

This article reports unique aspects of the management of hepatocellular carcinoma. The study aimed to determine if next‐generation sequencing of blood‐derived circulating tumor DNA from patients with hepatocellular carcinoma could identify actionable somatic molecular alterations. Illustrative examples of treated patients and of in silico molecular dynamic simulation to reveal genomic variant function are included.
Keywords: Hepatocellular carcinoma, Liquid biopsy, Circulating tumor DNA, Next‐generation sequencing
Abstract
Background.
Because imaging has a high sensitivity to diagnose hepatocellular carcinoma (HCC) and tissue biopsies carry risks such as bleeding, the latter are often not performed in HCC. Blood‐derived circulating tumor DNA (ctDNA) analysis can identify somatic alterations, but its utility has not been characterized in HCC.
Materials and Methods.
We evaluated 14 patients with advanced HCC (digital ctDNA sequencing [68 genes]). Mutant relative to wild‐type allele fraction was calculated.
Results.
All patients (100%) had somatic alterations (median = 3 alterations/patient [range, 1–8]); median mutant allele fraction, 0.29% (range, 0.1%–37.77%). Mutations were identified in several genes: TP53 (57% of patients), CTNNB1 (29%), PTEN (7%), CDKN2A (7%), ARID1A (7%), and MET (7%); amplifications, in CDK6 (14%), EGFR (14%), MYC (14%), BRAF (7%), RAF1 (7%), FGFR1 (7%), CCNE1 (7%), PIK3CA (7%), and ERBB2/HER2 (7%). Eleven patients (79%) had ≥1 theoretically actionable alteration. No two patients had identical genomic portfolios, suggesting the need for customized treatment. A patient with a CDKN2A‐inactivating and a CTNNB1‐activating mutation received matched treatment: palbociclib (CDK4/6 inhibitor) and celecoxib (COX‐2/Wnt inhibitor); des‐gamma‐carboxy prothrombin level decreased by 84% at 2 months (1,410 to 242 ng/mL [normal: ≤7.4 ng/mL]; alpha fetoprotein [AFP] low at baseline). A patient with a PTEN‐inactivating and a MET‐activating mutation (an effect suggested by in silico molecular dynamic simulations) received sirolimus (mechanistic target of rapamycin inhibitor) and cabozantinib (MET inhibitor); AFP declined by 63% (8,320 to 3,045 ng/mL [normal: 0–15 ng/mL]).
Conclusion.
ctDNA derived from noninvasive blood tests can provide exploitable genomic profiles in patients with HCC.
Implications for Practice.
This study reports that blood‐derived circulating tumor DNA can provide therapeutically exploitable genomic profiles in hepatocellular cancer, a malignancy that is known to be difficult to biopsy.
摘要
背景.因为影像学诊断肝细胞癌(HCC)的敏感度较高且组织活检有出血等风险, 所以通常不在HCC患者中进行组织活检。血液源性循环肿瘤DNA(ctDNA)分析可识别体细胞变异, 但其在HCC中的用途尚未描述。
材料和方法.我们评价了14例晚期HCC患者[数字ctDNA测序(68个基因)]。计算了相应的野生型等位基因部分的突变体。
结果.所有患者(100%)均有体细胞变异[中位数=3种变异/患者(范围为1‐8)];中位突变等位基因分数为0.29%(范围为0.1%–37.77%)。在若干基因中发现突变:TP53(57%的患者)、CTNNB1(29%)、PTEN(7%)、CDKN2A(7%)、ARID1A(7%)和 MET(7%);在若干基因中发现扩增:CDK6(14%)、EGFR(14%)、MYC(14%)、BRAF(7%)、RAF1(7%)、FGFR1(7%)、CCNE1(7%)、PIK3CA(7%)和 ERBB2/HER2(7%)。11例患者(79%)具有≥1个理论上可行的变异。未出现两例患者共有相同基因组组合的情况, 表明需要个性化治疗。携带CDKN2A失活和CTNNB1激活突变的患者接受了匹配治疗:帕博西尼(CDK4/6抑制剂)和塞来昔布(COX‐2/Wnt抑制剂);2个月时脱‐γ‐羧基凝血酶原水平下降84% [1 410至242 ng/mL(正常:≤7.4ng/mL);甲胎蛋白(AFP)于基线值时低]。携带PTEN失活和MET激活突变(计算机动态模拟提示的效应)的患者接受西罗莫司(雷帕霉素抑制剂的机制性靶标)和卡博替尼(MET抑制剂)治疗;AFP下降63% [8 320至3 045 ng/mL(正常值:0–15 ng/mL)]。
结论. 来自无创血检的ctDNA可为HCC患者提供可用的基因组谱。
对临床实践的提示:本研究报告, 血源性循环肿瘤DNA可提供治疗可用的肝细胞癌(已知难以进行活检的一种恶性肿瘤)基因组谱。
Introduction
Hepatocellular carcinoma (HCC) is a leading cause of cancer‐related death worldwide [1], [2]. Biopsy of HCC is often not performed to establish the diagnosis because the imaging is often characteristic and patients are at risk for complications from biopsy [3]. Treatment options for advanced disease are limited [4]. Sorafenib is the only systemic therapy approved by the U.S. Food and Drug Administration; it has demonstrated a median overall survival benefit of 2.3–2.8 months [5], [6]. Unfortunately, the median overall survival for advanced HCC is less than 1 year.
Molecular profiling is gaining popularity to identify genomic alterations in cancer. Traditionally, genomic sequencing is performed by extracting nucleic acids from tumor tissue. More recently, sequencing of circulating tumor DNA (ctDNA) has become available [7], [8]. Potential advantages of ctDNA sequencing include the following: (a) ctDNA can be extracted from a vial of blood, without an invasive tissue biopsy. (b) ctDNA can be interrogated serially. (c) ctDNA can potentially harbor genomic alterations derived from DNA shed from multiple tumor sites [9]. As ctDNA sequencing is an emerging technology, the utility of clinical‐grade testing in HCC has not been established. Chan and colleagues were the first to report analysis of ctDNA in HCC patients via shotgun sequencing [10]. Ono and colleagues extended these findings with polymerase chain reaction analysis of ctDNA and exosome sequencing in a single patient [11]. In this study, we aimed to determine if next‐generation sequencing (NGS) of ctDNA from patients with HCC could identify actionable somatic molecular alterations. We include illustrative examples of treated patients and of in silico molecular dynamic simulation to propose genomic variant function.
Materials and Methods
Patients
We prospectively evaluated 14 patients with advanced (unresectable) HCC (from January 2015 to June 2015) using a commercially available ctDNA sequencing panel (Guardant360; Guardant Health, Redwood City, CA). This study was conducted and consents obtained in accordance with University of California San Diego (UCSD) Moores Cancer Center Institutional Review Board requirements. When appropriate, patients were presented and reviewed at the UCSD Moores Cancer Center Molecular Tumor Board and UCSD Liver Cancer Group Tumor Board [12], [13]. Diagnostic radiographic findings, such as contrast‐enhanced computed tomography or magnetic resonance imaging that met the liver imaging reporting and data system (LI‐RADS) 5 criteria, and the presence of elevated tumor markers, such as alpha fetoprotein (AFP) or des‐gamma‐carboxy prothrombin (DCP), were considered in order to make a diagnosis of HCC.
Next‐Generation Sequencing
Guardant360 is an NGS panel of 68 oncogenes and tumor suppressor genes utilizing Digital Sequencing of cell‐free ctDNA. Complete exons are sequenced for all exons in 29 genes and critical exons (those reported as having somatic mutations in COSMIC) of 39 genes to detect and report single‐nucleotide variants (SNVs), copy number amplifications in 16 genes, ALK/RET/ROS1/NTRK1 fusions, and EGFR insertion/deletion mutations (supplemental online Fig. 1).
Two 10‐mL vials of whole blood were collected in Streck (La Vista, NE) Cell‐Free DNA Blood Collection tubes and shipped overnight at ambient temperature to the clinical laboratory improvement amendments (CLIA)‐laboratory. Cell‐free DNA (cfDNA) was extracted from plasma and genomic alterations were analyzed as described by Lanman et al [14].
Circulating cfDNA is mostly derived from leukocyte lysis (germline) and generally a much smaller amount of ctDNA is derived from cancer cell apoptosis/necrosis [15], [16]. All of the cfDNA fragments, both leukocyte and tumor‐derived, are simultaneously sequenced. The fractional concentration, or mutant allele frequency, for a given alteration is calculated as the fraction of ctDNA harboring that mutation in a background of wild‐type cfDNA. The analytic sensitivity reaches detection of 1–2 single mutant fragments from a 10‐mL blood sample (0.1% limit of detection) and analytic specificity is greater than 99.9999% (15,000× average coverage depth) [14].
Definition of Actionable Alteration
A potentially actionable molecular abnormality was defined as a genomic alteration that can be impacted by an antibody with the alteration being its primary target or at low 50% inhibitory concentration by a small molecular targeted agent, either directly or by modulating immediate downstream effectors [17].
Molecular Dynamics Simulations
The SEMA domain of MET protein has been extracted from pdb ID 1SHY [18]. The Y501C mutant was generated by replacing Tyr501 in the wild‐type (wt) structure with a cysteine. Mutated and wt protein, with disulphide bridges generated between relevant cysteine pairs, was solvated by an octahedral TIP3P water box containing ∼48,500 H2O molecules and 9 neutralizing potassium counterions. Molecular dynamics (MD) simulations were performed using the AMBER14 software package [19], [20], [21]; AMBER ff14SB force protein field and Joung/Cheatham ion parameters applied for the K+ counterions [22], [23]. After an initial unrestrained 10,000‐step minimization, each system was subjected to several simulation steps: (a) 5 ps gradual heating from 0 to 100 K (constant volume) with restraints on SEMA domain backbone; (b) 100 ps heating to 310 K (constant pressure) with restraints on SEMA domain backbone; (c) constant pressure simulation at 310 K without restraints. Analysis of the simulation trajectories was performed using the AMBER CPPTRAJ module [19], [20], [21], [22], [23]. The B‐factors computed from the simulations are directly proportional to the squared root mean square fluctuations of the atomic positions and therefore represent a quantification of the flexibility in the protein during the simulation.
Results
Clinical Characteristics
With or without tissue biopsy, all 14 patients were diagnosed as having HCC (Table 1). Median age was 62 years; 85.7% of the patients were men, and 50% had hepatitis C. Child‐Pugh B or C cirrhosis was noted in 64.3% of patients; 85.8% had an Eastern Cooperative Oncology Group performance status of 1 or 2 [24], [25]. The majority of patients were Barcelona Clinic Liver Cancer Staging (BCLC) C (57.2%) or D (28.6%), representing late‐stage disease populations [26].
Table 1. Demographic and baseline characteristics of 14 patients with hepatocellular carcinoma.
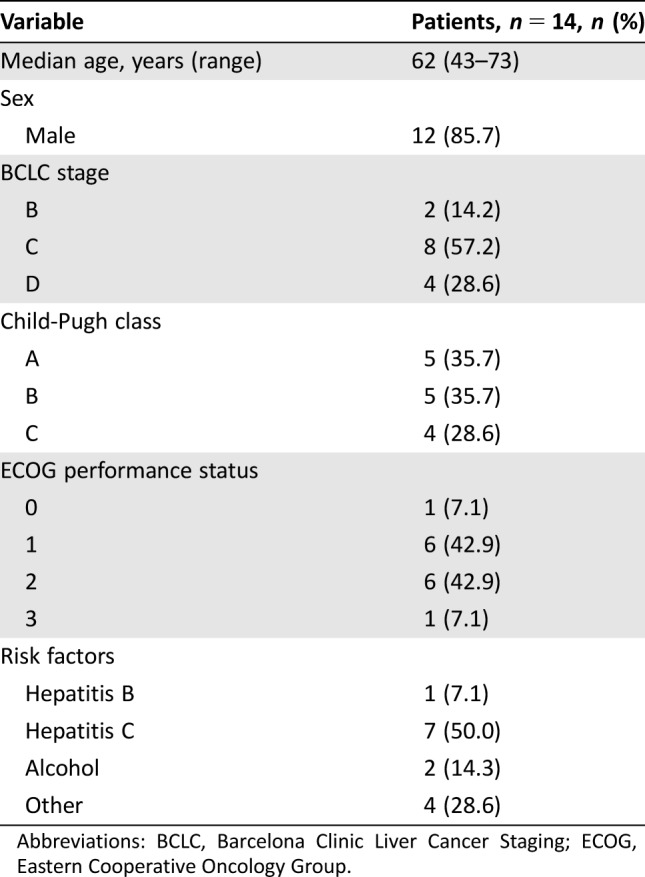
Abbreviations: BCLC, Barcelona Clinic Liver Cancer Staging; ECOG, Eastern Cooperative Oncology Group.
Genomic Profiling
NGS of ctDNA was successfully performed in all 14 patients (Fig. 1). All patients (100%) had somatic alterations identified (SNVs and/or amplifications; median = 3 alterations per patient [range, 1–8]) with a median mutant allele fraction of 0.29% (range, 0.10%–37.77%; analyses include variants of uncertain significance [VUS]). If VUS alterations were excluded, then 12/14 patients (85.7%) had somatic alterations identified (point mutations and/or amplifications; median = 2 alterations per patient [range, 0–5]) with a median mutant allele fraction of 1.18% (range, 0.13%–37.77%). Point mutations (excluding VUS) were identified in the following genes: TP53 (n = 8 patients; 57% of all patients), CTNNB1 (n = 4; 29% of all patients), and PTEN, CDKN2A, ARID1A, and MET (each n = 1; 7% of all patients); amplifications were identified in CDK6 (n = 2; 14% of all patients), EGFR (n = 2; 14% of all patients), MYC (n = 2; 14% of all patients), and BRAF, RAF1, FGFR1, CCNE1, PIK3CA, and ERBB2/HER2 (each n = 1; 7% of all patients). There was no distinct pattern based on HCC risk factors (Table 2). Importantly, no two patients had the same ctDNA‐derived somatic mutation pattern.
Figure 1.

Results of genomic profiling from circulating tumor DNA in advanced hepatocellular carcinoma.
Abbreviations: GA, genomic alterations; VUS, variant of uncertain significance.
Table 2. Risk factors and genomic alteration pattern in 14 patients with hepatocellular carcinoma.
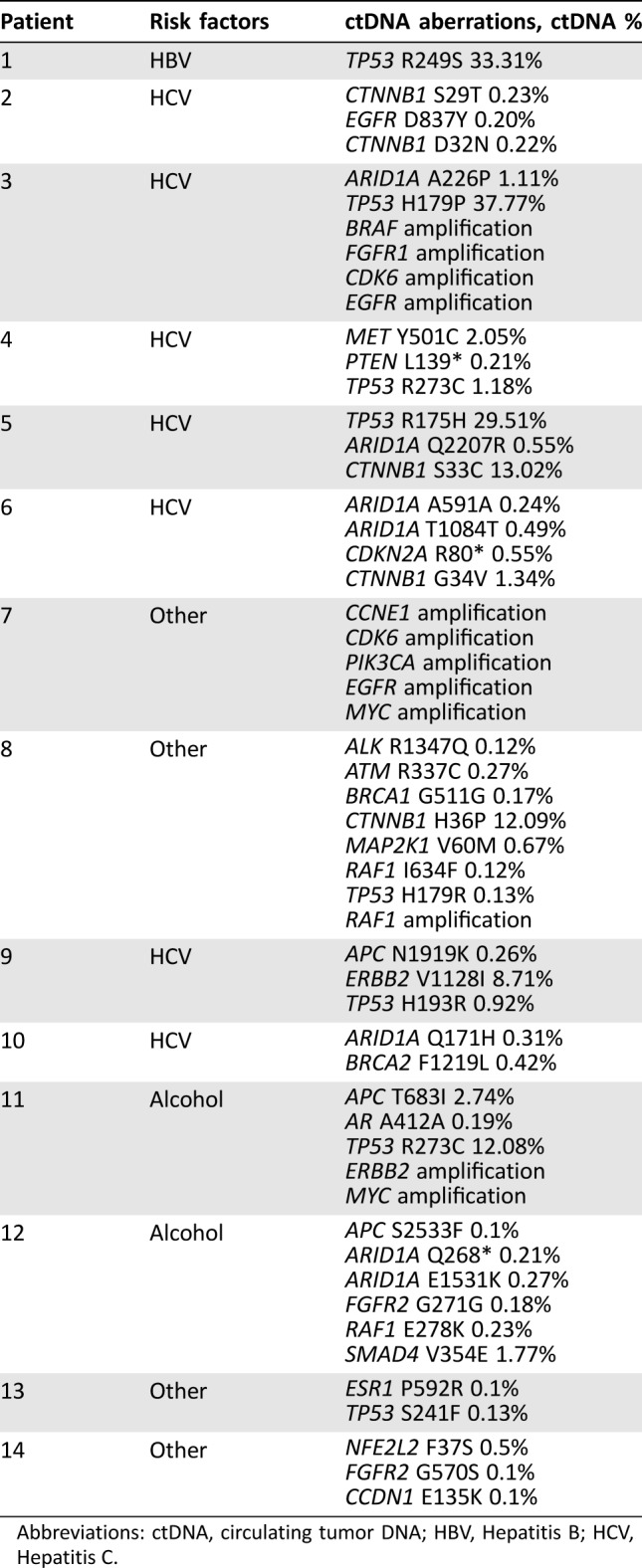
Abbreviations: ctDNA, circulating tumor DNA; HBV, Hepatitis B; HCV, Hepatitis C.
Therapy
Seventy‐nine percent of patients (12/14) had at least one potentially actionable genomic alteration. Two of five patients (40%) treated on the basis of the ctDNA results have shown evidence of salutary effects. These two patients are described in detail. The other three patients failed to benefit.
Case 1 (Patient 6; Table 2)
A 62‐year‐old man with chronic hepatitis C‐related cirrhosis was diagnosed with BCLC stage B hepatocellular carcinoma. He was treated with chemoembolization but developed metastatic disease approximately 2 years after the diagnosis. He was then treated with sorafenib for 2 years, after which disease progressed to BLCL stage D. Molecular profiling of ctDNA revealed the following genomic alterations: (a) CDKN2A R80* nonsense mutation in 0.55% of all cfDNA, and (b) CTNNB1 G34V missense mutation in 1.35% of cfDNA. His case was discussed in the multidisciplinary Molecular Tumor Board [12], [13]. The panel concluded that the CDKN2A R80* mutation is likely an inactivating mutation because it leads to truncation of the p16INK4a protein at amino acid 80 of 156 in the p16INK4a‐encoding transcript, which lacks the third and fourth ankyrin repeat that binds to CDK4, and that CDKN2A p16INK4a loss of function should lead to CDK4 upregulation [27]. Palbociclib is a CDK4/6 inhibitor that is available and can theoretically be used to target tumors where CDK4 or CDK6 is upregulated [27], [28]. CTNNB1 G34V is a missense mutation, which likely activates beta‐catenin and the Wnt pathway [29]. Preclinical studies have demonstrated that COX‐2 inhibitors, such as celecoxib, can decrease activity of the Wnt pathway by downregulating beta‐catenin expression [30]. Thus, per the Molecular Tumor Board discussion, the patient started combination treatment with palbociclib and celecoxib [12], [13].
Initially, celecoxib 200 mg by mouth twice daily was started. In 4 weeks, the DCP level declined by 31%, from 1,520 to 1,046 ng/mL (normal: 0.0–7.4 ng/mL; Fig 2A; AFP was normal at baseline). Dose‐reduced palbociclib (75 mg by mouth daily, 60% of full dose) was then added to celecoxib. In the next 4 weeks, the DCP further declined to 242, an 84% decline from the baseline. Unfortunately, he developed spontaneous bacterial peritonitis, a common complication of advanced cirrhosis, and this was complicated by upper gastrointestinal bleeding and acute kidney injury [31]. Subsequently, he elected to pursue comfort care and died.
Figure 2.

Clinical response to genomic alteration‐matched molecular targeted therapy in two patients with advanced hepatocellular carcinoma (HCC). (A): See Case 1 in Results. A 62‐year‐old man (Patient 6; Table 2) with chronic hepatitis C‐related HCC showed CDKN2A R80* nonsense mutation and CTNNB1 G34V in his circulating tumor DNA (ctDNA). Patient was started on palbociclib (CDK4/6 inhibitor; CDK2A loss of function upregulates CDK4/6) and celecoxib (inhibits CTNNB1‐activated pathway). Results show steep fall in levels of tumor marker des‐gamma carboxy prothrombin. (B): See Case 2 in Results. A 64‐year‐old woman (Patient 4; Table 2) with chronic hepatitis C‐related HCC, after liver transplant, showed MET Y501C missense, TP53 R273C missense, and PTEN L139* nonsense mutation in ctDNA. Patient had been on the mechanistic target of rapamycin inhibitor sirolimus as immunosuppression after liver transplant. Sirolimus suppresses the pathway activated by PTEN mutations. When the MET inhibitor cabozantinib was added, AFP showed a steep decline with stable imaging. MET Y501C ctDNA levels also disappeared at 8 weeks (Fig. 3).
Abbreviation: AFP, alpha fetoprotein.
Case 2 (Patient 4; Table 2)
A 64‐year‐old woman with chronic hepatitis C‐related cirrhosis was diagnosed with BCLC stage A HCC. Initially, she was treated with chemoembolization while waiting for liver transplant. After about 2.5 years, she received an orthotopic liver transplant. However, within 1 year, peritoneal carcinomatosis was discovered. Sorafenib was started, but she was not able to tolerate this agent due to severe facial skin rash. Her post‐transplant immunosuppression was switched from tacrolimus to sirolimus because of progressive disease. Analysis of ctDNA (done at the time that the patient was classified as BLCL stage C) and liver tumor tissue were performed (the latter by Foundation Medicine, NGS of 315 genes; www.foundationmedicine.com). All the 68 genes included in the ctDNA assay are assessed in the Foundation Medicine tissue NGS panel (315 genes). Tissue biopsy NGS showed a single TP53 R273C missense mutation. Liquid biopsy (ctDNA) revealed the following genomic alterations: (a) MET Y501C missense mutation in 2.05% of cfDNA; (b) TP53 R273C missense mutation in 1.18% of cfDNA; and (c) PTEN L139* nonsense mutation in 0.21% of cfDNA. TP53 R273C localizes within the DNA binding domain and is likely an inactivating mutation [32]. MET Y501C locates within the Sema domain in the extracellular region of the MET protein [18]. Polyphen score (http://genetics.bwh.harvard.edu/pph2/), which predicts functional importance of mutations based on three‐dimensional conformation change and functional consequence, was 0.9 (range, 0.0–1.0; 1.0 indicates high likelihood of changing the function), and it is likely that the MET Y501C alteration is an activating mutation [33]. The fact that this MET gene variant is also the variant detected at the highest mutant allele fraction also supports the notion that it may be the primary driver mutation of this patient's tumor.
The patient was started on cabozantinib, a MET inhibitor, at a dose of 140 mg by mouth daily, which was later lowered to 60 mg by mouth daily due to side effects (tongue pain, fatigue, loss of appetite, and dehydration) [34]. Sirolimus (an inhibitor of mechanistic target of rapamycin, an effector downstream of PIK3CA, which is activated by PTEN mutations as observed in this patient) used for immunosuppression after liver transplant was continued at doses of 0.5 mg and 1.0 mg alternating every other day by mouth. Within 1 month, the AFP level declined by 63% (8,320 ng/mL to 3,045 ng/mL [normal: 0–15 ng/mL]; Fig. 2B). Imaging after 6 weeks demonstrated stable disease with some central necrosis of tumors. Repeated ctDNA analysis after 8 weeks on cabozantinib demonstrated disappearance of the MET Y501C mutation consistent with molecular response to therapy (Fig. 3).
Figure 3.

Genomic alteration pattern after molecular targeted therapy. The patient (see Fig. 2B) with MET Y501C was treated with cabozantinib (anti‐MET tyrosine kinase inhibitor). Repeated sequencing demonstrated reduction in levels of the MET Y501C mutation.
Abbreviations: Pre‐Tx, pretreatment; Post‐Tx, post‐treatment.
Flexibility Analysis (MET Y501C).
TYR501 is located at the C‐terminal of the SEMA domain of MET. It interacts with GLU75 of the N‐terminal of this domain [35] (Fig. 4A) and most likely contributes to stabilize the domain conformation. We hypothesized that mutation of this residue to cysteine will impair the interface connection between the N‐ and C‐terminals of the SEMA domain and would make it more flexible in some specific sites important either for dimerization or hepatocyte growth factor (HGF) binding. To explore this possibility, we conducted 300 ns MD simulations of both wt and Y501C SEMA domain of MET. We conducted analysis of flexibility of the wt and mutated proteins during MD and found that, in select sites, flexibility of the protein significantly increases due to the Y501C mutation (Fig. 4B). We found that flexibility increased in two sites at the HGF (the MET ligand) binding zone, around the residues 150 and 210. (Figure 4C, red dotted line) [35]. These regions might be involved in possible interaction of Y501C MET with HGF Figure 4C.
Figure 4.

Flexibility analysis of Sema domain of MET. (A): Scheme of Sema domain. Sema domain has a ring‐like shape with N and C‐ terminals interacting residues (lower part). The residue Y501 of the C‐terminal interacts with the residue E75 of the N‐terminal (blue small ovals) which probably contributes to keep this ring structure stable. Magnification of the interaction is shown on the top panel. HGF (shown as purple square) interacts with the Sema domain. The red arrows from Figure 4B to 4A indicate that the greatest increase of B‐factors corresponds to residues in the HGF interacting zone of the Sema domain. (B): Mass‐weighted B‐factors for SEMA domain backbone atoms, which corresponds to the flexibility of the protein regions. Black line represents B‐factor of wt MET, whereas red line represents B‐factor of MET Y501C mutant. (C): Model of HGF (purple) docked to 300 ns conformer from molecular dynamics simulation of Sema domain (brown). The residues involved in the docked interface are shown in stick presentation with atomic colors (red = oxygen; blue = nitrogen; green = carbon). This region is encompassed by the red dotted oval.
Abbreviations: HGF, hepatocyte growth factor; wt, wild‐type.
The importance of flexibility for protein‐protein binding was demonstrated by Levy and colleagues [36], who studied complexes and showed that inhibiting protein flexibility causes slower binding and that flexibility is a necessary element in protein‐protein interactions. According to the abovementioned, in our case, the increased flexibility of MET sites at the HGF binding zone might improve HGF binding. This is important because HGF stimulates the tyrosine kinase activity of c‐MET [37], as demonstrated by Naldini and colleagues.
Discussion
Deployment of “liquid biopsies” to detect ctDNA is an attractive concept because it circumvents the need for invasive biopsies. This tool is especially important for diseases such as HCC, which are often not biopsied because imaging can accurately diagnose these malignancies and because biopsy carries the risk of bleeding in patients who commonly have cirrhosis, as well as the risk of seeding of the biopsy track. However, without a biopsy, assessment of the genomic profile becomes a challenge, which can be potentially addressed by a noninvasive liquid biopsy.
Our study demonstrated that all 14 patients with HCC had genomic alterations as assessed by ctDNA. The high frequency of detectable ctDNA may be related to the highly vascular nature of HCC, or possibly to attenuation in the hepatic clearance of ctDNA, because the liver provides the primary mechanism of cfDNA removal [38]. Excluding variants of uncertain significance, the median number of aberrations per patients was 2 (range, 0–5), with 86% of patients having at least one alteration. The current NGS panel completely sequenced 556 exons in a limited number of genes (n = 68). Conceivably, higher numbers of alterations could be found with a liquid biopsy panel targeting a higher number of genes. The median number of potentially druggable alterations was 2 (range, 0–5), with 79% of patients having at least one theoretically actionable alteration. Therefore, our study suggests that interrogation of ctDNA in this setting could have clinical utility.
There are advantages and disadvantages to use of ctDNA in blood versus tissue DNA for genomic sequencing. Advantages of ctDNA include the following: (a) serial profiling is feasible with liquid biopsies, which may therefore be deployable as a response biomarker or for monitoring of clonal evolution; (b) acquiring a blood sample is noninvasive as compared with a tissue biopsy; and (c) theoretically, ctDNA may reflect tumor DNA shed from cancer cells at multiple metastatic sites, hence illuminating intra‐ and inter‐tumor heterogeneity [39]; this is in contrast to tissue DNA, where the sampling is limited to one part of one tumor. The latter may also explain differences between genomic profiles seen in ctDNA versus tissue (as illustrated in our patient [described in Results Case 2] who showed a MET Y501C in the ctDNA but not in the tissue). However, ctDNA has significant limitations as well: (a) ctDNA gene panels are currently more limited than tissue panels; (b) the amount of tumor DNA in the circulation may be quite low and therefore below the limit of detection, especially in early‐stage and indolent tumors; and (c) there may be differences in the propensity of tumors to shed DNA into the bloodstream, and the factors that affect this phenomenon are still being defined. For tumor DNA to appear in the circulation, cancer cells must be undergoing significant apoptosis and/or necrosis.
In this small series of patients, ctDNA was found in all individuals, suggesting that liquid biopsy is a viable option for detecting genomic alterations in people with advanced HCC. Genetic anomalies commonly reported in HCC, including TP53, CTNNB1, and ARID1A, were detected in this study [40]. Furthermore, most patients (79%) had at least one alteration that was, in theory, pharmacologically tractable. Of interest in this regard, early evidence of tumor response was observed in two of five patients treated based on matching the ctDNA results to the targeted agents (Figs. 2, 3). This strategy merits additional investigation.
CtDNA NGS enables noninvasive interrogation of the genomic variants that drive resistance to initial lines of treatment. Many of these variants are not identified in The Cancer Genome Atlas (TCGA) compendium because TCGA is composed of primarily stage I–II pretreated patients whose surgically excised biopsies are studied, and ctDNA is often performed on patients with advanced disease. Here we characterize a putative VUS in the MET gene, via molecular dynamics simulations and flexibility analysis, and suggest that MET Y501C has functional significance (Fig. 4). The clinical response to cabozantinib in this patient supports our modeling of MET Y501C as a functional driver mutation, although this tyrosine kinase inhibitor targets more pathways than just MET. Taken together, this appears to be an example of ctDNA enabling discovery of a potential new genomic target in the landscape of secondary resistance.
Conclusion
This study addresses the unique aspects of the management of advanced HCC. The treatment options for advanced HCC are limited, and tissue biopsy is not routinely performed. Current National Comprehensive Cancer Network guideline for Hepatobiliary Cancers (version 2.2015; http://www.nccn.org) states that, for patients with advanced disease, physicians may wish to consider molecular profiling to determine eligibility for clinical trials of new molecular targeted agents. However, because the liver is a highly vascular organ, and most patients with HCC are ill and have underlying cirrhosis, percutaneous needle biopsy involves a higher risk of bleeding than for most other organ biopsies and can be expensive [41], [42]. In contrast, noninvasive liquid biopsy may provide actionable genomic information without the risk of complications. An additional problem with needle biopsy‐based tumor sequencing is that these small samples may have insufficient numbers of tumor cells to permit genotyping. Indeed, a recent study found that 35% of needle biopsies provided insufficient material for genomic sequencing of pancreaticobiliary cancers, whereas ctDNA was successfully detected in 100% of those subjects [9]. These findings may prove especially helpful in certain populations, such as those in Asia and Africa, with a higher prevalence of HCC (related to endemic viral hepatitis) [2].
Our study had several limitations, mostly related to the small number of patients, which precluded determining if genomic patterns differed between groups. Larger studies will be needed to address this issue. Another limitation relates to the estimation of percentage of ctDNA for mutations. Given the inability of the assay to distinguish tumor‐derived wild‐type alleles from normal‐derived wild‐type alleles, the percent given does not reflect the mutant allele fraction that would be represented in a tissue biopsy. Finally, we lacked tissue biopsies for comparison, and, furthermore, it might be of interest to determine if hepatitis C‐bearing patients without HCC demonstrate ctDNA alterations; however, TCGA (https://cancergenome.nih.gov/) data have previously shown that some of the most common genes altered in HCC include TP53, CTNNB1, and ARID1A, as seen in our patients.
This report serves to also provide pilot evidence of response to biomarker‐matched therapies based on ctDNA assessment in HCC. Because no two patients had the same genomic portfolio, consistent with previous reports in other cancers, individualized therapy may be necessary for optimum results [43], [44], [45]. Additional clinical utility of ctDNA may lie in its use for serial interrogation to follow molecular response to treatment. This is illustrated in Figure 3, which shows reduction in percentage of ctDNA of a MET mutation after targeting with the MET inhibitor cabozantinib. Future studies may want to emphasize such serial blood draws in order to identify clonal evolution. ctDNA testing may therefore illuminate the atlas of genomic changes in these difficult‐to‐biopsy patients and provide exploitable information about therapeutic vulnerabilities. Further study of the merits of ctDNA analysis in a bigger HCC cohort is warranted.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
This work was funded in part by the Joan and Irwin Jacobs Fund (philanthropic fund [R. K.]), NIH K08 CA168999, R21 CA192072, and P30 CA023100 (J.K.S.).
Footnotes
For Further Reading: Jeffrey S. Ross, Kai Wang, Laurie Gay et al. New Routes to Targeted Therapy of Intrahepatic Cholangiocarcinomas Revealed by Next‐Generation Sequencing. The Oncologist 2014;19:235–242.
Implications for Practice: The recent translation of next‐generation DNA sequencing technology from the research laboratory to clinical practice has enabled oncologists to personalize therapy decisions for each patient by targeting the genomic alterations driving the disease. For tumors such as primary cholangiocarcinoma of the liver, this new ability to determine all of the major genomic alterations (base substitutions, short insertions and deletions, copy number changes, homozygous deletions, and gene fusions) on very small formalin‐fixed paraffin embedded clinical samples holds great promise that less toxic targeted therapies may be available for patients currently being treated with conventional “one size fits all” approaches.
Author Contributions
Conception/design: Sadakatsu Ikeda, Razelle Kurzrock
Financial support: Jason K. Sicklick, Razelle Kurzrock
Administrative support: Kimberly C. Banks, AmirAli Talasaz, Richard B. Lanman, Scott Lippman, Razelle Kurzrock
Collection and/or assembly of data: Sadakatsu Ikeda, Igor F. Tsigelny, Åge A. Skjevik, Yuko Kono, Michel Mendler, Alexander Kuo, Jason K. Sicklick, Gregory Heestand, Razelle Kurzrock
Data analysis and interpretation: Sadakatsu Ikeda, Igor F. Tsigelny, Jason K. Sicklick, Kimberly C. Banks, AmirAli Talasaz, Richard B. Lanman, Razelle Kurzrock
Manuscript writing: Sadakatsu Ikeda, Igor F. Tsigelny, Åge A. Skjevik, Kimberly C. Banks, Richard B. Lanman, Razelle Kurzrock
Final approval of manuscript: Sadakatsu Ikeda, Igor F. Tsigelny, Åge A. Skjevik, Yuko Kono, Michel Mendler, Alexander Kuo, Jason K. Sicklick, Gregory Heestand, Kimberly C. Banks, AmirAli Talasaz, Richard B. Lanman, Scott Lippman, Razelle Kurzrock
Disclosures
Igor F. Tsigelny: Guardant Health, Inc. (C/A); Jason K. Sicklick: Foundation Medicine, Novartis, Blueprint Medicines (RF), Sirtex (C/A). Kimberly C. Banks: Guardant Health, Inc. (E); AmirAli Talasaz: Guardant Health, Inc. (E); Richard B. Lanman: Guardant Health, Inc. (E). Razelle Kurzrock: Incyte, Genentech, Pfizer, Sequenom, Guardant, Foundation Medicine, Merck Serono (RF), LOXO Oncology, XBiotech, Actuate Therapeutics (C/A), CureMatch Inc. (OI), Roche (H). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29. [DOI] [PubMed] [Google Scholar]
- 2. Venook AP, Papandreou C, Furuse J et al. The incidence and epidemiology of hepatocellular carcinoma: A global and regional perspective. The Oncologist 2010;15(suppl 4):5–13. [DOI] [PubMed] [Google Scholar]
- 3.European Association for the Study of the Liver , European Organization for Research and Treatment of Cancer. EASL‐EORTC clinical practice guidelines: Management of hepatocellular carcinoma. J Hepatol 2012;56:908–943. [DOI] [PubMed] [Google Scholar]
- 4. Benson AB 3rd, D'Angelica MI, Abrams TA et al. Hepatobiliary cancers, version 2. J Natl Compr Canc Netw 2014;12:1152–1182. [DOI] [PubMed] [Google Scholar]
- 5. Llovet JM, Ricci S, Mazzaferro V et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378–390. [DOI] [PubMed] [Google Scholar]
- 6. Cheng AL, Kang YK, Chen Z et al. Efficacy and safety of sorafenib in patients in the Asia‐Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double‐blind, placebo‐controlled trial. Lancet Oncol 2009;10:25–34. [DOI] [PubMed] [Google Scholar]
- 7. Janku F, Angenendt P, Tsimberidou AM et al. Actionable mutations in plasma cell‐free DNA in patients with advanced cancers referred for experimental targeted therapies. Oncotarget 2015;6:12809–12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Janku F, Vibat CR, Kosco K et al. BRAF V600E mutations in urine and plasma cell‐free DNA from patients with Erdheim‐Chester disease. Oncotarget 2014;5:3607–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zill OA, Greene C, Sebisanovic D et al. Cell‐free DNA next‐generation sequencing in pancreatobiliary carcinomas. Cancer Discov 2015;5:1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chan KC, Jiang P, Zheng YW et al. Cancer genome scanning in plasma: Detection of tumor‐associated copy number aberrations, single‐nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin Chem 2013;59:211–224. [DOI] [PubMed] [Google Scholar]
- 11. Sugita Y, Ono T, Ohshima K et al. Brain surface spindle cell glioma in a patient with medically intractable partial epilepsy: A variant of monomorphous angiocentric glioma? Neuropathology 2008;28:516–520. [DOI] [PubMed] [Google Scholar]
- 12. Schwaederle M, Parker BA, Schwab RB et al. Molecular tumor board: The University of California San Diego Moores Cancer Center experience. The Oncologist 2014;19:631–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parker BA, Schwaederle M, Scur MD et al. Breast cancer experience of the molecular tumor board at the University of California, San Diego Moores Cancer Center. J Oncol Pract 2015;11:442–449. [DOI] [PubMed] [Google Scholar]
- 14. Lanman RB, Mortimer SA, Zill OA et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell‐free circulating tumor DNA. PLoS One 2015;10:e0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jahr S, Hentze H, Englisch S et al. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res 2001;61:1659–1665. [PubMed] [Google Scholar]
- 16. Suzuki N, Kamataki A, Yamaki J et al. Characterization of circulating DNA in healthy human plasma. Clin Chim Acta 2008;387:55–58. [DOI] [PubMed] [Google Scholar]
- 17. Vidwans SJ, Turski ML, Janku F et al. A framework for genomic biomarker actionability and its use in clinical decision making. Oncoscience 2014;1:614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kong‐Beltran M, Stamos J, Wickramasinghe D. The Sema domain of Met is necessary for receptor dimerization and activation. Cancer Cell 2004;6:75–84. [DOI] [PubMed] [Google Scholar]
- 19. Salomon‐Ferrer R, Gotz AW, Poole D et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J Chem Theory Comput 2013;9:3878–888. [DOI] [PubMed] [Google Scholar]
- 20. Gotz AW, Williamson MJ, Xu D et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J Chem Theory Comput 2012;8:1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Case DA, Babin V, Berryman JT et al. AMBER 14. University of California, San Francisco. [Google Scholar]
- 22. Joung IS, Cheatham TE 3rd Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B 2008;112:9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roe DR, Cheatham TE 3rd PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput 2013;9:3084–3095. [DOI] [PubMed] [Google Scholar]
- 24. Pugh RN, Murray‐Lyon IM, Dawson JL et al. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg 1973;60:646–649. [DOI] [PubMed] [Google Scholar]
- 25. Oken MM, Creech RH, Tormey DC et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 1982;5:649–655. [PubMed] [Google Scholar]
- 26. Llovet JM, Bru C, Bruix J. Prognosis of hepatocellular carcinoma: The BCLC staging classification. Semin Liver Dis 1999;19:329–338. [DOI] [PubMed] [Google Scholar]
- 27. Asghar U, Witkiewicz AK, Turner NC et al. The history and future of targeting cyclin‐dependent kinases in cancer therapy. Nat Rev Drug Discov 2015;14:130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Turner NC, Ro J, Andre F et al. Palbociclib in hormone‐receptor‐positive advanced breast cancer. N Engl J Med 2015;373:209–219. [DOI] [PubMed] [Google Scholar]
- 29. Kazakov DV, Sima R, Vanecek T et al. Mutations in exon 3 of the CTNNB1 gene (beta‐catenin gene) in cutaneous adnexal tumors. Am J Dermatopathol 2009;31:248–255. [DOI] [PubMed] [Google Scholar]
- 30. Boon EM, Keller JJ, Wormhoudt TA et al. Sulindac targets nuclear beta‐catenin accumulation and Wnt signalling in adenomas of patients with familial adenomatous polyposis and in human colorectal cancer cell lines. Br J Cancer 2004;90:224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heidelbaugh JJ, Sherbondy M. Cirrhosis and chronic liver failure: Part II. Complications and treatment. Am Fam Physician 2006;74:767–776. [PubMed] [Google Scholar]
- 32. Eldar A, Rozenberg H, Diskin‐Posner Y et al. Structural studies of p53 inactivation by DNA‐contact mutations and its rescue by suppressor mutations via alternative protein‐DNA interactions. Nucleic Acids Res 2013;41:8748–8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc Hum Genet 2013;Chapter 7:Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kurzrock R, Sherman SI, Ball DW et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol 2011;29:2660–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stamos J, Lazarus RA, Yao X et al. Crystal structure of the HGF beta‐chain in complex with the Sema domain of the Met receptor. EMBO J 2004;23:2325–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Levy Y, Cho SS, Onuchic JN et al. A survey of flexible protein binding mechanisms and their transition states using native topology based energy landscapes. J Mol Biol 2005;346:1121–1145. [DOI] [PubMed] [Google Scholar]
- 37. Naldini L, Vigna E, Narsimhan RP et al. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto‐oncogene c‐MET. Oncogene 1991;6:501–504. [PubMed] [Google Scholar]
- 38. Elshimali YI, Khaddour H, Sarkissyan M et al. The clinical utilization of circulating cell free DNA (CCFDNA) in blood of cancer patients. Int J Mol Sci 2013;14:18925–18958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gerlinger M, Rowan AJ, Horswell S et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schulze K, Imbeaud S, Letouze E et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 2015;47:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rockey DC, Caldwell SH, Goodman ZD et al. Liver biopsy. Hepatology 2009;49:1017–1044. [DOI] [PubMed] [Google Scholar]
- 42. Younossi ZM, Teran JC, Ganiats TG et al. Ultrasound‐guided liver biopsy for parenchymal liver disease: An economic analysis. Dig Dis Sci 1998;43:46–50. [DOI] [PubMed] [Google Scholar]
- 43. Patel SP, Schwaederle M, Daniels GA et al. Molecular inimitability amongst tumors: Implications for precision cancer medicine in the age of personalized oncology. Oncotarget 2015;6:32602–32609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wheler J, Lee JJ, Kurzrock R. Unique molecular landscapes in cancer: Implications for individualized, curated drug combinations. Cancer Res 2014;74:7181–7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wheler JJ, Parker BA, Lee JJ et al. Unique molecular signatures as a hallmark of patients with metastatic breast cancer: Implications for current treatment paradigms. Oncotarget 2014;5:2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.