines. Here, we describe the synthesis of 6-arylsulfonyl pyridopyrimidinones and their structure/function characteristics, which suggest that the cytotoxic activity of 6-arylsulfonyl pyridopyrimidinones is dependent on the nature and position of substitutions at C-2, C-6 and N-8—positions. Compound 7ao, with a 5-aminoindolyl group at the C-2 position, 2,4-difluorophenyl group at the C-6 position and methyl group at the N-8 position, showed optimum biological activity. Kinase profiling revealed that 7ao inhibited PLK2 with a high degree of specificity. 7ao could not inhibit the kinase activities of PLK1, PLK3, or PLK4 in vitro, even at concentrations as high as 10 µM. Incubation of human tumor cell lines with 7ao resulted in a dose-dependent increase in Caspase 3/7 activity suggesting that this compound activates programmed cell death in human tumor cells. In tubulin polymerization assays, 7ao did not inhibit tubulin polymerization, indicating that this compound’s mechanism of action is distinct from tubulin poisons such as paclitaxel and vincristine. 7ao is well tolerated in mice, and reduces tumor burden in mice carrying subcutaneous and orthotopic xenografts of human tumor cells. Together, these studies demonstrate that PLK2 may be an important cancer target and suggest that its inhibitors can be used in cancer therapy.
4. Experimental section
4.1. Chemistry
All reagents and solvents were obtained from commercial suppliers and used without further purification unless otherwise stated. Solvents were dried using standard procedures and reactions requiring anhydrous conditions were performed under N2 atmosphere. Reactions were monitored by Thin Layer Chromatography (TLC) on preloaded silica gel F254 plates (Sigma–Aldrich) with a UV indicator. Column chromatography was performed using Merck 70–230 mesh silica gel 60 Å. Yields were of purified product and were not optimized. Melting points were determined using an Electro thermal Mel-Temp 3.0 micro melting point apparatus and are uncorrected. 1H NMR spectra were obtained using a Bruker AVANCE 300, 400 and 600 MHz spectrometer. Chemical shifts are reported in parts per million (δ) downfield using tetramethylsilane (SiMe4) as internal standard. Spin multiplicities are given as s (singlet), d (doublet), t (triplet), dd (double doublet), br s (broad singlet), m (multiplet), and q (quartet). All LC/MS data were gathered on an Agilent 1200 LC with Agilent 6410 triple quadrupole mass spectrometer detectors. The compound solution was infused into the electrospray ionization source operating positive and negative modes in methanol/water/TFA (50:50:0.1% v/v) at 0.4 mL/min. The sample cone (declustering) voltage was set at 100 V. The instrument was externally calibrated for the mass range m/z 100 to m/z 1000. The purity of the final compounds was determined by HPLC and is 95% or higher unless specified otherwise. ZorbaxExlipse XDB C18 (150 × 4.6 mm, 5 µm particle size) using gradient elution of acetonitrile in water, 20–90% for 25 min at a flow rate of 1 mL/min with detection at 235 nm wavelength. For all samples 0.00154% AcONH-4 was added to water. The aldehydes (4a–g),11 and the active methylene compounds 10,12 13,13 16,14 and 1815 were prepared as per the reported procedures.
4.1.1. General procedure for the preparation of 2-(arylthio) acetic acid (9) (Scheme 4)
The following 2-(arylthio)acetic acids were prepared according to the procedure reported in the literature.12
4.1.1.1. 2-(Phenylthio)acetic acid (9a)
Alkylation of thiophenol with chloroacetic acid yielded 65% 2-(phenylthio)acetic acid (9a); mp 59–61 °C; 1H NMR (300 MHz, DMSO-d6): δ 3.58 (s, 2H), 7.08–7.15 (m, 3H), 7.30 (d, J = 8.1 Hz, 2H), 11.89 (br s, 1H). MS found (M+H)+ (m/z), 169.03; calcd for C8H8O2S m/z, 168.02.
4.1.1.2. 2-((4-Fluorophenyl)thio)acetic acid (9b)
Alkylation of 4-fluorothiophenol with chloroacetic acid yielded 63% 2-((4-fluorophenyl) thio)acetic acid (9b); mp 78–80 °C; 1H NMR (300 MHz, DMSO-d6): δ 3.46 (s, 2H), 6.68–6.73 (m, 2H), 6.89–6.93 (m, 2H), 12.20 (br s, 1H). MS found (M+H)+ (m/z), 187.03; calcd for C8H7-FO2S m/z, 186.02.
4.1.1.3. 2-((4-Chlorophenyl)thio)acetic acid (9c)
Alkylation of 4-chlorothiophenol with chloroacetic acid yielded 63% 2-((4-chlorophenyl)thio)acetic acid (9c); mp 96–99 °C; 1H NMR (300 MHz, DMSO-d6): δ 3.44 (s, 2H), 7.52 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.2 Hz, 2H), 11.96 (br s, 1H). MS found (M+H)+ (m/z), 202.98; calcd for C8H7ClO2S m/z, 201.99.
4.1.1.4. 2-((4-Bromophenyl)thio)acetic acid (9d)
Alkylation of 4-bromothiophenol with chloroacetic acid yielded 67% 2-((4-bromophenyl)thio)acetic acid (9d); mp 113–115 °C; 1H NMR (300 MHz, DMSO-d6): δ 3.57 (s, 2H), 7.48 (d, J = 8.0 Hz, 2H), 7.65 (d, J = 8.1 Hz, 2H), 12.45 (br s, 1H). MS found (M+H)+ (m/z), 246.97; calcd for C8H7BrO2S m/z, 245.94.
4.1.1.5. 2-((4-Hydroxyphenyl)thio)acetic acid (9e)
Alkylation of 4-mercaptophenol with chloroacetic acid yielded 55% 2-((4-hydroxyphenyl)thio)acetic acid (9e); mp 132–136 °C; 1H NMR (300 MHz, DMSO-d6): δ 3.51 (s, 2H), 4.35 (br s, 1H), 6.72 (d, J = 8.1 Hz, 2H), 7.23 (d, J = 8.1 Hz, 2H), 12.07 (br s, 1H). MS found (M+H)+ (m/z), 185.03; calcd for C8H8O3S m/z, 184.02.
4.1.1.6. 2-(p-Tolylthio)acetic acid (9f)
Alkylation of 4-methylthiophenol with chloroacetic acid yielded 65% 2-((p-tolylthio) acetic acid (9f); mp 77–78 °C; 1H NMR (300 MHz, DMSO-d6): δ 2.53 (s, 3H), 3.47 (s, 2H), 7.38 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 8.2 Hz, 2H), 12.44 (br s, 1H). MS found (M+H)+ (m/z), 183.04; calcd for C9H10O2S m/z, 182.04.
4.1.1.7. 2-((4-Methoxyphenyl)thio)acetic acid (9g)
Alkylation of 4-methoxythiophenol with chloroacetic acid yielded 64% 2-((4-methoxyphenyl)thio)acetic acid (9g); mp 68–70 °C; 1H NMR (300 MHz, DMSO-d6): δ 3.57 (s, 2H), 3.96 (s, 3H), 7.31 (d, J = 7.8 Hz, 2H), 7.47 (d, J = 7.8 Hz, 2H), 12.70 (br s, 1H). MS found (M+H)+ (m/z), 199.06; calcd for C9H10O3S m/z, 198.04.
4.1.1.8. 2-((4-Carboxyphenyl)thio)acetic acid (9h)
Alkylation of 4-mercaptobenzoic acid with chloroacetic acid yielded 60% 2-((4-carboxyphenyl)thio)acetic acid (9h); mp 274–276 °C; 1H NMR (300 MHz, DMSO-d6): δ 3.68 (s, 2H), 7.48 (d, J = 8.0 Hz, 2H), 7.92 (d, J = 8.0 Hz, 2H), 12.89 (br s, 2H). MS found (M+H)+ (m/z), 213.04; calcd for C9H8O4S m/z, 212.01.
4.1.1.9. 2-((2,4-Difluorophenyl)thio)acetic acid (9i)
Alkylation of 2,4-difluorothiophenol with chloroacetic acid yielded 60% 2-((2,4-difluorophenyl)thio)acetic acid (9i); mp 78–80 °C; 1H NMR (600 MHz, CDCl3): δ 3.57 (s, 2H), 6.86–6.87 (m, 2H), 7.49–7.50 (m, 1H). MS found (M+H)+ (m/z), 205.10; calcd for C8H6F2O2S m/z, 204.01.
4.1.1.10. 2-((2,4-Dichlorophenyl)thio)acetic acid (9j)
Alkylation of 2,4-dichlorothiophenol with chloroacetic acid yielded 60% 2-((2,4-dichlorophenyl)thio)acetic acid (9j); mp 110–112 °C; 1H NMR (600 MHz, CDCl3): δ 3.68 (s, 2H), 7.19–7.21 (dd, J = 8.4 and 2.4 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.42 (d, J = 2.4 Hz, 1H). MS found (M+H)+ (m/z), 236.95; calcd for C8H6Cl2O2S m/z, 235.95.
4.1.2. General procedure for the preparation of 2-(arylsulfonyl) acetic acid (10) (Scheme 4)
The following 2-(arylsulfonyl)acetic acids were prepared according to the procedure reported in the literature.12
4.1.2.1. 2-(Phenylsulfonyl)acetic acid (10a)
Oxidation of 2-(phenylthio)acetic acid 9a with 30% hydrogen peroxide yielded 83% of corresponding 2-(phenylsulfonyl)acetic acid (10a); mp 108–110 °C; 1H NMR (300 MHz, DMSO-d6): δ 4.38 (s, 2H), 7.64–7.72 (m, 3H), 7.89 (d, J = 7.8 Hz, 2H), 12.24 (br s, 1H). MS found (M+H)+ (m/z), 201.01; calcd for C8H8O4S m/z, 200.01.
4.1.2.2. 2-((4-Fluorophenyl)sulfonyl)acetic acid (10b)
Oxidation of 2-((4-fluorophenyl)-thio)acetic acid 9b with 30% hydrogen peroxide yielded 75% of corresponding 2-((4-fluorophenyl) sulfonyl)acetic acid (10b); mp 120–122 °C; 1H NMR (400 MHz, DMSO-d6): δ 4.54 (s, 2H), 7.48–7.52 (m, 2H), 7.99–8.02 (m, 2H), 13.13 (br s, 1H). MS found (M+H)+ (m/z), 219.02; calcd for C8H7FO4S m/z, 218.00.
4.1.2.3. 2-((4-Chlorophenyl)sulfonyl)acetic acid (10c)
Oxidation of 2-((4-chlorophenyl)-thio)acetic acid 9c with 30% hydrogen peroxide yielded 90% of corresponding 2-((4-chlorophenyl)sulfonyl)acetic acid (10c); mp 123–124 °C; 1H NMR (400 MHz, DMSO-d6): δ 4.53 (s, 2H), 7.71 (d, J = 8.1 Hz, 2H), 7.91 (d, J = 8.0 Hz, 2H), 13.30 (br s, 1H). MS found (M+H)+ (m/z), 234.97; calcd for C8H7ClO4S m/z, 233.98.
4.1.2.4. 2-((4-Bromophenyl)sulfonyl)acetic acid (10d)
Oxidation of 2-((4-bromophenyl)-thio)acetic acid 9d with 30% hydrogen peroxide yielded 86% of corresponding 2-((4-bromophenyl) sulfonyl)acetic acid (10d); mp 136–138 °C; 1H NMR (300 MHz, DMSO-d6): δ 4.46 (s, 2H), 7.63 (d, J = 8.4 Hz, 2H), 7.88 (d, J = 8.3 Hz, 2H), 13.12 (br s, 1H). MS found (M+H)+ (m/z), 278.95; calcd for C8H7BrO4S m/z, 277.92.
4.1.2.5. 2-((4-Hydroxyphenyl)sulfonyl)acetic acid (10e)
Oxidation of 2-((4-hydroxyphenyl)-thio)acetic acid 9e with 30% hydrogen peroxide yielded 70% of corresponding 2-((4-hydroxyphenyl) sulfonyl)acetic acid (10e); mp 182–184 °C; 1H NMR (300 MHz, DMSO-d6): δ 4.04 (s, 2H), 6.70 (d, J = 9.0 Hz, 2H), 6.21 (br s, 1H), 7.47 (d, J = 8.7 Hz, 2H), 11.59 (br s, 1H). MS found (M+H)+ (m/z), 217.01; calcd for C8H8O5S m/z, 216.01.
4.1.2.6. 2-Tosylacetic acid (10f)
Oxidation of 2-((4-tolyl)thio)acetic acid 9f with 30% hydrogen peroxide yielded 78% of corresponding 2-tosyl acetic acid (10f); mp 98–101 °C; 1H NMR (300 MHz, DMSO-d6): δ 2.58 (s, 3H), 4.39 (s, 2H), 7.03 (d, J = 8.3 Hz, 2H), 7.55 (d, J = 8.1 Hz, 2H), 12.57 (br s, 1H). MS found (M+H)+ (m/z), 215.03; calcd for C9H10O4S m/z, 214.03.
4.1.2.7. 2-((4-Methoxyphenyl)sulfonyl)acetic acid (10g)
Oxidation of 2-((4-methoxyphenyl)-thio)acetic acid 9g with 30% hydrogen peroxide yielded 83% of corresponding 2-((4-methoxyphenyl)sulfonyl)acetic acid (10g); mp 113–114 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.73 (s, 3H), 4.28 (s, 2H), 7.03 (d, J = 7.9 Hz, 2H), 7.72 (d, J = 8.1 Hz, 2H), 12.96 (br s, 1H). MS found (M+H)+ (m/z), 231.00; calcd for C9H10O5S m/z, 230.02.
4.1.2.8. 2-((4-Carboxyphenyl)sulfonyl)acetic acid (10h)
Oxidation of 2-((4-carboxyphenyl)-thio)acetic acid 9h with 30% hydrogen peroxide yielded 74% of corresponding 2-((4-carboxyphenyl) sulfonyl)acetic acid (10h); mp 163–165 °C; 1H NMR (300 MHz, DMSO-d6): δ 4.64 (s, 2H), 8.11 (d, J = 7.7 Hz, 2H), 8.20 (d, J = 7.8 Hz, 2H), 13.04 (br s, 2H). MS found (M+H)+ (m/z), 245.02; calcd for C9H8O6S m/z, 244.00.
4.1.2.9. 2-((2,4-Difluorophenyl)sulfonyl)acetic acid (10i)
Oxidation of 2-((2,4-difluoro-phenyl)thio)acetic acid 9i with 30% hydrogen peroxide yielded 70% of corresponding 2-((2,4-difluorophenyl) sulfonyl)acetic acid (10i); mp 120–122 °C; 1H NMR (400 MHz, DMSO-d6): δ 4.54 (s, 2H), 7.33–7.38 (m, 1H), 7.59–7.64 (m, 1H), 7.91–7.97 (m, 1H). MS found (M+H)+ (m/z), 237.00; calcd for C8H6F2O4S m/z, 236.00.
4.1.2.10. 2-((2,4-Dichlorophenyl)sulfonyl)acetic acid (10j)
Oxidation of 2-((2,4-dichloro-phenyl)thio)acetic acid 9j with 30% hydrogen peroxide yielded 73% of corresponding 2-((2,4-dichlorophenyl)sulfonyl)acetic acid (10j); mp 140–142 °C; 1H NMR (600 MHz, DMSO-d6): δ 4.62 (s, 2H), 7.71 (d, J = 8.4, 1H), 7.98–8.00 (m, 2H). MS found (M+H)+ (m/z), 268.94; calcd for C8H6 Cl2O4S m/z, 267.94.
4.1.3. General procedure for the preparation of benzylthioacetic acids (Scheme 5)
The following benzylthioacetic acids were prepared according to the procedure reported in the literature.13
4.1.3.1. 4-Fluorobenzylthioacetic acid (12a)
Condensation of 4-fluorobenzyl bromide 11a with mercaptoacetic acid yielded 96% of the corresponding 4-fluorobenzylthioacetic acid (12a); mp 41–42 °C; 1H NMR (600 MHz, CDCl3): δ 3.17 (s, 2H), 3.84 (s, 2H), 7.12–7.14 (m, 2H), 7.31–7.34 (m, 2H), 11.58 (br s, 1H). MS found (M+H)+ (m/z), 201.03; calcd for C9H9FO2S m/z, 200.03.
4.1.3.2. 2,4-Difluorobenzylthioacetic acid (12b)
Condensation of 2,4-difluorobenzyl bromide 11b with mercaptoacetic acid yielded 90% of the corresponding 2,4-difluorobenzylthioacetic acid (12b); mp 58–60 °C; 1H NMR (600 MHz, CDCl3): δ 3.19 (s, 2H), 3.88 (s, 2H), 6.83–6.89 (m, 2H), 7.32–7.36 (m, 1H), 11.44 (br s, 1H). MS found (M+H)+ (m/z), 219.03; calcd for C9H8F2O2S m/z, 218.02.
4.1.4. General procedure for the preparation of benzylsulfonylacetic acids (13) (Scheme 5)
The following benzylsulfonylacetic acids were prepared according to the procedure reported in the literature.13
4.1.4.1. 4-Fluorobenzylsulfonylacetic acid (13a)
Oxidation of 4-fluorobenzylthioacetic acid 12a with 30% hydrogen peroxide yielded 93% of the corresponding 4-fluorobenzylsulfonylacetic acid 13a; mp 158–160 °C; 1H NMR (600 MHz, CDCl3): δ 3.89 (s, 2H), 4.58 (s, 2H), 6.94–6.99 (m, 2H), 7.48–7.52 (m, 2H), 11.89 (br s, 1H). MS found (M+H)+ (m/z), 233.02; calcd for C9H9FO4S m/z, 232.02.
4.1.4.2. 2,4-Difluorobenzylsulfonylacetic acid (13b)
Oxidation of 2,4-difluorobenzylthioacetic acid 12b with 30% hydrogen peroxide yielded 90% of the corresponding 2,4-difluorobenzylsulfonylacetic acid 13b; mp 116–118 °C; 1H NMR (600 MHz, CDCl3): δ 3.99 (s, 2H), 4.61 (s, 2H), 6.93–7.00 (m, 2H), 7.51–7.55 (m, 1H), 11.48 (br s, 1H). MS found (M+H)+ (m/z), 251.00; calcd for C9H8F2O4S m/z, 250.01.
4.1.5. General procedure for the preparation of methyl 3-((aryl) amino)-3-oxopropanoate (15) (Scheme 6)
The following methyl 3-((aryl)amino)-3-oxopropanoates (15) were prepared according to the procedure reported in the literature.14
4.1.5.1. Methyl 3-((4-fluorophenyl)amino)-3-oxopropanoate (15a)
Reaction of methyl 3-chloro-3-oxopropionate with 4-fluoroaniline 14a produced methyl 3-((4-fluorophenyl)amino)-3-oxopropionate 15a in 76% yield. Mp 109–112 °C; 1H NMR (600 MHz, CDCl3): δ 3.50 (s, 2H), 3.82 (s, 3H), 7.03–7.05 (m, 2H), 7.52–7.54 (m, 2H), 9.22 (s, 1H). MS found (M+H)+ (m/z), 212.13; calcd for C10H10FNO3 m/z, 211.06.
4.1.5.2. Methyl 3-((2,4-difluorophenyl)amino)-3-oxopropanoate (15b)
Reaction of methyl 3-chloro-3-oxopropionate with 2,4-difluoroaniline 14b produced methyl 3-((2,4-difluoro-phenyl)amino)-3-oxopropionate 15b in 77% yield. Mp 128–130 °C; 1H NMR (600 MHz, DMSO-d6): δ 3.39 (s, 2H), 3.78 (s, 3H), 7.05–7.08 (m, 1H), 7.32–7.36 (m, 1H), 7.89–7.91 (m, 1H), 9.98 (s, 1H). MS found (M+H)+ (m/z), 230.18; calcd for C10H9F2NO3 m/z, 229.05.
4.1.6. General procedure for the preparation of 2-(arylcarbamoyl)acetic acid (16) (Scheme 6)
The following 2-(arylcarbamoyl)acetic acids (16) were prepared according to the procedure reported in the literature.14
4.1.6.1. 2-(4-Fluorophenylcarbamoyl)acetic acid (16a)
Hydrolysis of methyl 3-((4-fluorophenyl)amino)-3-oxopropionate 15a with 10% NaOH followed by neutralization with diluted HCl resulted 2-(4-fluorophenylcarbamoyl)acetic acid (16a) in 75% yield. Mp 175–176 °C; 1H NMR (600 MHz, DMSO-d6): δ 3.31 (s, 2H), 7.07–7.10 (m, 2H), 7.51–7.53 (m, 2H), 10.24 (s, 1H), 12.86 (br s, 1H). MS found (M+H)+ (m/z), 198.04; calcd for C9H8FNO3 m/z, 197.05.
4.1.6.2. 2-(2,4-Difluorophenylcarbamoyl)acetic acid (16b)
Hydrolysis of methyl 2-(2,4-difluoroarylcarbamoyl)acetate 15b with 10% NaOH followed by neutralization with diluted HCl resulted 2-(2,4-difluorophenylcarbamoyl)acetic acid (16b) in 73% yield. Mp 183–184 °C; 1H NMR (600 MHz, DMSO-d6): δ 3.36 (s, 2H), 7.01–7.04 (m, 1H), 7.30–7.33 (m, 1H), 7.85–7.87 (m, 1H), 9.95 (s, 1H), 12.86 (br s, 1H). MS found (M+H)+ (m/z), 216.08; calcd for C9H7F2NO3 m/z, 215.04.
4.1.7. General procedure for the preparation of ethyl phenylsulfamoyl acetate (17) (Scheme 7)
The following ethyl phenylsulfamoyl acetates (17) were prepared according to the procedure reported in the literature.15
4.1.7.1. Ethyl 2-(N-(4-fluorophenyl)sulfamoyl)acetate (17a)
Addition of ethyl 2-(chlorosulfonyl)acetate to 4-fluoroaniline 14a yielded the corresponding ethyl 2-(N-(4-fluorophenyl)sulfamoyl) acetate (17a) in 72%. Mp 65–66 °C; 1H NMR (300 MHz, CDCl3): δ 1.33 (t, 3H), 3.93 (s, 2H), 4.29 (q, 2H), 7.03–7.13 (m, 2H), 7.18 (br s, 1H, NH), 7.32–7.40 (m, 2H). MS found (M+H)+ (m/z): 262.05; Calcd for C10H12FNO4S m/z, 261.05.
4.1.7.2. Ethyl 2-(N-(2,4-difluorophenyl)sulfamoyl)acetate (17b)
Addition of ethyl 2-(chlorosulfonyl)acetate to 2,4-difluoroaniline 14b yielded the corresponding ethyl 2-(N-(2,4-difluorophenyl)sulfamoyl)acetate (17b) in 70%. Mp 44–46 °C; 1H NMR (300 MHz, DMSO-d6): δ 1H NMR (600 MHz, CDCl3): δ 1.33 (t, 3H), 4.03 (s, 2H), 4.28–4.31 (q, 2H), 6.92–6.95 (m, 2H), 7.03 (br s, 1H), 7.58–7.62 (m, 1H). MS found (M+H)+ (m/z), 280.04; calcd for C10H11F2NO4S m/z, 279.04.
4.1.8. General procedure for the preparation of phenylsulfamoyl acetic acid (18) (Scheme 7)
The following ethyl phenylsulfamoylacetic acids (18) were prepared according to the procedure reported in the literature.15
4.1.8.1. 2-(N-(4-Fluorophenyl)sulfamoyl)acetic acid (18a)
Hydrolysis followed by neutralization of ethyl 2-(N-(4-fluorophenyl)sulfamoyl)acetate 17a resulted in the corresponding 2-(N-(4-fluorophenyl)sulfamoyl)acetic acid (18a) in 80%. Mp 114–116 °C; 1H NMR (300 MHz, DMSO-d6): δ 4.05 (s, 2H), 7.17–7.28 (m, 4H), 10.04 (br s, 1H), 12.70 (br s, 1H). MS found (M+H)+ (m/z): 234.02; Calcd for C8H8FNO4S m/z, 233.02.
4.1.8.2. 2-(N-(2,4-Difluorophenyl)sulfamoyl)acetic acid (18b)
Hydrolysis followed by neutralization of ethyl 2-(N-(2,4-difluorophenyl) sulfamoyl)acetate 17b resulted in the corresponding 2-(N-(2,4-difluorophenyl)-sulfamoyl)acetic acid (18b) in 76%. Mp 138–139 °C; 1H NMR (300 MHz, DMSO-d6): δ 1H NMR (600 MHz, DMSO-d6): δ 4.01 (s, 2H), 6.95–6.98 (m, 2H), 7.56–7.58 (m, 1H), 10.08 (br s, 1H), 12.68 (br s, 1H). MS found (M+H)+ (m/z), 252.03; calcd for C8H7F2NO4S m/z, 251.01.
4.1.9. General procedure for the preparation of compounds 21 and 22 (Scheme 8)
Methyl thioglycolate (62.8 mmol) was added to the compound 19/20 (62.8 mmol) dissolved in tetrahydrofuran (30 mL) and stirred the reaction mixture for 2 min at room temperature. N,N-Diisopropylethylamine (75.4 mmol) dissolved in tetrahydrofuran (10 mL) was added drop-wise to the above reaction mixture at room temperature and continued the reaction for 15 h. The solvent was removed under reduced pressure, ice-cold water was added to the crude product and stirred the reaction mixture for 5 min. Filtered the solid separated, washed with water and dried to get pure product 21/22.
4.1.9.1. Methyl 2-((2-fluoro-4-nitrophenyl)thio)acetate (21)
The reaction of 3,4-difluoronitrobenzene 19 with methyl thioglycolate yielded methyl 2-(2-fluoro-4-nitrophenylthio)acetate 21 in 96% yield. Mp 68–70 °C; 1H NMR (600 MHz, CDCl3): δ 3.77 (s, 3H), 3.80 (s, 2H), 7.52 (t, 1H), 7.85–7.87 (m, 1H), 8.02–8.04 (m, 1H). MS found (M+H)+ (m/z), 246.02; calcd for C9H8FNO4S m/z, 245.02.
4.1.9.2. Methyl 2-((4-fluoro-2-nitrophenyl)thio)acetate (22)
The reaction of 2,5-difluoronitrobenzene 20 with methyl thioglycolate yielded methyl 2-(4-fluoro-2-nitrophenylthio)acetate 22 in 85% yield. Mp 62–64 °C; 1H NMR (600MHz, DMSO-d6): δ 3.62 (s, 3H), 4.45 (s, 2H), 7.82–7.84 (m, 1H), 8.01–8.02 (m, 1H), 8.17–8.19 (m, 1H). MS found (M+H)+ (m/z), 246.02; calcd for C9H8FNO4S m/z, 245.02.
4.1.10. General procedure for the preparation of the compounds 23 and 24 (Scheme 8)
The compounds 23–24 were prepared according to the procedure reported in the literature.13
4.1.10.1. Methyl 2-((2-fluoro-4-nitrophenyl)sulfonyl)acetate (23)
Oxidation of methyl 2-((2-fluoro-4-nitrophenyl)thio)acetate 21 gave methyl 2-((2-fluoro-4-nitrophenyl)sulfonyl)acetate 23 in 99% yield. Mp 94–96 °C; 1H NMR (600 MHz, CDCl3): δ 3.73 (s, 3H), 4.39 (s, 2H), 8.14–8.16 (m, 1H), 8.18–8.21 (m, 1H), 8.24–8.25 (m, 1H). MS found (M+H)+ (m/z), 278.01; calcd for C9H8FNO6S m/z, 277.01.
4.1.10.2. Methyl 2-((4-fluoro-2-nitrophenyl)sulfonyl)acetate (24)
Oxidation of methyl 2-((4-fluoro-2-nitrophenyl)thio)acetate 22 gave methyl 2-((4-fluoro-2-nitrophenyl)sulfonyl)acetate 24 in 90% yield. Mp 104–106 °C; 1H NMR (600 MHz, DMSO-d6): δ 3.64 (s, 3H), 4.87 (s, 2H), 7.85–7.87 (m, 1H), 8.17–8.19 (m, 1H), 8.23–8.24 (m, 1H). MS found (M+H)+ (m/z), 278.01; calcd for C9H8FNO6S m/z, 277.01.
4.1.11. General procedure for the preparation of the compounds 25 and 26 (Scheme 8)
10% Pd/C was added (500 mg) was added to the solution of the compound 23/24 (18 mmol) in methanol (100 mL) and then continued the reaction in the presence of hydrogen gas at room temperature for 3 h. After completion of the reaction (monitored by TLC), the reaction mixture was filtered on celite and washed the celite pad thoroughly with a mixture of methanol and chloroform. The solvent was removed under reduced pressure and the crude product was treated with 20% diethyl ether in hexanes. The separated solid was filtered, washed with 20% diethyl ether in hexanes and dried to get pure product 25/26.
4.1.11.1. Methyl 2-(4-amino-2-fluorophenylsulfonyl)acetate (25)
Methyl 2-((2-fluoro-4-nitrophenyl) sulfonyl)acetate 23 on reduction with iron produced methyl 2-(4-amino-2-fluoro-phenylsulfonyl) acetate 25 in 86% yield. Mp 99–101 °C; 1H NMR (600 MHz, CDCl3): δ 3.72 (s, 3H), 4.22 (s, 2H), 4.48 (s, 2H), 6.39–6.42 (m, 1H), 6.46–6.48 (m, 1H), 7.62 (m, 1H). MS found (M+H)+ (m/z), 248.06; calcd for C9H10FNO4S m/z, 247.03.
4.1.11.2. Methyl 2-(2-amino-4-fluorophenylsulfonyl)acetate (26)
Methyl 2-((4-fluoro-2-nitrophenyl) sulfonyl)acetate 24 on reduction with iron produced methyl 2-(2-amino-4-fluoro-phenylsulfonyl) acetate 26 in 93% yield. Mp 85–86 °C; 1H NMR (600 MHz, CDCl3): δ 3.74 (s, 3H), 4.16 (s, 2H), 5.21 (br s, 2H), 6.45–6.48 (m, 1H), 6.53–6.57 (m, 1H), 7.71–7.73 (m, 1H). MS found (M+H)+ (m/z), 248.09; calcd for C9H10FNO4S m/z, 247.03.
4.1.12. General procedure for 8-alkyl/cycloalkyl-2-methylsulfanyl-6-substitued-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidine (5)
Mixture of 4-alkylamino-2-methylsulfanyl-pyrimidine-5-carbaldehyde 4 (4.2 mmol),11 1.2 equiv of active methylene compound and catalytic amount of benzylamine was taken into acetic acid and refluxed for about 6 h. After completion of the reaction (checked with TLC), the reaction mixture was cooled to room temperature, the precipitated product was filtered. The solid was washed with saturated NaHCO3, water and dried over vacuum. The crude product was recrystallized in 2-propanol to get pure product (5).
4.1.12.1. 8-Methyl-2-(methylthio)-6-(phenylsulfonyl)pyrido [2,3-d]pyrimidin-7(8H)-one (5a)
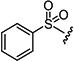
Starting from 4b and 2-(phenylsulfonyl)acetic acid (10a), 58% of 5a was obtained according to the method described for the synthesis of 5. Mp 232–236 °C; 1H NMR (400 MHz, DMSO-d6): δ 2.60 (s, 3H), 3.50 (s, 3H), 7.61–7.63 (m, 2H), 7.71–7.74 (m, 1H), 7.80–7.83 (m, 2H), 8.70 (s, 1H), 8.81 (s, 1H). MS found (M+H)+ (m/z), 348.06; calcd for C15H13N3O3S2 m/z, 347.04.
4.1.12.2. 6-((4-Fluorophenyl)sulfonyl)-8-methyl-2-(methylthio) pyrido[2,3-d]pyrimidin-7(8H)-one (5b)
Starting from 4b and 2-(4-fluorophenylsulfonyl)acetic acid (10b), 62% of 5b was obtained according to the method described for the synthesis of 5. Mp 182–185 °C; 1H NMR (400 MHz, CDC13): δ 2.54 (s, 3H), 3.61 (s, 3H), 7.32 (d, J = 8.2 Hz, 2H), 8.12 (d, J = 8.2 Hz, 2H), 8.63 (s, 1H), 8.77 (s, 1H). MS found (M+H)+ (m/z), 366.01; calcd for C15H12FN3O3S2 m/z, 365.03.
4.1.12.3. 6-((4-Chlorophenyl)sulfonyl)-8-methyl-2-(methylthio) pyrido[2,3-d]pyrimidin-7(8H)-one (5c)
Starting from 4b and 2-(4-chlorophenylsulfonyl)acetic acid (10c), 55% of 5c was obtained according to the method described for the synthesis of 5. Mp 296–298 °C; 1H NMR (400 MHz, CDC13): δ 2.57 (s, 3H), 3.60 (s, 3H), 7.25 (d, J = 7.8 Hz, 2H), 8.00 (d, J = 7.8 Hz, 2H), 8.64 (s, 1H), 8.73 (s, 1H). MS found (M+H)+ (m/z), 382.10; calcd for C15H12ClN3O3S2 m/z, 381.00.
4.1.12.4. 6-((4-Bromophenyl)sulfonyl)-8-methyl-2-(methylthio) pyrido[2,3-d]pyrimidin-7(8H)-one (5d)
Starting from 4b and 2-(4-bromophenylsulfonyl)acetic acid (10d), 58% of 5d was obtained according to the method described for the synthesis of 5. Mp 254–256 °C; 1H NMR (400 MHz, DMSO-d6): δ 2.62 (s, 3H), 3.51 (s, 3H), 7.86 (d, J = 8.0 Hz, 2H), 7.91 (d, J = 8.0 Hz, 2H), 9.03 (s, 1H), 9.20 (s, 1H). MS found (M+H)+ (m/z), 425.92; calcd for C15H12BrN3O3S2 m/z, 424.95.
4.1.12.5. 6-((4-Hydroxyphenyl)sulfonyl)-8-methyl-2-(methylthio) pyrido[2,3-d]pyrimidin-7(8H)-one (5e)
Starting from 4b and 2-(4-hydroxyphenylsulfonyl)acetic acid (10e), 42% of 5e was obtained according to the method described for the synthesis of 5. Mp 302–304 °C; 1H NMR (300 MHz, DMSO-d6): δ 2.56 (s, 3H), 3.48 (s, 3H), 6.90 (d, J = 8.7 Hz, 2H), 7.81 (d, J = 9.0 Hz, 2H), 8.90 (s, 1H), 9.12 (s, 1H). MS found (M+H)+ (m/z), 364.02; calcd for C15H13N3O4S2 m/z, 363.03.
4.1.12.6. 8-Methyl-2-(methylthio)-6-tosylpyrido[2,3-d]pyrimidin-7(8H)-one (5f)
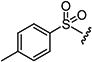
Starting from 4b and 2-tosylacetic acid (10f), 59% of 5f was obtained according to the method described for the synthesis of 5. Mp 228–230 °C; 1H NMR (400 MHz, DMSO-d6): δ 2.39 (s, 3H), 2.61 (s, 3H), 3.50 (s, 3H), 7.43 (d, J = 8.4 Hz, 2H), 7.89 (d, J = 8.4 Hz, 2H), 8.99 (s, 1H), 9.10 (s, lH). MS found (M+H)+ (m/z), 362.10; calcd for C16H15N3O3S2 m/z, 361.06.
4.1.12.7. 6-((4-Methoxyphenyl)sulfonyl)-8-methyl-2-(methylthio) pyrido[2,3-d]pyrimidin-7(8H)-one (5g)
Starting from 4b and 2-(4-methoxyphenylsulfonyl)acetic acid (10g), 61% of 5g was obtained according to the method described for the synthesis of 5. Mp 208–210 °C; 1H NMR (400 MHz, DMSO-d6): δ 2.60 (s, 3H), 3.51 (s, 3H), 3.84 (s, 3H), 7.14 (d, J = 8.8 Hz, 2H), 7.94 (d, J = 8.0 Hz, 2H), 8.96 (s, 1H), 9.17 (s, 1H). MS found (M+H)+ (m/z), 378.03; calcd for C16H15N3O4S2 m/z, 377.05.
4.1.12.8. 4-((8-Methyl-2-(methylthio)-7-oxo-7,8-dihydropyrido [2,3-d]pyrimidin-6-yl)sulfo-nyl)benzoic acid (5h)
Starting from 4b and 2-(4-carboxyphenylsulfonyl)acetic acid (10h), 58% of 5h was obtained according to the method described for the synthesis of 5. Mp 236–240 °C; 1H NMR (600 MHz, DMSO-d6): δ 2.60 (s, 3H), 3.48 (s, 3H), 8.03 (d, J = 8.7 Hz, 2H), 8.10 (d, J = 8.5 Hz, 2H), 9.04 (s, 1H), 9.19 (s, 1H). MS found (M+H)+ (m/z), 366.12; calcd for C16H13N3O5S2 m/z, 391.03.
4.1.12.9. 6-((2,4-Difluorophenyl)sulfonyl)-8-methyl-2-(methylthio) pyrido[2,3-d]pyrimidin-7(8H)-one (5i)
Starting from 4b and 2-(2,4-difluorophenylsulfonyl)acetic acid (10i), 60% of 5i was obtained according to the method described for the synthesis of 5. Mp 280–283 °C; 1H NMR (400 MHz, DMSO-d6): δ 2.61 (s, 3H), 3.50 (s, 3H), 7.40–7.56 (m, 2H), 8.12–8.19 (m, 1H), 9.07 (s, 1H), 9.24 (s, 1H). MS found (M+H)+ (m/z), 384.02; calcd for C15H11F2N3-O3S2 m/z, 383.02.
4.1.12.10. 6-((2,4-Dichlorophenyl)sulfonyl)-8-methyl-2-(methylthio) pyrido[2,3-d]pyrimidin-7(8H)-one (5j)
Starting from 4b and 2-(2,4-dichlorophenylsulfonyl)acetic acid (10j), 51% of 5j was obtained according to the method described for the synthesis of 5. Mp 118–122 °C; 1H NMR (600 MHz, DMSO-d6): δ 2.59 (s, 3H), 3.51 (s, 3H), 7.95 (s, 1H), 8.49 (d, J = 8.2 Hz, 1H), 8.57 (d, J = 8.0 Hz, 1H), 9.41 (s, 1H), 9.72 (s, 1H). MS found (M+H)+ (m/z), 415.95; calcd for C15H11Cl2N3O3S2 m/z, 414.96.
4.1.12.11. 6-((4-Fluoro-2-nitrophenyl)sulfonyl)-8-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5k)
Starting from 4b and methyl 2-(4-fluoro-2-nitrophenylsulfonyl) acetate (24), 52% of 5k was obtained according to the method described for the synthesis of 5. Mp 260–262 °C; 1H NMR (600 MHz, DMSO-d6): δ 2.62 (s, 3H), 3.54 (s, 3H), 7.87–7.90 (m, 1H), 8.13–8.14 (m, 1H), 8.47–8.49 (m, 1H), 8.93 (s, 1H), 9.28 (s, 1H). MS found (M+H)+ (m/z), 411.02; calcd for C15H11FN4O5S2 m/z, 410.02.
4.1.12.12. 6-((2-Amino-4-fluorophenyl)sulfonyl)-8-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5l)
Starting from 4b and methyl 2-(2-amino-4-fluorophenylsulfonyl) acetate (26), 67% of 5l was obtained according to the method described for the synthesis of 5. Mp 262–264 °C; 1H NMR (600 MHz, DMSO-d6): δ 2.67 (s, 3H), 3.96 (s, 3H), 7.28–7.30 (m, 1H), 7.36–7.38 (m, 1H), 8.05–8.07 (m, 1H), 9.08 (s, 1H), 9.13 (s, 1H). MS found (M+H)+ (m/z), 381.05; calcd for C15H13FN4O3S2 m/z, 380.04.
4.1.12.13. 6-((2-Fluoro-4-nitrophenyl)sulfonyl)-8-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5m)
Starting from 4b and methyl 2-(2–fluoro–4-nitrophenylsulfonyl)acetate (23), 45% of 5m was obtained according to the method described for the synthesis of 5. Mp 314–316 °C; 1H NMR (600 MHz, DMSO-d6): δ 2.62 (s, 3H), 3.51 (s, 3H), 8.28–8.39 (m, 3H), 9.15 (s, 1H), 9.26 (s, 1H). MS found (M+H)+ (m/z), 411.02; calcd for C15H11FN4O5S2 m/z, 410.02.
4.1.12.14. 6-((4-Amino-2-fluorophenyl)sulfonyl)-8-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5n)
Starting from 4b and methyl 2-(4-amino-2-fluorophenylsulfonyl) acetate (25), 32% of 5n was obtained according to the method described for the synthesis of 5. Mp 328–330 °C; 1H NMR (600 MHz, DMSO-d6): δ 2.61 (s, 3H), 3.51 (s, 3H), 6.26–6.28 (m, 1H), 6.47–6.49 (m, 1H), 6.52 (s, 2H), 7.62–7.64 (m, 1H), 8.91 (s, 1H), 9.21 (s, 1H). MS found (M+H)+ (m/z), 381.05; calcd for C15-H13FN4O3S2 m/z, 380.04.
4.1.12.15. 6-((4-Fluorobenzyl)sulfonyl)-8-methyl-2-(methylthio) pyrido[2,3-d]pyrimidin-7(8H)-one (5o)
Starting from 4b and 4-fluorobenzylsulfonylacetic acid (13a), 55% of 5o was obtained according to the method described for the synthesis of 5. Mp 170–174 °C; 1H NMR (400 MHz, CDCl3): δ 2.58 (s, 3H), 3.74 (s, 3H), 4.71 (s, 2H), 6.80–6.94 (m, 2H), 7.24–7.33 (m, 2H), 8.20 (s, 1H), 8.59 (s, 1H). MS found (M+H)+ (m/z), 380.10; calcd for C16H14FN3O3S2 m/z, 379.05.
4.1.12.16. 6-((2,4-Difluorobenzyl)sulfonyl)-8-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5p)
Starting from 4b and 2,4-fluorobenzylsulfonylacetic acid (13b), 46% of 5p was obtained according to the method described for the synthesis of 5. Mp: 264–266 °C; 1H NMR (600 MHz; DMSO-d6): δ 2.65 (s, 3H), 3.69 (s, 3H), 4.89 (s, 2H), 7.09–7.11 (m, 1H), 7.26–7.29 (m, 1H), 7.43–7.46 (m, 1H), 8.63 (s, 1H), 9.12 (s, 1H). MS found (M+H)+ (m/z), 398.08; calcd for C16H13F2N3O3S2 m/z, 397.04.
4.1.12.17. N-(4-Fluorophenyl)-8-methyl-2-(methylthio)-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidine-6-carboxamide (5q)
Starting from 4b and 2-(4-fluorophenylcarbamoyl)acetic acid (16a), 56% of 5q was obtained according to the method described for the synthesis of 5. Mp 240–242 °C; 1H NMR (600 MHz; CDCl3): δ 2.69 (s, 3H), 3.88 (s, 3H), 7.08 (t, 2H), 7.71–7.73 (m, 2H), 8.86 (s, 1H), 8.94 (s, 1H), 11.60 (s, 1H). MS found (M+H)+ (m/z), 345.08; calcd for C16H13FN4O2S m/z, 344.07.
4.1.12.18. N-(2,4-Difluorophenyl)-8-methyl-2-(methylthio)-7-oxo-7,8-dihydropyrido[2,3-d]-pyrimidine-6-carboxamide (5r)
Starting from 4b and 2-(2,4-difluorophenylcarbamoyl)acetic acid (17b), 53% of 5r was obtained according to the method described for the synthesis of 5. Mp 276–278 °C; 1H NMR (600 MHz; CDCl3): δ 2.66 (s, 3H), 3.74 (s, 3H), 7.15–7.16 (m, 1H), 7.45–7.47 (m, 1H), 8.45–8.46 (m, 1H), 9.04 (s, 1H), 9.25 (s, 1H), 11.92 (s, 1H). MS found (M+H)+ (m/z), 363.07; calcd for C16H12F2-N4O2S m/z, 362.06.
4.1.12.19. N-(4-Fluorophenyl)-8-methyl-2-(methylthio)-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidine-6-sulfonamide (5s)
Starting from 4b and 2-(4-fluorophenylsulfamoyl)acetic acid (18a), 62% of 5s was obtained according to the method described for the synthesis of 5. Mp 202–204 °C; 1H NMR (600 MHz, DMSO-d6): δ 2.59 (s, 3H), 3.61 (s, 3H), 7.02–7.06 (m, 2H), 7.08–7.11 (m, 2H), 8.71 (s, 1H), 9.06 (s, 1H), 10.31 (s, 1H). MS found (M+H)+ (m/z), 381.09; calcd for C15H13FN4O3S2 m/z, 380.04.
4.1.12.20. N-(2,4-Difluorophenyl)-8-methyl-2-(methylthio)-7-oxo-7,8-dihydropyrido[2,3-d]-pyrimidine-6-sulfonamide (5t)
Starting from 4b and 2-(2,4-difluorophenylsulfamoyl)acetic acid (18b), 51% of 5t was obtained according to the method described for the synthesis of 5. Mp 230–232 °C; 1H NMR (600 MHz, DMSO-d6): δ 2.63 (s, 3H), 3.66 (s, 3H), 6.99–7.01 (m, 1H), 7.25–7.29 (m, 2H), 8.57 (s, 1H), 9.06 (s, 1H), 10.02 (s, 1H). MS found (M+H)+ (m/z), 399.10; calcd for C15H12F2N4O3S2 m/z, 398.03.
4.1.12.21. 6-((2,4-Difluorophenyl)sulfonyl)-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5u)
Starting from 4a and 2-(2,4-difluorophenylsulfonyl)acetic acid (10i), 53% of 5u was obtained according to the method described for the synthesis of 5. Mp 338–340 °C; 1H NMR (600 MHz, DMSO-d6): δ 2.58 (s, 3H), 7.39–7.41 (m, 1H), 7.11–7.15 (m, 1H), 8.10–8.15 (m, 1H), 9.02 (s, 1H), 9.20 (s, 1H). MS found (M+H)+ (m/z), 370.02; calcd for C14H9F2N3O3S2 m/z, 369.01.
4.1.12.22. 6-((2,4-Difluorophenyl)sulfonyl)-8-ethyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5v)
Starting from 4c and 2-(2,4-difluorophenylsulfonyl)acetic acid (10i), 56% of 5v was obtained according to the method described for the synthesis of 5. Mp 215–217 °C; 1H NMR (CDCl3, 300 MHz): δ1.32–1.27 (t, 3H), 2.66 (s, 3H), 4.44–4.37 (q, 2H), 6.92–6.85 (m, 1H), 7.17–7.11 (m, 1H), 8.33–8.28 (m, 1H), 8.85 (s, 1H), 8.74 (s, 1H). MS found (M+H)+ (m/z): 398.10, Calcd for C16H13 F2N3O3S2 m/z: 397.04.
4.1.12.23. 6-((2,4-Difluorophenyl)sulfonyl)-2-(methylthio)-8-propylpyrido[2,3-d]pyrimidin-7(8H)-one (5w)
Starting from 4d and 2-(2,4-difluorophenylsulfonyl)acetic acid (10i), 64% of 5w was obtained according to the method described for the synthesis of 5. Mp 196–198 °C; 1H NMR (600 MHz, CDCl3): δ 0.93–0.96 (m, 3H), 1.68–1.71 (m, 2H), 2.64 (s, 3H), 4.25–4.32 (m, 2H), 6.86–6.89 (m, 1H), 7.11–7.15 (m, 1H), 8.29–8.33 (m, 1H), 8.77 (s, 1H), 8.84 (s, 1H). MS found (M+H)+ (m/z), 412.14; calcd for C17 H15F2N3O3S2 m/z, 411.05.
4.1.12.24. 8-Cyclopropyl-6-((2,4-difluorophenyl)sulfonyl)-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5x)
Starting from 4e and 2-(2,4-difluorophenylsulfonyl)acetic acid (10i), 55% of 5x was obtained according to the method described for the synthesis of 5. Mp 236–238 °C; 1H NMR (600 MHz, DMSO-d6): δ 0.75–0.77 (m, 2H), 1.12–1.14 (m, 2H), 2.62 (s, 3H), 2.80–2.81 (m, 1H), 7.40–7.54 (m, 2H), 8.14–8.16 (m, 1H), 9.01 (s, 1H), 9.18 (s, 1H). MS found (M+H)+ (m/z), 410.20; calcd for C17H13 F2N3O3S2 m/z, 409.04.
4.1.12.25. 8-Cyclopentyl-6-((2,4-difluorophenyl)sulfonyl)-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5y)
Starting from 4f and 2-(2,4-difluorophenylsulfonyl)acetic acid (10i), 60% of 5y was obtained according to the method described for the synthesis of 5. Mp 209–210 °C; 1H NMR (CDCl3, 300 MHz): δ 1.71–1.62 (m, 2H), 1.85–1.78 (m, 2H), 2.11–1.99 (m, 2H), 2.29–2.23 (m, 2H), 2.65 (s, 3H), 5.81–5.75 (m, 1H), 6.92–6.85 (m, 1H), 7.17–7.11 (m, 1H), 8.34–8.27 (m, 1H), 8.73 (s, 1H), 8.83 (s, 1H). MS found (M+H)+ (m/z): 438.10, Calcd for C19H17F2N3 O3S2 m/z: 437.07.
4.1.12.26. 8-Cyclohexyl-6-((2,4-difluorophenyl)sulfonyl)-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5z)
Starting from 4g and 2-(2,4-difluorophenylsulfonyl)acetic acid (10i), 55% of 5z was obtained according to the method described for the synthesis of 5. Mp 247–249 °C; 1H NMR (600 MHz, DMSO-d6): δ 1.12–1.32 (m, 4H), 1.55–1.64 (m, 4H), 1.78–1.80 (m, 2H), 2.49–2.50 (m, 1H), 2.63 (s, 3H), 7.41–7.44 (m, 1H), 7.53–7.56 (m, 1H), 8.12–8.16 (m, 1H), 9.03 (s, 1H), 9.21 (s, 1H). MS found (M+H)+ (m/z), 452.12; calcd for C20H19F2N3O3S2 m/z, 451.08.
4.1.13. General procedure for the synthesis of compounds (5aa–5ac) from 6-((2,4-difluorophenyl)sulfonyl)-2-(methylthio) pyrido[2,3-d]pyrimidin-7(8H)-one (5u) (Scheme 2)
The compound 5u (5 mmol) was taken into DMF and stirred at 50 °C for 10 min until to get the clear solution, and then K2CO3 (7.5 mmol) was added and stirred about 5 min. The reaction mixture was brought to room temperature and was added alkyliodide (6.5 mmol) and the stirring was continued for 1 h. After completion of the reaction, the reaction mixture was quenched with water and filtered the crude product. The crude product was purified with flash column chromatography using ethyl acetate and hexanes as eluents.
4.1.13.1. Methyl 2-(6-(2,4-difluorophenylsulfonyl)-2-(methylthio)-7-oxopyrido[2,3-d]pyrimidin-8(7H)yl)acetate (5aa)
Starting from 5u and methyl 2-bromoacetate, 55% of 5aa was obtained according to the method described for the synthesis of 5aa–5ac. Mp 225–227 °C; 1H NMR (600 MHz, DMSO-d6): δ 2.58 (s, 3H),3.73 (s, 3H), 5.06 (s, 2H), 6.90–6.93 (m, 1H), 7.09–7.16 (m, 1H), 8.25–8.32 (m, 1H), 8.83 (s, 1H), 8.87 (s, 1H). MS found (M+H)+ (m/z), 442.10; calcd for C17H13F2N3O5S2 m/z, 441.03.
4.1.13.2. 8-(Cyclopropylmethyl)-6-((2,4-difluorophenyl)sulfonyl)-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5ab)
Starting from 5u and (iodomethyl)cyclopropane, 60% of 5ab was obtained according to the method described for the synthesis of 5aa–5ac. Mp 204–205 °C; 1H NMR (CDCl3, 300 MHz): δ 0.28– 0.22 (m, 4H), 1.16–1.08 (m, 1H), 2.42 (s, 3H), 4.02 (d, 2H), 6.69–6.62 (m, 1H), 7.06–6.88 (m, 1H), 8.13–8.05 (m, 1H), 8.56 (s, 1H), 8.63 (s, 1H). MS found (M+H)+ (m/z): 424.10, Calcd for C18H15F2N3-O3S2 m/z: 423.05.
4.1.13.3. 8-(Cyclopentylmethyl)-6-((2,4-difluorophenyl)sulfonyl)-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (5ac)
Starting from 5u and (iodomethyl)cyclopentane, 60% of 5ac was obtained according to the method described for the synthesis of 5aa–5ac. Mp 190–191 °C; 1H NMR (CDCl3, 300 MHz): δ 1.30–1.26 (m, 2H), 1.55–1.49 (m, 2H), 1.70–1.60 (m, 4H), 2.35 (m, 1H), 2.73 (s, 3H), 4.33 (d, 2H),, 6.91–6.84 (m, 1H), 7.17–7.11 (m, 1H), 8.36–8.28 (m, 1H), 8.77 (s, 1H), 8.84 (s, 1H). MS found (M+H)+ (m/z): 452.10, Calcd for C20H19F2N3O3S2 m/z: 451.08.
4.1.14. General procedure for the synthesis of 8-substituted-2-methylsulfinyl-6-substituted-7-oxo-7,8-dihydro-pyrido[2,3-d] pyrimidines (6a–ac) (Schemes 1 and 2)
A solution of 8-substituted-2-methylsulfanyl-6-substitued-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidine 5a–ac (3.5 mmol), and 3-chloro-perbenzoic acid (m-CPBA) (4.4 mmol) in CH2Cl2 was stirred at room temperature for about 4 h. After completion of the reaction, the reaction mixture was washed with saturated NaHCO3, organic layer was dried over Na2SO4 and concentrated to produce the mixture of methylsulfoxide and methylsulfonyl product 6a–ac and was used for next step without further purification.
4.1.15. General procedure for 8-alkyl/cycloalkyl-2-(aryl/heteroarylamino)-6-substiuted-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidine (7) (Scheme 1)
The mixture of 8-alkyl-2-methylsulfinyl-6-substituted-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidine 6 (2 mmol) and aryl/heteroaryl amines (2.25 mmol) in toluene was stirred at 100 °C for overnight. The reaction mixture was cooled and solid was collected by filtration. The crude product was washed with toluene and purified by flash chromatography to get pure product.
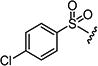
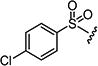
4.1.15.1. 2-((4-Chlorophenyl)amino)-6-((4-chlorophenyl)sulfonyl)-8-methylpyrido[2,3-d]-pyrimidin-7(8H)-one (7a)
Starting from 6c and 4-chlorobenzenamine, 72% of 7a was obtained as per the method described for the synthesis of 7; mp >300 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.48 (s, 3H), 7.41 (d, J = 8.8 Hz, 2H), 7.70 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.8 Hz, 2H), 8.88 (s, 1H), 9.10 (s, 1H), 10.40 (s, 1H). MS found (M+H)+ (m/z), 461.02; calcd for C20H14Cl2N4O3S m/z, 460.02.
4.1.15.2. 6-((4-Chlorophenyl)sulfonyl)-2-((4-methoxyphenyl)amino)-8-methylpyrido[2,3-d]-pyrimidin-7(8H)-one (7b)
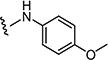
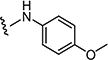
Starting from 6c and 4-methoxybenzenamine, 63% of 7b was obtained as per the method described for the synthesis of 7; mp 286–290 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.64 (s, 3H), 3.75 (s, 3H), 6.90 (d, J = 9.0 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 8.0 Hz, 2H), 7.72 (d, J = 8.8 Hz, 2H), 7.99 (s, 1H), 8.10 (s, 1H), 10.20 (s, 1H). MS found (M+H)+ (m/z), 457.06; calcd for C21H17ClN4 O4S m/z, 456.07.
4.1.15.3. 2-((4-Methoxyphenyl)amino)-8-methyl-6-tosylpyrido [2,3-d]pyrimidin-7(8H)-one (7c)
Starting from 6f and 4-methoxybenzenamine, 68% of 7c was obtained as per the method described for the synthesis of 7; mp 336–340 °C; 1H NMR (400 MHz; DMSO-d6): δ 2.39 (s, 3H), 3.47 (s, 3H), 3.75 (s, 3H), 6.41 (d, J = 8.0 Hz, 2H), 6.88 (d, J = 7.8 Hz, 2H), 7.41 (d, J = 8.0 Hz, 2H), 7.88 (d, J = 7.8 Hz, 2H), 8.80 (s, 1H), 9.05 (s, 1H), 10.20 (s, 1H). MS found (M+H)+ (m/z), 437.10; calcd for C22H20N4O4S m/z, 436.12.
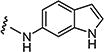
4.1.15.4. 2-((1H-Indol-5-yl)amino)-8-methyl-6-(phenylsulfonyl)pyrido[2,3-d]pyrimidin-7-(8H)-one (7d)
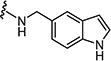
Starting from 6a and 1H-indol-5-amine, 62% of 7d was obtained as per the method described for the synthesis of 7. Mp 340–344 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.47 (s, 3H), 6.41 (s, 1H), 7.13–7.24 (m, 3H), 7.33–7.38 (m, 2H), 7.46–7.48 (m, 2H), 7.67–7.71 (m, 2H), 8.82 (s, 1H), 9.03 (s, 1H), 10.49 (s, 1H), 11.08 (s, 1H). MS found (M+H)+ (m/z), 432.08; calcd for C22H17N5O3S m/z, 431.11.
4.1.15.5. 2-((1H-Indol-6-yl)amino)-8-methyl-6-(phenylsulfonyl)pyrido[2,3-d]pyrimidin-7-(8H)-one (7e)
Starting from 6a and 1H-indol-6-amine, 65% of 7e was obtained as per the method described for the synthesis of 7; mp >350 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.29 (s, 3H), 6.12 (s, 1H), 6.90–7.04 (m, 3H), 7.21–7.44 (m, 3H), 7.74–7.94 (m, 3H), 8.59 (s, 1H), 8.81 (s, 1H), 10.38 (s, 1H), 10.90 (s, 1H). MS found (M+H)+ (m/z), 432.10; calcd for C22H17N5O3S m/z, 431.11.
4.1.15.6. 2-((1H-Pyrrolo[2,3-b]pyridin-5-yl)amino)-8-methyl-6-(phenylsulfonyl)pyrido[2,3-d]pyrimidin-7(8H)-one (7f)
Starting from 6a and 1H-pyrrolo[2,3-b]pyridin-5-amine, 68% of 7f was obtained as per the method described for the synthesis of 7; mp: 280–284 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.49 (s, 3H), 6.49 (s, 1H), 7.14–7.29 (m, 2H), 7.50–7.77 (m, 3H), 8.06–8.07 (m, 3H), 8.89 (s, 1H), 9.10 (s, 1H), 10.65 (s, 1H), 11.67 (s, 1H). MS found (M+H)+ (m/z), 433.12; calcd for C21H16N6O3S m/z, 432.10.
4.1.15.7. 8-Methyl-6-(phenylsulfonyl)-2-(quinolin-6-ylamino)pyrido[2,3-d]pyrimidin-7(8H)-one (7g)
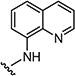
Starting from 6a and quinolin-6-amine, 65% of 7g was obtained as per the method described for the synthesis of 7; mp 348–350 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.50 (s, 3H), 6.80 (s, 1H), 7.42–7.49 (m, 2H), 7.62–7.71 (m, 3H), 7.82–7.86 (m, 3H), 8.10–8.23 (m, 2H), 8.65 (s, 1H), 8.83 (s, 1H), 11.18 (s, 1H). MS found (M+H)+ (m/z), 444.07; calcd for C23H17N5O3S m/z, 443.11.
4.1.15.8. 2-((1H-Indol-4-yl)amino)-6-((4-chlorophenyl)sulfonyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7h)
Starting from 6c and 1H-indol-4-amine, 54% of 7h was obtained as per the method described for the synthesis of 7; mp 320–324 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.30 (s, 3H), 6.44 (s, 1H), 6.80–7.08 (m, 4H), 7.59 (d, J = 8.4 Hz, 2H), 7.91 (d, J = 8.4 Hz, 2H), 8.75 (s, 1H), 8.98 (s, 1H), 10.30 (s, 1H), 11.07 (s, 1H). MS found (M+H)+ (m/z), 466.10; calcd for C22H16ClN5O3S m/z, 465.07.
4.1.15.9. 2-((1H-Indol-5-yl)amino)-6-((4-chlorophenyl)sulfonyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7i)
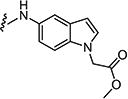
Starting from 6c and 1H-indol-5-amine, 58% of 7i was obtained as per the method described for the synthesis of 7; mp 316–320 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.40 (s, 3H), 6.40–6.43 (m, 1H), 7.29–7.30 (m, 3H), 7.36–7.37 (m, 2H), 8.01–8.18 (m, 3H), 8.80 (s, 1H), 9.03 (s, 1H), 10.30 (s, 1H), 11.07 (s, 1H). MS found (M+H)+ (m/z), 466.08; calcd for C22H16ClN5O3S m/z, 465.07.
4.1.15.10. 2-((1H-Pyrrolo[2,3-b]pyridin-5-yl)amino)-6-((4-chlorophenyl)sulfonyl)-8-methyl-pyrido[2,3-d]pyrimidin-7 (8H)-one (7j)
Starting from 6c and 1H-pyrrolo[2,3-b]pyridin-5-amine, 66% of 7j was obtained as per the method described for the synthesis of 7; mp 296–298 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.45 (s, 3H), 6.40 (s, 1H), 7.08–7.26 (m, 4H), 7.69–7.74 (m, 1H), 7.92–8.04 (m, 2H), 8.85 (s, 1H), 9.40 (s, 1H), 10.63 (s, 1H), 11.49 (s, 1H). MS found (M+H)+ (m/z), 467.11; calcd for C21H15ClN6O3S m/z, 466.06.
4.1.15.11. 6-((4-Chlorophenyl)sulfonyl)-8-methyl-2-(quinolin-3-ylamino)pyrido[2,3-d]pyrimidin-7(8H)-one (7k)
Starting from 6c and quinolin-3-amine, 61% of 7k was obtained as per the method described for the synthesis of 7; mp: 304–306 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.40 (s, 3H), 7.12 (s, 1H), 7.42 (s, 1H),7.50–7.51 (m, 1H), 7.55–7.56 (m, 1H), 7.62 (d, J = 8.0 Hz, 2H), 7.83 (s, 1H), 7.86 (s, 1H), 8.25 (s, 2H), 8.88 (s, 1H), 9.12 (s, 1H), 10.38 (s, 1H). MS found (M+H)+ (m/z), 478.05; calcd for C23H16ClN5O3S m/z, 477.07.
4.1.15.12. 2-((1H-Indol-5-yl)amino)-6-((4-bromophenyl)sulfonyl)-8-methylpyrido[2,3-d]-pyrimidin-7(8H)-one (7l)
Starting from 6d and 1H-indol-5-amine, 63% of 7l was obtained as per the method described for the synthesis of 7; mp: 316–320 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.53 (s, 3H), 6.47 (s, 1H), 7.38–7.52 (m, 3H), 7.88 (d, J = 8.8 Hz, 2H), 7.98 (d, J = 8.8 Hz, 2H), 8.08 (s, 1H), 8.87 (s, 1H), 9.09 (s, 1H), 10.58 (s, 1H), 11.14 (s, 1H). MS found (M+H)+ (m/z), 510.04; calcd for C22H16BrN5O3S m/z, 509.02.
4.1.15.13. 2-((1H-Indol-4-yl)amino)-6-((4-fluorophenyl)sulfonyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7m)
Starting from 6b and 1H-indol-4-amine, 57% of 7m was obtained as per the method described for the synthesis of 7; mp >350 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.93 (s, 3H), 6.61–6.65 (m, 1H), 7.08–7.14 (m, 3H), 7.41–7.56 (m, 3H), 8.04–8.08 (m, 2H), 8.80 (s, 1H), 9.07 (s, 1H), 10.35 (s, 1H), 11.16 (s, 1H). MS found (M+H)+ (m/z), 450.12; calcd for C22H16FN5O3S m/z, 449.10.
4.1.15.14. 2-((1H-Indol-5-yl)amino)-6-((4-fluorophenyl)sulfonyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7n)
Starting from 6b and 1H-indol-5-amine, 63% of 7n was obtained as per the method described for the synthesis of 7; mp: 278–282 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.58 (s, 3H), 6.51 (s, 1H), 7.42–7.47 (m, 3H), 7.53–7.57 (m, 2H), 8.15–8.19 (m, 3H), 8.90 (s, 1H), 9.10 (s, 1H), 10.50 (s, 1H), 11.10 (s, 1H). MS found (M+H)+ (m/z), 450.20; calcd for C22H16FN5O3S m/z, 449.10.
4.1.15.15. 2-((1H-Indol-6-yl)amino)-6-((4-fluorophenyl)sulfonyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7o)
Starting from 6b and 1H-indol-6-amine, 56% of 7o was obtained as per the method described for the synthesis of 7; mp >350 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.30 (s, 3H), 6.12 (s, 1H), 7.05–7.23 (m, 5H), 7.83–7.91 (m, 3H), 8.58 (s, 1H), 8.80 (s, 1H), 10.18 (s, 1H), 10.89 (s, 1H). MS found (M+H)+ (m/z), 450.10; calcd for C22 H16FN5O3S m/z, 449.10.
4.1.15.16. 2-((1H-Pyrrolo[2,3-b]pyridin-5-yl)amino)-6-((4-fluorophenyl)sulfonyl)-8-methyl-pyrido[2,3-d]pyrimidin-7(8H)-one (7p)
Starting from 6b and 1H-pyrrolo[2,3-b]pyridin-5-amine, 53% of 7p was obtained as per the method described for the synthesis of 7; mp 274–280 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.34 (s, 3H), 6.44–6.50 (m, 1H), 7.44–7.49 (m, 4H), 8.02–8.09 (m, 3H), 8.84 (s, 1H), 9.07 (s, 1H),10.70 (s, 1H), 11.50 (s, 1H). MS found (M+H)+ (m/z), 451.07; calcd for C21H15FN6O3S m/z, 450.09.
4.1.15.17. 2-((1H-Indazol-5-yl)amino)-6-((4-fluorophenyl)sulfonyl)-8-methylpyrido[2,3-d]-pyrimidin-7(8H)-one (7q)
Starting from 6b and 1H-indazol-5-amine, 65% of 7q was obtained as per the method described for the synthesis of 7; mp 160–165 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.35 (s, 3H), 7.31–7.35 (m, 4H), 7.91–7.94 (m, 4H), 8.69 (s, 1H), 8.92 (s, 1H),10.30 (s, 1H), 12.50 (s, 1H). MS found (M+H)+ (m/z), 451.10; calcd for C21 H15FN6O3S m/z, 450.09.
4.1.15.18. 2-((1H-Benzo[d]imidazol-5-yl)amino)-6-((4-fluorophenyl)sulfonyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7r)
Starting from 6b and 1H-benzo[d]imidazol-5-amine, 70% of 7r was obtained as per the method described for the synthesis of 7; mp: 320–324 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.61 (s, 3H), 5.38 (s, 1H), 6.70–6.72 (m, 1H), 6.84–6.85 (m, 1H), 7.28–7.30 (m, 3H), 6.84–6.85 (m, 1H), 8.09–8.11 (m, 3H), 9.08 (s, 1H), 9.40 (s, 1H). MS found (M+H)+ (m/z), 451.08; calcd for C21H15FN6O3S m/z, 450.09.
4.1.15.19. 2-(Benzofuran-5-ylamino)-6-((4-fluorophenyl)sulfonyl)-8-methylpyrido[2,3-d]-pyrimidin-7(8H)-one (7s)
Starting from 6b and benzofuran-5-amine, 68% of 7s was obtained as per the method described for the synthesis of 7; mp 296–298 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.48 (s, 3H), 6.96 (s, 1H), 7.44 (t, 2H), 7.57–7.66 (m, 2H), 7.96 (s, 1H), 8.05–8.06 (m, 2H), 8.13 (s, 1H), 8.83 (s, 1H), 9.06 (s, 1H), 10.66 (s, 1H). MS found (M+H)+ (m/z), 451.10; calcd for C22H15FN4O4S m/z, 450.08.
4.1.15.20. 2-((1H-Indol-5-yl)amino)-6-((4-hydroxyphenyl)sulfonyl)-8-methylpyrido[2,3-d]-pyrimidin-7(8H)-one (7t)
Starting from 6e and 1H-indol-5-amine, 58% of 7t was obtained as per the method described for the synthesis of 7; mp 288–290 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.55 (s, 3H), 6.39 (s, 1H), 6.89 (d, J = 8.0 Hz, 2H), 7.01–7.70 (m, 4H), 7.80 (d, J = 8.2 Hz, 2H), 8.72 (s, 1H), 9.08 (s, 1H), 10.55 (s, 1H), 11.29 (s, 1H). MS found (M+H)+ (m/z), 448.08; calcd for C22H17N5O4S m/z, 447.10.
4.1.15.21. Synthesis of 4-(2-(1H-indol-5-ylamino)-7,8-dihydro-8-methyl-7-oxopyrido[2,3-d]pyrimidin-6-ylsulfonyl)phenyl 5-(hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-6-yl)pentan-oate (7u)
EDC (1-ethyl-3-[(3-dimethylamino)propyl]-carbodiimide) (4 mmol) was added to the solution of 2-((1H-indol-5-yl)amino)-6-((4-hydroxyphenyl)sulfonyl)-8-methylpyrido[2,3-d]pyrimidin-7 (8H)-one 7t (2.15 mmol), biotin (2 mmol) and 4-dimethylaminopyridine (2 mmol) in DMF at °C and stirred the reaction for 24 h at room temperature. After completion of the reaction (monitored by LC–MS) the reaction mixture was filtered and the filtrate was concentrated. The crude product was purified using combiflash to get the pure product. Yield 45%, mp: 310–312 °C; 1H NMR (600 MHz; DMSO-d6): δ 1.35–1.62 (m, 6H), 2.20 (t, 2H), 2.57 (d, J = 12.4 Hz, 1H), 2.80–2.83 (m, 1H), 3.08–3.11 (m, 1H), 3.58 (s, 3H), 4.10–4.12 (m, 1H), 4.28–4.30 (m, 1H), 6.37 (br s, 1H), 6.37 (s, 1H), 6.45 (br s, 1H), 6.88 (d, J = 8.2 Hz, 2H), 7.08–7.75 (m, 4H), 7.84 (d, J = 8.2 Hz, 2H), 8.73 (s, 1H), 9.11 (s, 1H), 10.58 (s, 1H), 11.27 (s, 1H). MS found (M+H)+ (m/z), 674.18; calcd for C32H31N7O6S2 m/z, 673.18.
4.1.15.22. 2-((1H-Indol-4-yl)amino)-8-methyl-6-tosylpyrido [2,3-d]pyrimidin-7(8H)-one (7v)
Starting from 6f and 1H-indol-4-amine, 70% of 7v was obtained as per the method described for the synthesis of 7; mp 295–300 °C; 1H NMR (400 MHz; DMSO-d6): δ 2.38 (s, 3H), 3.53 (s, 3H), 6.47 (s, 1H), 7.39–7.54 (m, 5H), 7.94 (d, J = 8.0 Hz, 2H), 8.09 (s, 1H), 8.85 (s, 1H), 9.08 (s, 1H), 10.54 (s, 1H), 11.13 (s, 1H). MS found (M+H)+ (m/z), 446.10; calcd for C23H19N5O3S m/z, 445.12.
4.1.15.23. 2-((1H-Indol-5-yl)amino)-8-methyl-6-tosylpyrido[2,3-d]pyrimidin-7(8H)-one (7w)
Starting from 6f and 1H-indol-5-amine, 67% of 7w was obtained as per the method described for the synthesis of 7; mp 310–314 °C; 1H NMR (400 MHz; DMSO-d6): δ 2.39 (s, 3H), 3.42 (s, 3H), 6.67–6.69 (m, 1H), 7.12–7.42 (m, 5H), 7.70 (s, 1H), 7.90 (d, J = 7.2 Hz, 1H), 8.84 (s, 1H), 9.07 (s, 1H), 10.36 (s, 1H), 11.18 (s, 1H). MS found (M+H)+ (m/z), 446.11; calcd for C23H19N5O3S m/z, 445.12.
4.1.15.24. 2-((1H-Indol-6-yl)amino)-8-methyl-6-tosylpyrido[2,3-d]pyrimidin-7(8H)-one (7x)
Starting from 6f and 1H-indol-6-amine, 70% of 7x was obtained as per the method described for the synthesis of 7; mp 334–336 °C; 1H NMR (400 MHz; DMSO-d6): δ 2.38 (s, 3H), 3.54 (s, 3H), 6.38 (s, 1H), 7.04–7.49 (m, 5H), 7.90–8.01 (m, 2H), 8.17 (s, 1H), 8.85 (s, 1H), 9.05 (s, 1H), 10.58 (s, 1H), 11.15 (s, 1H). MS found (M+H)+ (m/z), 446.08; calcd for C23H19N5O3S m/z, 445.12.
4.1.15.25. 2-((1H-Pyrrolo[2,3-b]pyridin-5-yl)amino)-8-methyl-6-tosylpyrido[2,3-d]pyrimidin-7(8H)-one (7y)
Starting from 6f and 1H-pyrrolo[2,3-b]pyridin-5-amine, 62% of 7y was obtained as per the method described for the synthesis of 7; mp 252–255 °C; 1H NMR (400 MHz; DMSO-d6): δ 2.43 (s, 3H), 3.46 (s, 3H), 6.60 (s, 1H), 7.25–7.37 (m, 2H), 7.54–7.59 (m, 2H), 7.98–8.03 (m, 3H), 8.89 (s, 1H), 9.08 (s, 1H), 10.55 (s, 1H), 11.23 (s, 1H). MS found (M+H)+ (m/z), 447.13; calcd for C22H18N6O3S m/z, 446.12.
4.1.15.26. 8-Methyl-2-((2-oxo-2,3-dihydro-1H-pyrrolo[2,3-b] pyridin-5-yl)amino)-6-tosyl-pyrido[2,3-d]pyrimidin-7(8H)-one (7z)
Starting from 6f and 5-amino-1H-pyrrolo[2,3-b]pyridin-2(3H)-one, 62% of 7z was obtained as per the method described for the synthesis of 7; mp 280 °C; 1H NMR (400 MHz; DMSO-d6): δ 2.39 (s, 3H), 3.47 (s, 3H), 3.69 (s, 2H), 6.31–6.67 (m, 2H), 7.03–7.48 (m, 3H), 7.61–7.74 (m, 2H), 8.67 (s, 1H), 8.90 (s, 1H), 9.91 (s, 1H), 10.27 (s, 1H). MS found (M+H)+ (m/z), 463.07; calcd for C22H18N6 O4S m/z, 462.11.
4.1.15.27. 8-Methyl-2-(quinolin-8-ylamino)-6-tosylpyrido[2,3-d]pyrimidin-7(8H)-one (7aa)
Starting from 6f and quinolin-8-amine, 58% of 7aa was obtained as per the method described for the synthesis of 7; mp >350 °C; 1H NMR (400 MHz; DMSO-d6): δ 2.22 (s, 3H), 3.50 (s, 3H), 7.40–7.43 (m, 4H), 7.55–7.56 (m, 2H), 7.74 (d, J = 8.0 Hz, 2H), 8.25–8.32 (m, 2H), 8.68 (s, 1H), 8.82 (s, 1H), 10.20 (s, 1H). MS found (M+H)+ (m/z), 458.10; calcd for C24H19N5O3S m/z, 457.12.
4.1.15.28. 8-Methyl-2-(quinolin-6-ylamino)-6-tosylpyrido[2,3-d]pyrimidin-7(8H)-one (7ab)
Starting from 6f and quinolin-6-amine, 59% of 7ab was obtained as per the method described for the synthesis of 7; mp 325–330 °C; 1H NMR (400 MHz; DMSO-d6): δ 2.34 (s, 3H), 3.40 (s, 3H), 6.80 (s, 1H), 7.42–7.43 (m, 3H), 7.48–7.49 (m, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.82–7.83 (m, 1H), 8.15–8.25 (m, 2H), 8.60 (s, 1H), 8.81 (s, 1H), 10.60 (s, 1H). MS found (M+H)+ (m/z), 458.04; calcd for C24H19N5O3S m/z, 457.12.
4.1.15.29. 2-((1H-Indol-4-yl)amino)-6-((4-methoxyphenyl)sulfonyl)-8-methylpyrido[2,3-d]-pyrimidin-7(8H)-one (7ac)
Starting from 6g and 1H-indol-4-amine, 62% of 7ac was obtained as per the method described for the synthesis of 7; mp 289–292 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.31 (s, 3H), 3.79 (s, 3H), 6.60 (s, 1H), 7.03–7.50 (m, 6H), 7.87–7.90 (m, 2H), 8.75 (s, 1H), 9.01 (s, 1H), 10.28 (s, 1H), 11.10 (s, 1H). MS found (M+H)+ (m/z), 462.15; calcd for C23H19N5O4S m/z, 461.12.
4.1.15.30. 2-((1H-Indol-5-yl)amino)-6-((4-methoxyphenyl)sulfonyl)-8-methylpyrido[2,3-d]-pyrimidin-7(8H)-one (7ad)
Starting from 6g and 1H-indol-5-amine, 63% of 7ad was obtained as per the method described for the synthesis of 7; mp 260–262 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.25 (s, 3H), 3.60 (s, 3H), 6.17 (s, 1H), 6.74–7.38 (m, 6H), 7.62–7.79 (m, 2H), 8.53 (s, 1H), 8.78 (s, 1H), 10.23 (s, 1H), 10.84 (s, 1H). MS found (M+H)+ (m/z), 462.11; calcd for C23H19N5O4S m/z, 461.12.
4.1.15.31. 4-((2-((1H-Indol-5-yl)amino)-8-methyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidin-6-yl)sulfonyl)benzoic acid (7ae)
Starting from 6h and 1H-indol-5-amine, 69% of 7ae was obtained as per the method described for the synthesis of 7; mp 306–310 °C; 1H NMR (600 MHz, DMSO-d6): δ 3.87 (s, 3H), 6.39 (s, 1H), 7.35–7.46 (m, 4H), 8.01–8.12 (m, 4H), 8.84 (s, 1H), 9.03 (s, 1H), 10.53 (s, 1H), 11.07 (s, 1H). MS found (M+H)+ (m/z), 476.12; calcd for C23H17N5O5S m/z, 475.10.
4.1.15.32. 2-((1H-Indol-5-yl)amino)-6-((2,4-dichlorophenyl)sulfonyl)-8-methylpyrido[2,3-d]-pyrimidin-7(8H)-one (7af)
Starting from 6j and 1H-indol-5-amine, 69% of 7af was obtained as per the method described for the synthesis of 7; mp 323–326 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.46 (s, 3H), 6.41 (s, 1H), 7.34–7.49 (m, 4H), 7.76–7.83 (m, 2H), 8.03–8.26 (m, 2H), 8.88 (s, 1H), 9.08 (s, 1H), 10.59 (s, 1H), 11.09 (s, 1H). MS found (M+H)+ (m/z), 500.90; calcd for C22H15Cl2N5O3S m/z, 499.03.
4.1.15.33. 6-((2,4-Dichlorophenyl)sulfonyl)-8-methyl-2-((1-methyl-1H-indol-5-yl)amino)pyrido[2,3-d]pyrimidin-7(8H)-one (7ag)
Starting from 6j and 1-methyl-1H-indol-5-amine, 6a% of 7ag was obtained as per the method described for the synthesis of 7;mp 290–292 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.44 (s, 3H), 3.76 (s, 3H), 6.39 (s, 1H), 7.30 (s, 1H), 7.41 (d, J = 9.0 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.75 (d, J = 8.4 Hz, 1H), 7.82 (s, 1H), 7.99 (s, 1H), 8.24 (d, J = 9.0 Hz, 1H), 8.87 (s, 1H), 9.07 (s, 1H), 10.60 (s, 1H). MS found (M+H)+ (m/z), 514.02; calcd for C23H17Cl2N5O3S m/z, 513.04.
4.1.15.34. 2-((1H-Indol-5-yl)amino)-6-((4-fluoro-2-nitrophenyl)-sulfonyl)-8-methylpyrido-[2,3-d]pyrimidin-7(8H)-one (7ah)
Starting from 6k and 1H-indol-5-amine, 62% of 7ah was obtained as per the method described for the synthesis of 7; mp 290–292 °C; 1H NMR: (600 MHz, DMSO-d6): δ 3.51 (s, 3H), 6.41 (s, 1H), 7.33–7.47 (m, 3H), 7.85 (s, 1H), 8.03–8.09 (m, 2H), 8.47 (s, 1H), 8.68 (s, 1H), 9.11 (s, 1H), 10.62 (s, 1H), 11.08 (s, 1H). MS found (M+H)+ (m/z), 495.10; calcd for C22H15FN6O5S m/z, 494.08.
4.1.15.35. 2-((1H-Indol-5-yl)amino)-6-((2-amino-4-fluorophenyl)sulfonyl)-8-methylpyrido-[2,3-d]pyrimidin-7(8H)-one (7ai)
Starting from 6l and 1H-indol-5-amine, 63% of 7ai was obtained as per the method described for the synthesis of 7; mp 330–332 °C; 1H NMR (600 MHz, DMSO-d6): δ 3.94 (s, 3H), 6.45 (s, 1H), 7.21–7.51 (m, 5H), 8.01–8.07 (m, 2H), 8.89 (s, 1H), 9.04 (s, 1H), 10.66 (s, 1H), 11.11 (s, 1H). MS found (M+H)+ (m/z), 465.11; calcd for C22H17FN6 O3S m/z, 464.11.
4.1.15.36. 2-((1H-Indol-5-yl)amino)-6-((2-fluoro-4-nitrophenyl)-sulfonyl)-8-methylpyrido-[2,3-d]pyrimidin-7(8H)-one (7aj)
Starting from 6m and 1H-indol-5-amine, 67% of 7aj was obtained as per the method described for the synthesis of 7; mp 338–340 °C; 1H NMR (600 MHz, DMSO-d6): δ 3.46 (s, 3H),6.41 (s, 1H), 7.34–7.38 (m, 2H), 7.47–7.49 (m, 1H), 8.03 (s, 1H), 8.30–8.36 (m, 3H), 8.91 (s, 1H), 9.10 (s, 1H), 10.62 (s, 1H), 11.08 (s, 1H). MS found (M+H)+ (m/z), 495.08; calcd for C22H15FN6O5S m/z, 494.08.
4.1.15.37. 2-((1H-Indol-5-yl)amino)-6-((4-amino-2-fluorophenyl)sulfonyl)-8-methylpyrido-[2,3-d]pyrimidin-7(8H)-one (7ak)
Starting from 6n and 1H-indol-5-amine, 69% of 7ak was obtained as per the method described for the synthesis of 7; mp 292–294 °C; 1H NMR (600 MHz, DMSO-d6): δ 3.49 (s, 3H),6.27 (m, 1H), 6.39–6.41 (m, 3H), 6.47 (d, J = 8.4 Hz, 1H), 7.33–7.37 (m, 2H), 7.47–7.49 (m, 1H), 7.61–7.64 (m, 1H), 8.03 (s, 1H), 8.71 (s, 1H), 9.04 (s, 1H), 10.41 (s, 1H), 11.04 (s, 1H). MS found (M+H)+ (m/z), 465.11; calcd for C22H17FN6O3S m/z, 464.11.
4.1.15.38. 6-((2,4-Difluorophenyl)sulfonyl)-8-methyl-2-(pyridin-2-ylamino)pyrido[2,3-d]-pyrimidin-7(8H)-one (7al)
Starting from 6i and 2-aminopyridine, 68% of 7al was obtained as per the method described for the synthesis of 7; mp >350 °C; 1H NMR (600 MHz, DMSO-d6): δ 3.50 (s, 3H), 7.17 (d, J = 8.4 Hz, 1H), 7.23–7.25 (m, 1H), 7.38–7.40 (m, 1H), 7.52–7.54 (m, 2H), 8.11–8.12 (m, 2H), 8.26 (d, J = 8.4 Hz, 1H), 8.97 (s, 1H), 9.21 (s, 1H), 10.87 (s, 1H). MS found (M+H)+ (m/z), 430.07; calcd for C19H13F2N5 O3S m/z, 429.07.
4.1.15.39. 6-(2,4-difluorophenylsulfonyl)-8-methyl-2-(pyrimidin-4-ylamino)pyrido[2,3-d]-pyrimidin-7(8H)-one (7am)
Starting from 6i and 2-aminopyrimidine, 60% of 7am, was obtained as per the method described for the synthesis of 7; mp >330 °C; 1H NMR (600 MHz, DMSO-d6): δ 3.52 (s, 3H), 6.68–6.74 (m, 2H), 7.27–7.29 (m, 1H), 7.35–7.48 (m, 1H), 7.54–7.57 (m, 1H), 8.21 (d, J = 7.8 Hz, 1H), 8.95 (s, 1H), 9.25 (s, 1H), 10.89 (s, 1H). MS found (M+H)+ (m/z), 431.07; calcd for C18H12F2N6O3S m/z, 430.07.
4.1.15.40. 2-((1H-Indol-4-yl)amino)-6-((2,4-difluorophenyl)sulfonyl)-8-methylpyrido[2,3-d]-pyrimidin-7(8H)-one (7an)
Starting from 6i and 1H-indol-4-amine, 71% of 7an was obtained as per the method described for the synthesis of 7; mp 334–336 °C; 1H NMR (600 MHz, DMSO-d6): δ 3.34 (s, 3H), 6.62–6.75 (m, 1H), 7.09–7.58(m, 6H), 8.12–8.15 (m, 1H), 8.89 (s, 1H), 9.12 (s, 1H), 10.45 (s, 1H), 11.17 (s, 1H). MS found (M+H)+ (m/z), 468.09; calcd for C22H15F2N5O3S m/z, 467.09.
4.1.15.41. 2-((1H-Indol-5-yl)amino)-6-((2,4-difluorophenyl)sulfonyl)-8-methylpyrido[2,3-d]-pyrimidin-7(8H)-one (7ao)
Starting from 6i and 1H-indol-5-amine, 86% of 7ao was obtained as per the method described for the synthesis of 7; mp >350 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.60 (s, 3H), 6.50 (s, 1H), 7.20–8.40 (m, 7H), 8.80 (s, 1H), 9.10 (s, 1H), 10.50 (s, 1H), 11.30 (s, 1H). MS found (M+H)+ (m/z), 468.10; calcd for C22H15F2 N5O3S m/z, 467.09.
4.1.15.42. 2-((1H-Indol-6-yl)amino)-6-((2,4-difluorophenyl)sulfonyl)-8-methylpyrido[2,3-d]-pyrimidin-7(8H)-one (7ap)
Starting from 6i and 1H-indol-6-amine, 75% of 7ap was obtained as per the method described for the synthesis of 7; mp >350 °C; 1H NMR (600 MHz, DMSO-d6): δ 3.54 (s, 3H), 6.37 (s, 1H), 7.30–7.32 (m, 2H), 7.38–7.41 (m, 1H), 7.47–7.52 (m, 2H), 8.13–8.17 (m, 2H), 8.88 (s, 1H), 9.10 (s, 1H), 10.67 (s, 1H), 11.16 (s, 1H). MS found (M+H)+ (m/z), 468.09; calcd for C22H15F2N5O3S m/z, 467.09.
4.1.15.43. 2-((1H-Indol-5-yl)amino)-6-((4-fluorobenzyl)sulfonyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7aq)
Starting from 6o and 1H-indol-5-amine, 68% of 7aq was obtained as per the method described for the synthesis of 7; mp 260–262 °C; 1H NMR (400 MHz; DMSO-d6): δ 3.66 (s, 3H), 4.81 (s, 2H), 6.44 (s, 1H), 7.16 (t, 2H), 7.30–7.33 (m, 5H), 7.35–7.40 (m, 1H), 8.37 (s, 1H), 8.92 (s, 1H), 10.25 (s, 1H), 11.10 (s, 1H). MS found (M+H)+ (m/z), 464.10; calcd for C23H18FN5O3S m/z, 463.11.
4.1.15.44. 2-((1H-Indol-5-yl)amino)-6-((2,4-difluorobenzyl)sulfonyl)-8-methylpyrido[2,3-d]-pyrimidin-7(8H)-one (7ar)
Starting from 6p and 1H-indol-5-amine, 77% of 7ar was obtained as per the method described for the synthesis of 7; mp 228–230 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.65 (s, 3H), 4.84 (s, 2H), 6.44 (s, 1H), 7.10–7.49 (m, 6H), 8.04–8.05 (m, 1H), 8.40 (s, 1H), 8.94 (s, 1H), 10.55 (s, 1H), 11.11 (s, 1H). MS found (M+H)+ (m/z), 482.10; calcd for C23H17F2N5O3S m/z, 481.10.
4.1.15.45. 2-((1H-Indol-5-yl)amino)-N-(4-fluorophenyl)-8-methyl-7-oxo-7,8-dihydropyrido-[2,3-d]pyrimidine-6-carboxamide (7as)
Starting from 6q and 1H-indol-5-amine, 72% of 7as was obtained as per the method described for the synthesis of 7; mp 284–286 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.69 (s, 3H), 6.43 (s, 1H), 7.20–7.49 (m, 5H), 7.75–8.06 (m, 3H), 8.81 (s, 1H), 9.04 (s, 1H), 10.45 (s, 1H), 11.08 (s, 1H), 11.73 (s, 1H). MS found (M+H)+ (m/z), 429.14; calcd for C23H17FN6O2 m/z, 428.14.
4.1.15.46. 2-((1H-Indol-5-yl)amino)-N-(2,4-difluorophenyl)-8-methyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidine-6-carboxamide (7at)
Starting from 6r and 1H-indol-5-amine, 64% of 7at was obtained as per the method described for the synthesis of 7; mp 322–324 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.68 (s, 3H), 6.43 (s, 1H), 7.11–7.49 (m, 5H), 8.05–8.06 (m, 1H), 8.47–8.48 (m, 1H), 8.83 (s, 1H), 9.05 (s, 1H), 10.47 (s, 1H), 11.09 (s, 1H), 12.02 (s, 1H). MS found (M+H)+ (m/z), 447.13; calcd for C23H16F2N6O2 m/z, 446.13.
4.1.15.47. 2-((1H-Indol-5-yl)amino)-N-(4-fluorophenyl)-8-methyl-7-oxo-7,8-dihydropyrido-[2,3-d]pyrimidine-6-sulfonamide (7au)
Starting from 6s and 1H-indol-5-amine, 75% of 7au was obtained as per the method described for the synthesis of 7; mp 324–326 °C; 1H NMR (600 MHz, DMSO-d6): δ 3.55 (s, 3H), 6.39 (s, 1H), 7.05–7.12 (m, 4H), 7.32–7.43 (m, 3H), 7.99 (s, 1H), 8.52 (s, 1H), 8.90 (s, 1H), 10.10 (s, 1H), 10.41 (s, 1H), 11.06 (s, 1H). MS found (M+H)+ (m/z), 465.12; calcd for C22H17FN6O3S m/z, 464.11.
4.1.15.48. 2-((1H-Indol-5-yl)amino)-N-(2,4-difluorophenyl)-8-methyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidine-6-sulfonamide (7av)
Starting from 6t and 1H-indol-5-amine, 65% of 7av was obtained as per the method described for the synthesis of 7; mp 332–334 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.62 (s, 3H), 6.42 (s, 1H), 7.00–7.48 (m, 6H), 8.04 (s, 1H), 8.38 (s, 1H), 8.89 (s, 1H), 9.74 (s, 1H), 10.43 (s, 1H), 11.08 (s, 1H). MS found (M+H)+ (m/z), 483.10; calcd for C22H16F2N6O3S m/z, 482.10.
4.1.15.49. 2-((1H-Indazol-5-yl)amino)-6-((2,4-difluorophenyl)-sulfonyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7aw)
Starting from 6i and 1H-indazol-5-amine, 68% of 7aw was obtained as per the method described for the synthesis of 7; mp 284–286 °C; 1H NMR (300 MHz; DMSO-d6): δ 3.51 (s, 3H), 7.35–7.53 (m, 3H), 7.61–7.66 (m, 1H), 7.93–8.24 (m, 3H), 8.87 (s, 1H), 9.17 (s, 1H), 10.71 (s, 1H), 13.02 (s, 1H). MS found (M+H)+ (m/z), 469.10; calcd for C21H14F2N6O3S m/z, 468.08.
4.1.15.50. 2-(Benzo[b]thiophen-5-ylamino)-6-((2,4-difluorophenyl)sulfonyl)-8-methylpyrido-[2,3-d]pyrimidin-7(8H)-one (7ax)
Starting from 6i and benzo[b]thiophenl-5-amine, 61% of 7ax was obtained as per the method described for the synthesis of 7; mp 320–322 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.50 (s, 3H), 7.39–7.50 (m, 3H), 7.71–7.75 (m, 2H), 7.97 (d, J = 8.0 Hz, 1H), 8.13–8.14 (m, 1H), 8.41 (s, 1H), 8.90 (s, 1H), 9.14 (s, 1H), 10.79 (s, 1H). MS found (M+H)+ (m/z), 485.10; calcd for C22H14F2N4O3S2 m/z, 484.05.
4.1.15.51. 2-(Benzofuran-5-ylamino)-6-((2,4-difluorophenyl)-sulfonyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7ay)
Starting from 6i and benzofuran-5-amine, 64% of 7ay was obtained as per the method described for the synthesis of 7; mp 318–320 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.47 (s, 3H), 6.97 (s, 1H), 7.38 (t, 1H), 7.49 (t, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.66 (s, 1H), 7.97 (d, J = 1.8 Hz, 1H), 8.09–8.14 (m, 2H), 8.88 (s, 1H), 9.12 (s, 1H), 10.72 (s, 1H). MS found (M+H)+ (m/z), 469.10; calcd for C22 H14F2N4O4S m/z, 468.07.
4.1.15.52. 6-((2,4-Difluorophenyl)sulfonyl)-8-methyl-2-((1-methyl-1H-indol-5-yl)amino)pyrido[2,3-d]pyrimidin-7(8H)-one (7az)
Starting from 6i and 1-methyl-1H-indol-5-amine, 72% of 7az was obtained as per the method described for the synthesis of 7; mp 330–332 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.45 (s, 3H), 3.76 (s, 3H), 6.39 (s, 1H), 7.30–7.49 (m, 5H), 7.99 (s, 1H), 8.11–8.13 (m, 1H), 8.84 (s, 1H), 9.07 (s, 1H), 10.57 (s, 1H). MS found (M+H)+ (m/z), 482.20; calcd for C23H17F2N5O3S m/z, 481.10.
4.1.15.53. 6-((2,4-Difluorophenyl)sulfonyl)-8-methyl-2-((2-methyl-1H-indol-5-yl)amino)pyrido[2,3-d]pyrimidin-7(8H)-one (7aaa)
Starting from 6i and 2-methyl-1H-indol-5-amine, 70% of 7aaa was obtained as per the method described for the synthesis of 7; mp 288–290 °C; 1H NMR (300 MHz; DMSO-d6): δ 2.36 (s, 3H), 3.46 (s, 3H), 6.10 (s, 1H), 7.23–8.15 (m, 6H), 8.85 (s, 1H), 9.07 (s, 1H), 10.52 (s, 1H), 10.90 (s, 1H). MS found (M+H)+ (m/z), 482.07; calcd for C23H17F2N5O3S m/z, 481.10.
4.1.15.54. 2-(((1H-Indol-5-yl)methyl)amino)-6-((2,4-difluorophenyl)sulfonyl)-8-methylpyrido[2,3-d]pyrimidin-7(8H)-one (7aab)
Starting from 6i and (1H-indol-5yl)methanamine, 75% of 7aab was obtained as per the method described for the synthesis of 7; mp 216–220 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.42 (s, 3H), 4.64 (d, J = 6.0 Hz, 2H), 7.06–7.51 (m, 6H), 8.09 (t, 1H), 8.76 (s, 1H), 8.93 (s, 1H), 9.02 (s, 1H), 9.10 (t, 1H), 11.01 (s, 1H). MS found (M+H)+ (m/z), 482.20; calcd for C23H17F2N5O3S m/z, 481.10.
4.1.15.55. Methyl 2-(5-((6-((2,4-difluorophenyl)sulfonyl)-8-methyl-7-oxo-7,8-dihydropyrido-[2,3-d]pyrimidin-2-yl)amino)-1H-indol-1-yl)acetate (7aac)
Starting from 6i and methyl 2-(5-amino-1H-indol-1yl)acetate, 65% of 7aac was obtained as per the method described for the synthesis of 7; mp 296–298 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.48 (s, 3H), 3.80 (s, 3H), 5.05 (s, 2H), 6.46 (s, 1H), 7.35–7.59 (m, 5H), 8.03–8.16 (m, 2H), 8.85 (s, 1H), 9.10 (s, 1H), 10.62 (s, 1H). MS found (M+H)+ (m/z), 540.11; calcd for C25H19F2N5O5S m/z, 539.11.
4.1.15.56. 5-((6-((2,4-Difluorophenyl)sulfonyl)-8-methyl-7-oxo-7,8-dihydropyrido[2,3-d]-pyrimidin-2-yl)amino)indoline-2,3-dione (7aad)
Starting from 6i and methyl 5-aminoindolin-2,3-dione, 64% of 7aad was obtained as per the method described for the synthesis of 7; mp 312–314 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.55 (s, 3H), 3.80 (s, 3H), 6.61–6.70 (m, 4H), 6.71–6.81 (m, 1H), 7.27–7.34 (m, 1H), 8.20 (s, 1H), 8.91 (s, 1H), 10.56 (s, 1H), 10.96 (s, 1H). MS found (M+H)+ (m/z), 498.10; calcd for C22H13F2N5O5S m/z, 497.06.
4.1.15.57. 6-((2,4-Difluorophenyl)sulfonyl)-2-((8-hydroxyquinolin-2-yl)amino)-8-methyl-pyrido[2,3-d]pyrimidin-7(8H)-one (7aae)
Starting from 6i and 2-aminoquinolin-8-ol, 61% of 7aae was obtained as per the method described for the synthesis of 7; mp 320–322 °C. 1H NMR (600 MHz; DMSO-d6): δ 3.49 (s, 3H), 6.71–6.81 (m, 1H), 6.93–6.96 (m, 2H), 7.15–7.82 (m, 4H), 8.20 (d, J = 8.2 Hz, 1H), 9.06 (s, 1H), 9.18 (s, 1H), 10.60 (s, 1H), 10.64 (s, 1H). MS found(M+H)+ (m/z), 496.09; calcd for C23H15F2N5O4Sm/z, 495.08.
4.1.15.58. 6-((2,4-Difluorophenyl)sulfonyl)-8-methyl-2-((2-oxoindolin-5-yl)amino)pyrido-[2,3-d]pyrimidin-7(8H)-one (7aaf)
Starting from 6i and 5-aminoindolin-2-one, 67% of 7aaf was obtained as per the method described for the synthesis of 7; mp 308–310 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.42 (s, 2H), 3.48 (s, 3H), 6.53–6.79 (m, 2H), 6.53–6.79 (m, 4H), 9.25 (s, 1H), 9.42 (s, 1H), 10.28 (s, 1H), 11.12 (s, 1H). MS found (M+H)+ (m/z), 484.05; calcd for C22H15F2N5O4S m/z, 483.08.
4.1.15.59. 5-((6-((2,4-Difluorophenyl)sulfonyl)-8-methyl-7-oxo-7,8-dihydropyrido[2,3-d]-pyrimidin-2-yl)amino)-1H-indole-2-carboxylic acid (7aag)
Starting from 6i and 5-amino-1H-indole-2-carboxylic acid, 62% of 7aag was obtained as per the method described for the synthesis of 7; mp 240–244 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.46 (s, 3H), 6.68–6.76 (m, 2H), 7.07–7.13 (m, 2H), 7.37–7.59 (m, 2H), 8.13 (s, 1H), 8.85 (s, 1H), 9.08 (s, 1H), 10.60 (s, 1H), 11.25 (s, 1H), 11.73 (s, 1H). MS found (M+H)+ (m/z), 512.03; calcd for C23H15F2N5O5S m/z, 511.08.
4.1.15.60. 6-((2,4-Difluorophenyl)sulfonyl)-8-methyl-2-((4-(4-methylpiperazin-1-yl)phenyl)-amino)pyrido[2,3-d]pyrimidin-7 (8H)-one (7aah)
Starting from 6i and 4-(4-methylpiperazin-1-yl)benzenamine, 74% of 7aah was obtained to method described for the synthesis of 7; mp 246–250 °C; 1H NMR (600 MHz; DMSO-d6): δ 2.21 (s, 3H), 2.40–2.44 (m, 4H), 3.07–3.10 (m, 4H), 3.44 (s, 3H), 6.93 (d, J = 8.4 Hz, 2H), 7.38–7.64 (m, 4H), 8.11–8.14 (m, 1H), 8.84 (s, 1H), 9.05 (s, 1H), 10.50 (s, 1H). MS found (M+H)+ (m/z), 527.73; calcd for C25H24F2N6O3S m/z, 526.16.
4.1.15.61. 2-((1H-Pyrrolo[2,3-b]pyridin-5-yl)amino)-6-((2,4-difluorophenyl)sulfonyl)-8-methylpyrido[2,3-d]pyrimidin-7 (8H)-one (7aai)
Starting from 6i and 1H-pyrrolo[2,3-b]-pyridine-5-amine, 74% of 7aai was obtained as per the method described for the synthesis of 7; mp 320–322 °C; 1H NMR (600 MHz; DMSO-d6): δ 3.42 (s, 3H), 6.43 (s, 1H), 7.37–7.47 (m, 3H), 8.12–8.51 (m, 3H), 8.87 (s, 1H), 9.10 (s, 1H), 10.64 (s, 1H), 11.60 (s, 1H). MS found (M+H)+ (m/z), 469.09; calcd for C21H14F2N6O3S m/z, 468.08.
4.1.15.62. 2-((1H-Indol-5-yl)amino)-6-((2,4-difluorophenyl)sulfonyl)pyrido[2,3-d]pyrimidin-7(8H)-one (7aaj)
Starting from 6u and 1H-indol-5-amine, 59% of 7aaj was obtained as per the method described for the synthesis of 7; mp >350 °C; 1H NMR (600 MHz; DMSO-d6): δ 6.38 (s, 1H), 7.31–7.58 (m, 5H), 8.09–8.25 (m, 2H), 8.81 (s, 1H), 9.03 (s, 1H), 10.41 (s, 1H), 11.04 (s, 1H), 12.55 (s, 1H). MS found (M+H)+ (m/z), 454.07; calcd for C21H13F2N5O3S m/z, 453.07.
4.1.15.63. 2-(1H-Indol-5-ylamino)-6-(2,4-difluorophenylsulfonyl)-8-ethylpyrido[2,3-d]pyrimidin-7(8H)-one (7aak)
Starting from 6v and 1H-indol-5-amine, 65% of 7aak was obtained as per the method described for the synthesis of 7; mp 278–280 °C; 1H NMR (DMSO-d6, 300 MHz): δ1.19 (t, 3H), 4.19 (br s, 2H), 6.40 (br s, 1H),7.54–7.47 (m, 5H), 8.18–8.09 (m, 2H), 8.86 (s, 1H), 9.09 (s, 1H), 10.57 (br s, 1H), 11.09 (br s, 1H). MS found (M−H)+ (m/z), 480.10; Calcd for C23H17F2N5O3S m/z, 481.10.
4.1.15.64. 2-((1H-Indol-5-yl)amino)-6-((2,4-difluorophenyl)sulfonyl)-8-propylpyrido[2,3-d]-pyrimidin-7(8H)-one (7aal)
Starting from 6w and 1H-indol-5-amine, 64% of 7aal was obtained as per the method described for the synthesis of 7; mp 218–220 °C; 1H NMR: (600 MHz, DMSO-d6): δ 0.85–0.92 (m, 3H), 1.55–1.62 (m, 2H), 3.95–4.09 (m, 2H), 6.37 (s, 1H), 7.23–7.52 (m, 5H), 7.95–8.12 (m, 2H), 8.85 (s, 1H), 9.08 (s, 1H), 10.58 (s, 1H), 11.09 (s, 1H). MS found (M+H)+ (m/z), 496.12; calcd for C24H19F2 N5O3S m/z, 495.12.
4.1.15.65. Methyl 2-(2-(1H-indol-5-ylamino)-6-(2,4-difluorophenylsulfonyl)-7-oxopyridio-[2,3-d]pyrimidin-8(7H)-yl)acetate (7aam)
Starting from 6aa and 1H-indol-5-amine, 64% of 7aam was obtained as per the method described for the synthesis of 7; mp 270–272 °C; 1H NMR (DMSO-d6, 300 MHz): δ 3.67 (s, 3H), 4.92 (br s, 2H), 6.38 (br s, 1H), 7.43–7.37 (m, 4H), 7.56–7.50 (m, 1H), 7.91 (br s, 1H), 8.16–8.08 (m, 1H), 8.96 (s, 1H), 9.13 (s, 1H), 10.68 (br s, 1H), 11.09 (br s, 1H). MS found (M−H)+ (m/z), 524.10; Calcd for C24H17F2N5O5S m/z, 525.09.
4.1.15.66. 2-((1H-Indol-5-yl)amino)-8-cyclopropyl-6-((2,4-difluorophenyl)sulfonyl)pyrido-[2,3-d]pyrimidin-7(8H)-one (7aan)
Starting from 6x and 1H-indol-5-amine, 73% of 7aan was obtained as per the method described for the synthesis of 7; mp 262–264 °C; 1H NMR: (600 MHz, DMSO-d6): δ 0.77–0.79 (m, 2H), 1.17–1.18 (m, 2H), 2.79–2.81 (m, 1H), 6.40 (s, 1H), 7.32–7.50 (m, 5H), 8.12–8.14 (m, 1H), 8.30 (s, 1H), 8.79 (s, 1H), 9.03 (s, 1H), 10.51 (s, 1H), 11.06 (s, 1H). MS found (M+H)+ (m/z), 494.11; calcd for C24H17F2N5O3S m/z, 493.10.
4.1.15.67. 2-(1H-Indol-5-ylamino)-6-(2,4-difluorophenylsulfonyl)-8-cyclopentylpyrido[2,3-d]pyrimidin-7(8H)-one (7aao)
Starting from 6y and 1H-indol-5-amine, 70% of 7aao was obtained as per the method described for the synthesis of 7; mp 241–242 °C; 1H NMR (DMSO-d6, 300 MHz): δ 1.36 (br s, 2H), 1.62–1.54 (m, 4H), 2.09–2.06 (m, 2H), 5.62–5.56 (br s, 1H), 6.37 (br s, 1H), 7.55–7.24 (m, 5H), 7.85 (br s, 1H), 8.16–8.08 (m, 1H), 8.82 (s, 1H), 9.07 (s, 1H), 10.45 (br s, 1H), 11.09 (br s, 1H). MS found (M+H)+ (m/z), 522.10; Calcd for C26H21F2N5O3S m/z, 521.13.
4.1.15.68. 2-((1H-Indol-5-yl)amino)-8-cyclohexyl-6-((2,4-difluorophenyl)sulfonyl)pyrido-[2,3-d]pyrimidin-7(8H)-one (7aap)
Starting from 6z and 1H-indol-5-amine, 60% of 7aap was obtained as per the method described for the synthesis of 7; mp 282–284 °C; 1H NMR: (600 MHz, DMSO-d6): δ 1.12–1.43 (m, 4H), 1.48–1.61 (m, 4H), 1.70–1.81 (m, 2H), 2.27–2.30 (m, 1H), 6.36 (s, 1H), 7.20–7.52 (m, 6H), 8.10–8.12 (m, 1H), 8.80 (s, 1H), 9.06 (s, 1H), 10.86 (s, 1H), 11.10 (s, 1H). MS found (M+H)+ (m/z), 536.17; calcd for C27H23F2N5O3S m/z, 535.15.
4.1.15.69. 2-(1H-Indol-5-ylamino)-6-(2,4-difluorophenylsulfonyl)-8-(cyclopropylmethyl)-pyrido[2,3-d]pyrimidin-7(8H)-one (7aaq)
Starting from 6ab and 1H-indol-5-amine, 67% of 7aaq was obtained as per the method described for the synthesis of 7; mp 210–212 °C; 1H NMR (DMSO-d6, 300 MHz): δ 0.13–0.06 (m, 4H), 1.02–0.93 (m, 1H), 3.81 (br s, 2H), 6.14 (m, 1H), 7.26–7.12 (m, 5H), 7.91–7.84 (m, 2H), 8.62 (s, 1H), 8.85 (s, 1H), 10.32 (br s, 1H), 10.84 (br s, 1H). MS found (M+H)+ (m/z), 508.10; Calcd for C25H19 F2N5O3S m/z, 507.12.
4.1.15.70. 2-(1H-Indol-5-ylamino)-6-(2,4-difluorophenylsulfonyl)-8-(cyclopentylmethyl)-pyrido[2,3-d]pyrimidin-7(8H)-one (7aar)
Starting from 6ac and 1H-indol-5-amine, 70% of 7aar was obtained as per the method described for the synthesis of 7; mp 230–232 °C; 1H NMR (DMSO-d6, 300 MHz): δ 1.23 (br s, 2H),1.32 (br s, 2H), 1.52 (br s, 4H), 2.44 (m, 1H), 4.13 (d, 2H),, 6.37 (br s, 1H), 7.53–7.34 (m, 5H), 8.18–8.09 (m, 2H), 8.85 (s, 1H), 9.08 (s, 1H), 10.56 (br s, 1H), 11.08 (br s, 1H). MS found (M+H)+ (m/z), 536.10; Calcd for C27H23F2N5O3S m/z, 535.15.
5. Biology
5.1. Materials and methods
Unless otherwise stated, all chemicals used in these experiments were either from Sigma–Aldrich Co, St. Louis, MO or Fisher Scientific, Fair Lawn, NJ.
5.1.1. Cell culture
K562 and GRANTA-519 cells were maintained in RPMI medium 1640 (CellGro) supplemented with 10% FBS (Cellgeneration, CO) and 1 unit/mL penicillin–streptomycin (Invitrogen). All other cells were maintained in DMEM (CellGro) supplemented with 10% FBS and 1 unit/mL penicillin–streptomycin. Cells were incubated at 37 °C under humidified conditions and 5% CO2.
5.1.2. Growth inhibition and GI50 determination
Cells were grown in the presence of increasing concentrations of 7n, 7t, 7u, or 7ao for 72 h. Cell viability was measured by trypan blue exclusion. Growth inhibition and GI50 determination were performed as previously described.19
5.1.3. Kinase assays and IC50 determination
10 ng recombinant PLK2 (Life Technologies, Grand Island, NY) was diluted with kinase buffer (20 Mm Tris pH 7.5, 10 mM MgCl2, 0.01% NP-40, 2 mM DTT) and incubated with the indicated concentrations of each inhibitor at room temperature for 30 min. The kinase reactions were initiated by the addition of 1 µg (1.5 µM) of dephosphorylated bovine alpha-casein, 5 µM ATP and 10 µCi γ-32P-ATP. The reactions were incubated at 30 °C for 20 min, terminated by the addition of 3% phosphoric acid, spotted onto PE-30 filter mat, washed, dried and taken for scintillation counts. The data were plotted (after background subtraction) as a function of log-drug concentration using Prism 4 Graph pad software and IC50 values determined by plotting sigmoidal non-linear regression curves with variable slopes.
5.1.4. In vitro tubulin polymerization assay
Tubulin polymerization was performed as previously described20 using MAP-rich bovine tubulin 1.5 mg/mL (Cytoskeleton Inc., Denver, CO) reconstituted in a buffer containing 80 mM PIPES pH 6.9, 1 mM GTP, 0.5 mM EGTA, and 2 mM MgCl2. Reaction mixtures were briefly incubated in 1.5 mL tubes with DMSO, compounds 7n, 7t and 7u, 7ao or Vincristine, transferred to UV-cuvettes and placed in a preheated (37 °C) spectrophotometer to analyze polymerization. Polymer formation was measured by monitoring changes in absorbance at 340 nm and recorded every 2 min over a 30 min period (n = 3).
5.1.5. Apoptosis assays
U2OS cells were plated at a density of 1–3 × 104 cells/well in Optilux black-walled clear-bottom tissue culture microtiter plates (BD Biosciences, San Jose, CA). After an overnight incubation, the cells were treated with increasing concentrations of compound 7n or 7ao for 24 h. DMSO was used as the control. Cell viability was measured using the Cell Titer Blue reagent (Promega Corporation, Madison, WI). Induction of apoptotic effect or CASPASEs 3/7 activity was assayed using the Caspase-Glo reagent (Promega Corporation, Madison, WI) using the fluorescence and luminescence modes, respectively, of Promega’s Glomax Multi detection System.
5.1.6. Nude mouse xenograft studies
1 × 107 MDAMB-231 cells were subcutaneously implanted into the right flank of 6–8 week old NCR nu/nu female mice. When the tumors reached an average volume of 75 mm3, the mice were weighed and randomized into treatment groups (n = 10) and treated Q2D with either placebo, or the indicated amounts of 7n or 7ao. Tumors volumes were measured using electronic Vernier calipers and calculated by the formula 0.4 (short2 × long).21
5.1.7. Molecular modeling
7ao small molecule was generated using ChemDraw (PerkinElmer). Molecule was then imported and prepared in Maestro (Schrödinger suite). Binding positions of 7ao were predicted using GLIDE22–24 and the coordinates of the X-ray co-crystal structure of PLK2-1C8 (4I6H). The ligand of the co-crystal (1C8) was used for grid generation. Multiple poses were generated. Binding position with high docking score was visualized and graphed using PyMOL. The PLK1 kinase domain (2RKU) was superimposed on PLK2 and analyzed using PyMOL.
5.1.8. Differential scanning fluorimetry (DSF)
Binding of 7ao to PLK2 or PLK1 kinase domain was assessed by incubating different concentrations of the drug with 1 µg of protein for 5 min and analyzing drug induced thermal shifts by DSF assays.16,17
Supplementary Material
Acknowledgments
This work was supported by Grants from the NIH (1RO1 CA 158209) and Onconova Therapeutics Inc. We are thankful to Dr. Stacey Baker for editorial assistance.
Footnotes
Kinase inhibition profile of 7ao (Reaction Biology Corp) and Kinome map for 7ao. This material is available free of charge via the Internet at http://sciencedirect.com. Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2015.11.045.
References
- 1.Weinstein IB, Joe A. Nat. Clin. Pract. Oncol. 2006;3:448. doi: 10.1038/ncponc0558. [DOI] [PubMed] [Google Scholar]
- 2.Druker BJ. Nat. Med. 2009;15:1149. doi: 10.1038/nm1009-1149. [DOI] [PubMed] [Google Scholar]
- 3.Lowery DM, Lim D, Yaffe MB. Oncogene. 2005;24:248. doi: 10.1038/sj.onc.1208280. [DOI] [PubMed] [Google Scholar]
- 4.Dai W. Oncogene. 2005;24:214. doi: 10.1038/sj.onc.1208270. [DOI] [PubMed] [Google Scholar]
- 5.Archambault V, Glover DM. Nat. Rev. Mol. Cell Biol. 2009;10:265. doi: 10.1038/nrm2653. [DOI] [PubMed] [Google Scholar]
- 6.Strebhardt K. Nat. Rev. Drug Disc. 2010;9:643. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 7.Simmons DL, Neel BG, Stevens R, Evett G, Erikson RL. Mol. Cell Biol. 1992;12:4164. doi: 10.1128/mcb.12.9.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warnke S, Kemmler S, Hames RS, Tsai HL, Hoffmann-Rohrer U, Fry AM, Hoffmann I. Curr. Biol. 2004;14:1200. doi: 10.1016/j.cub.2004.06.059. [DOI] [PubMed] [Google Scholar]
- 9.Cizmecioglu O, Krause A, Bahtz R, Ehret L, Malek N, Hoffmann I. J. Cell Sci. 2012;125:981. doi: 10.1242/jcs.095075. [DOI] [PubMed] [Google Scholar]
- 10.Burns TF, Fei P, Scata KA, Dicker DT, El-Deiry WS. Mol. Cell Biol. 2003;23:5556. doi: 10.1128/MCB.23.16.5556-5571.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reddy MVR, Akula B, Cosenza CS, Divaker SA, Mallireddigari MR, Pallela VR, Billa VK, Subbaiah DRC, Bharathi VE, Padgaonkar A, Baker SJ, Reddy EP. J. Med. Chem. 2014;57:578. doi: 10.1021/jm401073p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reddy MVR, Mallireddigari MR, Pallela VR, Venkatapuram P, Boominathan R, Bell SC, Reddy EP. Bioorg. Med. Chem. 2005;13:1715. doi: 10.1016/j.bmc.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 13.(a) Reddy MVR, Mallireddigari MR, Cosenza SC, Pallela VR, Iqbal NM, Robell KA, Kang AD, Reddy EP. J. Med. Chem. 2008;51:86. doi: 10.1021/jm701077b. [DOI] [PubMed] [Google Scholar]; (b) Reddy MVR, Reddy S. Acta Chim. Hung. 1984;115:269. [Google Scholar]; (c) Reddy DB, Reddy NS, Reddy MVR, Balasubramanyam S. Org. Prep. Proced. Int. 1988;20:205. [Google Scholar]
- 14.Reddy NS, Gumireddy K, Mallireddigari MR, Cosenza SC, Venkatapuram P, Bell SC, Reddy EP, Reddy MVR. Bioorg. Med. Chem. 2005;13:3141. doi: 10.1016/j.bmc.2005.02.051. [DOI] [PubMed] [Google Scholar]
- 15.Reddy MVR, Mallireddigari MR, Pallela VR, Cosenza CS, Billa VK, Akula B, Subbaiah DRC, Bharathi VE, Padgaonkar A, Hua LV, Gallo JM, Reddy EP. J Med. Chem. 2013;56:5562. doi: 10.1021/jm400575x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, Salemme FR. J. Biomol. Screening. 2001;6:429. doi: 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- 17.Niesen FH, Berglund H, Vedadi M. Nat. Protoc. 2007;2:2212. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 18.Danielle LA, Roy KH, Marc A, Robert A, Galemmo J, Simeon B, Anh PT, Hu P, Paul B, Jeffrey RN, Nanhua Y, May L, George T, Heather Z, Michael P, Bova ZR, Danny T, Lany R, Jeanne B, Linnea D, Kent F, Jennifer H, Ruth M, Donald F, Pearl T, Michael D, Jacek J, Wayman C, Andrei WK, Lee L, Yong LZ, Hing LS, John PA, Marcelle B, Dean RA. Chem. Med. Chem. 2013;8:1295. [Google Scholar]
- 19.Jatiani SS, Cosenza SC, Reddy MVR, Ha JH, Baker SJ, Samanta AK, Olnes MJ, Pfannes L, Sloand EM, Arlinghaus RB, Reddy EP. Genes Cancer. 2010;1:331. doi: 10.1177/1947601910371337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shelanski ML, Gaskin F, Cantor C. Proc. Natl. Acad. Sci. U.S.A. 1973;1973:765. doi: 10.1073/pnas.70.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sendo F, Miyake T, Fuyama S, Arai S. J. Natl. Cancer Inst. 1978;60:385. doi: 10.1093/jnci/60.2.385. [DOI] [PubMed] [Google Scholar]
- 22.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J. Med. Chem. 2006;49:6177. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 23.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. J. Med. Chem. 2004;47:1750. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 24.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shaw DE, Shelley M, Perry JK, Francis P, Shenkin PS. J. Med. Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.