

Abstract
Our recent studies have shown that chronic kidney disease (CKD) affects the pharmacokinetics (PKs) of cytochrome P450 (CYP)2D6‐metabolized drugs, whereas effects were less evident on CYP3A4/5. Therefore, the effect of CKD on the disposition of CYP1A2‐metabolized, CYP2C8‐metabolized, CYP2C9‐metabolized, CYP2C19‐metabolized, and organic anion‐transporting polypeptide (OATP)‐transported drugs was investigated. We identified dedicated CKD studies with 6, 5, 6, 4, and 12 “model” substrates for CYP1A2, CYP2C8, CYP2C9, CYP2C19, and OATP, respectively. Our analyses suggest that clearance of OATP substrates decreases as kidney function declines. Similar trends were seen for CYP2C8; but overlap between some CYP2C8 and OATP substrates highlights that their interplay needs further investigation. In contrast, the effect of CKD on CYP1A2, CYP2C9, and CYP2C19 was variable and modest compared to CYP2C8 and OATP. This improved understanding of elimination‐pathway‐dependency in CKD is important to inform the need and conduct of PK studies in these patients for nonrenally eliminated drugs.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Our recent studies indicate that CKD affects the PK of drugs metabolized by CYP2D6, whereas effects on CYP3A4/5 drugs are variable and limited. However, there is a lack of systematically evaluated data on other metabolic and transport pathways.
WHAT QUESTION DID THE STUDY ADDRESS?
☑ We investigated elimination‐pathway‐dependency in the effect of CKD on several nonrenal clearance pathways. Specifically, we assessed the effect of CKD on the PK of model drug substrates of CYP1A2, CYP2C8, CYP2C9, CYP2C19, and OATP.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Despite the limited data available, a consistent decrease in clearance of multiple CYP2C8 and OATP model substrate drugs with increasing severity of CKD was observed. Conversely, only minimal effects on the clearance of CYP1A2, CYP2C9, and CYP2C19 model substrate drugs were observed in patients with CKD.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ It will facilitate the overall mechanistic characterization of the effect of CKD on individual nonrenal clearance pathways and guide PK study design in these patients.
Chronic kidney disease (CKD) is a major cause of morbidity and mortality and a significant contributor to the burden of chronic disease.1, 2 CKD has an estimated worldwide prevalence of 8–16%, and as such is considered an important global public health issue.3 Despite the existence of many guidelines related to dose adjustment of renally cleared medications in patients with CKD, impaired kidney function continues to be associated with an increased risk of adverse drug events.4 Thus, it is critical to adequately assess the effect of impaired kidney function on systemic exposure and pharmacokinetics (PK) of drugs to optimize drug usage in this tenuous patient population.
Renal impairment not only alters drug elimination by the kidneys, but also affects drug metabolism and transport in other organs (e.g., liver) that may lead to clinically relevant changes in nonrenal clearance.5, 6 For example, repaglinide, which is primarily metabolized in the liver after organic anion transporting polypeptide (OATP)‐mediated uptake, had a nearly fourfold increase in terminal half‐life and threefold increase in area under the concentration‐time curve (AUC) in patients with severe and endstage renal disease (ESRD) compared with subjects with normal renal function.7 The US Food and Drug Administration (FDA) and European Medicines Agency recently published guidances to recommend performing clinical studies to assess the effect of renal impairment on the PK of both renally and nonrenally eliminated drugs.8, 9 However, dosing adjustments to account for nonrenal clearance changes are not common.
The mechanisms that underlie CKD‐mediated changes in the PK of nonrenally eliminated drugs are complex and not well understood. CKD may change hepatic drug clearance by either a direct impact on hepatic enzymes or through alterations in other factors, such as drug absorption, protein binding, hepatic uptake, or accumulation of metabolites.10 Among them, increasing evidence has demonstrated that uremic toxins in patients with CKD may reduce metabolism or active uptake/efflux mechanisms through either direct inhibition or through transcriptional down regulation of proteins,1, 5, 6, 11, 12, 13 including cytochrome P450 (CYP) and UDP‐glucuronosyltransferase enzymes and membrane transporters.14, 15, 16 This uremia‐mediated impact on enzymes and transporters is supported by findings in experimental animal models of ESRD.5 Several of these preclinical studies demonstrated that uremia leads to decreased function and expression of metabolizing enzymes and transporters in the intestines and liver.17, 18, 19, 20 However, direct measurement of protein levels and activities in human patients with CKD may be needed to confirm the actual mechanism.
To this point, PK modeling and simulation studies using clinically observed data have been used to predict the effect of impaired kidney function on various elimination pathways for several model drugs.10, 21 Recently, we compiled the available data to systematically evaluate the relationship between CKD and PK of model substrate drugs for two metabolic pathways, CYP2D6 and CYP3A4/5.22 The analysis showed that CYP2D6‐mediated clearance generally decreases as kidney function declines. Conversely, no relationship between the level of CKD and clearance of CYP3A4/5 substrates was observed, suggesting potential pathway‐dependent effects of CKD on metabolism. Additionally, the effect of CKD on the PK of drugs that are actively transported in the liver has only been recently assessed.15, 23, 24 Quantitative and systematic investigations of specific clearance pathways in patients with kidney disease will improve our understanding of the effect of CKD on the PK of nonrenally eliminated drugs and will inform whether there is a need to perform further clinical studies to guide dose adjustments in CKD.
Here, we compiled the available clinical data and assessed the effect of renal impairment on CYP1A2‐mediated, CYP2C8‐mediated, CYP2C9‐mediated, CYP2C19‐mediated, and OATP‐mediated clearance pathways. These nonrenal pathways were selected because of their important role in drug disposition of many therapeutic drugs.
RESULTS
Clinical CKD studies for CYP1A2, CYP2C8, CYP2C9, CYP2C19, and OATP model substrate drugs
Of a total of 1,067 drugs, 6 CYP1A2, 5 CYP2C8, 6 CYP2C9, 4 CYP2C19, and 12 OATP model substrate drugs were identified in the analysis that had 7, 11, 9, 10, and 20 dedicated CKD studies, respectively (Figure 1 and Table 1). The PK parameters for these model substrate drugs along with the individual or mean clearance ratios of CKD groups to the healthy control group are summarized in Tables 1 and 2. In 30–50% of the selected CKD studies, protein binding was measured or PK parameters were reported using unbound concentrations. Model substrate drugs and their respective CKD studies are listed in Table 3.
Figure 1.

Overview of the workflow of clinical chronic kidney disease (CKD) data collection for cytochrome P450 (CYP)1A2, CYP2C8, CYP2C9, CYP2C19, and organic anion transporting polypeptide (OATP) model substrate drugs. AUCR, area under the concentration‐time curve ratio; DDI, drug‐drug interaction; DIDB, The University of Washington Metabolism and Transport Drug Interaction Database; fe,urine, fraction of the dose eliminated into urine unchanged; PGx, pharmacogenetics; USFDA, US Food and Drug Administration.
Table 1.
Effect of CKD on pharmacokinetics of model substrate drugs
| Drugs | Parameters | Ratios of parameters with CKD | ||||
|---|---|---|---|---|---|---|
| Mild | Moderate | Severe | ESRDa | ESRDb | ||
| CYP1A2 model substrate drugs | ||||||
| R_CLunbound | ||||||
| Duloxetine | CLoral/fu | – | – | – | 0.38 | – |
| Ropivacaine | CLoral/fu | – | 0.91 | 0.88 | – | – |
| Tasimelteon | CLoral/fu | – | – | 0.62 | – | 0.90 |
| CLoral/fu | – | – | 0.68 | – | 1.01 | |
| R_CLtotal | ||||||
| Duloxetine | CLoral | – | – | – | 0.46 | – |
| Lidocaine | CLiv | – | 0.82 | 0.51 | 0.93 | – |
| Pirfenidone | CLoral | 0.70 | 0.67 | 0.87 | – | – |
| Ramelteon | CLoral | 0.97 | 1.17 | 0.26 | – | 1.64 |
| Ropivacaine | CLoral | – | 0.95 | 0.80 | – | |
| Tasimelteon | CLoral | – | – | 0.70 | – | 0.91 |
| CLoral | – | – | 0.68 | – | 1.01 | |
| CYP2C8 model substrate drugs | ||||||
| R_CLunbound | ||||||
| Cerivastatin | CLoral/fu | 1.07 | 0.64 | 0.58 | – | – |
| CLoral/fu | 0.54 | 0.39 | 0.39 | – | – | |
| CLoral/fu | 0.58 | 0.47 | 0.43 | – | – | |
| Dasabuvir | CLoral/fu | – | – | 0.35c | – | – |
| Pioglitazone | CLoral/fu | – | – | 1.09c | – | – |
| Repaglinide | CLoral/fu | 0.55 | – | 0.37 | 0.37 | 0.27 |
| CLoral/fu | – | – | 0.31c | – | – | |
| Rosiglitazoned | CLoral/fu | 0.91 | 0.94 | 0.90 | – | – |
| R_CLtotal | ||||||
| Cerivastatin | CLoral | 1.03 | 0.59 | 0.71 | – | – |
| CLoral | 0.64 | 0.43 | 0.60 | – | – | |
| CLoral | – | – | – | – | 0.86 | |
| CLoral | 0.67 | 0.49 | 0.65 | – | – | |
| Dasabuvir | CLoral | 0.30 | 0.58 | 0.41 | ||
| Pioglitazone | CLoral | – | 1.20 | 1.29 | – | – |
| Repaglinide | CLoral | 0.55 | – | 0.37 | 0.37 | 0.27 |
| CLoral | 1.14 | – | 0.70 | – | – | |
| Rosiglitazoned | CLoral | 0.91 | 0.88 | 1.24 | – | – |
| CLoral | – | – | – | – | 0.57 | |
| CLoral | – | – | – | 1.09 | 1.21 | |
| CYP2C9 model substrate drugs | ||||||
| R_CLunbound | ||||||
| Meloxicam | CLoral/fu | – | – | – | 1.04 | – |
| CLoral/fu | 0.84 | 0.94 | – | – | – | |
| Tenoxicam | CLoral/fu | 1.15 | 0.75 | – | – | – |
| R_CLtotal | ||||||
| Flubiprofen | CLoral | – | – | – | – | 4.56 |
| CLoral | – | – | – | – | 1.65 | |
| Lesinuradd | CLoral | 0.77 | 0.58 | – | – | – |
| CLoral | – | 0.67 | 0.47 | – | – | |
| Losartand | CLoral | – | 0.60 | – | – | – |
| Meloxicam | CLoral | – | – | – | 3.12 | – |
| CLoral | 1.00 | 1.62 | – | – | – | |
| Phenytoind | CLiv | – | – | – | 1.08 | – |
| Tenoxicam | CLoral | 1.15 | 1.15 | – | – | – |
| CYP2C19 model substrate drugs | ||||||
| R_CLunbound | ||||||
| Lansoprazole | CLoral/fu | 0.75 | 1.09 | 1.14 | – | – |
| CLoral/fu | – | – | – | 0.97 | – | |
| CLoral/fu | – | – | – | – | 1.35 | |
| Voriconazole | CLoral/fu | 0.61 | 0.65 | 1.41 | – | – |
| R_CLtotal | ||||||
| Citalopramd | CLoral | – | 0.89 | 0.71 | 0.91 | – |
| CLoral | – | – | – | 0.92 | – | |
| Lansoprazole | CLoral | 0.89 | 1.35 | 1.68 | – | – |
| CLoral | 0.48 | 0.81 | 0.96 | – | – | |
| CLoral | – | – | – | 1.84 | – | |
| CLoral | – | – | – | – | 2.58 | |
| Rabeprazole | CLoral | – | – | – | 1.66 | 1.45 |
| Voriconazole | CLoral | 0.60 | 0.69 | 1.54 | – | – |
| CLiv | – | 1.57 | – | – | – | |
| OATP model substrate drugs | ||||||
| R_CLunbound | ||||||
| Atorvastatin | CLoral/fu | – | – | 0.58c | – | – |
| Bosentan | CLoral/fu | – | – | 0.93c | – | – |
| Cerivastatin | CLoral/fu | 1.07 | 0.64 | 0.58 | – | – |
| CLoral/fu | 0.54 | 0.39 | 0.39 | – | – | |
| CLoral/fu | 0.58 | 0.47 | 0.43 | – | – | |
| Erythromycin | (CLiv‐CLr)/fu | – | – | – | 0.63 | – |
| CLoral/fu | – | – | – | 0.39 | – | |
| Fluvastatin | CLoral/fu | – | – | 0.72c | – | – |
| Imatinib | CLoral/fu | – | 0.74 | 0.72 | – | – |
| Pitavastatin | CLoral/fu | – | – | 0.74 | – | – |
| CLoral/fu | – | 0.57 | – | 0.40 | – | |
| Repaglinide | CLoral/fu | 0.55 | – | 0.37 | 0.37 | |
| CLoral/fu | – | – | 0.31c | – | ||
| Rosuvastatin | CLoral/fu | – | – | 0.27c | – | – |
| Torsemide | CLiv,NR/fu | – | – | 1.03c | – | – |
| R_CLtotal | ||||||
| Atorvastatin | CLoral | – | 1.00 | 0.70 | – | – |
| Bosentan | CLoral | – | – | 1.13 | – | – |
| Cerivastatin | CLoral | 1.03 | 0.59 | 0.71 | – | – |
| CLoral | 0.64 | 0.43 | 0.60 | – | – | |
| CLoral | – | – | – | – | 0.86 | |
| CLoral | 0.67 | 0.49 | 0.65 | – | – | |
| Erythromycin | CLiv–CLr | – | – | – | 0.70 | – |
| CLoral | – | – | – | 0.43 | – | |
| CLoral | 0.87 | 0.76 | – | 0.75 | – | |
| CLoral | – | – | – | 0.28 | – | |
| Fexofenadine | CLoral | – | – | – | 0.37 | – |
| Fluvastatin | CLoral | 1.03 | 1.18 | 0.86 | 0.66 | 0.65 |
| Imatinib | CLoral | – | 0.73 | 0.54 | 0.56 | – |
| Pitavastatin | CLoral | – | – | 0.74 | – | – |
| CLoral | – | 0.56 | – | 0.54 | – | |
| Repaglinide | CLoral | 0.55 | – | 0.37 | 0.37 | 0.27 |
| CLoral | 1.14 | – | 0.70 | – | – | |
| Rosuvastatin | CLoral | 0.71 | 0.93 | 0.32 | – | – |
| Simeprevir | CLoral | – | – | – | 0.58 | – |
| Torsemide | CLiv,NR | – | – | 1.24 | – | – |
CKD, chronic kidney disease; CYP, cytochrome P450; ESRD, endstage renal disease; CLiv, i.v. clearance; CLoral, oral clearance; fu, fraction unbound in plasma; R_CLtotal, ratio of clearance calculated with total (bound plus unbound) concentration between CKD and healthy control group; R_CLunbound, ratio of unbound clearance between CKD and healthy control group.
aESRD subjects on regular dialysis but studied at off‐dialysis periods. bESRD subjects on dialysis during the study period. cEstimated based on averaged plasma albumin level change for organic anion‐transporting polypeptide/CY2C8 model drugs missing unbound information for severe patients. See Methods for detail. References are listed in Supplementary Table S3. dDrugs with 2 ≤ area under the concentration‐time curve ratio <3.
Table 2.
Mean and range of R_CLunbound or R_CLtotal from CKD studies
| Mild CKD | Moderate CKD | Severe CKD | ESRDa | ESRDb | ||
|---|---|---|---|---|---|---|
| CYP1A2 |
R_CLunbound
(3 drugsc) |
– |
0.92 (n = 1) |
0.73 (0.62–0.88; n = 3) |
0.38 (n = 1) |
0.95 (n = 2) |
|
R_CLtotal
(6 drugsd) |
0.83 (0.70–0.97; n = 2) |
0.90 (0.67–1.17; n = 4) |
0.64 (0.23–0.80; n = 6) |
0.70 (0.46–0.93; n = 2) |
1.15 (0.91–1.64; n = 3) |
|
| CYP2C8 |
R_CL unbound
(3 drugsc) |
0.73 (0.54–1.07; n = 5) |
0.61 (0.39–0.94; n = 4) |
0.53 (0.37–0.90; n = 5) |
0.37 (n = 1) |
0.27 (n = 1) |
|
R_CLtotal
(5 drugsd) |
0.75 (0.30–1.14; n = 7) |
0.69 (0.43–1.20; n = 6) |
0.75 (0.37–1.29; n = 8) |
0.73 (0.37–1.09; n = 2) |
0.73 (0.27–1.21; n = 4) |
|
| CYP2C9 |
R_CLunbound
(2 drugsc) |
1.0 (0.84–1.15; n = 2) |
0.84 (0.75–0.93; n = 2) |
– |
1.04 (n = 1) |
– |
|
R_CLtotal
(6 drugsd) |
0.97 (0.77–1.15; n = 3) |
0.92 (0.58–1.62; n = 5) |
0.47 (n = 1) |
2.10 (1.08–3.12; n = 2) |
3.10 (1.65–4.56; n = 2) |
|
| CYP2C19 |
R_CLunbound
(2 drugsc) |
0.68 (0.61–0.75; n = 2) |
0.87 (0.65–1.09; n = 2) |
1.28 (0.59–1.41; n = 2) |
0.97 (n = 1) |
1.35 (n = 1) |
|
R_CLtotal
(4 drugsd) |
0.66 (0.48–0.89; n = 3) |
1.06 (0.69–1.57; n = 5) |
1.04 (0.71–1.68; n = 4) |
1.33 (0.91–1.84; n = 4) |
2.01 (1.45–2.58; n = 2) |
|
| OATP |
R_CLunbound
(5 drugsc) |
0.68 (0.55–1.07; n = 4) |
0.56 (0.39–0.74; n = 5) |
0.54 (0.37–0.72; n = 6) |
0.45 (0.37–0.63; n = 4) |
0.27 (n = 1) |
|
R_CLtotal
(12 drugsd) |
0.83 (0.55–1.14; n = 8) |
0.74 (0.43–1.18; n = 9) |
0.71 (0.32–1.24; n = 12) |
0.52 (0.28–0.75; n = 10) |
0.59 (0.27–0.86; n = 3) |
|
| Theoretical lowest R_CLe | 0.88 | 0.79 | 0.73 | 0.67 | 0.67 | |
CKD, chronic kidney disease; CYP, cytochrome P450; OATP, organic anion‐transporting polypeptide; R_CLtotal, ratio of clearance calculated with total (bound plus unbound) concentration between CKD and healthy control group; R_CLunbound, ratio of unbound clearance between CKD and healthy control group; n, number of CKD studies in each categrory.
Mean and range of reported R_CLunbound or R_CLtotal in each CKD category were summarized.
aESRD subjects on regular dialysis but studied at off‐dialysis periods. bESRD subjects on dialysis during the study period. cAll drugs that have R_CLunbound data with at least one of the four CKD groups summarized in this table. dAll drugs that have R_CLtotal data with at least one of the four CKD groups summarized in this table. eTheoretical lowest R_CL values were calculated assuming no changes in nonrenal clearance, as described in the Method section.
Table 3.
PK parameters of model substrate drugs that have PK study reports in patients with CKD
| Drugs | fm,CYP1A2 | 1‐fm,CYP1A2 | fe,urine a | F | DDI or PGx with maximum AUCR | |
|---|---|---|---|---|---|---|
| AUCR | Inhibitors or PGx | |||||
| CYP1A2 model substrate drugs | ||||||
| Duloxetine | 0.82 | 0.18 | <0.01b | 0.43 | 5.60 | Fluvoxamine |
| Lidocaine | 0.67 | 0.33 | 0.03b | 0.35 | 3.05 | Fluvoxamine |
| Pirfenidone | 0.85 | 0.15 | – | – | 6.81 | Fluvoxamine |
| Ramelteon | 0.99 | 0.01 | 0b | 0.02 | 189.86 | Fluvoxamine |
| Ropivacaine | 0.73 | 0.27 | 0.01 | – | 3.69 | Fluvoxamine |
| Tasimelteon | 0.86 | 0.15 | <0.01b | 0.38 | 6.87 | Fluvoxamine |
| CYP2C8 model substrate drugs | ||||||
| Cerivastatin | 0.77 | 0.23 | <0.02b | 0.6 | 4.36 | Gemfibrozil |
| Dasabuvir | 0.91 | 0.09 | 0.003b | 0.46 | 11.3 | Gemfibrozil |
| Pioglitazone | 0.79 | 0.21 | 0b | – | 4.66 | Gemfibrozil |
| Repaglinide | 0.88 | 0.12 | 0.001b | 0.63 | 8.26 | Gemfibrozil |
| Rosiglitazone | 0.56 | 0.44 | 0.0001b | 0.99 | 2.29 | Gemfibrozil |
| CYP2C9 model substrate drugs | ||||||
| Flurbiprofen | 0.67 | 0.33 | <0.03b | 0.96 | 3.03 | Fluconazole |
| Lesinurad | 0.53 | 0.47 | 0.31b | 1 | 2.11 | PM vs. EM |
| Losartan | 0.59 | 0.41 | 0.12 | 0.33 | 2.42 | PM vs. EM |
| Meloxicam | 0.88 | 0.12 | 0.002b | 0.89 | 8.22 | PM vs. EM |
| Phenytoin | 0.77 | 0.23 | 0.02b | 0.86 | 4.26 | PM vs. EM |
| Tenoxicam | 0.60 | 0.40 | <0.01b | 1 | 2.51 | PM vs. EM |
| CYP2C19 model substrate drugs | ||||||
| Citalopram | 0.54 | 0.46 | 0.1 | 0.8 | 2.19 | PM vs. EM |
| Lansoprazole | 0.88 | 0.12 | 0b | 0.91 | 8.56 | PM vs. EM |
| Rabeprazole | 0.81 | 0.19 | 0b | 0.52 | 5.26 | PM vs. EM |
| Voriconazole | 0.75 | 0.25 | <0.02 | 0.96 | 4.05 | PM vs. EM |
| Drugs | OATP model substrate drugs | |||||||
|---|---|---|---|---|---|---|---|---|
| fe,urine a | f | AUCR | ft | Inhibitors | AUCR | ft | PGx | |
| Atorvastatin | <0.02b | 0.77 | 12 | 0.92 | Rifampin | 2.45 | 0.59 | NF vs. PF |
| Bosentan | <0.03b | 0.5 | 5.22 c | 0.81 | Lopinavir & ritonavirc | – | – | – |
| Cerivastatin | <0.02b | 0.6 | 4.75 | 0.79 | Cyclosporine | – | – | – |
| Erythromycin | 0.12 | – | – | – | – | 2.60f | 0.62 | NF vs. PF |
| Fexofenadine | 0.20b | 0.35 | 4.14d | 0.76 | Lopinavir and ritonavird | – | – | – |
| Fluvastatin | 0.21b | 0.24 | 3.55 | 0.72 | Cyclosporine | 2.39 | 0.58 | NF vs. PF |
| Imatinib | 0.05b | 0.98 | – | – | – | 2.58f | 0.61 | NF vs. PF |
| Pitavastatin | 0.06b | 0.51 | 6.66 | 0.85 | Rifampin | 3.85 | 0.74 | NF vs. PF |
| Simeprevir | 0.15b | 0.62 | 5.80 | 0.83 | Cyclosporine | – | – | – |
| Repaglinide | 0.001b | 0.63 | 2.60e | 0.62 | Cyclosporinee | 3.13 | 0.68 | NF vs. PF |
| Rosuvastatin | 0.28 | – | 7.08 | 0.86 | Cyclosporine | 2.19 | 0.54 | NF vs. PF |
| Torsemide | 0.28 | 0.8 | – | – | – | 2.51 f | 0.60 | NF vs. PF |
AUCR, area under the concentration‐time curve ratio; CL, clearance; CYP, cytochrome P450; DDI, drug‐drug interaction; EM, extensive metabolizer; F, absolute bioavailability after oral administration; fe,urine, fraction eliminated into urine as an unchanged drug; fm,CYP1A2, estimated fraction metabolized by CYP1A2; fm,CYP2C8, estimated fraction metabolized by CYP2C8; fm,CYP2C9, estimated fraction metabolized by CYP2C9; fm,CYP2C19, estimated fraction metabolized by CYP2C19; ft = 1‐1/AUCR for transporter; NF, normal function; PGx, pharmacogenetics; PF, poor function; PM, poor metabolizer.
afe,urine after intravenous administration or fe,urine after oral administration divided by absolute bioavailability, if not indicated otherwise. bfe,urine after oral administration. cMaximum AUCR = 1.97 with inhibitor cyclosporine. dNo DDI studies available with inhibitors cyclosporine/rifampin. eMaximum AUCR = 8.26 with inhibitor gemfibrozil. fMaximum ratio of 1/CL or F/CL. References are listed in Supplementary Table S4. The availability of both PGx and DDI data for the same OATP substrate was limited to allow any direct comparison and conclusions.
Impact of CKD on the clearance of CYP1A2, CYP2C8, CYP2C9, CYP2C19, and OATP model substrate drugs
The total clearance ratios (R_CLtotal) observed between various CKD and healthy control groups from each CKD study, as well as the unbound clearance ratios (R_CLunbound) for drugs with available protein binding data, are shown in Figures 2 and 3 and Table 1. Average and range of these ratios are listed in Table 2.
Figure 2.
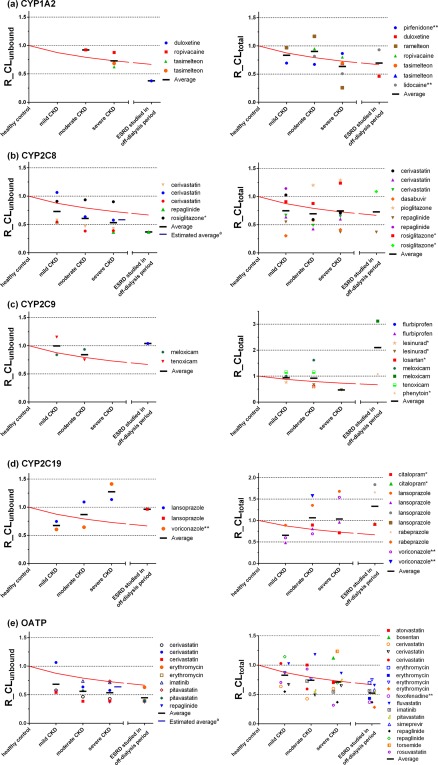
Comparison of observed clearance ratio (R_CL) and theoretical lowest R_CL without changes in nonrenal clearance for cytochrome P450 (CYP)1A2 (a), CYP2C8 (b), CYP2C9 (c), CYP2C19 (d), and organic anion transporting polypeptide (OATP) (e) model substrate drugs. Symbols represent R_CLunbound in each chronic kidney disease (CKD) group of a clinical CKD study for drugs with protein binding information in healthy control and CKD groups, or R_CLtotal for all drugs, including those without protein binding information in CKD studies. Solid red lines represent the theoretical lowest ratio assuming no changes in nonrenal clearance (the values are 0.88, 0.79, 0.73, and 0.69 for the mild CKD, moderate CKD, severe CKD, and the endstage renal disease (ESRD) patient groups, respectively). aEstimated unbound clearance ratios based on averaged plasma albumin level change for OATP/CYP2C8 model drugs missing protein binding information for severe patients. See Methods section for details. R_CLunbound, unbound clearance ratio between CKD patient groups and the healthy control group; R_CLtotal, ratio of clearance calculated with total (bound plus unbound) concentration between CKD groups and the healthy control group. *Drugs with 2 ≤ AUCR <3. **Drugs fu ≥ 0.3.
Figure 3.
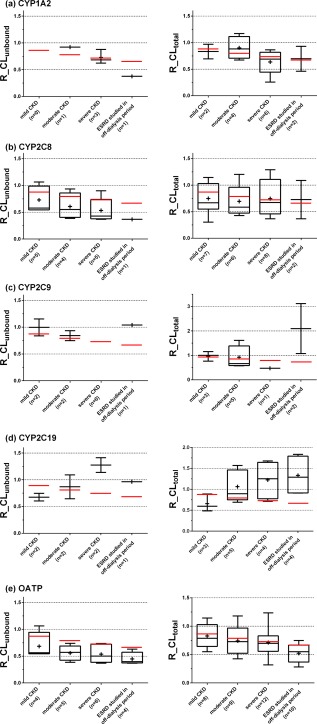
Effect of chronic kidney disease (CKD) on cytochrome P450 (CYP)1A2 (a), CYP2C8 (b), CYP2C9 (c), CYP2C19 (d) and organic anion transporting polypeptide (OATP) (e) model substrate drugs. The box‐and‐whisker plots represent interquartile range of unbound clearance ratio between CKD patient groups and the healthy control group (R_CLunbound) for drugs with protein binding information, or total clearance ratio between CKD patient groups and the healthy control group (R_CLtotal) for all drugs with CKD studies. The “+” symbols represents the mean value of R_CL, and red lines represent the theoretical lowest ratio assuming no CKD effect on nonrenal clearance (the values are 0.88, 0.79, 0.73, and 0.69 for the mild CKD, moderate CKD, severe CKD, and the endstage renal disease (ESRD) groups, respectively). n, number of CKD studies in each group.
Using method 1, the theoretical lowest values of the clearance ratios assuming no change in nonrenal clearance were calculated to be 0.88, 0.79, 0.73, and 0.67 with mild CKD, moderate CKD, severe CKD, and ESRD, respectively. As shown in Figures 2 and 3, CYP2C8 and OATP model substrate drugs showed a consistently lower R_CLunbound than the theoretical lowest ratios. The ratios for CYP2C8 were 0.73, 0.61, 0.53, and 0.37 and for OATP were 0.68, 0.56, 0.54, and 0.45 for the mild CKD, moderate CKD, severe CKD, and ESRD (on off‐dialysis day), respectively. In contrast, the R_CLunbound for CYP1A2, CYP2C9, and CYP2C19 did not show a consistent trend.
The results were similar using method 2 (calculated with individually estimated fm shown in Table 3). The average R_CLunbound for CYP2C8 model substrate drugs were 0.69, 0.57, 0.54, and 0.49, respectively, for the mild CKD, moderate CKD, severe CKD, and ESRD (on off‐dialysis day) (Supplementary Figures S1 and S2).
Using method 3 (based on fe,urine, see Methods section), the results were comparable to methods 1 and 2 (Supplementary Figures S3 and S4). Average R_CLunbound were 0.73, 0.61, 0.54, and 0.37 for CYP2C8 model drugs in the mild CKD, moderate CKD, severe CKD, and ESRD, respectively. Average R_CLunbound for OATP model drugs were 0.69, 0.57, 0.54, and 0.49 in mild CKD, moderate CKD, severe CKD, and ESRD, respectively. These average unbound R_CLCYP or R_CLOATP values for CYP2C8 and OATP model drugs were again smaller than the theoretical value of 1 (assuming unchanged nonrenal clearance).
Review of plasma protein binding data (Supplementary Table S2) indicated that ratios of nonprotein bound fractions between CKD and control groups increased with the severity of CKD: 1.06, 1.07, 1.16, and 1.55 for patients with mild CKD, moderate CKD, severe CKD, and ESRD, respectively (Figure 4). This increased trend seems more apparent for drugs that are highly protein bound (fu < 0.01; Figure 4).
Figure 4.
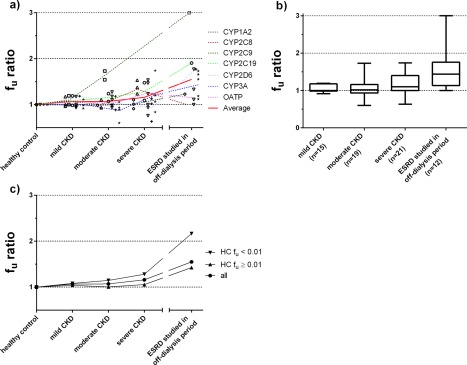
Observed chronic kidney disease (CKD) effect on the plasma unbound fraction fu ratio (CKD groups with respect to healthy control) for cytochrome P450 (CYP)1A2 (⋄), CYP2C8 (Δ), CYP2C9 (□), CYP2C19 (○), CYP2D6 (▽), CYP3A (+), and organic anion transporting polypeptide (OATP) (*) model substrate drugs. (a) Symbols represent observed fu ratios, dashed lines represent the average of the model drugs for each CYP enzyme and transporter, and solid lines represent the averaged fu ratios from all the model drugs investigated. (b) The box‐and‐whisker plots represent interquartile range of fu ratios. (c) Unbound fraction ratios increase with CKD for drugs with the relatively low fu (fu < 0.01) vs. relatively high fu (fu ≥ 0.01). The 2D6 and 3A model drugs from a previous study22 were also included. ESRD, endstage renal disease; fu, unbound fraction; HC, healthy control; n, number of CKD studies in each category; fu ratio, ratio of fu between CKD groups and the healthy control group.
DISCUSSION
We evaluated the effect of CKD on the clearance of several CYP1A2, CYP2C8, CYP2C9, CYP2C19, and OATP model substrate drugs, extending our previous work to develop generalizable rules concerning the conduct of PK studies for nonrenally cleared drugs in patients with CKD.
OATP model drugs showed a consistent reduction in unbound OATP‐mediated clearance with increasing CKD severity with a 46% reduction on average in the severe CKD group (the highest value was 63% in case of repaglinide). Only 5 of 12 OATP model drugs had originally reported plasma binding in CKD. We extended the dataset and estimated fu for drugs with missing protein binding information (based on averaged albumin changes in CKD). Our calculated unbound clearance ratios in severe CKD (0.62) were in agreement with ratios observed (0.54) for drugs with available fu data. Collectively, these data corroborate previous observations in animals5, 25 and in humans, suggesting that OATP transporter function is decreased in patients with kidney disease.15, 23, 24
CYP2C8 model drugs also showed a consistent decrease in unbound CYP2C8‐mediated clearance with increasing CKD severity, with an average 44% reduction within the severe CKD group. The highest reduction in clearance (63%) was seen in the case of repaglinide, followed by dasabuvir and cerivastatin. However, interpretation of CYP2C8 trends is challenging for multiple reasons. Repaglinide and cerivastatin were selected as CYP2C8 model drugs based on drug‐drug interactions (DDIs) with gemfibrozil; however, both parent and metabolite of gemfibrozil also inhibit OATP1B1.26, 27 Although dasabuvir (a non‐OATP1B substrate) showed a pronounced reduction in clearance in severe CKD, dasabuvir is a P‐glycoprotein and breast cancer resistance protein substrate, which may confound the observed finding.28 In contrast, the CYP2C8 model drug rosiglitazone did not show a clear decrease in clearance with an increasing severity of CKD. Thus, the observed decreases in CYP2C8‐mediated clearance in CKD may need to be further confirmed by either performing additional clinical studies in patients with CKD with protein binding data or with specific inhibitors of OATP or CYP2C8 to differentiate their relative contributions. Quantifying the rate‐determining step and contribution of metabolic clearance and hepatic uptake (e.g., via OATP1B1) to drug disposition is a complex and challenging task.29 One possibility is to incorporate changes in the activity of individual enzymes or transporters observed in CKD based on a specific probe substrate (e.g., pitavastatin for OATP1B130) in physiologically based pharmacokinetic (PBPK) models.
Compared to CYP2C8 and OATP model drugs, the PK of CYP1A2 model drugs showed relatively smaller changes with CKD (an average 8% and 27% decrease in unbound clearance within the moderate and severe groups, respectively) and were comparable to the theoretical lowest values. It is worth mentioning that using method 3, CYP1A2 showed lower unbound clearance ratios than the theoretical R_CL (Supplementary Figure S2). Considering the very limited data points available and the relatively small changes with CKD compared to CYP2C8 and OATP, further study is needed to clarify the effect of CKD on CYP1A2 model drugs. For ramelteon, there was a large reduction in R_CLtotal within the severe CKD group. However, the raw data showed that one subject (out of a total of seven) in this group had a markedly higher AUC than others,31 and excluding that individual would bring the average R_CLtotal closer to the theoretical lowest clearance ratio. Overall, both observed and calculated clearance ratios were, on average, comparable to theoretical lowest values, suggesting a limited effect of CKD on CYP1A2.
Similar to CYP1A2, the PK of CYP2C9 model drugs showed relatively small change with CKD (an average 0% and 16% decrease in unbound clearance within the mild and moderate groups, respectively) and were, on average, comparable to theoretical lowest values. These findings suggest that there is a limited effect of CKD on the activity of CYP2C9.
For CYP2C19, an increased clearance of many model drugs was seen across all CKD groups and was greater than the theoretical lowest line when no change in nonrenal clearance is assumed. However, the effect seems to be more prominent when clearance was calculated with total concentration (R_CLtotal), which suggests a potential effect of CKD on plasma protein binding. Therefore, although unbound clearance may be unchanged, total clearance could be affected in CKD due to an increase in the unbound drug fraction. For instance, there was a moderate increase of the total drug clearance for the highly bound drug lansoprazole in the severe CKD or ESRD groups, whereas minimal change was seen in its unbound clearance.
In general, plasma protein binding was decreased with increasing CKD severity and increases in unbound fractions were seen for most drugs investigated in our current and previous studies.22 This trend seems to be more apparent for model drugs with relatively low fu (fu < 0.01). It is also worth mentioning that the impact of CKD on plasma protein binding varies by drug.10, 32, 33 Despite a comprehensive analysis of the dataset of CKD studies, less than half of the drugs had sufficient protein binding information available. Thus, it is crucial to evaluate protein binding changes for each drug studied in patients with CKD.
Most subjects with ESRD, particularly those who received dialysis during the study period, exhibited an increased clearance. This likely occurs because “uremic toxins” are continuously removed during dialysis and some portion of the drug may also be removed (“cleared”) during dialysis. Some patients with ESRD who regularly received dialysis but were off‐dialysis during the study period also demonstrated a higher clearance than patients with ESRD not receiving dialysis, but less than those receiving dialysis. Studying patients with ESRD receiving chronic dialysis may not represent the “worst case” scenario as compared with other groups of patients with CKD.8 However, all the ESRD groups in this study were either on regular dialysis during the study period or off dialysis but were on regular dialysis before the study period, consistent with the typical standard of care.34
One aim of this work was to quantitatively assess and interpret observed data in patients with CKD groups for a multitude of nonrenal clearance pathways. We used three methods to determine if CKD may affect individual pathways. Method 1 compared observed R_CL to theoretical lowest clearance ratios assuming that renal elimination comprised a maximum of 33.3% of systemic clearance. Method 1 may not correctly identify the impact of CKD on nonrenal clearance of drugs that have a much higher fm than 66.7%, resulting in a “theoretical lowest R_CL” value that is higher than our current assumption. In addition to method 1, methods 2 and 3 considered the actual maximum area under the concentration‐time curve ratio (AUCR) for each individual pathway and fraction eliminated into urine as unchanged drug fe,urine, respectively, to calculate the clearance ratios. Overall, the calculated ratios R_CLCYP using fm (=1‐1/AUCR) or R_CLOATP using ft (=1‐1/AUCR) are similar to those calculated using fe,urine for most of the individual pathways of interest. Some discrepancies existed, as seen in the cases of rosiglitazone, flurbiprofen, and citalopram (AUCR ranging between 2 and 3 for these drugs; Table 3). For example, the calculated unbound rosiglitazone R_CLCYP using fm (method 2) were 1.13, 1.67, and 1.45, in contrast to the calculated unbound R_CLCYP using fe,urine (method 3; 0.91, 0.94, and 0.90, for mild, moderate, and severe CKD in the patient groups, respectively). Current analysis suggests that the calculated R_CLCYP using method 2 is more representative if the inhibitor is a specific strong inhibitor resulting in a large AUCR for a pathway of interest. However, there are many cases in which DDI studies with a strong inhibitor are not available or the inhibitor is not specific, in particular for studies other than CYP3A‐ or CYP2D6‐mediated DDIs. The results from method 2 may be biased if imperfect inhibitor/pharmacogenetics (PGx) data are used, even though method 2 gave very similar results as method 1 in this study. Method 3 using fe,urine assumed all nonrenal elimination was attributed to the pathway of interest, which may not hold true for all the drugs in our dataset. The limitations of each method need to be considered when drawing conclusions regarding the effects of CKD on various elimination pathways.
It is noteworthy that limited information is available for many pathways we examined in this study (i.e., for many clearance pathways there are a limited number of drugs that have PK data for patients with CKD and information on plasma protein binding in these patients). For example, although data were available for all three (mild, moderate, and severe) CKD groups in dedicated studies for 10 drugs, only 4 of these drugs with 6 studies included unbound drug information. To overcome the missing information on plasma protein binding, we predicted fu in severe CKD for acidic drugs that primarily bind to albumin using the average albumin concentration in this population.10 Although this approach was acceptable for the analysis of acidic drugs in the current dataset, it highlighted the necessity to measure plasma protein binding in different CKD groups to fully distinguish the CKD effect on metabolic/transporter elimination pathways from changes in plasma protein binding. Based on the fact that many of our drugs were similar with respect to their extent of binding to plasma proteins, performing PBPK sensitivity analysis on the impact of fu changes over a broader range of plasma protein binding may provide additional quantitative insight to potential PK changes in patients with CKD. Last, high variability was observed in the clinical PK data curated (Figures 2 and 3), which makes it difficult to draw robust conclusions for some pathways.
In conclusion, this work suggests that the level of decrease in clearance observed in patients with CKD was consistent among multiple OATP substrate drugs, and was larger than the estimated decrease when the change in nonrenal clearance was assumed to be nil. One of our future applications is to incorporate the observed changes in OATP activity into PBPK models to quantitatively predict the effect of renal failure on the PKs of other OATP substrates or in different groups of patients with CKD. A similar trend in the reduction of clearance in CKD was also seen for CYP2C8 model drugs. Some of these are dual CYP2C8‐OATP substrates highlighting importance of further investigation of the rate‐determining step in disposition of those drugs and changes imposed by CKD. In contrast, the effect of CKD on CYP1A2, CYP2C9, and CYP2C19 was variable and modest by comparison to CYP2C8 and OATP. Further investigation of the potential effect of CKD on plasma protein binding, other nonrenal clearance pathways, and the interplay of transporter and enzyme‐mediated clearance are necessary to establish a comprehensive understanding of the elimination‐routine dependency in CKD effects. These findings are useful to identify nonrenally cleared drugs, which may benefit from additional PK or PBPK analysis to optimize their use in patients with CKD, and, ultimately, may facilitate optimal drug selection and dosing in these patients.
METHODS
Selection of CYP1A2, CY2C8, CYP2C9, and CYP2C19 model substrate drugs with existing PK data in patients with CKD
Model substrate drugs of CYP1A2, CYP2C8, CYP2C9, and CYP2C19 were selected using the approach applied previously in the assessment of CYP2D6 and CYP3A.22 Briefly, the University of Washington Metabolism and Transport Drug Interaction Database (UW DIDB) and the FDA's new drug application reviews (Drugs@FDA) were searched to identify a comprehensive list of potential model substrate drugs for each elimination pathway (Figure 1). A CYP model substrate drug was defined as one that is predominantly eliminated by a specific CYP enzyme in vivo (e.g., fm ≥ 0.67) based on observed AUCR from either DDI or pharmacogenetic studies. New drug applications approved between 2014 and October 2016 were also curated in order to include drugs that were not yet incorporated into the UW DIDB when the data were originally curated in December 2014. A total of 981 drugs were selected as potential CYP substrate drugs for investigation.
Typical inhibitors assessed in this study were: fluvoxamine, ciprofloxacin, enoxacin, zafirlukast, methoxsalen, and mexiletine for CYP1A2; clopidogrel, gemfibrozil, deferasirox, and teriflunomide for CYP2C8; amiodarone, felbamate, fluconazole, miconazole, and piperine for CYP2C9; and fluconazole, fluvoxamine, and ticlopidine for CYP2C19. If a drug exhibited a predefined AUCR of ≥3 between the presence and absence of a typical inhibitor, then it was considered a potential model substrate drug for the corresponding enzymes. Similarly, if a drug exhibited an AUCR of ≥3 between poor and extensive metabolizers of each enzyme, then the drug was considered a potential model substrate drug of that enzyme. The AUCR ≥3 criterion was used to ensure the list of selected drugs was comprised of substrates with a high contribution (≥67%) of CYP1A2, CYP2C8, CYP2C9, or CYP2C19‐mediated clearance to their systemic clearance. For CYP2C8, CYP2C9, and CYP2C19, one, three, and one substrate drugs, respectively, with AUCR between two and three were also included in the analysis (see method 2). Studies reporting collateral DDIs due to overlapping inhibitor and substrate specificity were not considered (Supplementary Table S1). The UW DIDB, PubMed, and FDA documents (new drug application reviews and study reports) were searched for PK studies in patients with CKD for the potential model substrate drugs of CYP1A2, CYP2C8, CYP2C9, or CYP2C19. Population PK studies using historical values as healthy controls were not included. Clearance ratios between each CKD group and controls were calculated, and classification of CKD groups was carried out, as previously described.22
Selection of OATP model substrate drugs with existing PK data in patients with CKD
The in vitro or in vivo substrates of OATP1B1 and OATP1B3 were identified from the UW DIDB (Figure 1). A total of 86 compounds were identified as potential substrate drugs after excluding compounds that are endogenous, not approved for clinical use, or known metabolites. As above, the UW DIDB, PubMed, and FDA documents (new drug application reviews and study reports) were searched further to identify dedicated clinical studies in patients with CKD for each of the 86 drugs.
Forty‐three of 86 drugs had CKD studies reported. The fe,urine values were calculated to estimate the contribution of renal elimination pathway, either as fe,urine for intravenously administered drugs or fe,urine divided by absolute bioavailability for orally administered drugs. For the selected OATP model drugs, DDI studies with the potent inhibitors cyclosporine and rifampin were collected; if data were not available then alternative inhibitors were considered and pharmacogenetic studies (SLCO1B1 521 T>C) were also examined. OATP model substrate drugs were defined as those having fe,urine <33.3% (assuming all the nonrenal clearance was mediated by OATP).
Interpretation of the observed ratios of clearance in the CKD group vs. the control group with normal kidney function
In method 1, theoretical lowest values in clearance ratios (R_CL) of the patients with CKD groups over healthy controls were calculated, assuming CKD has no effect on respective nonrenal pathways. Comparison with observed ratios of clearance (R_CLobs) is shown in Figure 2 and Table 2. If observed clearance ratios were lower than the theoretical lowest ratio assuming no change in nonrenal clearance, the data then indicated an effect of CKD on nonrenal clearance. Theoretical lowest R_CL was calculated using a fixed minimum fm or maximum fe for all drugs. For example, for a model substrate for a particular enzyme showing AUCR of ≥3 in the presence of a specific inhibitor, the assumed minimum fm for that pathway was 0.667. Assuming the rest of its clearance is renal clearance, then at most one third of systemic clearance is via renal elimination. For the purpose of our analyses, we also assumed that renal clearance decreased in parallel with glomerular filtration rate (GFR) in patients with different degrees of CKD (i.e., as kidney function declines), irrespective of the influence of active tubular secretion or reabsorption.21
In method 2, changes in CYP1A2, CYP2C8, CYP2C9, and CYP2C19 metabolizing pathways were evaluated using our previously published approach,22 as follows:
Therefore,
where R_CLCYP is the theoretical ratio of clearance mediated by the respective enzyme. R_CLr is the renal clearance ratio of each CKD patient group, as described below. It was assumed that (1) all elimination pathways except CYP1A2, CYP2C8, CYP2C9, and CYP2C19 decrease in parallel with GFR, and (2) the absorption of model drugs are not altered by CKD.
Similarly, a change in OATP‐mediated transport was calculated as follows:
In method 3, the fraction of drugs eliminated unchanged into urine (fe,urine) calculated either as fe,urine for intravenously administered drugs or fe,urine divided by absolute bioavailability for orally administered drugs (Table 3). The R_CLCYP was then calculated using fe,urine:
Similarly, R_CLOATP was calculated using the fraction eliminated unchanged into urine fe,urine for OATP transporter model drugs:
For both methods 2 and 3, R_CLCYP or R_CLOATP different from 1 (e.g., <1), would suggest that CKD may have an effect (i.e., impair) on the particular CYP or transporter‐mediated pathway.
In all methods, we used GFR of 0 mL/min and 120 mL/min for ESRD groups and healthy controls, respectively, and maximum and minimum averages for each patients with CKD groups (i.e., 74.5, 44.5, and 22.5 mL/min for mild CKD, moderate CKD, and severe CKD, respectively) to calculate R_CLr, which were 0.625, 0.375, 0.188, and 0 for mild CKD, moderate CKD, severe CKD, and ESRD, respectively.
Estimation of unbound fraction in plasma for OATP/CYP2C8 drugs with missing observed plasma protein binding data
For acidic drugs, the decreased plasma protein binding is generally related to the reduced albumin levels in plasma.5, 10 The missing fu was estimated for severe CKD group using the information on the changed albumin levels in this group relative to healthy control, using the following equation10:
where pop is the healthy population, [P]pop is the mean concentration of albumin in healthy subjects (44.9 g/L for men and 41.8 g/L for women) and [P] is the mean concentration in patients with severe CKD (37.6 g/L for men and 35 g/L for women), respectively.10 The gender differences in albumin concentration were accounted for during the calculations based on the corresponding clinical CKD study designs for each substrate drugs. This prediction method assumes that albumin is the major plasma protein contributing to binding and that CKD does not cause any conformational changes in albumin binding sites.
This method was applied to calculate fu for all OATP1B1 substrates with missing binding information in severe CKD because they are mostly acidic drugs. Considering overlap in CYP2C8 and OATP1B1 substrates, the same approach was applied for CYP2C8 substrates. The calculated fu values are listed in Supplementary Table S2 and the corresponding calculated R_CLunbound using the calculated fu is shown in Figure 2.
AUTHOR CONTRIBUTIONS
M.‐L.T., K.Y., P.Z., L.Z., A.G., T.D.N., M.P.‐M., and S.‐M.H. wrote the manuscript. M.‐L.T., K.Y., P.Z., L.Z., and S.‐M.H. designed the research. M.‐L.T. and K.Y. performed the research. M.‐L.T., K.Y., P.Z., L.Z., A.G., T.D.N., M. P.‐M., and S.‐M.H. analyzed the data.
SOURCE OF FUNDING
This research was supported by the US Food and Drug Administration's (FDA's) Medical Countermeasures Initiative. Drs Ming‐Liang Tan, Kenta Yoshida, Micheline Piquette‐Miller, Thomas D. Nolin and Aleksandra Galetin were supported in part by an appointment to the Research Participation Program at the Center for Drug Evaluation and Research, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the US Department of Energy and the FDA. Dr Nolin is supported in part by the National Institutes of Health, National Institute of General Medical Sciences (R01‐GM107122).
CONFLICT OF INTEREST/DISCLOSURE
S.‐M.H. and M.P.‐M., Associate Editors of Clinical Pharmacology & Therapeutics, were not involved in the review and decision making for this article. The other authors declared no conflicts of interest.
DISCLAIMER
The contents of this manuscript reflect the views of the authors and should not be construed to represent the FDA's views or policies. No official support or endorsement by the FDA is intended or should be inferred. The mention of commercial products, their sources, or their use in connection with material reported herein is not to be construed as either an actual or implied endorsement of such products by the FDA.
Supporting information
Supplementary Table S1 List of drugs with CKD studies not included as model substrate drugs. These drugs had DDI studies with the typical inhibitors used for the CYP enzymes in this study or PGx studies.
Supplementary Table S2 Mean ± SD, if available, plasma unbound fraction information.
Supplementary Table S3 References for Table 1 in the main text.
Supplementary Table S4 References for Table 3 in the main text.
Supplementary Figure S1 Comparison of calculated R_CL using maximum AUCR (method 2) and normalized theoretical R_CL for CYP1A2 (a), CYP2C8 (b), CYP2C9 (c), CYP2C19 (d), and OATP (e) pathways.
Supplementary Figure S2 Effect of CKD on CYP1A2 (a), CYP2C8 (b), CYP2C9 (c), CYP2C19 (d), and OATP (e) mediated clearance calculated using maximum AUCR (method 2).
Supplementary Figure S3 Comparison of calculated R_CL using fe,urine and normalized theoretical R_CL (method 3) for CYP1A2 (a), CYP2C8 (b), CYP2C9 (c), CYP2C19 (d), and OATP (e) pathways.
Supplementary Figure S4 Effect of CKD on CYP1A2 (a), CYP2C8 (b), CYP2C9 (c), CYP1A2 (d), and OATP (e) mediated clearance calculated using fe,urine (method 3).
ACKNOWLEDGMENTS
The authors thank Drs Darrell Abernethy, Lauren Brum, Rajnikanth Madabushi, Kellie Reynolds, Martina Sahre, Robert Schuck, Aliza Thompson, and Issam Zineh (U.S. Food and Drug Administration) for their invaluable comments.
References
- 1. Ladda, M.A. & Goralski, K.B. The effects of CKD on cytochrome P450‐mediated drug metabolism. Adv. Chronic Kidney Dis. 23, 67–75 (2016). [DOI] [PubMed] [Google Scholar]
- 2. Jha, V. et al Chronic kidney disease: global dimension and perspectives. Lancet 382, 260–272 (2013). [DOI] [PubMed] [Google Scholar]
- 3. Weiner, D.E. Public health consequences of chronic kidney disease. Clin. Pharmacol. Ther. 86, 566–569 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen, Y.C. et al Risk factors associated with adverse drug events among older adults in emergency department. Eur. J. Intern. Med. 25, 49–55 (2014). [DOI] [PubMed] [Google Scholar]
- 5. Nolin, T.D. , Naud, J. , Leblond, F.A. & Pichette, V. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin. Pharmacol. Ther. 83, 898–903 (2008). [DOI] [PubMed] [Google Scholar]
- 6. Yeung, C.K. , Shen, D.D. , Thummel, K.E. & Himmelfarb, J. Effects of chronic kidney disease and uremia on hepatic drug metabolism and transport. Kidney Int. 85, 522–528 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marbury, T.C. et al Pharmacokinetics of repaglinide in subjects with renal impairment. Clin. Pharmacol. Ther. 67, 7–15 (2000). [DOI] [PubMed] [Google Scholar]
- 8. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) . Draft Guidance / Guidance for Industry. Pharmacokinetics in Patients with Impaired Renal Function — Study Design, Data Analysis, and Impact on Dosing and Labeling. <http://www.fda.gov/downloads/Drugs/Guidances/UCM204959.pdf, http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm064982.htm> (2010). Accessed 26 March 2017.
- 9. European Medicines Agency . Draft guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function. <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/02/WC500162133.pdf> (2014). Accessed 26 March 2017.
- 10. Rowland Yeo, K. , Aarabi, M. , Jamei, M. & Rostami‐Hodjegan, A. Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev. Clin. Pharmacol. 4, 261–274 (2011). [DOI] [PubMed] [Google Scholar]
- 11. Mutsaers, H.A. et al Uremic toxins inhibit transport by breast cancer resistance protein and multidrug resistance protein 4 at clinically relevant concentrations. PLoS One 6, e18438 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nigam, S.K. , Wu, W. , Bush, K.T. , Hoenig, M.P. , Blantz, R.C. & Bhatnagar, V. Handling of drugs, metabolites, and uremic toxins by kidney proximal tubule drug transporters. Clin. J. Am. Soc. Nephrol. 10, 2039–2049 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsueh, C.H. et al Identification and quantitative assessment of uremic solutes as inhibitors of renal organic anion transporters, OAT1 and OAT3. Mol. Pharm. 13, 3130–3140 (2016). [DOI] [PubMed] [Google Scholar]
- 14. Nolin, T.D. , Appiah, K. , Kendrick, S.A. , Le, P. , McMonagle, E. & Himmelfarb, J. Hemodialysis acutely improves hepatic CYP3A4 metabolic activity. J. Am. Soc. Nephrol. 17, 2363–2367 (2006). [DOI] [PubMed] [Google Scholar]
- 15. Nolin, T.D. et al ESRD impairs nonrenal clearance of fexofenadine but not midazolam. J. Am. Soc. Nephrol. 20, 2269–2276 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barnes, K.J. , Rowland, A. , Polasek, T.M. & Miners, J.O. Inhibition of human drug‐metabolising cytochrome P450 and UDP‐glucuronosyltransferase enzyme activities in vitro by uremic toxins. Eur. J. Clin. Pharmacol. 70, 1097–1106 (2014). [DOI] [PubMed] [Google Scholar]
- 17. Leblond, F.A. , Guévin, C. , Demers, C. , Pellerin, I. , Gascon‐Barré, M. & Pichette, V. Downregulation of hepatic cytochrome P450 in chronic renal failure. J. Am. Soc. Nephrol. 12, 326–332 (2001). [DOI] [PubMed] [Google Scholar]
- 18. Leblond, F.A. , Petrucci, M. , Dubé, P. , Bernier, G. , Bonnardeaux, A. & Pichette, V. Downregulation of intestinal cytochrome p450 in chronic renal failure. J. Am. Soc. Nephrol. 13, 1579–1585 (2002). [DOI] [PubMed] [Google Scholar]
- 19. Naud, J. et al Down‐regulation of intestinal drug transporters in chronic renal failure in rats. J. Pharmacol. Exp. Ther. 320, 978–985 (2007). [DOI] [PubMed] [Google Scholar]
- 20. Naud, J. , Michaud, J. , Leblond, F.A. , Lefrancois, S. , Bonnardeaux, A. & Pichette, V. Effects of chronic renal failure on liver drug transporters. Drug Metab. Dispos. 36, 124–128 (2008). [DOI] [PubMed] [Google Scholar]
- 21. Sayama, H. , Takubo, H. , Komura, H. , Kogayu, M. & Iwaki, M. Application of a physiologically based pharmacokinetic model informed by a top‐down approach for the prediction of pharmacokinetics in chronic kidney disease patients. AAPS J. 16, 1018–1028 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yoshida, K. et al Systematic and quantitative assessment of the effect of chronic kidney disease on CYP2D6 and CYP3A4/5. Clin. Pharmacol. Ther. 100, 75–87 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thomson, B.K. et al Effect of CKD and dialysis modality on exposure to drugs cleared by nonrenal mechanisms. Am. J. Kidney Dis. 65, 574–582 (2015). [DOI] [PubMed] [Google Scholar]
- 24. Joy, M.S. et al In vivo alterations in drug metabolism and transport pathways in patients with chronic kidney diseases. Pharmacotherapy 34, 114–122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Naud, J. et al Effects of chronic renal failure on kidney drug transporters and cytochrome P450 in rats. Drug Metab. Dispos. 39, 1363–1369 (2011). [DOI] [PubMed] [Google Scholar]
- 26. Fujino, H. , Shimada, S. , Yamada, I. , Hirano, M. , Tsunenari, Y. & Kojima, J. Studies on the interaction between fibrates and statins using human hepatic microsome. Arzneimittelforschung 53, 701–707 (2003). [DOI] [PubMed] [Google Scholar]
- 27. Shitara, Y. , Hirano, M. , Sato, H. & Sugiyama, Y. Gemfibrozil and its glucuronide inhibit the organic anion transporting polypeptide 2 (OATP2/OATP1B1:SLC21A6)‐mediated hepatic uptake and CYP2C8‐mediated metabolism of cerivastatin: analysis of the mechanism of the clinically relevant drug‐drug interaction between cerivastatin and gemfibrozil. J. Pharmacol. Exp. Ther. 311, 228–236 (2004). [DOI] [PubMed] [Google Scholar]
- 28. Polepally, A.R. et al Drug‐drug interactions between the anti‐hepatitis C virus 3D regimen of ombitasvir, paritaprevir/ritonavir, and dasabuvir and eight commonly used medications in healthy volunteers. Clin. Pharmacokinet. 55, 1003–1014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gertz, M. , Tsamandouras, N. , Säll, C. , Houston, J.B. & Galetin, A. Reduced physiologically‐based pharmacokinetic model of repaglinide: impact of OATP1B1 and CYP2C8 genotype and source of in vitro data on the prediction of drug‐drug interaction risk. Pharm. Res. 31, 2367–2382 (2014). [DOI] [PubMed] [Google Scholar]
- 30. Prueksaritanont, T. et al Pitavastatin is a more sensitive and selective organic anion‐transporting polypeptide 1B clinical probe than rosuvastatin. Br. J. Clin. Pharmacol. 78, 587–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) . Drugs@FDA: FDA Approved Drug Product. <http://www.accessdata.fda.gov/scripts/cder/daf/>.
- 32. Vanholder, R. , Van Landschoot, N. , De Smet, R. , Schoots, A. & Ringoir, S. Drug protein binding in chronic renal failure: evaluation of nine drugs. Kidney Int. 33, 996–1004 (1988). [DOI] [PubMed] [Google Scholar]
- 33. Yamasaki, K. , Chuang, V.T. , Maruyama, T. & Otagiri, M. Albumin‐drug interaction and its clinical implication. Biochim. Biophys. Acta 1830, 5435–5443 (2013). [DOI] [PubMed] [Google Scholar]
- 34. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) . Transcript from the March 17, 2010, Meeting of the Pharmaceutical Science and Clinical Pharmacology Advisory Committee. <http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM210803.pdf> (2010). Accessed 2 April 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1 List of drugs with CKD studies not included as model substrate drugs. These drugs had DDI studies with the typical inhibitors used for the CYP enzymes in this study or PGx studies.
Supplementary Table S2 Mean ± SD, if available, plasma unbound fraction information.
Supplementary Table S3 References for Table 1 in the main text.
Supplementary Table S4 References for Table 3 in the main text.
Supplementary Figure S1 Comparison of calculated R_CL using maximum AUCR (method 2) and normalized theoretical R_CL for CYP1A2 (a), CYP2C8 (b), CYP2C9 (c), CYP2C19 (d), and OATP (e) pathways.
Supplementary Figure S2 Effect of CKD on CYP1A2 (a), CYP2C8 (b), CYP2C9 (c), CYP2C19 (d), and OATP (e) mediated clearance calculated using maximum AUCR (method 2).
Supplementary Figure S3 Comparison of calculated R_CL using fe,urine and normalized theoretical R_CL (method 3) for CYP1A2 (a), CYP2C8 (b), CYP2C9 (c), CYP2C19 (d), and OATP (e) pathways.
Supplementary Figure S4 Effect of CKD on CYP1A2 (a), CYP2C8 (b), CYP2C9 (c), CYP1A2 (d), and OATP (e) mediated clearance calculated using fe,urine (method 3).