

The conformation of the title diaminopyrimidine sulfanly acetamides, (I) and (II), have similar conformations, with the pyrimidine ring being inclined to the pyridine ring in (I) by 71.10 (9) °, and by 62.93 (15) ° to the pyrazine ring in (II).
Keywords: crystal structure; 4,6-diaminopyrimidine; sulfanyl; acetamide; pyridine; pyrazine; hydrogen bonding; offset π-π interactions; Hirshfeld surface
Abstract
In the title compounds, C11H12N6OS (I) and C10H11N7OS (II), the diaminopyrimidine ring makes dihedral angles of 71.10 (9)° with the pyridine ring in (I) and 62.93 (15)° with the pyrazine ring in (II). The ethanamine group, –CH2–C(=O)–NH– lies in the plane of the pyridine and pyrazine rings in compounds (I) and (II), respectively. In both compounds, there is an intramolecular N—H⋯N hydrogen bond forming an S(7) ring motif and a short C—H⋯O interaction forming an S(6) loop. In the crystals of both compounds, molecules are linked by pairs of N—H⋯N hydrogen bonds, forming inversion dimers with R
2
2(8) ring motifs. In (I), the dimers are linked by N—H⋯O and N—H⋯N hydrogen bonds, forming layers parallel to (1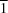 ). The layers are linked by offset π–π interactions [intercentroid distance = 3.777 (1) Å], forming a three-dimensional supramolecular structure. In (II), the dimers are linked by N—H⋯O, N—H⋯N and C—H⋯O hydrogen bonds, also forming a three-dimensional supramolecular structure.
). The layers are linked by offset π–π interactions [intercentroid distance = 3.777 (1) Å], forming a three-dimensional supramolecular structure. In (II), the dimers are linked by N—H⋯O, N—H⋯N and C—H⋯O hydrogen bonds, also forming a three-dimensional supramolecular structure.
Chemical context
An important property of diaminopyrimidine derivatives is their inhibition potential against cancer targets. Because of the limited capacity of drugs that can cure cancer, there is always an urgent requirement for new chemotherapeutics. 2,4-Diaminopyrimidine derivatives combined with arylthiazole derivatives have shown to possess significant anti-proliferation properties against breast cancer cell lines (Zhou et al., 2015 ▸). 2,4-Diaminopyrimidine derivatives have shown significant inhibitory activity against influenza viruses (Kimura et al., 2006 ▸). A series of 2,4- diaminopyrimidine derivatives were evaluated against Bacillus anthracis, which showed inhibition (Nammalwar et al., 2012 ▸). Dihydrofolate reductase inhibitor drugs such as pyrimethamine, trimetrexate and piritrexim (Nelson & Rosowsky, 2001 ▸) and the antibiotics iclaprim and trimethoprim all include diaminopyrimidine as the fundamental structural motif. Diaminopyrimidine derivatives have also shown anti-retroviral activity (Hocková et al., 2004 ▸), antibacterial (Kandeel et al., 1994 ▸) and potential anti-microbial properties (Holla et al., 2006 ▸). As part of our own studies in this area, we report herein on the syntheses, crystal structures and Hirshfeld surface analyses of the title compounds, 2-[(4,6-diaminopyrimidin-2-yl)sulfanyl]-N-(pyridin-2-yl)acetamide (I) and 2-[(4,6-diaminopyrimidin-2-yl)sulfanyl]-N-(pyrazin-2-yl)acetamide (II).
Structural commentary
The molecular structure of compounds (I) and (II) are shown in the Figs. 1 ▸ and 2 ▸, respectively. Compound (I) crystallizes in the triclinic space group P
and compound (II) crystallizes in the monoclinic space group P21/c. In both the compounds, there is an intramolecular N—H⋯N hydrogen bond forming an S(7) ring motif and a short C—H⋯O interaction forming an S(6) loop; see Tables 1 ▸ and 2 ▸ for details of the hydrogen bonding. The nitrogen atoms N1 and N2 lie in the plane of the pyrimidine ring to which they are attached [deviations are −0.0269 (17) and 0.0521 (16) Å, respectively, for compound (I), and 0.0350 (28) and 0.0284 (28) Å, respectively, for compound (II)]. The diaminopyrimidine ring makes a dihedral angle of 71.10 (9)° with the pyridine ring in compound (I) and a dihedral angle of 62.93 (15)° with the pyrazine ring in compound (II). In (I) the ethanamine group (N5/O1/C6/C5) and the pyridine ring are coplanar, as evidenced by torsion angle C7—N5—C6—C5 = 179.1 (2)°. In (II) the ethanamine group (N5/O1/C6/C5) and pyrazine ring also lie in a plane [C7—N5—C6—C5 = 177.6 (3)°]. Bond lengths C4—S1 [1.768 (2) Å] and C5—S1 [1.802 (2) Å] for compound (I), and C4—S1 [1.768 (3) Å] and C5—S1 [1.795 (3) Å] for compound (II), are comparable with values reported for similar compounds (see Section 4. Database survey).
Figure 1.

The molecular structure of the compound (I), showing the atom labelling and displacement ellipsoids drawn at the 50% probability level. The intramolecular N—H⋯N and C—H⋯O hydrogen bonds (see Table 1 ▸) are shown as dashed lines.
Figure 2.

The molecular structure of the compound (II), showing the atom labelling and displacement ellipsoids drawn at the 50% probability level. The intramolecular N—H⋯N and C—H⋯O hydrogen bonds (see Table 2 ▸) are shown as dashed lines.
Table 1. Hydrogen-bond geometry (Å, °) for (I) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N5—H5⋯N3 | 0.86 (2) | 2.18 (2) | 2.975 (2) | 154 (2) |
| C8—H8⋯O1 | 0.93 | 2.31 | 2.894 (2) | 121 |
| N2—H2B⋯N4i | 0.88 (2) | 2.20 (2) | 3.082 (2) | 178 (2) |
| N1—H1A⋯N6ii | 0.86 (2) | 2.38 (2) | 3.174 (2) | 155 (2) |
| N2—H2A⋯O1iii | 0.86 (2) | 2.13 (2) | 2.956 (2) | 159 (2) |
Symmetry codes: (i) 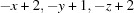 ; (ii)
; (ii) 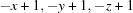 ; (iii)
; (iii) 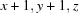 .
.
Table 2. Hydrogen-bond geometry (Å, °) for (II) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N5—H5⋯N3 | 0.82 (3) | 2.25 (3) | 2.993 (4) | 151 (3) |
| C8—H8⋯O1 | 0.93 | 2.24 | 2.854 (4) | 123 |
| N2—H2B⋯N4i | 1.00 (3) | 2.11 (3) | 3.092 (4) | 169 (3) |
| N1—H1A⋯O1ii | 0.86 (3) | 2.06 (4) | 2.904 (4) | 167 (3) |
| N2—H2A⋯N7iii | 0.85 (3) | 2.41 (3) | 3.235 (4) | 164 (3) |
| C9—H9⋯O1iv | 0.93 | 2.56 | 3.368 (4) | 145 |
Symmetry codes: (i) 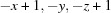 ; (ii)
; (ii) 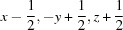 ; (iii)
; (iii) 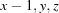 ; (iv)
; (iv) 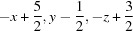 .
.
Supramolecular features
The crystal packing in compound (I) is illustrated in Fig. 3 ▸, and that for compound (II) in Fig. 4 ▸. Details of the hydrogen-bonding geometry in compound (I) are given in Table 1 ▸ and in Table 2 ▸ for (II). In the crystals of both compounds, molecules are linked by pairs of N2—H2B⋯N4i hydrogen bonds, forming inversion dimers with 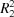 (8) ring motifs (Figs. 3 ▸ and 4 ▸, respectively).
(8) ring motifs (Figs. 3 ▸ and 4 ▸, respectively).
Figure 3.

A view normal to the (1
) plane of the crystal packing of compound (I). The hydrogen bonds (see Table 1 ▸) are shown as dashed lines and C-bound H atoms have been omitted for clarity.
Figure 4.

A view along the a axis of the crystal packing of compound (II). The hydrogen bonds (see Table 2 ▸) are shown as dashed lines, and C-bound H atoms have been omitted for clarity.
In the crystal of (I), the dimers are linked by N2—H2A⋯O1iii hydrogen bonds, forming ribbons along [010], enclosing 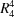 (18) ring motifs. Adjacent ribbons are linked by N1—H1A⋯N6ii hydrogen bonds, forming sheets lying parallel to the (1
) plane, see Fig. 3 ▸. The layers are linked by offset π–π interactions, forming a three-dimensional supramolecular structure [Cg⋯Cg
v = 3.777 (1) Å, interplanar distance = 3.483 (1) Å, slippage = 1.459 Å, Cg is the centroid of the pyridine ring (N6/C7–C11); symmetry code: (v) −x + 1, −y, −z + 1)].
(18) ring motifs. Adjacent ribbons are linked by N1—H1A⋯N6ii hydrogen bonds, forming sheets lying parallel to the (1
) plane, see Fig. 3 ▸. The layers are linked by offset π–π interactions, forming a three-dimensional supramolecular structure [Cg⋯Cg
v = 3.777 (1) Å, interplanar distance = 3.483 (1) Å, slippage = 1.459 Å, Cg is the centroid of the pyridine ring (N6/C7–C11); symmetry code: (v) −x + 1, −y, −z + 1)].
In the crystal of (II), the dimers are linked by N1—H1A⋯Oii, N2—H2A⋯N7iii and C9—H9⋯O1iv hydrogen bonds (Table 2 ▸), forming a three-dimensional supramolecular structure (Fig. 4 ▸). In contrast, in the crystal of (II) there are no π–π interactions present.
Database survey
A search of the Cambridge Structure Database (Version 5.39, last update February 2018; Groom et al., 2016 ▸) for [(4,6-diaminopyrmidin-2-yl)sulfanyl]acetamide yielded nine hits, eight of which have a substituted phenyl substituent in place of the pyridine ring in (I) and the pyrazine ring in (II), and one a naphthalene group (JARPOK; Subasri et al., 2017a ▸). They include the following analogues: 3-nitrophenyl (ARAROC; Subasri et al., 2016 ▸), 2-chlorophenyl (ARARUI; Subasri et al., 2016 ▸), 2-methylphenyl (GOKWIO; Subasri et al., 2014 ▸), 4-fluorophenyl (JARPUQ; Subasri et al., 2017a ▸), 2,4-dimethylphenyl (JAXFIA; Choudhury et al., 2017 ▸), 3-methoxyphenyl (JAXFOG; Choudhury et al., 2017 ▸), 4-chlorophenyl (KAPQIE; Subasri et al., 2017b ▸), and 3-chlorophenyl (KAPQOK; Subasri et al., 2017b ▸).
In these eight compounds, the diaminopyrimidine and benzene rings are inclined to one another by dihedral angles varying from ca 42.25 to 78.33°. The dihedral angle between the diaminopyrimidine and the pyridine ring in (I) is 71.10 (9)° and with the pyrazine ring in (II) is 62.93 (15)°, well within these limits. As in the title compounds, there is also an intramolecular N—H⋯N hydrogen bond present in all eight compounds, stabilizing the folded conformation of each molecule. In the crystals of all but two compounds (ARAROC and JARPUQ), molecules are linked by pairs of N—H⋯N hydrogen bonds, involving the 4,6-diaminopyrimidine moieties, forming inversion dimers with (8) ring motifs, as for compounds (I) and (II).
Hirshfeld surface analysis
In Figs. 5 ▸ and 6 ▸, the ball and stick model of the front and back views of the compounds (I) and (II), respectively, and the intermolecular contacts are shown by conventional mapping of d norm on the molecular Hirshfeld surfaces, where the red-spot areas denote intermolecular contacts involved in the hydrogen-bonding interactions (McKinnon et al., 2007 ▸). The electrostatic potential is mapped on the Hirshfeld surface using the STO-3G basis set at the Hartree–Fock theory over the range of ±0.025 a.u. The positive electrostatic potential (blue region) over the surface shows hydrogen-donor potential, and the hydrogen-bond acceptors are shown by negative electrostatic potential (red regions); see Figs. 5 ▸ and 6 ▸. The two-dimensional fingerprint plots [Fig. 7 ▸ for (I) and Fig. 8 ▸ for (II)] are deconvoluted to highlight atom-pair close contacts by which different atomic types, overlapping the full fingerprint can be separated based on different interaction types. For compound (I), intermolecular H⋯H contacts of 39.1% are the most significant, followed by 17.7% for N⋯H/H⋯N, 12% for C⋯H/H⋯C, 9.3% for O⋯H/H⋯O, 8.4% for S⋯H/H⋯S and 4.1% for C⋯C contacts. In contrast, for compound (II) the H⋯H contacts at 28.2% are significantly lower than in (I), while the N⋯H/H⋯N contacts at 27% are significantly higher than in (I). The C⋯C contacts at only 1.9% are much lower than in (I) where offset π–π interactions are observed in the crystal structure.
Figure 5.

Ball and stick, Hirshfeld surface and electrostatic potential surface diagrams for compound (I).
Figure 6.

Ball and stick, Hirshfeld surface and electrostatic potential surface diagrams for compound (II).
Figure 7.

The 2D fingerprint plot for all the intermolecular contacts for compound (I).
Figure 8.

The 2D fingerprint plot for all the intermolecular contacts for compound (II).
Synthesis and crystallization
Compound (I): To a solution of 4, 6-diamino-pyrimidine-2-thiol (0.5 g; 3.52 mmol) in 25 ml of ethanol, (0.2g; 3.52 mmol) potassium hydroxide was added and refluxed for about 30 min. Then an equimolar quantity of 2-chloro-N-(pyridin-2-yl)acetamide (3.52 mmol) was added to the above reaction mixture and it was refluxed for 5 h. Evaporation of the organic layer under vacuum provided compound (I). After purification, the compound was crystallized from ethanol solution by slow evaporation of the solvent giving yellow block-like crystals.
Compound (II): To a solution of 4, 6-diamino-pyrimidine-2-thiol (0.5 g; 3.52 mmol) in 25 ml of ethanol, (0.2g; 3.52 mmol) potassium hydroxide was added and refluxed for about 30 min. Then an equimolar quantity of 2-chloro-N-(pyrazin-2-yl)acetamide (3.52 mmol) was added to the above reaction mixture and it was refluxed for 5.5 h. Evaporation of the organic layer under vacuum resulted in compound (II). After purification, the compound was crystallized from ethanol solution by slow evaporation of the solvent giving yellow block-like crystals.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 3 ▸. For both compounds, the NH2 and NH H atoms were located in difference-Fourier maps and freely refined, and the C-bound H atoms were placed in calculated positions and refined in the riding model: C—H = 0.93–0.97 Å with U iso(H) = 1.2U eq(C).
Table 3. Experimental details.
| (I) | (II) | |
|---|---|---|
| Crystal data | ||
| Chemical formula | C11H12N6OS | C10H11N7OS |
| M r | 276.33 | 277.32 |
| Crystal system, space group | Triclinic, P
|
Monoclinic, P21/n |
| Temperature (K) | 293 | 293 |
| a, b, c (Å) | 7.2341 (2), 9.3852 (2), 9.7971 (2) | 12.1333 (5), 8.1561 (3), 12.8442 (5) |
| α, β, γ (°) | 95.820 (1), 91.116 (1), 105.682 (1) | 90, 94.307 (3), 90 |
| V (Å3) | 636.33 (3) | 1267.48 (9) |
| Z | 2 | 4 |
| Radiation type | Mo Kα | Mo Kα |
| μ (mm−1) | 0.26 | 0.26 |
| Crystal size (mm) | 0.30 × 0.25 × 0.20 | 0.28 × 0.25 × 0.20 |
| Data collection | ||
| Diffractometer | Bruker SMART APEXII area-detector | Bruker SMART APEXII area-detector |
| Absorption correction | Multi-scan (SADABS; Bruker, 2008 ▸) | Multi-scan (SADABS; Bruker, 2008 ▸) |
| T min, T max | 0.742, 0.841 | 0.723, 0.863 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 9447, 2605, 2160 | 11968, 3124, 1320 |
| R int | 0.020 | 0.084 |
| (sin θ/λ)max (Å−1) | 0.626 | 0.667 |
| Refinement | ||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.034, 0.094, 1.05 | 0.054, 0.126, 0.94 |
| No. of reflections | 2605 | 3124 |
| No. of parameters | 192 | 192 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.23, −0.20 | 0.20, −0.23 |
Supplementary Material
Crystal structure: contains datablock(s) global, I, II. DOI: 10.1107/S2056989018005704/su5430sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989018005704/su5430Isup4.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S2056989018005704/su5430IIsup5.hkl
Supporting information file. DOI: 10.1107/S2056989018005704/su5430Isup4.cml
Supporting information file. DOI: 10.1107/S2056989018005704/su5430IIsup5.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors thank the TBI X-ray facility, CAS in Crystallography and Biophysics, University of Madras, India, for the data collection
supplementary crystallographic information
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyridin-2-yl)acetamide (I) . Crystal data
| C11H12N6OS | Z = 2 |
| Mr = 276.33 | F(000) = 288 |
| Triclinic, P1 | Dx = 1.442 Mg m−3 |
| a = 7.2341 (2) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 9.3852 (2) Å | Cell parameters from 2605 reflections |
| c = 9.7971 (2) Å | θ = 2.1–26.4° |
| α = 95.820 (1)° | µ = 0.26 mm−1 |
| β = 91.116 (1)° | T = 293 K |
| γ = 105.682 (1)° | Block, yellow |
| V = 636.33 (3) Å3 | 0.30 × 0.25 × 0.20 mm |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyridin-2-yl)acetamide (I) . Data collection
| Bruker SMART APEXII area-detector diffractometer | 2160 reflections with I > 2σ(I) |
| Radiation source: X-ray | Rint = 0.020 |
| ω and φ scans | θmax = 26.4°, θmin = 2.1° |
| Absorption correction: multi-scan (SADABS; Bruker, 2008) | h = −9→9 |
| Tmin = 0.742, Tmax = 0.841 | k = −11→11 |
| 9447 measured reflections | l = −10→12 |
| 2605 independent reflections |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyridin-2-yl)acetamide (I) . Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.034 | Hydrogen site location: mixed |
| wR(F2) = 0.094 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.05 | w = 1/[σ2(Fo2) + (0.0443P)2 + 0.1669P] where P = (Fo2 + 2Fc2)/3 |
| 2605 reflections | (Δ/σ)max < 0.001 |
| 192 parameters | Δρmax = 0.23 e Å−3 |
| 0 restraints | Δρmin = −0.20 e Å−3 |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyridin-2-yl)acetamide (I) . Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyridin-2-yl)acetamide (I) . Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.47564 (6) | 0.24187 (5) | 0.97878 (4) | 0.04443 (15) | |
| O1 | 0.1201 (2) | −0.07191 (14) | 0.81504 (14) | 0.0637 (4) | |
| N1 | 0.4212 (3) | 0.58093 (19) | 0.63223 (17) | 0.0509 (4) | |
| H1A | 0.470 (3) | 0.652 (2) | 0.584 (2) | 0.059 (6)* | |
| H1B | 0.299 (3) | 0.546 (2) | 0.641 (2) | 0.066 (7)* | |
| N2 | 1.0373 (2) | 0.60541 (19) | 0.83751 (19) | 0.0500 (4) | |
| H2A | 1.087 (3) | 0.696 (2) | 0.820 (2) | 0.059 (6)* | |
| H2B | 1.092 (3) | 0.590 (2) | 0.914 (2) | 0.071 (7)* | |
| N3 | 0.45530 (19) | 0.42030 (14) | 0.78589 (14) | 0.0378 (3) | |
| N4 | 0.75997 (18) | 0.44133 (14) | 0.89781 (13) | 0.0374 (3) | |
| N5 | 0.2555 (2) | 0.10689 (16) | 0.67916 (14) | 0.0395 (3) | |
| H5 | 0.304 (3) | 0.201 (2) | 0.6830 (18) | 0.046 (5)* | |
| N6 | 0.3287 (2) | 0.11118 (16) | 0.45436 (15) | 0.0458 (4) | |
| C1 | 0.5404 (2) | 0.53494 (17) | 0.71413 (16) | 0.0373 (4) | |
| C2 | 0.7366 (2) | 0.59919 (18) | 0.72509 (17) | 0.0398 (4) | |
| H2 | 0.795008 | 0.672763 | 0.670694 | 0.048* | |
| C3 | 0.8435 (2) | 0.55043 (16) | 0.81970 (16) | 0.0367 (4) | |
| C4 | 0.5717 (2) | 0.38421 (16) | 0.87394 (15) | 0.0351 (4) | |
| C5 | 0.2265 (2) | 0.17793 (19) | 0.91965 (17) | 0.0452 (4) | |
| H5A | 0.149123 | 0.139634 | 0.994486 | 0.054* | |
| H5B | 0.185101 | 0.261192 | 0.891905 | 0.054* | |
| C6 | 0.1943 (2) | 0.05740 (18) | 0.80001 (18) | 0.0415 (4) | |
| C7 | 0.2500 (2) | 0.02603 (17) | 0.55029 (17) | 0.0369 (4) | |
| C8 | 0.1697 (3) | −0.12615 (19) | 0.5237 (2) | 0.0506 (4) | |
| H8 | 0.118824 | −0.182978 | 0.593511 | 0.061* | |
| C9 | 0.1674 (3) | −0.1909 (2) | 0.3911 (2) | 0.0603 (5) | |
| H9 | 0.113148 | −0.292746 | 0.369861 | 0.072* | |
| C10 | 0.2447 (3) | −0.1056 (2) | 0.2905 (2) | 0.0574 (5) | |
| H10 | 0.243138 | −0.147400 | 0.200125 | 0.069* | |
| C11 | 0.3246 (3) | 0.0435 (2) | 0.32694 (19) | 0.0551 (5) | |
| H11 | 0.379420 | 0.101358 | 0.258857 | 0.066* |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyridin-2-yl)acetamide (I) . Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0510 (3) | 0.0393 (2) | 0.0344 (2) | −0.00414 (19) | 0.00358 (18) | 0.00918 (17) |
| O1 | 0.0848 (10) | 0.0348 (7) | 0.0585 (8) | −0.0084 (6) | 0.0102 (7) | 0.0109 (6) |
| N1 | 0.0466 (10) | 0.0522 (10) | 0.0532 (10) | 0.0079 (8) | −0.0012 (8) | 0.0183 (8) |
| N2 | 0.0377 (8) | 0.0444 (9) | 0.0648 (11) | 0.0003 (7) | 0.0006 (7) | 0.0222 (8) |
| N3 | 0.0399 (7) | 0.0308 (7) | 0.0382 (7) | 0.0019 (6) | 0.0022 (6) | 0.0041 (5) |
| N4 | 0.0404 (8) | 0.0310 (7) | 0.0369 (7) | 0.0020 (6) | 0.0018 (6) | 0.0064 (5) |
| N5 | 0.0438 (8) | 0.0288 (7) | 0.0397 (8) | −0.0008 (6) | 0.0032 (6) | 0.0044 (6) |
| N6 | 0.0523 (9) | 0.0425 (8) | 0.0418 (8) | 0.0106 (7) | 0.0039 (7) | 0.0070 (6) |
| C1 | 0.0443 (9) | 0.0331 (8) | 0.0332 (8) | 0.0084 (7) | 0.0034 (7) | 0.0030 (6) |
| C2 | 0.0427 (9) | 0.0349 (8) | 0.0411 (9) | 0.0058 (7) | 0.0085 (7) | 0.0127 (7) |
| C3 | 0.0395 (9) | 0.0281 (8) | 0.0397 (9) | 0.0042 (6) | 0.0063 (7) | 0.0040 (6) |
| C4 | 0.0427 (9) | 0.0266 (7) | 0.0311 (8) | 0.0022 (6) | 0.0056 (7) | −0.0004 (6) |
| C5 | 0.0451 (10) | 0.0417 (9) | 0.0412 (9) | −0.0018 (7) | 0.0134 (8) | 0.0050 (7) |
| C6 | 0.0385 (9) | 0.0354 (9) | 0.0458 (10) | 0.0003 (7) | 0.0037 (7) | 0.0087 (7) |
| C7 | 0.0319 (8) | 0.0358 (8) | 0.0419 (9) | 0.0079 (6) | −0.0010 (7) | 0.0038 (7) |
| C8 | 0.0531 (11) | 0.0375 (9) | 0.0546 (11) | 0.0026 (8) | 0.0031 (9) | 0.0009 (8) |
| C9 | 0.0629 (13) | 0.0441 (11) | 0.0672 (13) | 0.0099 (9) | −0.0011 (10) | −0.0111 (9) |
| C10 | 0.0611 (12) | 0.0643 (13) | 0.0488 (11) | 0.0266 (10) | −0.0007 (9) | −0.0100 (10) |
| C11 | 0.0636 (12) | 0.0607 (12) | 0.0436 (10) | 0.0204 (10) | 0.0081 (9) | 0.0073 (9) |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyridin-2-yl)acetamide (I) . Geometric parameters (Å, º)
| S1—C4 | 1.7682 (15) | N6—C7 | 1.332 (2) |
| S1—C5 | 1.8021 (18) | N6—C11 | 1.338 (2) |
| O1—C6 | 1.2124 (19) | C1—C2 | 1.381 (2) |
| N1—C1 | 1.348 (2) | C2—C3 | 1.384 (2) |
| N1—H1A | 0.86 (2) | C2—H2 | 0.9300 |
| N1—H1B | 0.86 (2) | C5—C6 | 1.512 (2) |
| N2—C3 | 1.358 (2) | C5—H5A | 0.9700 |
| N2—H2A | 0.86 (2) | C5—H5B | 0.9700 |
| N2—H2B | 0.88 (2) | C7—C8 | 1.384 (2) |
| N3—C4 | 1.324 (2) | C8—C9 | 1.376 (3) |
| N3—C1 | 1.358 (2) | C8—H8 | 0.9300 |
| N4—C4 | 1.328 (2) | C9—C10 | 1.365 (3) |
| N4—C3 | 1.3570 (19) | C9—H9 | 0.9300 |
| N5—C6 | 1.354 (2) | C10—C11 | 1.368 (3) |
| N5—C7 | 1.400 (2) | C10—H10 | 0.9300 |
| N5—H5 | 0.856 (19) | C11—H11 | 0.9300 |
| C4—S1—C5 | 102.83 (8) | C6—C5—S1 | 111.72 (12) |
| C1—N1—H1A | 118.3 (14) | C6—C5—H5A | 109.3 |
| C1—N1—H1B | 117.3 (15) | S1—C5—H5A | 109.3 |
| H1A—N1—H1B | 124 (2) | C6—C5—H5B | 109.3 |
| C3—N2—H2A | 117.2 (13) | S1—C5—H5B | 109.3 |
| C3—N2—H2B | 117.6 (15) | H5A—C5—H5B | 107.9 |
| H2A—N2—H2B | 110 (2) | O1—C6—N5 | 124.47 (16) |
| C4—N3—C1 | 114.94 (13) | O1—C6—C5 | 121.07 (15) |
| C4—N4—C3 | 115.04 (13) | N5—C6—C5 | 114.46 (14) |
| C6—N5—C7 | 129.23 (14) | N6—C7—C8 | 123.05 (16) |
| C6—N5—H5 | 114.6 (12) | N6—C7—N5 | 112.92 (13) |
| C7—N5—H5 | 116.2 (12) | C8—C7—N5 | 124.02 (15) |
| C7—N6—C11 | 116.91 (15) | C9—C8—C7 | 118.03 (18) |
| N1—C1—N3 | 115.65 (15) | C9—C8—H8 | 121.0 |
| N1—C1—C2 | 122.72 (15) | C7—C8—H8 | 121.0 |
| N3—C1—C2 | 121.63 (15) | C10—C9—C8 | 120.00 (18) |
| C1—C2—C3 | 117.79 (14) | C10—C9—H9 | 120.0 |
| C1—C2—H2 | 121.1 | C8—C9—H9 | 120.0 |
| C3—C2—H2 | 121.1 | C9—C10—C11 | 117.86 (18) |
| N4—C3—N2 | 116.08 (15) | C9—C10—H10 | 121.1 |
| N4—C3—C2 | 121.51 (14) | C11—C10—H10 | 121.1 |
| N2—C3—C2 | 122.39 (15) | N6—C11—C10 | 124.13 (18) |
| N3—C4—N4 | 128.88 (14) | N6—C11—H11 | 117.9 |
| N3—C4—S1 | 119.16 (12) | C10—C11—H11 | 117.9 |
| N4—C4—S1 | 111.95 (12) | ||
| C4—N3—C1—N1 | −174.86 (14) | C7—N5—C6—O1 | −0.5 (3) |
| C4—N3—C1—C2 | 5.2 (2) | C7—N5—C6—C5 | 179.12 (15) |
| N1—C1—C2—C3 | 175.39 (15) | S1—C5—C6—O1 | 105.06 (17) |
| N3—C1—C2—C3 | −4.7 (2) | S1—C5—C6—N5 | −74.58 (17) |
| C4—N4—C3—N2 | −176.87 (14) | C11—N6—C7—C8 | 1.2 (2) |
| C4—N4—C3—C2 | 1.5 (2) | C11—N6—C7—N5 | −178.11 (15) |
| C1—C2—C3—N4 | 1.2 (2) | C6—N5—C7—N6 | −178.11 (16) |
| C1—C2—C3—N2 | 179.38 (15) | C6—N5—C7—C8 | 2.5 (3) |
| C1—N3—C4—N4 | −2.5 (2) | N6—C7—C8—C9 | −1.7 (3) |
| C1—N3—C4—S1 | 177.30 (10) | N5—C7—C8—C9 | 177.57 (17) |
| C3—N4—C4—N3 | −0.8 (2) | C7—C8—C9—C10 | 0.7 (3) |
| C3—N4—C4—S1 | 179.39 (10) | C8—C9—C10—C11 | 0.7 (3) |
| C5—S1—C4—N3 | 3.32 (14) | C7—N6—C11—C10 | 0.2 (3) |
| C5—S1—C4—N4 | −176.85 (11) | C9—C10—C11—N6 | −1.2 (3) |
| C4—S1—C5—C6 | 87.65 (13) |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyridin-2-yl)acetamide (I) . Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N5—H5···N3 | 0.86 (2) | 2.18 (2) | 2.975 (2) | 154 (2) |
| C8—H8···O1 | 0.93 | 2.31 | 2.894 (2) | 121 |
| N2—H2B···N4i | 0.88 (2) | 2.20 (2) | 3.082 (2) | 178 (2) |
| N1—H1A···N6ii | 0.86 (2) | 2.38 (2) | 3.174 (2) | 155 (2) |
| N2—H2A···O1iii | 0.86 (2) | 2.13 (2) | 2.956 (2) | 159 (2) |
Symmetry codes: (i) −x+2, −y+1, −z+2; (ii) −x+1, −y+1, −z+1; (iii) x+1, y+1, z.
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyrazin-2-yl)acetamide (II) . Crystal data
| C10H11N7OS | F(000) = 576 |
| Mr = 277.32 | Dx = 1.453 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| a = 12.1333 (5) Å | Cell parameters from 3124 reflections |
| b = 8.1561 (3) Å | θ = 2.2–28.3° |
| c = 12.8442 (5) Å | µ = 0.26 mm−1 |
| β = 94.307 (3)° | T = 293 K |
| V = 1267.48 (9) Å3 | Block, yellow |
| Z = 4 | 0.28 × 0.25 × 0.20 mm |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyrazin-2-yl)acetamide (II) . Data collection
| Bruker SMART APEXII area-detector diffractometer | 1320 reflections with I > 2σ(I) |
| Radiation source: X-ray | Rint = 0.084 |
| ω and φ scans | θmax = 28.3°, θmin = 2.2° |
| Absorption correction: multi-scan (SADABS; Bruker, 2008) | h = −16→12 |
| Tmin = 0.723, Tmax = 0.863 | k = −10→9 |
| 11968 measured reflections | l = −17→17 |
| 3124 independent reflections |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyrazin-2-yl)acetamide (II) . Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.054 | Hydrogen site location: mixed |
| wR(F2) = 0.126 | H atoms treated by a mixture of independent and constrained refinement |
| S = 0.94 | w = 1/[σ2(Fo2) + (0.0452P)2] where P = (Fo2 + 2Fc2)/3 |
| 3124 reflections | (Δ/σ)max = 0.001 |
| 192 parameters | Δρmax = 0.20 e Å−3 |
| 0 restraints | Δρmin = −0.23 e Å−3 |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyrazin-2-yl)acetamide (II) . Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyrazin-2-yl)acetamide (II) . Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.77107 (6) | 0.20394 (11) | 0.50448 (6) | 0.0525 (3) | |
| O1 | 1.06221 (18) | 0.2503 (3) | 0.55054 (17) | 0.0779 (8) | |
| N1 | 0.6951 (3) | 0.2961 (4) | 0.8740 (2) | 0.0651 (9) | |
| H1A | 0.651 (3) | 0.297 (4) | 0.923 (3) | 0.084 (13)* | |
| H1B | 0.760 (3) | 0.342 (4) | 0.881 (3) | 0.079 (13)* | |
| N2 | 0.4369 (2) | 0.0046 (4) | 0.6423 (3) | 0.0635 (9) | |
| H2A | 0.391 (2) | −0.015 (4) | 0.687 (2) | 0.060 (11)* | |
| H2B | 0.423 (2) | −0.044 (4) | 0.571 (3) | 0.080 (11)* | |
| N3 | 0.72886 (19) | 0.2473 (3) | 0.70403 (17) | 0.0452 (7) | |
| N4 | 0.59901 (19) | 0.1011 (3) | 0.58901 (17) | 0.0463 (7) | |
| N5 | 0.9712 (2) | 0.1889 (3) | 0.69294 (19) | 0.0494 (7) | |
| H5 | 0.912 (2) | 0.197 (4) | 0.719 (2) | 0.057 (11)* | |
| N6 | 1.0164 (2) | 0.0445 (3) | 0.84291 (19) | 0.0545 (7) | |
| N7 | 1.2266 (2) | −0.0273 (4) | 0.7815 (2) | 0.0659 (8) | |
| C1 | 0.6580 (3) | 0.2321 (4) | 0.7813 (2) | 0.0450 (8) | |
| C2 | 0.5579 (2) | 0.1540 (4) | 0.7649 (2) | 0.0466 (8) | |
| H2 | 0.509877 | 0.146453 | 0.817830 | 0.056* | |
| C3 | 0.5306 (2) | 0.0872 (4) | 0.6680 (2) | 0.0438 (8) | |
| C4 | 0.6909 (2) | 0.1845 (4) | 0.6132 (2) | 0.0421 (7) | |
| C5 | 0.8779 (2) | 0.3427 (4) | 0.5525 (2) | 0.0497 (9) | |
| H5A | 0.897729 | 0.412594 | 0.495784 | 0.060* | |
| H5B | 0.849528 | 0.412281 | 0.605640 | 0.060* | |
| C6 | 0.9796 (3) | 0.2561 (4) | 0.5981 (2) | 0.0499 (9) | |
| C7 | 1.0498 (2) | 0.0968 (4) | 0.7528 (2) | 0.0440 (8) | |
| C8 | 1.1544 (3) | 0.0605 (4) | 0.7222 (3) | 0.0613 (10) | |
| H8 | 1.174535 | 0.098921 | 0.658172 | 0.074* | |
| C9 | 1.1922 (3) | −0.0808 (4) | 0.8710 (3) | 0.0610 (10) | |
| H9 | 1.239366 | −0.144894 | 0.914481 | 0.073* | |
| C10 | 1.0895 (3) | −0.0446 (4) | 0.9011 (2) | 0.0598 (9) | |
| H10 | 1.069630 | −0.083866 | 0.965011 | 0.072* |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyrazin-2-yl)acetamide (II) . Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0478 (5) | 0.0731 (6) | 0.0376 (4) | −0.0116 (5) | 0.0092 (3) | −0.0006 (4) |
| O1 | 0.0459 (15) | 0.134 (3) | 0.0572 (14) | 0.0005 (14) | 0.0236 (12) | 0.0208 (14) |
| N1 | 0.062 (2) | 0.091 (2) | 0.0445 (17) | −0.021 (2) | 0.0163 (16) | −0.0161 (17) |
| N2 | 0.0446 (19) | 0.090 (3) | 0.0569 (19) | −0.0240 (17) | 0.0124 (16) | −0.0020 (18) |
| N3 | 0.0437 (16) | 0.0552 (19) | 0.0379 (13) | −0.0049 (12) | 0.0104 (12) | −0.0024 (12) |
| N4 | 0.0372 (15) | 0.0617 (18) | 0.0408 (14) | −0.0088 (14) | 0.0095 (12) | −0.0002 (13) |
| N5 | 0.0379 (18) | 0.069 (2) | 0.0428 (15) | −0.0018 (16) | 0.0162 (13) | 0.0043 (14) |
| N6 | 0.0478 (17) | 0.072 (2) | 0.0447 (15) | 0.0030 (15) | 0.0119 (13) | 0.0055 (14) |
| N7 | 0.0512 (19) | 0.079 (2) | 0.0690 (19) | 0.0126 (17) | 0.0169 (15) | 0.0064 (17) |
| C1 | 0.049 (2) | 0.046 (2) | 0.0402 (16) | 0.0041 (16) | 0.0087 (15) | −0.0021 (15) |
| C2 | 0.042 (2) | 0.058 (2) | 0.0416 (17) | −0.0005 (17) | 0.0110 (14) | 0.0027 (15) |
| C3 | 0.0338 (19) | 0.049 (2) | 0.0493 (18) | 0.0015 (16) | 0.0076 (15) | 0.0061 (16) |
| C4 | 0.0395 (19) | 0.047 (2) | 0.0410 (16) | 0.0036 (16) | 0.0083 (14) | 0.0024 (15) |
| C5 | 0.047 (2) | 0.055 (2) | 0.0479 (18) | −0.0136 (16) | 0.0103 (15) | 0.0067 (15) |
| C6 | 0.044 (2) | 0.061 (2) | 0.0449 (18) | −0.0142 (17) | 0.0067 (16) | 0.0004 (16) |
| C7 | 0.0338 (19) | 0.053 (2) | 0.0463 (18) | −0.0016 (16) | 0.0105 (15) | −0.0048 (16) |
| C8 | 0.053 (2) | 0.076 (3) | 0.057 (2) | 0.007 (2) | 0.0187 (18) | 0.0088 (19) |
| C9 | 0.054 (2) | 0.073 (3) | 0.057 (2) | 0.012 (2) | 0.0051 (17) | 0.0020 (19) |
| C10 | 0.061 (3) | 0.069 (3) | 0.0508 (19) | 0.002 (2) | 0.0107 (18) | 0.0063 (19) |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyrazin-2-yl)acetamide (II) . Geometric parameters (Å, º)
| S1—C4 | 1.768 (3) | N6—C7 | 1.325 (3) |
| S1—C5 | 1.795 (3) | N6—C10 | 1.331 (4) |
| O1—C6 | 1.213 (3) | N7—C8 | 1.326 (4) |
| N1—C1 | 1.346 (4) | N7—C9 | 1.326 (4) |
| N1—H1A | 0.86 (3) | C1—C2 | 1.374 (4) |
| N1—H1B | 0.88 (3) | C2—C3 | 1.375 (4) |
| N2—C3 | 1.341 (4) | C2—H2 | 0.9300 |
| N2—H2A | 0.85 (3) | C5—C6 | 1.502 (4) |
| N2—H2B | 1.00 (3) | C5—H5A | 0.9700 |
| N3—C4 | 1.325 (3) | C5—H5B | 0.9700 |
| N3—C1 | 1.367 (3) | C7—C8 | 1.389 (4) |
| N4—C4 | 1.323 (3) | C8—H8 | 0.9300 |
| N4—C3 | 1.364 (3) | C9—C10 | 1.364 (4) |
| N5—C6 | 1.347 (4) | C9—H9 | 0.9300 |
| N5—C7 | 1.398 (4) | C10—H10 | 0.9300 |
| N5—H5 | 0.82 (3) | ||
| C4—S1—C5 | 102.17 (14) | N4—C4—S1 | 111.4 (2) |
| C1—N1—H1A | 118 (2) | N3—C4—S1 | 119.0 (2) |
| C1—N1—H1B | 120 (2) | C6—C5—S1 | 112.8 (2) |
| H1A—N1—H1B | 122 (3) | C6—C5—H5A | 109.0 |
| C3—N2—H2A | 121 (2) | S1—C5—H5A | 109.0 |
| C3—N2—H2B | 120.4 (17) | C6—C5—H5B | 109.0 |
| H2A—N2—H2B | 118 (3) | S1—C5—H5B | 109.0 |
| C4—N3—C1 | 114.1 (2) | H5A—C5—H5B | 107.8 |
| C4—N4—C3 | 114.7 (3) | O1—C6—N5 | 124.1 (3) |
| C6—N5—C7 | 128.4 (3) | O1—C6—C5 | 120.5 (3) |
| C6—N5—H5 | 118 (2) | N5—C6—C5 | 115.4 (3) |
| C7—N5—H5 | 114 (2) | N6—C7—C8 | 121.7 (3) |
| C7—N6—C10 | 115.5 (3) | N6—C7—N5 | 114.4 (3) |
| C8—N7—C9 | 116.0 (3) | C8—C7—N5 | 124.0 (3) |
| N1—C1—N3 | 114.9 (3) | N7—C8—C7 | 122.1 (3) |
| N1—C1—C2 | 123.3 (3) | N7—C8—H8 | 119.0 |
| N3—C1—C2 | 121.9 (3) | C7—C8—H8 | 119.0 |
| C1—C2—C3 | 118.2 (3) | N7—C9—C10 | 121.9 (3) |
| C1—C2—H2 | 120.9 | N7—C9—H9 | 119.1 |
| C3—C2—H2 | 120.9 | C10—C9—H9 | 119.1 |
| N2—C3—N4 | 114.2 (3) | N6—C10—C9 | 122.9 (3) |
| N2—C3—C2 | 124.3 (3) | N6—C10—H10 | 118.5 |
| N4—C3—C2 | 121.4 (3) | C9—C10—H10 | 118.5 |
| N4—C4—N3 | 129.6 (3) | ||
| C4—N3—C1—N1 | −179.7 (3) | C7—N5—C6—O1 | −2.8 (5) |
| C4—N3—C1—C2 | 1.7 (4) | C7—N5—C6—C5 | 177.6 (3) |
| N1—C1—C2—C3 | −177.3 (3) | S1—C5—C6—O1 | 105.2 (3) |
| N3—C1—C2—C3 | 1.3 (5) | S1—C5—C6—N5 | −75.2 (3) |
| C4—N4—C3—N2 | 179.0 (3) | C10—N6—C7—C8 | −0.1 (5) |
| C4—N4—C3—C2 | −0.9 (4) | C10—N6—C7—N5 | 179.5 (3) |
| C1—C2—C3—N2 | 178.4 (3) | C6—N5—C7—N6 | −178.8 (3) |
| C1—C2—C3—N4 | −1.7 (5) | C6—N5—C7—C8 | 0.8 (5) |
| C3—N4—C4—N3 | 4.6 (5) | C9—N7—C8—C7 | 1.1 (5) |
| C3—N4—C4—S1 | −177.3 (2) | N6—C7—C8—N7 | −0.4 (5) |
| C1—N3—C4—N4 | −5.0 (5) | N5—C7—C8—N7 | 180.0 (3) |
| C1—N3—C4—S1 | 177.1 (2) | C8—N7—C9—C10 | −1.3 (5) |
| C5—S1—C4—N4 | 172.4 (2) | C7—N6—C10—C9 | −0.1 (5) |
| C5—S1—C4—N3 | −9.3 (3) | N7—C9—C10—N6 | 0.9 (5) |
| C4—S1—C5—C6 | 93.4 (2) |
2-[(4,6-Diaminopyrimidin-2-yl)sulfanyl]-N-(pyrazin-2-yl)acetamide (II) . Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N5—H5···N3 | 0.82 (3) | 2.25 (3) | 2.993 (4) | 151 (3) |
| C8—H8···O1 | 0.93 | 2.24 | 2.854 (4) | 123 |
| N2—H2B···N4i | 1.00 (3) | 2.11 (3) | 3.092 (4) | 169 (3) |
| N1—H1A···O1ii | 0.86 (3) | 2.06 (4) | 2.904 (4) | 167 (3) |
| N2—H2A···N7iii | 0.85 (3) | 2.41 (3) | 3.235 (4) | 164 (3) |
| C9—H9···O1iv | 0.93 | 2.56 | 3.368 (4) | 145 |
Symmetry codes: (i) −x+1, −y, −z+1; (ii) x−1/2, −y+1/2, z+1/2; (iii) x−1, y, z; (iv) −x+5/2, y−1/2, −z+3/2.
Funding Statement
This work was funded by Council of Scientific and Industrial Research grant SRF fellowship to M. Choudhury.
References
- Bruker (2008). APEX2, SAINT and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Choudhury, M., Viswanathan, V., Timiri, A. K., Sinha, B. N., Jayaprakash, V. & Velmurugan, D. (2017). Acta Cryst. E73, 996–1000. [DOI] [PMC free article] [PubMed]
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Hocková, D., Holý, A. N., Masoj\?ídková, M., Andrei, G., Snoeck, R., De Clercq, E. & Balzarini, J. (2004). Bioorg. Med. Chem. 12, 3197–3202. [DOI] [PubMed]
- Holla, B. S., Mahalinga, M., Karthikeyan, M. S., Akberali, P. M. & Shetty, N. S. (2006). Bioorg. Med. Chem. 14, 2040–2047. [DOI] [PubMed]
- Kandeel, M., El-Meligie, S., Omar, R., Roshdy, S. & Youssef, K. (1994). J. Pharm. Sci. 3, 197–205.
- Kimura, H., Katoh, T., Kajimoto, T., Node, M., Hisaki, M., Sugimoto, Y., Majima, T., Uehara, Y. & Yamori, T. (2006). Anticancer Res. 26, 91–97. [PubMed]
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst. 41, 466–470.
- McKinnon, J. J., Fabbiani, F. P. & Spackman, M. A. (2007). Cryst. Growth Des. 7, 755–769.
- Nammalwar, B., Bunce, R. A., Berlin, K. D., Bourne, C. R., Bourne, P. C., Barrow, E. W. & Barrow, W. W. (2012). Eur. J. Med. Chem. 54, 387–396. [DOI] [PMC free article] [PubMed]
- Nelson, R. G. & Rosowsky, A. (2001). Antimicrob. Agents Chemother. 45, 3293–3303. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Subasri, S., Kumar, T. A., Sinha, B. N., Jayaprakash, V. & Velmurugan, D. (2014). Acta Cryst. E70, o850. [DOI] [PMC free article] [PubMed]
- Subasri, S., Kumar, T. A., Sinha, B. N., Jayaprakash, V., Viswanathan, V. & Velmurugan, D. (2017a). Acta Cryst. E73, 306–309. [DOI] [PMC free article] [PubMed]
- Subasri, S., Kumar, T. A., Sinha, B. N., Jayaprakash, V., Viswanathan, V. & Velmurugan, D. (2017b). Acta Cryst. E73, 467–471. [DOI] [PMC free article] [PubMed]
- Subasri, S., Timiri, A. K., Barji, N. S., Jayaprakash, V., Vijayan, V. & Velmurugan, D. (2016). Acta Cryst. E72, 1171–1175. [DOI] [PMC free article] [PubMed]
- Zhou, W., Huang, A., Zhang, Y., Lin, Q., Guo, W., You, Z., Yi, Z., Liu, M. & Chen, Y. (2015). Eur. J. Med. Chem. 96, 269–280. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I, II. DOI: 10.1107/S2056989018005704/su5430sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989018005704/su5430Isup4.hkl
Structure factors: contains datablock(s) II. DOI: 10.1107/S2056989018005704/su5430IIsup5.hkl
Supporting information file. DOI: 10.1107/S2056989018005704/su5430Isup4.cml
Supporting information file. DOI: 10.1107/S2056989018005704/su5430IIsup5.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report