

Abstract
Agranulocytosis is a serious, although rare, adverse reaction to sulfasalazine, which is used to treat inflammatory joint and bowel disease. We performed a genome‐wide association study comprising 9,380,034 polymorphisms and 180 HLA alleles in 36 cases of sulfasalazine‐induced agranulocytosis and 5,170 population controls. Sulfasalazine‐induced agranulocytosis was significantly associated with the HLA region on chromosome 6. The top hit (rs9266634) was located close to HLA‐B, odds ratio (OR) 5.36 (95% confidence interval (CI) (2.97, 9.69) P = 2.55 × 10−8). We HLA‐sequenced a second cohort consisting of 40 cases and 142 treated controls, and confirmed significant associations with HLA‐B*08:01, OR = 2.25 (95% CI (1.02, 4.97) P = 0.0439), in particular the HLA‐B*08:01 haplotype HLA‐DQB1*02:01‐DRB1*03:01‐B*08:01‐C*07:01, OR = 3.79 (95% CI (1.63, 8.80) P = 0.0019), and with HLA‐A*31:01, OR = 4.81 (95% CI (1.52, 15.26) P = 0.0077). The number needed to test for HLA‐B*08:01 and HLA‐A*31:01 to avoid one case was estimated to be 1,500. We suggest that intensified monitoring or alternative treatment should be considered for known carriers of HLA‐B*08:01 or HLA‐A*31:01.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Agranulocytosis is a serious, although rare, adverse reaction to sulfasalazine‐a drug used to treat inflammatory joint and bowel disease. The risk is most pronounced during the first 3 months of treatment, and agranulocytosis should be suspected in patients with unexplained fever or nonspecific illness during this period. There are no good predictors for sulfasalazine‐induced agranulocytosis.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The aim was to identify genetic variants predisposing to sulfasalazine‐induced agranulocytosis in a European population.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ We performed a genome‐wide association study (GWAS) and identified genetic variants in the HLA region that increased the risk of sulfasalazine‐induced agranulocytosis significantly.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ We suggest that known carriers of HLA‐B*08:01 or HLA‐A*31:01 should be placed under intensified monitoring when starting sulfasalazine or be offered an alternative drug.
Agranulocytosis is a blood dyscrasia characterized by an absolute neutrophil count below 0.5 × 109/L (500/μL).1 The vast majority of cases are drug‐related, either through direct dose‐dependent cytotoxicity or idiosyncratic reactions to at least 125 nonchemotherapy drugs.2, 3, 4 Idiosyncratic agranulocytosis leads to a rapid decline in neutrophils that causes a high risk of overwhelming infection, with an average mortality rate of about 5%.5, 6, 7 The onset of drug‐induced idiosyncratic agranulocytosis is usually within months of starting therapy, and the reaction is generally believed to be immune‐mediated.4 In the few instances where genetic studies have been performed, associations with human leukocyte antigen (HLA) genes or other genes associated with the immune response have been found.4 This provides further evidence for an immune‐mediated mechanism for drug‐induced idiosyncratic agranulocytosis.
Sulfasalazine (Salazopyrin, Pfizer, Groton, CT), a drug developed in the 1930s for the treatment of inflammatory joint and bowel disease, was reported to cause fatal idiosyncratic agranulocytosis already in 1942.8 In nonfatal cases, patients usually recover within weeks of drug withdrawal.7 The incidence was estimated to be 1 in 2,400 patients during the first month of treatment, 1 in 700 during the second and third months (0.14%), and 1 in 11,200 after more than 3 months of treatment in a Swedish population.9 In the same population, the background incidence of agranulocytosis was approximately 1 in 150,000 inhabitants.9
Sulfasalazine‐induced agranulocytosis is believed to be a host immune response to a reactive metabolite of sulfasalazine.4 Sulfasalazine is first split into 5‐amino salicylic acid and sulfapyridine by intestinal bacteria.10 Sulfapyridine is in contrast to 5‐amino salicylic acid rapidly absorbed into the bloodstream. It is further metabolized in the liver into N‐acetyl‐sulfapyridine by the enzyme N‐acetyltransferase 2 (NAT2).11, 12 N‐acetyl‐sulfapyridine is primarily bioactivated by the enzyme CYP2C9 into a cytotoxic hydroxylamine that is subsequently excreted as a glucuronic acid conjugate.13 Inherited variants of the drug‐metabolizing enzymes NAT2 or CYP2C9 could in theory shunt the metabolism towards more toxic metabolites.14 It has been speculated that slow NAT2 acetylators treated with sulfonamides may be predisposed to idiosyncratic toxicity, such as severe skin reactions and systemic lupus erythematosus.15, 16, 17, 18 In a previous study, we investigated whether coding variants in the NAT2 gene influenced the risk of sulfasalazine‐induced agranulocytosis.19 A higher proportion of slow acetylators was seen among 39 cases of sulfasalazine‐induced agranulocytosis (69%) compared with 75 sulfasalazine‐tolerant patients (45%). However, no difference in slow acetylator frequency was seen between the 39 cases and 448 population‐based control subjects. We further genotyped these cases and treated controls for CYP2C9*2 and *3, but no association between agranulocytosis and genotype was seen (unpublished results). A subsequent study investigated the possible association between sulfasalazine‐induced agranulocytosis and mutations in the human neutrophil elastase gene (ELANE) that causes severe congenital neutropenia (SCN).20 None of the mutations previously reported to cause SCN were found in 36 patients with agranulocytosis induced by sulfasalazine. No other studies on candidate genes for sulfasalazine‐induced agranulocytosis have to our knowledge been published. The European Drug‐induced Agranulocytosis Consortium (EuDAC, see Acknowledgments) was initiated with the aim to perform a genome‐wide association study (GWAS) to identify novel genetic variants predisposing to agranulocytosis. We here present the results on sulfasalazine‐induced agranulocytosis.
RESULTS
Characteristics of 36 cases of agranulocytosis induced by sulfasalazine and 5,170 population controls collected in Sweden, Germany, France, and Spain are shown in Supplementary Table S1. Comparisons of demographic and clinical factors between the 30 cases recruited in Sweden and their 183 matched controls are shown in Table 1.
Table 1.
Characteristics of cases and matched controls recruited in Sweden for the discovery cohort
| Cases (n = 30) | Matched controls (n = 183) | P | |
|---|---|---|---|
| Gender, male [%] | 14 [46.7] | 80 [43.7] | 0.92 |
| Age, years | at agranulocytosis [%] | at diagnosis [%] | |
| <25 | 1 [3.3] | 6 [3.3] | 0.49 |
| 25‐29 | 2 [6.7] | 10 [5.5] | |
| 30‐34 | 2 [6.7] | 4 [2.2] | |
| 35‐39 | 2 [6.7] | 7 [3.8] | |
| 40‐44 | 1 [3.3] | 12 [6.6] | |
| 45‐49 | 1 [3.3] | 15 [8.2] | |
| 50‐54 | 5 [16.7] | 14 [7.7] | |
| 55‐59 | 7 [23.3] | 43 [23.5] | |
| 60‐64 | 5 [16.7] | 25 [13.7] | |
| 65‐69 | 2 [6.7] | 27 [14.8] | |
| 70‐74 | 0 | 14 [7.7] | |
| >74 | 2 [6.7] | 6 [3.3] | |
| Diagnosisa | [% of cases] | [% of controls] | |
| Rheumatoid arthritis | 14 [46.7] | 75 [41.0] | 0.56 |
| Crohn's disease | 3 [10.0] | 41 [22.4] | 0.15 |
| Ulcerative colitis | 3 [10.0] | 48 [26.2] | 0.06 |
| Psoriasis arthritis | 5 [16.7] | 13 [7.1] | 0.15 |
| Ankylosing spondylitis | 1 [3.3] | 5 [2.7] | 1.00 |
| Arthritis unspecified | 5 [16.7] | 8 [4.4] | 0.02 |
| Sjögren's syndrome | 1 [3.3] | 12 [6.6] | 0.70 |
| Total number of diagnosis | 32 | 202 |
Cases included one of Swedish–Italian, one of Swedish–Finnish, and one of Finnish origin. Matched controls all had a diagnosis that is an indication for sulfasalazine treatment.
Two cases had two diagnosis: rheumatoid arthritis and psoriasis arthritis, and Crohn's disease and unspecified arthritis. Among the controls, 17 had two diagnosis and one had three diagnosis.
Tests used: chi2 test for comparisons of age, otherwise Fisher's exact test.
Genome‐wide association analyses
Sulfasalazine‐induced agranulocytosis was associated with the major histocompatibility complex (MHC) region (HLA region) on chromosome 6 on a genome‐wide level after adjusting according to sex and genetic principal components 1–4, P < 5 × 10−8 (Figure 1, Supplementary Figure S1). The single nucleotide polymorphism (SNP) with the best evidence for association was rs9266634, which is located in an intergenic region 22,000 bases from HLA‐B, odds ratio (OR) 5.36 (95% confidence interval (CI) 2.97, 9.69) P = 2.55 × 10−8, (Table 2, Supplementary Table S2, Supplementary Figure S1). There was also a significant association with the intergenic insertion rs202233001 positioned in a repeat region close to the paired box 1 gene (PAX1) on chromosome 20, OR = 6.76 (95% CI 3.41, 13.38), P = 4.19 × 10−8. When the data were adjusted for the top hit rs9266634, the chromosome 20 SNP rs202233001 was no longer significantly associated.
Figure 1.
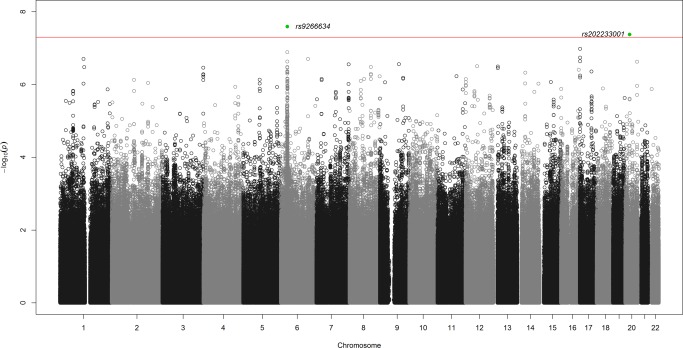
Manhattan plot of the genome‐wide association analysis. Analysis of 36 cases induced by sulfasalazine vs. all controls with 9,380,034 SNPs after imputation, adjusted by sex and genetic principal components 1–4. The red line shows the threshold for genome‐wide significance of 5 × 10−8. The top SNP was rs9266634 marked in green is located in an intergenic region close to HLA‐B on chromosome 6. There was also a significant association with an intergenic insertion/deletion on chromosome 20, rs202233001, marked in green. The closest protein coding gene to this variant is the transcription factor gene Paired Box 1 (PAX1). After adjustment for rs9266634, the intronic SNP rs111876221 in the gene serine incorporator 5 (SERINC5) on chromosome 5, reached genome‐wide significance. After adjustment for both rs9266634 and rs111876221, rs113887891 in a nonprotein coding RNA gene (LINC01762) on chromosome 1 was significant on a genome‐wide level. After adjusting also for this SNP, the intron variant rs12082628 in the gene cholinergic receptor, muscarinic 3 (CHRM3) on chromosome 1 gave a significant result. SNP = single nucleotide polymorphism, HLA = human leukocyte antigen, RNA = ribonucleic acid.
Table 2.
Top genome‐wide associations and HLA associations with agranulocytosis induced by sulfasalazine in the discovery cohort
| Associations | CHR | SNP | BP | Alleles (major/minor) | N | MAF case | MAF ‘control | OR [95% CI] | P | Nearby gene |
|---|---|---|---|---|---|---|---|---|---|---|
| a. Genome‐wide | ||||||||||
| Adjusted by sex and | 6 | rs9266634 | 31346978 | A/T | 5,183 | 0.81 | 0.44 | 5.37 [2.97, 9.69] | 2.55 × 10−8 | HLA‐B |
| genetic PC 1‐4 | 20 | rs202233001 | 21735353 | C/CACA | 5,081 | 0.14 | 0.03 | 6.76 [3.41, 13.38] | 4.19 × 10−8 | PAX1 |
| Adjusted by sex, genetic PC 1‐4 and rs9266634 | 5 | rs111876221 | 79535200 | A/G | 5,152 | 0.10 | 0.02 | 12.65 [5.15, 31.1] | 3.22 × 10−8 | SERINC5 |
| Adjusted by sex, genetic PC 1‐4, rs9266634 and rs111876221 | 1 | rs113887891 | 117006870 | G/T | 5,041 | 0.13 | 0.02 | 9.52 [4.24, 21.37] | 4.78 × 10−8 | LINC01762 |
| Adjusted by sex, genetic PC 1‐4, rs9266634, rs111876221 and rs113887891 | 1 | rs12082628 | 239797706 | A/G | 4,872 | 0.09 | 0.01 | 14.99 [5.75, 39.04] | 3.00 × 10−8 | CHRM3 |
| b. HLA | ||||||||||
| 6 | HLA‐C*14:02 | 31346171 | A/P | 5,206 | 0.07 | 0.01 | 7.72 [2.90, 20.58] | 4.36 × 10−5 | ||
| Adjusted by sex and | 6 | HLA‐B*08:01 | 31431272 | A/P | 5,206 | 0.26 | 0.11 | 2.92 [1.71, 4.99] | 8.37 × 10−5 | |
| PC 1‐4 | 6 | HLA‐C*02:06 | 31346171 | A/P | 5,206 | 0.01 | 0.00 | 92.68 [8.02, 1071] | 2.86 × 10−4 | |
| 6 | HLA‐B*08:01 | 31431272 | A/P | 5,206 | 0.26 | 0.11 | 3.24 [1.88‐5.57] | 2.26 × 10−5 | ||
| Adjusted by sex, PC 1‐4 | 6 | HLA‐C*07:01 | 31346171 | A/P | 5,206 | 0.29 | 0.14 | 2.72 [1.62, 4.57] | 1.49 × 10−4 | |
| and HLA‐C*14:02 | 6 | HLA‐A*31:01 | 30019970 | A/P | 5,206 | 0.10 | 0.03 | 3.72 [1.63, 8.47] | 1.74 × 10−3 | |
| Adjusted by sex, PC 1‐4, | 6 | HLA‐C*02:02 | 31346171 | A/P | 5,206 | 0.15 | 0.06 | 3.52 [1.81, 6.84] | 2.09 × 10−4 | |
| HLA‐C*14:02 | 6 | HLA‐A*31:01 | 30019970 | A/P | 5,206 | 0.10 | 0.03 | 4.24 [1.83, 9.82] | 7.57 × 10−4 | |
| and B*08:01 | 6 | HLA‐C*07:02 | 31346171 | A/P | 5,206 | 0.25 | 0.15 | 2.67 [1.50, 4.77] | 8.51 × 10−4 | |
| Adjusted by sex, PC 1‐4, | 6 | HLA‐B*07:02 | 31431272 | A/P | 5,206 | 0.24 | 0.15 | 3.57 [1.90, 6.72] | 7.74 × 10−5 | |
| HLA‐C*14:02, B*08:01 | 6 | HLA‐C*07:02 | 31346171 | A/P | 5,206 | 0.25 | 0.15 | 3.16 [1.74, 5.75] | 1.67 × 10−4 | |
| and C*02:02 | 6 | HLA‐A*31:01 | 30019970 | A/P | 5,206 | 0.10 | 0.03 | 3.87 [1.64, 9.12] | 1.99 × 10−3 | |
| Adjusted by sex, PC 1‐4, | 6 | HLA‐A*3101 | 30019970 | A/P | 5,206 | 0.10 | 0.03 | 4.42 [1.83, 10.68] | 9.48 × 10−4 | |
| HLA‐C*14:02, B*08:01, | 6 | HLA‐B*5101 | 31431272 | A/P | 5,206 | 0.14 | 0.05 | 3.18 [1.25, 8.11] | 1.55 × 10−2 | |
| C*02:02 and B*07:02 | 6 | HLA‐B*5501 | 31431272 | A/P | 5,206 | 0.03 | 0.01 | 5.48 [1.23, 24.31] | 2.53 × 10−2 |
a. Top GWAS results based on 9,380,034 SNPs after imputation in 36 patients with sulfasalazine‐induced agranulocytosis vs. all 5,170 controls. All results were adjusted for sex and four genetic principal components (PC). No genome‐wide significant signals were left after adjusting for four SNP variants. b. Odds ratios for the top HLA alleles for patients with sulfasalazine‐induced agranulocytosis. The effect was modeled per increase of one present HLA allele. Chromosomal location is according to the Genome Reference Consortium human assembly GRCh37. Complete lists of associated SNPs and HLA types are available in the appendix.
GWAS = genome‐wide association study, CHR = chromosome, PC = genetic principal components, SNP = single nucleotide polymorphism, BP = base pair, N = number, MAF = minor allele frequency, OR [95% CI] = odds ratio with 95% confidence interval. When referring to HLA allele: A = absent, P = present.
Follow‐up analyses were performed adjusting for each top hit in a sequential manner (Table 2). After adjusting for rs9266634 on chromosome 6, rs111876221 on chromosome 5 reached genome‐wide significance. This SNP is located in intron 1 of the serine incorporator 5 gene (SERINC5). After adjusting for the two independently associated rs9266634 and rs111876221, the SNP rs113887891 on chromosome 1 was significant on a genome‐wide level. This intron variant is in a repeat region in the gene for the nonprotein‐coding transcript LINC01762. After adjusting also for rs113887891, the SNP rs12082628 on chromosome 1 was significant on a genome‐wide level. This variant is in a repeat region in an intron of the cholinergic receptor, muscarinic 3 gene (CHRM3). All associated variants outside the HLA‐region were infrequent, and none of them had strong evidence of being functional in primary B cells from peripheral blood.21 After adjusting for rs9266634, rs111876221, rs113887891, and rs12082628, no genome‐wide significant association remained.
Analysis of classical HLA alleles at second field resolution
The HLA alleles HLA‐C*14:02 and HLA‐B*08:01 were associated with sulfasalazine‐induced agranulocytosis on an HLA‐wide level (P < 2.8 × 10−4), OR = 7.72 (95% CI (2.90, 20.58) P = 4.36 × 10−5) and 2.92 (95% CI (1.71, 4.98) P = 8.37 × 10−5), respectively (Table 2, Supplementary Table S3). The top genome‐wide SNP rs9266634 was in linkage disequilibrium (LD) with both HLA‐C*14:02 (D′ = 0.87, r2 = 0.01, P < 2.22 × 10−16) and HLA‐B*08:01 (D′ = 0.99, r2 = 0.16, P < 2.22 × 10−16). A multiple analysis, including all associated HLA alleles and the top SNP, shows that rs9266634 had a small independent effect (Supplementary Figure S2). Top HLA types obtained by sequential adjustment for each significantly associated type are shown in Table 2. The association with HLA‐B*08:01 was strengthened after adjusting for HLA‐C*14:02, OR = 3.24 (95% CI (1.988 5.57) P = 2.26 × 10−5). HLA‐A*31:01 consistently had odds ratios around 4, but P‐values were above the defined HLA‐wide significance level.
We corrected for possible confounding by indication and population stratification by considering the Swedish cohort separately (Table 1). The odds ratio for HLA‐C*14:02 was 11.08 (95% CI (4.10, 29.89) P = 2.05 × 10−6) when Swedish cases and population controls were compared, and 10.30 (95% CI (2.44, 43.52) P = 1.51 × 10−3) when Swedish cases were compared with controls matched for diagnosis of inflammatory disease (Figure 2). The odds ratio for HLA‐B*08:01 was 3.15 (95% CI (1.78, 5.57) P = 7.83 × 10−5) compared with population controls, and 5.43 (95% CI (2.40, 12.27) P = 4.82 × 10−5) compared with matched controls. The corresponding odds ratios for HLA‐A*31:01 were 3.70 (95% CI (1.54, 8.90) P = 3.39 × 10−3), and 4.97 (95% CI (1.54, 16.01) P = 7.25 × 10−3) (Figure 2).
Figure 2.
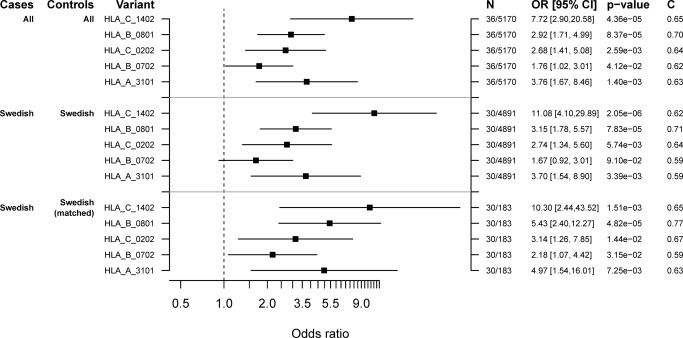
Forest plot of associations with HLA in the discovery cohort. Estimated odds ratio (OR) with 95% confidence intervals (CI), P‐value and C‐statistic for the independently associated HLA types HLA‐C*14:02, HLA‐B*08:01, HLA‐C*02:02, HLA‐B*07:02, and HLA‐A*31:01 in 1) all discovery cases and controls, 2) Swedish discovery cases and controls, and 3) Swedish discovery cases with matched controls. Matched controls were individuals who had been treated with sulfasalazine and/or had been diagnosed with inflammatory joint or bowel disease. The effect is adjusted for sex and genetic principal components 1–4. HLA = human leukocyte antigen.
Replication of SNPs and HLA alleles
An independent cohort recruited in Sweden was used for replication of the association with HLA (Table 3). We sequenced HLA‐A, HLA‐B, HLA‐C, HLA‐DRB1, HLA‐DQB1, and HLA‐DPB1 in 40 cases of sulfasalazine‐induced agranulocytosis and 142 controls treated with sulfasalazine for at least 90 days. Due to correction for multiple testing of the five top HLA types shown in Table 2, the cutoff for significance was P < 0.01. HLA‐B*08:01 was nominally associated with agranulocytosis, OR = 2.25 (95% CI (1.02, 4.97) P = 0.0439, C‐statistic = 0.57) (Table 4). Thirteen cases (32.5%) were heterozygous for HLA‐B*08:01, as compared to 25 controls (17.6%), and none were homozygous. We hypothesized that these patients carried the most frequent HLA‐B*08:01 haplotype, HLA‐DQB1*02:01‐DRB1*03:01‐B*08:01‐C*07:01, which is present in 7.4% of Europeans.22 Notably, all 13 cases with HLA‐B*08:01 carried HLA‐C*07:01, DQB1*02:01, and DRB1*03:01, and thus probably had the common haplotype. In comparison, only 16 of 25 sulfasalazine‐treated controls with HLA‐B*08:01 carried this combination of HLA alleles. The odds ratio for carrying HLA‐DQB1*02:01‐DRB1*03:01‐B*08:01‐C*07:01 was 3.79 (95% CI (1.63, 8.80) P = 0.0019, C‐statistic = 0.61), which was statistically significant (Table 4). Furthermore, the association with HLA‐A*31:01 was statistically significant, OR = 4.81 (95% CI (1.52, 15.26) P = 0.0077, C‐statistic = 0.57) (Table 4). The allele frequency of HLA‐A*31:01 was high among cases (9%) compared with the expected frequency 2.7% in Europeans.22 HLA‐C*14:02 was only present in one control, and therefore too rare to be assessed.
Table 3.
Characteristics of the patients in the replication cohort collected in Sweden
| Cases (n = 40) | Controls treated ≥ 90 days (n = 142) | P | |
|---|---|---|---|
| Gender, male [%] | 18 [45.0] | 64 [45.1] | 1.00 |
| Age, years | at agranulocytosis [%] | at enrolment [%] | |
| <25 | 4 [10.3] | 8 [5.6] | 0.21 |
| 25‐29 | 0 | 7 [4.9] | |
| 30‐34 | 1 [2.6] | 14 [9.9] | |
| 35‐39 | 2 [5.1] | 11 [7.7] | |
| 40‐44 | 2 [5.1] | 14 [9.9] | |
| 45‐49 | 6 [15.4] | 16 [11.3] | |
| 50‐54 | 4 [10.3] | 20 [14.1] | |
| 55‐59 | 2 [5.1] | 14 [9.9] | |
| 60‐64 | 4 [10.3] | 11 [7.7] | |
| 65‐69 | 5 [12.8] | 7 [4.9] | |
| 70‐74 | 6 [15.4] | 8 [5.6] | |
| >74 | 3 [7.7] | 12 [8.5] | |
| Diagnosisa | [% of cases] | [% of controls] | |
| Rheumatoid arthritis | 20 [50.0] | 71 [50.0] | 1.00 |
| Crohn's disease | 1[2.5] | 3 [2.1] | 1.00 |
| Colitis, ulcerative or unspecified | 10 [25.0] | 5 [3.5] | 1.25 × 10−4 |
| Psoriasis arthritis | 2 [5.0] | 18 [12.7] | 0.25 |
| Ankylosing spondylitis | 0 | 24 [16.9] | 0.07 |
| Arthritis unspecified | 6 [15.0] | 22 [15.5] | 1.00 |
| Plantar fasciitis | 1 [2.5] | 0 | 0.22 |
| Total number of diagnosis | 40 | 143 | |
| Mean time to onset in days [range] | 43 [16‐90] | — | |
| Mean lowest neutrophil count x109cells/l [range] | 0.03 [0‐0.5] | — | |
| Mean daily dose in mgb [range] | 2062 [500‐4000] | 1963 [500‐3000] | 0.40 |
| Cases with co‐suspected drugs [%] | 0 | — |
Controls were patients treated with sulfasalazine for at least 90 days without reacting with agranulocytosis.
One control had two diagnosis: ulcerative colitis and arthritis.
Dose missing for four controls.
Tests used: chi2 test for comparisons of age, t‐test for mean daily dose, otherwise Fisher's exact test.
Table 4.
Replication of HLA types associated with sulfasalazine‐induced agranulocytosis
| HLA allele | N | N Case | N Control | MAF Case | MAF Control | OR | 95% CI | C Stat | P |
|---|---|---|---|---|---|---|---|---|---|
| C*14:02 | 182 | 40 | 142 | 0.00 | 0.00 | N/A | |||
| B*08:01 | 182 | 40 | 142 | 0.16 | 0.09 | 2.25 | [1.02, 4.97] | 0.574 | 0.0439 |
| Full haplotype | 182 | 40 | 142 | 0.16 | 0.06 | 3.79 | [1.63, 8.80 | 0.606 | 0.0019 |
| C*02:02 | 182 | 40 | 142 | 0.09 | 0.09 | 0.99 | [0.39, 2.50] | 0.501 | 0.9876 |
| B*07:02 | 182 | 40 | 142 | 0.19 | 0.12 | 1.67 | [0.84, 3.28] | 0.571 | 0.1406 |
| A*31:01 | 182 | 40 | 142 | 0.09 | 0.02 | 4.81 | [1.52, 15.26] | 0.566 | 0.0077 |
The HLA types were selected by sequential adjustment for each significant association in the discovery cohort. Associated HLA types were compared between replication cases and controls treated 90 days or more using logistic regression. The effect was modeled per increase of one present HLA type. The full haplotype is HLA‐DQB1*02:01‐DRB1*03:01‐B*08:01‐C*07:01.
N = number, OR = odds ratio, 95% CI = 95% confidence interval, C Stat = C statistic, N/A = not available.
Predictive ability and clinical implications
The specificity to predict risk of agranulocytosis increased from 82.4% to 88.7% when using HLA‐DQB1*02:01‐DRB1*03:01‐B*08:01‐C*07:01 instead of HLA‐B*08:01. This indicates that fewer patients would be falsely predicted as being at risk when using the full haplotype. The sensitivity was 32.5% with either HLA‐B*08:01 or the full haplotype, but when combining HLA‐B*08:01 with HLA‐A*31:01, the sensitivity increased to 47.5%. With a sensitivity of 47.5% and an incidence of 0.0014 (1 in 700 patients receiving treatment),9 we could theoretically reduce the incidence of agranulocytosis to 0.000735 (0.0014–0.475*0.0014) by screening for HLA‐B*08:01 and HLA‐A*31:01. The number needed to genotype (NNG) for HLA‐B*08:01 and HLA‐A*31:01 to avoid one case was estimated to be about 1,500, which is the reciprocal of the absolute risk reduction, 1/(0.0014*0.475).
DISCUSSION
The sulphonamide sulfasalazine is known to carry a high risk of agranulocytosis,8 and the relative risk is estimated to be almost 10 times higher than for the sulphonamide sulfamethoxazole.23 The characteristics of our cases (53% women, median age 55–59 years, median treatment time to onset 50 days) were similar to those found in a previous study on the epidemiology of sulfasalazine‐induced agranulocytosis in Sweden.9 In the present study, sulfasalazine‐induced agranulocytosis was mainly associated with the MHC region on chromosome 6 that encodes HLA genes. Agranulocytosis induced by other drugs has previously been associated with HLA.4, 24, 25, 26 We found an association between sulfasalazine‐induced agranulocytosis and HLA‐B*08:01 that was replicated with similar ORs (2.92 vs. 2.25). All HLA sequenced cases carrying HLA‐B*08:01 also carried DQB1*02:01, DRB1*03:01, and HLA‐C*07:01, and the full haplotype conferred a higher OR (3.79). This indicates that the HLA‐DQB1*02:01‐DRB1*03:01‐B*08:01‐C*07:01 haplotype could be a better predictor of agranulocytosis than HLA‐B*08:01. However, the sensitivity did not change when adding the full haplotype, while it was higher when combining HLA‐B*08:01 with HLA‐A*31:01.
Notably, HLA‐B*08:01 and HLA‐A*31:01 have both been shown to increase the risk of carbamazepine hypersensitivity.27, 28 In a European study on carbamazepine, individuals homozygous for HLA‐B*08:01 were more likely to suffer severe hypersensitivity than those carrying at least one copy of the common HLA‐B*07:02 allele.22, 27 Several studies have shown that HLA‐A*31:01 increases the risk of carbamazepine hypersensitivity.28 There is no apparent chemical structural similarity between sulfasalazine and carbamazepine. We did not replicate our association with the rare HLA‐C*14:02 allele in the HLA sequenced cohort. HLA‐C*14:02 has an expected allele frequency of only 1.3% in Europeans, but is more common in Asia, with allele frequencies up to 8.2%.22 HLA‐C*14:02 has previously been associated with serious skin reactions due to phenytoin in patients of Thai ancestry.29
There are three main proposed hypotheses through which HLA can interact with a drug to trigger an inappropriate immune response.28 According to the first model, the drug binds covalently to a peptide fragment that is presented on the MHC molecule, while the second model proposes that the drug binds directly to the T‐cell receptor or MHC molecule. In the third model, the drug binds noncovalently to the antigen‐binding cleft of the HLA protein, which alters the repertoire of peptides it can bind.28 This last model has been demonstrated for abacavir, a drug strongly associated with acute hypersensitivity syndrome among carriers of the HLA‐B*57:01 allele.30 It is not known whether any of the proposed mechanisms explain agranulocytosis induced by sulfasalazine. There is evidence that another sulphonamide, sulfamethoxazole, triggers an immune response by binding both covalently to proteins including MHC, and noncovalently to MHC/peptide complexes.31
Although we found associations between sulfasalazine‐induced agranulocytosis and classical HLA alleles, we cannot exclude influence from nearby noncoding regulatory variants. In the HLA‐region, it is generally difficult to identify an actual causative variant due to a high degree of gene density and polymorphism, and extended linkage disequilibrium.32, 33 Apart from our top hit close to HLA‐B, a genome‐wide significant association was observed in an intergenic region on chromosome 20 close to PAX1, a member of the paired box family of transcription factors. This infrequent variant is located in a repeat region and has no evidence of being functional in primary B cells from peripheral blood.21 After correcting for the top HLA hit, we found an association with a rare intronic variant in SERINC5, which encodes a protein that restricts human immunodeficiency virus (HIV) infectivity.34 Based on imputed data from the Roadmap Epigenome project, the SERINC5 SNP is located in a weak enhancer region in primary B cells from peripheral blood characterized by histone 3 being monomethylated at lysine 4 (H3K4me1) and acetylated at lysine 27 (H3K27ac).21 Lastly, after sequentially adjusting for the other top hits, two infrequent SNPs on chromosome 1 were independently associated on a genome‐wide level. However, these associations were likely to be spurious since both SNPs are in repeat regions and have no evidence of being regulatory variants in B‐cells.21
There are several limitations of this study. First, sulfasalazine‐induced agranulocytosis is a rare event, and it is difficult to identify a large enough number of cases. For the present study, cases were collected over a period of 20 years, which resulted in a total of 76 cases. Second, HLA sequencing could not be performed in the discovery phase since we did not have access to DNA from the controls. In the replication phase all cases and controls were HLA‐sequenced, thus making the results more reliable. Third, several polymorphisms in the HLA loci are known to confer genetic susceptibility to inflammatory arthritis or bowel disease.32 Due to the small sample size we did not stratify by disease; however, the distribution of diseases was similar among cases and matched controls. We therefore believe that there was no confounding by indication. Fourth, the most frequent disease in our cohort, rheumatoid arthritis, is strongly associated with a subgroup of HLA‐DRB1*04 alleles, and to a lesser extent seronegative rheumatoid arthritis is associated with HLA‐B*08.35 We did not discriminate between seropositive and seronegative rheumatoid arthritis, and could not test whether serotype influenced the detected association with HLA‐B*08:01. Fifth, the number needed to screen (NNG) for HLA‐B*08:01 and HLA‐A*31:01 to avoid one case was high, 1,500. HLA‐A*31:01 also increases the risk of carbamazepine‐induced hypersensitivity or skin reaction.28 Since HLA‐A*31:01 is prevalent throughout the world, and the reactions are common, only 15–43 patients starting carbamazepine need to be screened for HLA‐A*31:01 to avoid one case. There are other examples where preemptive genotyping is recommended despite a high NNG, although this is unusual.28, 36 HLA‐B*15:02 is a strong predictor of the serious skin reactions Stevens–Johnson's syndrome and toxic epidermal necrolysis in East Asia, where this HLA type is prevalent.28 Before starting carbamazepine, patients originating from East Asia should be tested for HLA‐B*15:02. Even in Asia, 10,000–24,665 patients need to be screened to avoid one case, due to the rarity of these serious skin reactions.
Strengths of this study are that it has a fairly large and homogenous discovery cohort, and that the finding was replicated in a second cohort. In general, pharmacogenomic associations are robust and more likely to be translated into clinical practice than genetic associations with complex traits.37 There are several reasons. First, genetic variants associated with drug‐related phenotypes usually have larger effect sizes than those associated with complex disease risk. Second, pharmacogenomic variants tend to have higher allele frequencies than variants associated with complex traits. This is possibly due to the fact that complex traits have been subject to negative selection over the course of human evolution, while pharmacogenomic variants only are deleterious in the presence of a certain drug, and drug treatment is a relatively new intervention. Third, pharmacogenomic variants are more likely to be actionable, since it is easier to select an alternative drug than to modify risk factors for complex disease, such as lifestyle. Fourth, it has been predicted that before long patients will have their pharmacogenome readily available in their medical records, thus removing the necessity to order the test.38
In conclusion, we have confirmed that sulfasalazine‐induced agranulocytosis is associated with HLA‐B*08:01 and HLA‐A*31:01 in Europeans. The association was stronger for the HLA‐DQB1*02:01‐DRB1*03:01‐B*08:01‐C*07:01 haplotype than for HLA‐B*08:01 alone, but the NNG did not change when using the full haplotype. Whether to use genetics in translational medicine to adapt drug treatment depends on the severity of the expected adverse drug reaction (ADR) and the existence of other treatment options.36 We believe that the drug sulfasalazine fulfils these criteria, although the high NNG makes testing challenging. The solution could be to include HLA‐B*08:01 and HLA‐A*31:01 into preemptive screening programs, where multiple tests are performed at a low cost. Known carriers of HLA‐B*08:01 or HLA‐A*31:01 starting sulfasalazine could then be placed under intensified monitoring or be offered an alternative drug. This individualization would be a further step towards personalized treatment of inflammatory disease.
METHODS
Ethical statement
The study was approved by the local Ethics Committees (2008/213 and 2010/231, Uppsala, Sweden; Dec. 22, 2014, Málaga, Spain; RTF011, Barcelona, Spain; Charité‐Universitätsmedizin Berlin, Germany; CPP Sud‐Ouest et Outre‐Mer I No. 1‐09‐24, Toulouse, France; and for recruitment of Swedish controls Stockholm Dnr 2007‐644‐31 and 2011/463‐32). Research was carried out in accordance with the latest update of the Declaration of Helsinki. Written informed consent was obtained from all participants. The study protocol has been indexed in the European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP) register at http://www.encepp.eu (“The EuDAC Study”). Part of the replication cohort that had previously been collected for genetic studies of sulfasalazine‐induced adverse reactions was approved by the Ethics Committee of the Medical Faculty, Uppsala University, Sweden (95‐200 and 97‐312).
Sample description
EuDAC consists of a network of investigators in Sweden, Spain, France, and Germany. The basis for case recruitment in Sweden (http://www.swedegene.se) and France was through nationwide spontaneous ADR reports sent from healthcare professionals to the respective national drug regulatory authority. In Spain, cases were recruited both from spontaneous ADR reports and through active surveillance at 17 hospitals in Barcelona. German cases were recruited through active surveillance at 50 hospitals in Berlin, as described.2
Each included subject was at least 18 years of age and able to give informed consent. We defined cases as patients who had developed an absolute neutrophil count of 0.5 × 109/L or less (≤500/μL) during drug therapy or within 7 days of stopping medication. Each case was required to exhibit complete recovery after cessation of the drug with an absolute neutrophil count >1.0 × 109/L (>1000/μL) or a compatible bone marrow aspirate or biopsy. Additional inclusion and exclusion criteria have been described previously.39 We collected clinical data (demographics, medical history, drug treatment history, laboratory data, and ancestry) through interviews using a standardized questionnaire, and by obtaining and reviewing medical records. At each center, cases were evaluated by at least one senior investigator, and a final adjudication of the complete dataset was performed by a specialist in hematology. Causality assessment was according to the WHO standard algorithm.40
Overall, 243 cases were collected for the study. Seven Swedish cases and two Spanish cases were excluded after adjudication due to either exposure to chemotherapy, negative rechallenge, unknown white blood cell count, diagnosis of chronic lymphatic leukemia, or missing clinical data. No German or French case was excluded. Out of the 234 cases of drug‐induced agranulocytosis fulfilling all requirements, 36 were associated with sulfasalazine, which is the focus of this study. These cases originated from Sweden (n = 30), Germany (n = 3), and France (n = 3), while Spain had no case induced by sulfasalazine (Supplementary Table S1).
Consenting population controls were available from Sweden, Spain, and Germany. In total, 5,170 controls were utilized: 4,891 unrelated individuals from the Swedish Twin Registry,41 183 Spanish, and 96 German individuals.2 Of the 183 Spanish controls, 147 had been recruited in a previous study of upper gastrointestinal bleeding,42 while the remaining 36 were healthy control subjects. Controls with a known history of agranulocytosis or neutropenia from any cause were excluded. Matching of controls from the Swedish Twin Registry was performed by linking with individual data from the Swedish National Patient Register, and the Swedish Prescribed Drug Register. Matched controls were those with a hospital diagnosis that is an indication for sulfasalazine treatment (data available from 1964). Diseases included were Crohn's disease, ulcerative colitis, rheumatoid arthritis, ankylosing spondylitis, psoriasis arthritis, unspecified arthritis, and Sjögren's syndrome. Some of these controls had also collected prescriptions of sulfasalazine (data available from July 2005). Patient characteristics were compared between discovery cases and controls using Fisher's exact and chi2 tests (Table 1).
For the replication, we used 40 cases of sulfasalazine‐induced agranulocytosis and 142 controls who had been treated with sulfasalazine for a minimum of 3 months (90 days) without developing agranulocytosis (Table 3). Twenty‐nine of the cases and all the controls had been collected previously in Sweden for genetic studies of sulfasalazine‐induced adverse reactions.19, 20 In addition, 11 new cases of sulfasalazine‐induced agranulocytosis collected in Sweden were included in the replication. All patients and controls had been treated with sulfasalazine for inflammatory joint disease, inflammatory bowel disease, or Sjögren's syndrome. Patient characteristics were compared between replication cases and controls using Fisher's exact, chi2 and t‐tests.
Power calculation
Given a genome‐wide significance level of 5 × 10−8, an ADR incidence of 0.14% (9), and using an additive genetic model, our sample size was powered to detect common genetic variants with effect sizes of clinical utility.43 For example, with 36 cases and 5,000 controls, we had 80% power to detect an OR of 4 for variants with a minor allele frequency (MAF) of 40%, and 80% power to detect OR slightly above 5 for variants with an MAF of 20% (Supplementary Figure S3).
Genome‐wide array data and analyses
DNA was extracted from peripheral venous blood. Thirty‐three cases recruited in Sweden and France, and 147 Spanish controls were genotyped with the Illumina HumanOmni 2.5M chip (Illumina, San Diego, CA; Figure 3). The remaining 36 Spanish controls had been genotyped with the Illumina HumanOmni1‐Quad 1M chip. Three cases and 96 controls from Germany were genotyped with the Illumina HumanOmniExpress 700K array, as were 4,891 controls from the Swedish Twin Registry. Genotype calls were generated using the Genome Studio software from Illumina and the Genome Reference Consortium human assembly GRCh37.
Figure 3.
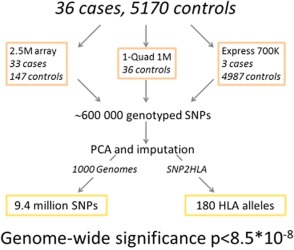
Study design. Design of the EuDAC study on sulfasalazine‐induced agranulocytosis.
GWAS quality control (QC) and data management was performed using PLINK v. 1.9. The resulting merged data included 596,010 SNPs. Imputation of genotypes was performed using PhaseIT44 and Impute v. 245 with the 1000 Genomes Project reference set (phase III, v. October 2014). The total number of SNPs after imputation was 9,380,034. All cases and controls were within the European cluster according to genetic principal component analysis (PCA) (Supplementary Figures S4, S5). Due to cases and controls being from different countries of Europe, sensitivity analyses were performed by reanalyzing each top finding in the largest group of cases and controls, which was from Sweden. See Supplementary Methods for additional details on QC, PCA, and imputation.
All genome‐wide analyses were adjusted for sex and the first four principal components. SNP effects were modeled only as additive. The conventional genome‐wide significance threshold P < 5 × 10−8 was used to correct for multiple testing.46 Results are presented as Manhattan plots. When genome‐wide significant signals were found, analyses were performed sequentially by adjusting for each genome‐wide significant signal until none were left. Logistic regression was used to estimate univariate and multiple models. Genome‐wide statistical analyses was performed using PLINK v. 1.947, 48 and individual SNPs were analyzed statistically, including LD calculation, using R 3.3.1 and the R packages rms and genetics (R Foundation for Statistical Computing, Vienna, Austria). The Q‐Q plot is shown in Supplementary Figure S6.
HLA allele imputation and analysis of the discovery cohort
Imputation to first and second field resolution of 180 classical HLA alleles, amino acid residues, and individual SNPs was performed on the nonimputed merged and quality‐controlled genome‐wide data using the software SNP2HLA with a reference panel of 5,225 individuals.49 Logistic regression was used to test the association between HLA types and agranulocytosis. The HLA‐wide significance level was set to 0.05/180 = 2.8 × 10−4. When HLA‐wide significant signals were found, analyses were performed sequentially by adjusting for each HLA‐wide significant signal until none remained. To avoid confounding by indication, top HLA signals were tested using a cohort of cases and controls matched for sulfasalazine treatment and/or inflammatory joint or bowel disease that was available from Sweden.
HLA sequencing of the replication cohort
For the replication cohort, HLA sequencing of HLA A, B, C, DP, DQ, and DR was performed on MiSeq (Illumina) as previously described by Cereb et al.50 The top HLA types were obtained by sequential adjustment for each significant HLA type in the discovery cohort. These five HLA types were tested for statistical associations, thus correction for five multiple tests was performed (P < 0.01).
Analysis of predictive ability
The predictive ability of logistic regression models for selected HLA analyses was expressed as the C‐statistic (equivalent to the area under the receiver operating characteristic curve), which is a measure of the ability of the model to discriminate between cases and controls. A C‐statistic of 0.5 indicates predictive ability similar to chance (a 50:50 likelihood of predicting the outcome), while 1 indicates perfect discrimination. The number needed to genotype (NNG) to avoid one case was estimated from the reciprocal of the predicted absolute risk reduction, where the absolute risk reduction is calculated as the prevalence (adverse drug reaction in the population) × the sensitivity of the diagnostic test.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
M.W., N.E., R.K., L.I., M.I.L., M.M., P.K.E.M., M.B., and P.H. wrote the article; M.W., N.E., R.K., E.B‐G., L.I., A.C., M.I.L., M.M., Q‐Y.Y., and P.H. designed the research; M.W., N.E., R.K., E.B‐G., L.I., A.C., M.I.L., E.S.P., M.M., J.M., T.A., H.K., Q‐Y.Y., P.K.E.M., M.B., and P.H. performed the research; M.W., N.E., M.B., and P.H. analyzed the data.
Supporting information
SUPPLEMENTARY MATERIAL is linked to the online version of the article at http://www.cpt-journal.com
Supporting Information
ACKNOWLEDGMENTS
EuDAC collaborators: Maryse Lapeyre‐Mestre, Jean Louis Montastruc (Service de Pharmacologie Médicale et Clinique, Centre Hospitalier Universitaire, Faculté de Médecine de l'Université de Toulouse, UMR Inserm 1027, CIC 1436, Toulouse, France); Edeltraut Garbe (Leibniz‐Institute for Prevention Research and Epidemiology – BIPS, Bremen, Germany ); Lourdes Vendrell (Fundació Institut Català de Farmacologia, Hospital Universitari Vall d'Hebron, Barcelona, Spain); Ramon Puig Treserra (Fundació Institut Català de Farmacologia, Barcelona, Spain); Jose Luis Caro (Banc de Sang i Teixits, Barcelona, Spain); Maria Sainz Gil, Maria‐Isabel Jimenez Serrania, Inés Salado (Centro de Estudios sobre la Seguridad de los Medicamentos. Universidad de Valladolid, Spain); Paul McKeigue (University of Edinburgh Medical School, UK); Erik Eliasson (Clinical Pharmacology, Karolinska Institutet, Sweden); Håkan Melhus, Ulrica Ramqvist, Elisabet Stjernberg, Sofie Collin, Eva Prado Lopez, Agnes Wadelius, Martha Wadelius and Agnes Kataja Knight (Department of Medical Sciences, Clinical Pharmacology, Uppsala University, Uppsala, Sweden); Daniel Garwicz (Department of Medical Sciences, Clinical Chemistry, Uppsala University, Uppsala, Sweden); Bruno Stricker (Erasmus Medical Center, The Netherlands); Julia Ruiz‐Nuñez, Camilla Stephens (S Farmacologia Clinica, IBIMA, H Universitario Virgen de la Victoria, Universidad de Málaga, CIBERehd, Madrid, Spain), Inger Öhman (Medical Products Agency, Uppsala, Sweden), Barbro Sandin (Swedish Twin Registry, MEB, Karolinska Institutet, Stockholm, Sweden). We thank Marco Cavalli and Claes Wadelius (Department of Immunology, Genetics and Pathology, Medical Genetics, Uppsala University, Uppsala, Sweden) for input on the functionality of associated SNPs. We are grateful to Ulla Lindqvist (Department of Medical Sciences, Rheumatology, Uppsala University, Uppsala, Sweden) for recruiting sulfasalazine treated replication patients at Uppsala University Hospital, Sweden. Computations were performed on resources provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX). This research was supported by the Swedish Research Council (Medicine 521‐2011‐2440 and 521‐2014‐3370), Swedish Heart and Lung Foundation (20120557 and 20140291), Selander's foundation, Thuréus' foundation, Clinical Research Support (ALF) at Uppsala University (MW), the German Federal Institute for Drugs and Medical Devices (BfArM) (RK), the European Commission and the National Institute for Health Research (NIHR) Biomedical Research Centre at Guy's and St Thomas' NHS Foundation Trust and King's College London (MM), Carlos III Spanish Health Institute (FIS10/02632) (LI), and cofunded by the European Regional Development Fund – FEDER (FIS10/02632) (LI) and (FIS12/00378) (MIL). In addition, CIBERedh is funded by Carlos III Spanish Health Institute. T.A. reports grants from the Swedish Research Council, Science for Life laboratory, and Uppsala University, and R.K. obtained personal fees from Bayer Pharma AG during the course of the study. The funding agencies played no role in the writing of the article or the decision to submit it for publication. The views expressed are those of the authors/collaborators and not necessarily those of the national health services or regulatory agencies in the respective countries. Finally, we thank all study participants.
References
- 1. Andres, E. & Maloisel, F. Idiosyncratic drug‐induced agranulocytosis or acute neutropenia. Curr. Opin. Hematol. 15, 15–21 (2008). [DOI] [PubMed] [Google Scholar]
- 2. Huber, M. et al Drug‐induced agranulocytosis in the Berlin case‐control surveillance study. Eur. J. Clin. Pharmacol. 70, 339–345 (2014). [DOI] [PubMed] [Google Scholar]
- 3. Andersohn, F. , Konzen, C. & Garbe, E. Systematic review: agranulocytosis induced by nonchemotherapy drugs. Ann. Intern. Med. 146, 657–665 (2007). [DOI] [PubMed] [Google Scholar]
- 4. Johnston, A. & Uetrecht, J. Current understanding of the mechanisms of idiosyncratic drug‐induced agranulocytosis. Expert Opin. Drug Metab. Toxicol. 11, 243–257 (2015). [DOI] [PubMed] [Google Scholar]
- 5. Garbe, E. Non‐chemotherapy drug‐induced agranulocytosis. Expert Opin. Drug Saf. 6, 323–335 (2007). [DOI] [PubMed] [Google Scholar]
- 6. Tesfa, D. , Keisu, M. & Palmblad, J. Idiosyncratic drug‐induced agranulocytosis: possible mechanisms and management. Am. J. Hematol. 84, 428–434 (2009). [DOI] [PubMed] [Google Scholar]
- 7. Capell, H.A. , Pullar, T. & Hunter, J.A. Comparison of white blood cell dyscrasias during sulphasalazine therapy of rheumatoid arthritis and inflammatory bowel disease. Drugs 32 Suppl 1, 44–48 (1986). [DOI] [PubMed] [Google Scholar]
- 8. Svartz, N. Salazopyrin, a new sulfanilamide preparation. Acta Med. Scand. CX, 577–598 (1942). [Google Scholar]
- 9. Keisu, M. & Ekman, E. Sulfasalazine associated agranulocytosis in Sweden 1972‐1989. Clinical features, and estimation of its incidence. Eur. J. Clin. Pharmacol. 43, 215–218 (1992). [DOI] [PubMed] [Google Scholar]
- 10. Svartz, N. , Kallner, S. & Helander, S. Sönderdelas salazopyrin snabbt i organismen? Nord. Med. 25, 211–212 (1945). [Google Scholar]
- 11. Schroder, H. & Evans, D.A. The polymorphic acetylation of sulphapyridine in man. J. Med. Genet. 9, 168–171 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meyer, U.A. , Zanger, U.M. , Skoda, R.C. , Grant, D. & Blum, M. Genetic polymorphisms of drug metabolism. Prog. Liver Dis. 9, 307–323 (1990). [PubMed] [Google Scholar]
- 13. Pirmohamed, M. , Coleman, M.D. , Hussain, F. , Breckenridge, A.M. & Park, B.K. Direct and metabolism‐dependent toxicity of sulphasalazine and its principal metabolites towards human erythrocytes and leucocytes. Br. J. Clin. Pharmacol. 32, 303–310 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spielberg, S.P. Pharmacogenetics and blood dyscrasias. Eur. J. Haematol. Suppl. 60, 93–97 (1996). [DOI] [PubMed] [Google Scholar]
- 15. Shear, N.H. , Spielberg, S.P. , Grant, D.M. , Tang, B.K. & Kalow, W. Differences in metabolism of sulfonamides predisposing to idiosyncratic toxicity. Ann. Intern. Med. 105, 179–184 (1986). [DOI] [PubMed] [Google Scholar]
- 16. Rieder, M.J. , Shear, N.H. , Kanee, A. , Tang, B.K. & Spielberg, S.P. Prominence of slow acetylator phenotype among patients with sulfonamide hypersensitivity reactions. Clin. Pharmacol. Ther. 49, 13–17 (1991). [DOI] [PubMed] [Google Scholar]
- 17. Wolkenstein, P. et al A slow acetylator genotype is a risk factor for sulphonamide‐induced toxic epidermal necrolysis and Stevens‐Johnson syndrome. Pharmacogenetics 5, 255–258 (1995). [DOI] [PubMed] [Google Scholar]
- 18. Gunnarsson, I. , Kanerud, L. , Pettersson, E. , Lundberg, I. , Lindblad, S. & Ringertz, B. Predisposing factors in sulphasalazine‐induced systemic lupus erythematosus. Br. J. Rheumatol. 36, 1089–1094 (1997). [DOI] [PubMed] [Google Scholar]
- 19. Wadelius, M. , Stjernberg, E. , Wiholm, B.E. & Rane, A. Polymorphisms of NAT2 in relation to sulphasalazine‐induced agranulocytosis. Pharmacogenetics 10, 35–41 (2000). [DOI] [PubMed] [Google Scholar]
- 20. Jacobson, A. , Melhus, H. & Wadelius, M. Can mutations in ELA2, neutrophil elastase expression or differential cell toxicity explain sulphasalazine‐induced agranulocytosis? BMC Blood Disord. 4, 5 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kasowski, M. et al Extensive variation in chromatin states across humans. Science 342, 750–752 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gragert, L. , Madbouly, A. , Freeman, J. & Maiers, M. Six‐locus high resolution HLA haplotype frequencies derived from mixed‐resolution DNA typing for the entire US donor registry. Hum. Immunol. 74, 1313–1320 (2013). [DOI] [PubMed] [Google Scholar]
- 23. Keisu, M. , Ekman, E. & Wiholm, B.E. Comparing risk estimates of sulphonamide‐induced agranulocytosis from the Swedish Drug Monitoring System and a case‐control study. Eur. J. Clin. Pharmacol. 43, 211–214 (1992). [DOI] [PubMed] [Google Scholar]
- 24. Chen, P.L. et al Genetic determinants of antithyroid drug‐induced agranulocytosis by human leukocyte antigen genotyping and genome‐wide association study. Nat. Commun. 6, 7633 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tamai, H. et al Association between the DRB1*08032 histocompatibility antigen and methimazole‐induced agranulocytosis in Japanese patients with Graves disease. Ann. Intern. Med. 124, 490–494 (1996). [DOI] [PubMed] [Google Scholar]
- 26. Cheung, C. et al HLA‐B*38:02:01 predicts carbimazole/methimazole‐induced agranulocytosis. Clin. Pharmacol. Ther. 99, 555–561 (2016). [DOI] [PubMed] [Google Scholar]
- 27. Alfirevic, A. , Jorgensen, A.L. , Williamson, P.R. , Chadwick, D.W. , Park, B.K. & Pirmohamed, M. HLA‐B locus in Caucasian patients with carbamazepine hypersensitivity. Pharmacogenomics 7, 813–818 (2006). [DOI] [PubMed] [Google Scholar]
- 28. Ghattaoraya, G.S. , Middleton, D. , Santos, E.J. , Dickson, R. , Jones, A.R. & Alfirevic, A. Human leucocyte antigen‐adverse drug reaction associations: from a perspective of ethnicity. Int. J. Immunogenet. 44, 7–26 (2017). [DOI] [PubMed] [Google Scholar]
- 29. Tassaneeyakul, W. et al Associations between HLA class I and cytochrome P450 2C9 genetic polymorphisms and phenytoin‐related severe cutaneous adverse reactions in a Thai population. Pharmacogenet. Genomics 26, 225–234 (2016). [DOI] [PubMed] [Google Scholar]
- 30. Illing, P.T. et al Immune self‐reactivity triggered by drug‐modified HLA‐peptide repertoire. Nature 486, 554–558 (2012). [DOI] [PubMed] [Google Scholar]
- 31. Weltzien, H.U. , Dötze, A. , Gamerdinger, K. , Hellwig, S. & Thierse, H.J. Molecular recognition of haptens by T cells: More than one way to tickle the receptor In: Madame Curie Bioscience Database [Internet], Vol. Accessed March 8, 2017 (Landes Bioscience, Austin (TX), 2000–2013). [Google Scholar]
- 32. Klein, J. & Sato, A. The HLA system. First of two parts. N. Engl. J. Med. 343, 702–709 (2000). [DOI] [PubMed] [Google Scholar]
- 33. Erlich, H.A. , Opelz, G. & Hansen, J. HLA DNA typing and transplantation. Immunity 14, 347–356 (2001). [DOI] [PubMed] [Google Scholar]
- 34. Usami, Y. , Wu, Y. & Gottlinger, H.G. SERINC3 and SERINC5 restrict HIV‐1 infectivity and are counteracted by Nef. Nature 526, 218–223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Terao, C. , Raychaudhuri, S. & Gregersen, P.K. Recent advances in defining the genetic basis of rheumatoid arthritis. Annu Rev Genomics Hum Genet 17, 273–301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bohm, R. & Cascorbi, I. Pharmacogenetics and predictive testing of drug hypersensitivity reactions. Front. Pharmacol. 7, 396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maranville, J.C. & Cox, N.J. Pharmacogenomic variants have larger effect sizes than genetic variants associated with other dichotomous complex traits. Pharmacogenomics J. 16, 388–392 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Relling, M.V. & Evans, W.E. Pharmacogenomics in the clinic. Nature 526, 343–350 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hallberg, P. et al Genetic variants associated with antithyroid drug‐induced agranulocytosis: a genome‐wide association study in a European population. Lancet Diabetes Endocrinol. 4, 507–516 (2016). [DOI] [PubMed] [Google Scholar]
- 40. The Uppsala Monitoring Centre . The use of the WHO‐UMC system for standardised case causality assessment. < http://who-umcorg/Graphics/24734pdf>(2015).
- 41. Magnusson, P.K. et al The Swedish Twin Registry: establishment of a biobank and other recent developments. Twin Res. Hum. Genet. 16, 317–329 (2013). [DOI] [PubMed] [Google Scholar]
- 42. Figueiras, A. et al CYP2C9 variants as a risk modifier of NSAID‐related gastrointestinal bleeding: a case‐control study. Pharmacogenet. Genomics 26, 66–73 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Purcell, S. , Cherny, S.S. & Sham, P.C. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19, 149–150 (2003). [DOI] [PubMed] [Google Scholar]
- 44. Delaneau, O. , Zagury, J.F. & Marchini, J. Improved whole‐chromosome phasing for disease and population genetic studies. Nat. Methods 10, 5–6 (2013). [DOI] [PubMed] [Google Scholar]
- 45. Howie, B.N. , Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet. 5, e1000529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sham, P.C. & Purcell, S.M. Statistical power and significance testing in large‐scale genetic studies. Nat. Rev. Genet. 15, 335–346 (2014). [DOI] [PubMed] [Google Scholar]
- 47. Chang, C.C. , Chow, C.C. , Tellier, L.C. , Vattikuti, S. , Purcell, S.M. & Lee, J.J. Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Purcell, S.M. & Chang, C.C. PLINKv1.9. < https://www.cog-genomics.org/plink2>Accessed 12 Oct 2016.
- 49. Jia, X. et al Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One 8, e64683 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cereb, N. , Kim, H.R. , Ryu, J. & Yang, S.Y. Advances in DNA sequencing technologies for high resolution HLA typing. Hum. Immunol. 76, 923–927 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL is linked to the online version of the article at http://www.cpt-journal.com
Supporting Information