

Abstract
Objective
Mavrilimumab, a human monoclonal antibody, targets granulocyte–macrophage colony‐stimulating factor receptor α. We undertook to determine the long‐term safety and efficacy of mavrilimumab in rheumatoid arthritis patients in 2 phase IIb studies (1071 and 1107) and in 1 open‐label extension study (ClinicalTrials.gov identifier: NCT01712399).
Methods
In study 1071, patients with an inadequate response to disease‐modifying antirheumatic drugs (DMARDs) received mavrilimumab (30, 100, or 150 mg) or placebo every other week plus methotrexate. In study 1107, patients with an inadequate response to anti–tumor necrosis factor agents and/or DMARDs received 100 mg mavrilimumab every other week or 50 mg golimumab every 4 weeks plus methotrexate. Patients entering the open‐label extension study received 100 mg mavrilimumab every other week plus methotrexate. Long‐term safety and efficacy of mavrilimumab were assessed.
Results
A total of 442 patients received mavrilimumab (14 of 245 patients from study 1071, 9 of 70 patients from study 1107, and 52 of 397 patients from the open‐label extension study discontinued mavrilimumab treatment throughout the studies). The cumulative safety exposure was 899 patient‐years; the median duration of mavrilimumab treatment was 2.5 years (range 0.1–3.3 years). The most common treatment‐emergent adverse events (AEs) were nasopharyngitis (n = 69; 7.68 per 100 patient‐years) and bronchitis (n = 51; 5.68 per 100 patient‐years). At weeks 74 and 104, 3.5% and 6.2% of patients, respectively, demonstrated reduction in forced expiratory volume in 1 second, while 2.9% and 3.4% of patients, respectively, demonstrated reduction in forced vital capacity (>20% reduction from baseline to <80% predicted). Most pulmonary changes were transient and only infrequently associated with AEs. Mavrilimumab at 100 mg every other week demonstrated sustained efficacy; at week 122, 65.0% of patients achieved a Disease Activity Score in 28 joints using the C‐reactive protein level (DAS28‐CRP) of <3.2, and 40.6% of patients achieved a DAS28‐CRP of <2.6.
Conclusion
Long‐term treatment with mavrilimumab maintained response and was well‐tolerated with no increased incidence of treatment‐emergent AEs. Safety data were comparable with those from both phase IIb qualifying studies.
Rheumatoid arthritis (RA) is associated with a high prevalence of comorbidities 1, including increased risk of serious infections 2 and malignancies 1. This is because of the immune system dysregulation intrinsic to the disease and prolonged use of conventional immunosuppressive therapies and targeted biologic agents 2, 3, 4.
Granulocyte–macrophage colony‐stimulating factor (GM‐CSF) is a proinflammatory cytokine involved in RA pathogenesis 5, 6. GM‐CSF binds to GM‐CSF receptor α (GM‐CSFRα), thereby reducing macrophage and neutrophil numbers and function in rheumatoid inflammatory lesions 7, 8. GM‐CSF has a well‐established role in hematopoiesis in vitro and in vivo, supporting neutrophilic granulocyte proliferation and differentiation 9, 10; however, there is little deficiency in resting hematopoiesis in gene‐targeted mice lacking GM‐CSF or its receptor 11. This suggests that GM‐CSF is not required in vivo for resting hematopoiesis. Patients with RA may still have anemia 12 and occasionally neutropenia 13. In these cases, or in the presence of infection, it is possible that GM‐CSF is necessary for adequate granulocyte production, and GM‐CSF inhibition may induce neutropenia 14.
GM‐CSF is involved in lung homeostasis and promotes alveolar macrophage proliferation 15, 16, which has a critical role in lung defense and surfactant homeostasis 17 through catabolism of surfactant lipids and proteins 18. Pulmonary alveolar proteinosis is a rare disease characterized by alveolar surfactant accumulation 19. Polyclonal anti–GM‐CSF autoantibodies and mutations in surfactant proteins or GM‐CSFR are considered responsible for most cases of pulmonary alveolar proteinosis.
Mavrilimumab is a human monoclonal antibody that blocks GM‐CSFRα, acting as a competitive antagonist of GM‐CSF signaling 20. It has previously demonstrated efficacy and an acceptable safety profile in RA patients in a 12‐week phase IIa study 21 and in 24‐week phase IIb studies 22, 23 at dosages up to 150 mg every other week.
To evaluate the sustained safety and efficacy of mavrilimumab (including the longitudinal assessment of pulmonary function), we report the long‐term (up to >3 years) safety profile in RA patients who participated in 2 phase IIb studies: study 1071 22 and study 1107 23. Irrespective of initial treatment allocation, patients were subsequently enrolled in an open‐label extension study (study 1109 [ClinicalTrials.gov identifier: NCT01712399]) and received mavrilimumab at 100 mg every other week.
Patients and methods
Study design. This report includes data from a phase II open‐label extension study (study 1109 [ClinicalTrials.gov identifier: NCT01712399]) and 2 qualifying phase IIb studies. Studies 1071 (EARTH EXPLORER 1 [ClinicalTrials.gov identifier: NCT01706926]) 22 and 1107 (EARTH EXPLORER 2 [ClinicalTrials.gov identifier: NCT01712399]) 23 were randomized, double‐blind, multicenter studies. In study 1071, 326 patients with moderate‐to‐severe RA who had inadequate responses to disease‐modifying antirheumatic drugs (termed DMARD inadequate responders) received subcutaneous (SC) mavrilimumab (30, 100, or 150 mg) or placebo every other week plus standard therapy (methotrexate at 7.5–25.0 mg/week and folic acid at ≥5 mg/week). In study 1107, 138 patients who had a previous inadequate response to DMARDs and/or DMARD inadequate responders for whom 1 or 2 anti–tumor necrosis factor (anti‐TNF) agents (excluding golimumab) had failed received SC mavrilimumab at 100 mg every other week or golimumab at 50 mg every 4 weeks plus standard therapy (Figure 1). In study 1107, placebo was administered every other week to patients receiving golimumab to maintain masking.
Figure 1.
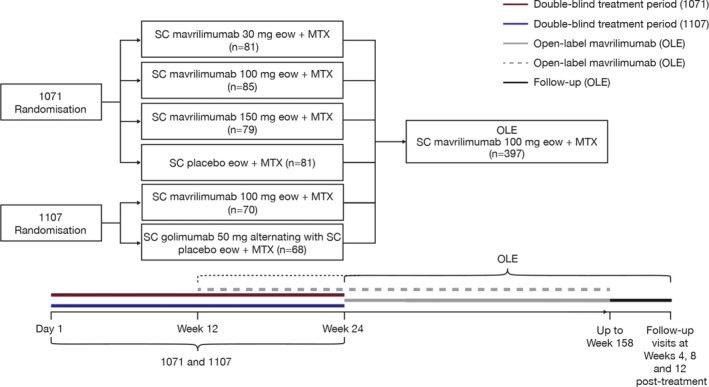
Designs of the phase IIb studies (1071 and 1107) and the open‐label extension study (OLE). After week 24, patients enrolled in study 1071 or study 1107 were eligible for the open‐label extension study and received mavrilimumab for up to a maximum of 3.3 years. After week 12, patients enrolled in study 1071 or study 1107 who were not responding adequately to blinded study treatment were allowed to enter the open‐label extension study to receive mavrilimumab. Studies 1071 and 1107 were not conducted simultaneously. SC = subcutaneous; eow = every other week; MTX = methotrexate.
Patients who completed studies 1071 or 1107 or who had inadequate clinical responses after week 12 were eligible to enter the open‐label extension study. All patients enrolled in the open‐label extension study received SC mavrilimumab at 100 mg every other week plus standard therapy (Figure 1). Patients received mavrilimumab at 100 mg as this was the highest, most efficacious dose in a previous phase IIa study 21. At initiation of the open‐label extension study, no data from study 1071 were available demonstrating improved efficacy and an acceptable safety profile with mavrilimumab at 150 mg versus 100 mg.
All 3 studies were conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation Guidelines for Good Clinical Practice, and all were approved by the appropriate institutional review board or independent ethics committee at each study site. All patients provided written informed consent.
Inclusion and exclusion criteria for patients. Patients were ages 18–80 years with moderate‐to‐severe adult‐onset RA according to the American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) 2010 classification criteria 24, a Disease Activity Score in 28 joints 25 using the C‐reactive protein level (DAS28‐CRP) of ≥3.2 at screening, a DAS28 using the erythrocyte sedimentation rate of ≥3.2 26 on day 1, and ≥4 swollen joints at screening and on day 1. Detailed inclusion and exclusion criteria for studies 1071 and 1107 are included in Supplementary Methods, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40420/abstract.
Patients who completed the treatment period of the qualifying study (1071 or 1107), or who did not have an adequate response to the blinded investigational product at a predefined time point in the qualifying study despite their initial randomization, were eligible to enroll in the open‐label extension study. However, ≤12 weeks had to elapse since their completion/withdrawal visit in the qualifying study. Other key inclusion criteria for the open‐label extension study were receipt of a stable dose of conventional DMARDs (chloroquine, hydroxychloroquine, parenteral gold, sulfasalazine, or leflunomide) ≥4 weeks prior to the first mavrilimumab dose, if patients were receiving treatment with these drugs, and no evidence of clinically uncontrolled respiratory disease (confirmed by a pulmonologist who reviewed patient data from respiratory assessments, including a chest radiograph and pulmonary function tests performed at screening; the patient must have had forced expiratory volume in 1 second/forced vital capacity [FEV1/FVC] ≥60% of predicted values). Key exclusion criteria for the open‐label extension study included permanent discontinuation of investigational product in either qualifying study, receipt of B cell–depleting therapies during or after discontinuation from the qualifying studies, and discontinuation of non–cell depleting biologic DMARDs within 8 weeks of day 1 of the open‐label extension study. Stable dosages of oral corticosteroids, analgesics, and nonsteroidal antiinflammatory drugs were permitted.
Analysis sets. The overall phase as‐treated population included all mavrilimumab‐treated patients in studies 1071 or 1107 or in the open‐label extension study. The open‐label as‐treated population included all 100 mg mavrilimumab–treated patients in the open‐label extension study.
Safety analysis. Long‐term safety of mavrilimumab was assessed by evaluating treatment‐emergent adverse events (AEs), treatment‐emergent serious AEs (SAEs), treatment‐emergent AEs of special interest (events of scientific and medical interest specific to understanding of the investigational product and requiring rapid communication with the sponsor [hepatic function abnormality, acute and delayed hypersensitivity reactions, clinically relevant pulmonary abnormality, neutropenia {absolute neutrophil count <1.0 × 109/liter}, malignancy, and infection]), and laboratory measurements in the overall as‐treated population. The severity of all events was determined by the investigator.
Lung function abnormalities and pulmonary AEs were assessed in the overall as‐treated population. Pulmonary monitoring was conducted throughout all studies, including serial standardized lung function testing (FEV1/FVC), assessments of dyspnea (using the Borg Dyspnea Index 27), and chest radiographs. While assessment of diffusing capacity for carbon monoxide (Dlco) was performed in most patients included in the open‐label extension study, this test was not available at the start of the 2 qualifying randomized studies due to initial standardization difficulties. Therefore, baseline Dlco % predicted data were only collected in 48 patients from study 1107 and in no patients from study 1071, which limited full interpretation of the results. A blinded Independent Pulmonary Evaluation Committee (IPEC) adjudicated any patient with a pulmonary abnormality (pulmonary AE/SAE and/or abnormalities on pulmonary function tests and Dlco considered clinically relevant) for the presence of pulmonary alveolar proteinosis.
Efficacy analysis. The long‐term clinical efficacy of mavrilimumab was evaluated as an exploratory objective. Efficacy end points included proportions of patients achieving a DAS28‐CRP of <3.2 and <2.6 (overall as‐treated population), proportions of patients meeting the ACR 20% improvement criteria (achieving an ACR20 response) 28 or achieving an ACR50 or ACR70 response at weeks 74, 98, and 122 (as‐treated population in the open‐label extension study), Clinical Disease Activity Index (CDAI) 29 remission rates at weeks 74, 98, and 122 (as‐treated population in the open‐label extension study), radiographic progression assessed by change from baseline in the modified Sharp/van der Heijde score (SHS) 30 through week 74 (as‐treated population, only including patients from study 1071), and patient‐reported outcomes (overall as‐treated population), including the Health Assessment Questionnaire disability index 31, Short Form 36 health survey 32, and Functional Assessment of Chronic Illness Therapy–Fatigue subscale 33.
Biomarker analysis. For a complete understanding of mavrilimumab's molecular mechanism of action, biomarkers related to the GM‐CSF pathway were analyzed in all patients participating in study 1107 who enrolled in the open‐label extension study. GM‐CSF is believed to drive production of thymus and activation–regulated chemokine (TARC)/CCL17 34 and macrophage‐derived chemokine (MDC)/CCL22 35, and serum concentrations of CCL17 and CCL22 were suppressed by mavrilimumab but not by golimumab in the qualifying 1107 study 36. Serum CCL17/TARC and CCL22/MDC concentrations were measured by enzyme‐linked immunosorbent assay on days 1, 8, 29, 85, 169, 225, 253, 337, and 393 and are reported herein.
Statistical analysis. Safety and patient‐reported outcome data were summarized descriptively. Exposure‐corrected incidence rates for AEs and AEs of special interest were used, expressed as number of patients experiencing an event per 100 patient‐years.
Proportions of patients achieving a DAS28‐CRP of <3.2 and <2.6 and ACR20/ACR50/ACR70 response rates were calculated using the number of patients with an assessment at each visit as the denominator and the number of responders as the numerator. The CDAI was defined as the sum of the tender joint count (using 28 joints), the swollen joint count (using 28 joints), the patient's global assessment of disease activity (on a 0–10‐cm visual analog scale [VAS]), and the physician's global assessment of disease activity (on a 0–10‐cm VAS). CDAI remission was defined as a score of ≤2.8. Change in SHS is presented using a cumulative incidence plot at week 74.
Results
Patients. Randomization for study 1071 began September 11, 2012; the last patient was evaluated January 29, 2014. In study 1107, patients were randomized from March 19, 2013; the last patient was evaluated February 6, 2015. The first patient entered the open‐label extension study February 11, 2013; the last patient visit was completed December 30, 2015.
Across the 3 studies, 442 patients received mavrilimumab (overall as‐treated population). Of these, 409 (92.5%) consented to participate in the open‐label extension study; 12 were ineligible (primarily due to inclusion/exclusion criteria) and did not receive treatment. Therefore, the open‐label as‐treated population included 397 patients, of whom 345 (86.9%) were being treated at study closure. Throughout the studies, 14 of 245 patients from study 1071, 9 of 70 patients from study 1107, and 52 of 397 patients from the open‐label extension study discontinued mavrilimumab treatment (see Supplementary Figure 1, http://onlinelibrary.wiley.com/doi/10.1002/art.40420/abstract). A drug survival curve is shown in Supplementary Figure 2, http://onlinelibrary.wiley.com/doi/10.1002/art.40420/abstract. Baseline demographic and disease characteristics of the patients are summarized in Table 1.
Table 1.
Baseline demographic and disease characteristics of the patients (as‐treated population)a
| Open‐label extension study (n = 397)b | |
| Demographic characteristicsc | |
| Age, mean ± SD years | 51.1 ± 11.2 |
| Female, no. (%) | 339 (85.4) |
| Race, no. (%) | |
| White | 364 (91.7) |
| American Indian/Alaska Native | 29 (7.3) |
| Otherd | 4 (1.0) |
| Disease characteristicsc | |
| RA duration, mean ± SD years | 7.9 ± 6.8 |
| Prior biologic DMARD therapy, no. (%)e | 100 (25.1) |
| Stopped because of loss of efficacy | 47 (11.8) |
| Stopped because of safety/otherf | 53 (13.4) |
| MTX dosage, mean ± SD mg/week | 14.8 ± 3.8 |
| Corticosteroid dosage, mean ± SD mg/weekg | 5.4 ± 1.5 |
| DAS28‐CRP, mean ± SD | 4.3 ± 1.6 |
| HAQ DI score, mean ± SD | 1.2 ± 0.6 |
| Swollen joint count, mean ± SD | 7.2 ± 7.9 |
| Tender joint count, mean ± SD | 14.0 ± 13.8 |
| CRP, geometric mean mg/liter (CV %) | 4.7 (170) |
| ESR, geometric mean mm/hour (CV %) | 27.9 (62) |
| Studies 1071 and 1107 (n = 442) | |
| Disease characteristics | |
| DAS28‐CRP, mean ± SD | 5.8 ± 0.8 |
| HAQ DI score, mean ± SD | 1.6 ± 0.5 |
| Swollen joint count, mean ± SD | 15.7 ± 8.4 |
| Tender joint count, mean ± SD | 26.3 ± 12.6 |
| CRP, geometric mean mg/liter (CV %) | 6.0 (181) |
| ESR, geometric mean mm/hour (CV %) | 36.8 (52) |
| Pulmonary disease characteristics | |
| Concomitant pulmonary diseases, no. (%)h | |
| Asthma | 17 (3.8) |
| COPD | 8 (1.8) |
| Other | 21 (4.8) |
| Ever smoked, no. (%) | 134 (30.3) |
| Current smokers, no. (%) | 76 (17.2) |
| RF and ACPA positive, no. (%) | 359 (81.2) |
| FEV1, mean ± SD % predicted | 93.9 ± 14.7 |
| FVC, mean ± SD % predicted | 94.0 ± 14.6 |
| Dlco, mean ± SD % predicted (n)i | 72.4 ± 9.3 (48) |
| Borg Dyspnea Index score, mean ± SEM | 0.4 ± 0.0 |
RA = rheumatoid arthritis; MTX = methotrexate; DAS28‐CRP = Disease Activity Score in 28 joints using the C‐reactive protein level; HAQ DI = Health Assessment Questionnaire disability index; CV = coefficient of variation; ESR = erythrocyte sedimentation rate; COPD = chronic obstructive pulmonary disease; RF = rheumatoid factor; ACPA = anti–citrullinated protein antibody; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity.
An 8‐week washout period occurred prior to the open‐label extension study. Patients who did not have a response to mavrilimumab were also included in the open‐label extension study.
Baseline values were obtained from predosing in studies 1071 and 1107.
Black, Asian, African American, Native Hawaiian, or other Pacific Islander.
Patients were counted only once for each disease‐modifying antirheumatic drug (DMARD).
Other reasons include discontinuation because of lack of initial efficacy, expense of medication, or because the medication was given only in a clinical trial.
Derived from 234 of 397 patients (59%) who received concomitant corticosteroids.
Clinically relevant uncontrolled pulmonary disease was an exclusion criterion for all 3 studies.
Diffusing capacity for carbon monoxide (Dlco) data were not collected during study 1071 and were not collected in all patients in study 1107; baseline values were available for 48 of 397 patients in the open‐label extension study.
Safety analysis findings. Across the 3 studies, patients had a cumulative mavrilimumab safety exposure of 899 patient‐years and a median treatment duration of 2.5 years (range 0.1–3.3 years). The majority of AEs were mild or moderate, with 44 patients (10.0%) in the overall as‐treated population reporting a treatment‐emergent AE of grade ≥3 severity. The most frequently reported treatment‐emergent AEs and treatment‐emergent SAEs in the overall phase are shown in Table 2. There were no reports of monocytopenia. Neutropenia was reported in 4 patients (2 with grade 1, 1 with grade 2, 1 with grade 3; 0.45 per 100 patient‐years), and 14 patients reported serious infections (1.56 per 100 patient‐years). One event of neutropenia (not recorded as an AE of special interest) was considered serious and was associated with an SAE of urinary tract infection that resolved with standard therapy. Pulmonary events of special interest occurred in 83 patients (a full breakdown is shown in Supplementary Table 1, http://onlinelibrary.wiley.com/doi/10.1002/art.40420/abstract). The most frequent was bronchitis; 2 cases of active pulmonary tuberculosis infection were reported (0.22 per 100 patient‐years), and 1 was bacteriologically confirmed. Neither patient had active or latent tuberculosis at screening or had previously received anti‐TNF agents for RA treatment. There were no cases of pulmonary alveolar proteinosis or findings suggestive of pulmonary alveolar proteinosis (confirmed by the IPEC).
Table 2.
Summary of treatment‐emergent AEs, treatment‐emergent SAEs, and treatment‐emergent AEs of special interest (as‐treated population for the overall phase) in patients receiving 100 mg mavrilimumab every other week (n = 442), with a cumulative mavrilimumab safety exposure of 899 patient‐yearsa
| Summary of treatment‐emergent AEsb | |
| Patients reporting ≥1 treatment‐emergent AE, no. (%) | 335 (75.8) |
| Treatment‐emergent AEs in ≥15 patients, no. (rate per 100 patient‐years) | |
| Nasopharyngitis | 69 (7.68) |
| Bronchitis | 51 (5.68) |
| Hypertension | 38 (4.23) |
| Rheumatoid arthritis | 44 (4.90) |
| Upper respiratory tract infection | 38 (4.23) |
| Headache | 31 (3.45) |
| Urinary tract infection | 40 (4.45) |
| Influenza | 29 (3.23) |
| Pharyngitis | 20 (2.23) |
| Osteoarthritis | 17 (1.89) |
| Diarrhea | 21 (2.34) |
| Back pain | 15 (1.67) |
| Gastroenteritis | 15 (1.67) |
| Summary of treatment‐emergent SAEsb | |
| Patients reporting ≥1 treatment‐emergent SAE, no. (%) | 60 (13.6) |
| Treatment‐emergent SAEs in ≥2 patients, no. (rate per 100 patient‐years) | |
| Osteoarthritis | 4 (0.45) |
| Bronchitis | 4 (0.45) |
| Rheumatoid arthritis | 4 (0.45) |
| Anemia | 3 (0.33) |
| Pulmonary tuberculosis | 2 (0.22) |
| Gastroenteritis | 2 (0.22) |
| Pneumonia | 2 (0.22) |
| Urinary tract infection | 2 (0.22) |
| Cardiopulmonary failure | 2 (0.22) |
| Myocardial infarction | 2 (0.22) |
| Cholelithiasis | 2 (0.22) |
| Uterine leiomyoma | 2 (0.22) |
| Deaths, no. (%) | 3 (0.7) |
| Summary of treatment‐emergent AEs of special interestb | |
| Patients reporting ≥1 treatment‐emergent AE of special interest, no. (%) | 114 (25.8) |
| Treatment‐emergent AEs of special interest, no. (rate per 100 patient‐years) | |
| Hepatic function abnormalities | 2 (0.22) |
| Hypersensitivity reactions | 13 (1.45) |
| Serious infections | 14 (1.56) |
| Malignancies | 5 (0.56) |
| Neutropenia | 4 (0.45) |
| Pulmonary eventsc | 83 (9.24) |
Includes all patients exposed to mavrilimumab in studies 1071 and 1107 or in the open‐label extension study. AEs = adverse events; SAEs = serious AEs.
Includes AEs with onset after the first mavrilimumab dosage.
All were reviewed by an Independent Pulmonary Evaluation Committee.
Treatment‐emergent AEs leading to discontinuation were reported in 12 patients (3.0%) in the open‐label as‐treated population (see Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.40420/abstract). Three deaths were reported during the open‐label extension study, 1 due to a car accident and 2 due to cardiopulmonary failure. One of the 2 patients with cardiopulmonary failure was hospitalized for a few days before death (apparently to treat sepsis subsequent to a biliary tract infection); the other patient died suddenly. None of these deaths was considered related to mavrilimumab treatment. There were no clinically relevant shifts in laboratory values for the overall phase.
Pulmonary function and long‐term pulmonary safety analysis findings. The mean Borg Dyspnea Index score in the overall as‐treated population remained unchanged at week 134 compared with week 74 (Table 3). Similar dyspnea scores from baseline through week 134 were observed in the populations of patients who switched to 100 mg mavrilimumab from 30 mg mavrilimumab, 150 mg mavrilimumab, 50 mg golimumab, or placebo.
Table 3.
Borg Dyspnea Index score and reductions from baseline in pulmonary function test results (as‐treated population for the overall phase) in patients receiving 100 mg mavrilimumab every other week (n = 442)a
| Borg Dyspnea Index score (measure of breathlessness), mean ± SEM (n) | |
| Week 12b | NA |
| Week 74 | 0.3 ± 0.0 (279) |
| Week 134 | 0.3 ± 0.0 (58) |
| Pulmonary function test resultsc | |
| >20% reduction from baseline to <80% predicted FEV1, no./total no. (%) | |
| Week 12b | 2/298 (0.7) |
| Week 74 | 8/231 (3.5) |
| Week 104 | 11/178 (6.2) |
| Week 130 | 1/29 (3.4) |
| >20% reduction from baseline to <80% predicted FVC, no./total no. (%) | |
| Week 12b | 2/298 (0.7) |
| Week 74 | 7/239 (2.9) |
| Week 104 | 6/177 (3.4) |
| Week 130 | 0/32 (0.0) |
Includes all patients who received mavrilimumab in studies 1071 and 1107 or in the open‐label extension study. NA = not available; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity.
Between weeks 12 and 24, 3 patients receiving 150 mg mavrilimumab (3.8%), 8 patients receiving 100 mg mavrilimumab (9.4%), 12 patients receiving 30 mg mavrilimumab (14.8%), and 37 patients receiving placebo (45.7%) transferred from study 1071 to the open‐label extension study because of lack of efficacy. Between weeks 12 and 24, 2 patients receiving 100 mg mavrilimumab (2.9%) and no patients receiving 50 mg golimumab (0.0%) transferred from study 1107 to the open‐label extension study because of lack of efficacy.
Reported events do not necessarily overlap between time points.
A small proportion of patients experienced an event defined as clinically relevant (>20% decrease from baseline and <80% predicted) reduction in lung function (FEV1 and FVC). At weeks 74 and 104, 3.5% and 6.2% of patients, respectively, demonstrated reduction in FEV1, while 2.9% and 3.4% of patients, respectively, demonstrated reduction in FVC (>20% reduction from baseline to <80% predicted) (Table 3). In most cases, these events occurred at a single time point and were not associated with a respiratory AE. Mean FEV1, FVC, and Dlco values remained within 5% of the mean baseline value for patients treated during the randomized phases of the phase IIb studies (Figure 2); however, baseline Dlco values were measured in a minority of patients (48 patients and 0 patients in studies 1107 and 1071, respectively).
Figure 2.
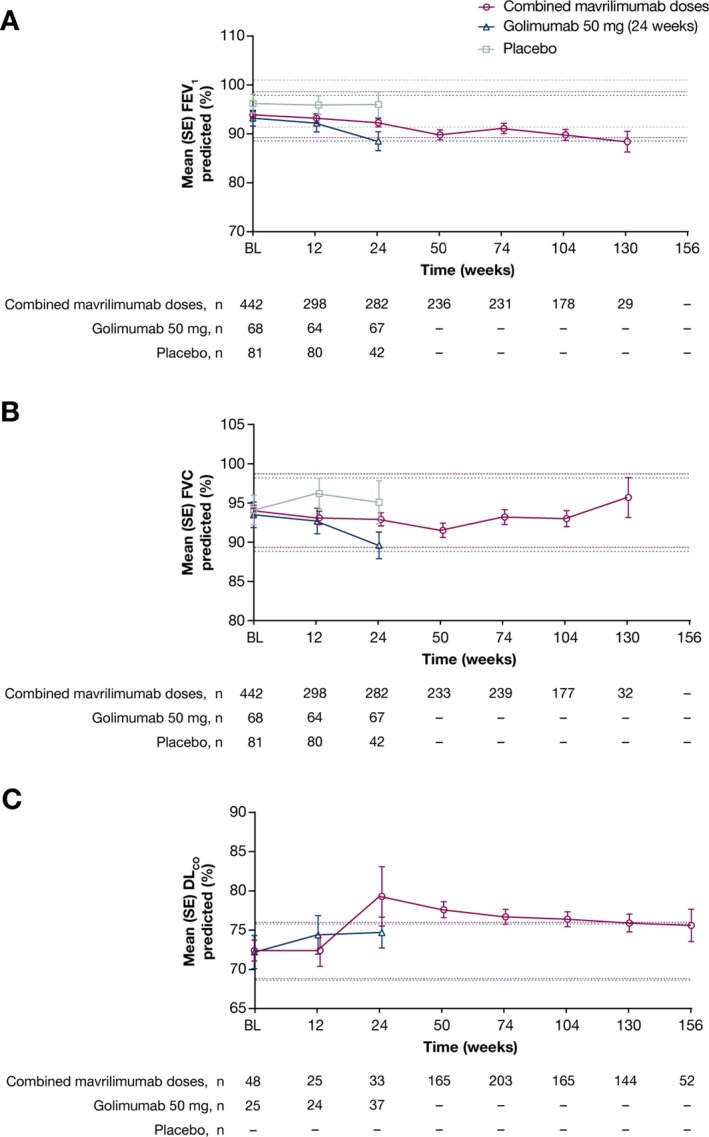
Pulmonary function values over time (as‐treated population for the overall phase). A, Forced expiratory volume in 1 second (FEV 1). B, Forced vital capacity (FVC). C, Diffusing capacity for carbon monoxide (DL co). Dotted lines represent 5% above and below baseline values for FEV1, FVC, and DLco. Between weeks 12 and 24, 3 patients receiving 150 mg mavrilimumab (3.8%), 8 patients receiving 100 mg mavrilimumab (9.4%), 12 patients receiving 30 mg mavrilimumab (14.8%), and 37 patients receiving placebo (45.7%) transferred from study 1071 to the open‐label extension study (study 1109) because of lack of efficacy (<20% improvement in both the swollen and tender joint counts compared with day 1). Between weeks 12 and 24, 2 patients receiving 100 mg mavrilimumab (2.9%) and no patients receiving 50 mg golimumab (0.0%) transferred from study 1107 to study 1109 because of lack of efficacy. Following the decision of the sponsor to discontinue study 1109, patients’ exposure to a study drug/placebo ranged from 2 to 156 weeks, depending on their date of entry and reason for withdrawal from the studies. For final time points in which n ≤ 5, results were not shown because the number of patients was too low to enable meaningful interpretation. DL co data were not collected during study 1071, and during study 1107 baseline (BL) values were collected in only 48 patients.
Efficacy analysis findings. In exploratory analyses, patients demonstrated sustained efficacy with mavrilimumab treatment. At week 122, a total of 117 patients (65.0%) achieved low disease activity with a DAS28‐CRP of <3.2, and 73 patients (40.6%) achieved a DAS28‐CRP of <2.6 (overall as‐treated population). DAS28‐CRP <3.2 and <2.6 response rates are shown in Figures 3A and B. Patients treated with 100 mg mavrilimumab also demonstrated sustained ACR20/ACR50/ACR70 responses (Figure 3C). There was an increase in the number of patients with CDAI remission over time in the open‐label extension study (see Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.40420/abstract). After 74 weeks, 54% of patients demonstrated no radiographic progression (≤0.5‐point change in SHS compared with baseline values) (see Supplementary Figure 3, http://onlinelibrary.wiley.com/doi/10.1002/art.40420/abstract). The mean change in SHS from baseline to week 74 was 1.40. Improvements from baseline in patient‐reported outcome end points were also observed and maintained with mavrilimumab treatment (see Supplementary Table 4, http://onlinelibrary.wiley.com/doi/10.1002/art.40420/abstract).
Figure 3.

A and B, Proportions of patients with a Disease Activity Score in 28 joints using the C‐reactive protein level (DAS28‐CRP) of <3.2 (A) and proportions of patients with a DAS28‐CRP of <2.6 (B) (data split by original randomized treatment; as‐treated population for the overall phase). C, Proportions of patients meeting the American College of Rheumatology 20% improvement criteria (achieving an ACR20 response) or achieving an ACR50 or ACR70 response at weeks 74, 98, and 122 (as‐treated population for the open‐label extension [OLE] study). All visits with at least 5 patients are shown. From week 24, initial treatment was switched to 100 mg mavrilimumab every other week (eow). Q4W = every 4 weeks.
Biomarker analysis findings. In study 1107, serum TARC/CCL17 and MDC/CCL22 concentrations were suppressed in patients treated with 100 mg mavrilimumab, but not in patients treated with 50 mg golimumab (see Supplementary Figure 4, http://onlinelibrary.wiley.com/doi/10.1002/art.40420/abstract). These results were observed in patients with an inadequate response to DMARDs (≥1 failed regimen) and in those with an inadequate response to anti‐TNF (1–2 failed regimens, excluding golimumab). After an 8‐week washout period prior to the open‐label extension study, TARC/CCL17 and MDC/CCL22 concentrations returned to baseline. However, in the open‐label extension study, mavrilimumab treatment suppressed TARC/CCL17 and MDC/CCL22 concentrations regardless of treatment received in study 1107.
Discussion
We evaluated the safety of long‐term mavrilimumab treatment in patients with RA and demonstrated that it is well tolerated, with most treatment‐emergent AEs mild or moderate in severity. Low incidences of neutropenia (0.90%; 0.45 per 100 patient‐years) and serious infections (3.17%; 1.56 per 100 patient‐years) were observed in the overall as‐treated population; there were no reports of monocytopenia. Both reported deaths due to cardiopulmonary failure were considered unrelated to mavrilimumab treatment. Previous studies of mavrilimumab evaluated treatment for ≤24 weeks; herein, we have shown that long‐term mavrilimumab treatment generated safety data comparable with those from the phase IIb qualifying studies 22, 23.
RA affects pulmonary function, and lung disease is a major contributor to patient morbidity and mortality 37, 38. Disease may affect all lung areas, and the majority of patients demonstrate low total lung capacity and FVC 39. Pulmonary comorbidities may arise because of chronic immune activation, increased susceptibility to infections (often related to immunomodulatory medications), or direct toxicity from DMARDs or biologic therapies 40; however, there are few long‐term data concerning changes in pulmonary function with progressive RA. Minor alterations in pulmonary function have been observed in patients receiving long‐term low‐dose methotrexate 41; however, another study demonstrated that mean changes in FEV1, FVC, and Dlco after 2 years of treatment with low‐dose oral methotrexate were comparable with those observed with placebo 42.
We showed that mean FEV1, FVC, and Dlco values remained within 5% of mean baseline values for the overall phase; therefore, pulmonary deterioration was not evident with long‐term mavrilimumab treatment in addition to standard therapy. Full Dlco data that included baseline values were only available for a limited proportion of patients. While patients with uncontrolled pulmonary disease were excluded from all studies analyzed, those with less severe disease, such as asthma and chronic obstructive pulmonary disease, were permitted. Hence, although these studies may not completely represent the RA patient population, these data are representative of the majority of patients with RA.
Mavrilimumab competitively antagonizes GM‐CSF signaling through GM‐CSFRα blockade. Abrogation of GM‐CSF signaling in alveolar macrophages may cause surfactant metabolism abnormalities 43, accumulation of which could cause pulmonary alveolar proteinosis, a rare disease with potentially life‐threatening consequences 44. Our adjudicated individual data demonstrate that long‐term treatment with 100 mg mavrilimumab every other week with concomitant methotrexate was not associated with pulmonary alveolar proteinosis. In the qualifying studies, no clinically relevant differences in pulmonary function were observed between different mavrilimumab dosages and placebo up to week 24 in study 1071, and between mavrilimumab and golimumab up to week 24 in study 1107. Long‐term controlled comparisons of the treatment regimens cannot be made beyond this time point since all patients included in the open‐label extension study were treated with 100 mg mavrilimumab every other week; however, it should be noted that a clinically relevant and generally transient reduction in FEV1 or FVC was only experienced by a small number of patients beyond week 24. Furthermore, discontinuation due to pulmonary events in the open‐label phase was low.
We observed clinically meaningful long‐term efficacy in patients receiving mavrilimumab for up to >3 years across many disease activity parameters. Biomarker analyses demonstrated sustained suppression of 2 GM‐CSF pathway–related protein markers following mavrilimumab treatment, regardless of previous treatment received. These results support data from previous studies 22, 23, confirming that TARC/CCL17 and MDC/CCL22 specifically relate to the GM‐CSF pathway and indicating a potential benefit of inhibiting this pathway in RA treatment.
This analysis had a number of limitations. The extension phase was not randomized or controlled; therefore, we were unable to evaluate the long‐term safety and efficacy of mavrilimumab in comparison with a reference population of patients with RA. Furthermore, the population was reduced because of discontinuations from the 2 qualifying studies and ineligibility of patients who did not meet inclusion criteria. However, the analysis conformed to the EULAR recommendations for reporting rheumatology clinical trial extension studies 45. The open‐label extension study dosage of 100 mg mavrilimumab every other week was selected to be consistent with the highest and most efficacious dosage in a previous phase IIa study 21. However, data analysis from study 1071 (conducted while the open‐label extension study was ongoing) demonstrated that 150 mg mavrilimumab was more effective in DMARD inadequate responders 22, which suggests that a suboptimal dosage was used in the open‐label extension study. Consequently, the study was discontinued.
To our knowledge, this study is unique, as multiple pulmonary function tests were performed in a systematic, long‐term, longitudinal manner. No new safety signals or evidence of clinically meaningful lung function deterioration were seen with long‐term mavrilimumab treatment. Long‐term mavrilimumab treatment was also associated with clear and sustained benefits in measures of RA disease outcomes. In light of the results from study 1071 22, we advocate phase III studies with mavrilimumab (at 150 mg every other week) in RA.
Author contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Burmester had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Burmester, McInnes, Kremer, Godwood, Michaels, Guo, Close, Weinblatt.
Acquisition of data
Miranda, Vencovský, Godwood, Albulescu, Guo, Close.
Analysis and interpretation of data
Burmester, McInnes, Kremer, Vencovský, Godwood, Albulescu, Michaels, Guo, Close, Weinblatt.
Role of the study sponsor
The study was funded by MedImmune. As many of the authors are present or former employees of MedImmune, MedImmune had a substantial role in the study design, collection of the data, analysis and interpretation of the data, and the decision to submit the manuscript for publication. MedImmune funded medical writing support provided by QXV Comms, an Ashfield Company, part of UDG Healthcare. Publication of this article was not contingent upon approval by MedImmune.
Additional disclosures
Author Godwood is an employee of Heptares Therapeutics. Author Close is an employee of AstraZeneca.
Supporting information
Acknowledgments
The authors would like to thank the study investigators and the members of the IPEC (Professors Ian Bruce, Hal Collard, Jonathan Goldin, Manny Kavuru, and Robert Wise). The authors would like to acknowledge Wendy White (MedImmune, Gaithersburg, MD) for her contribution to the biomarker analysis and Kate Middleton (MedImmune, Cambridge, UK) for assistance with reviewing the pulmonary and general data. The authors would like to acknowledge Rebecca Plant, MSc, of QXV Comms, an Ashfield Company, part of UDG Healthcare, for medical writing support that was funded by MedImmune.
ClinicalTrials.gov identifier: NCT01712399.
Supported by MedImmune.
Drs. Close and Weinblatt contributed equally to this work.
Dr. Burmester has received consulting fees and/or speaking fees from AbbVie, Bristol‐Myers Squibb, Eli Lilly and Company, MSD, MedImmune, Novartis, Pfizer, and Roche (less than $10,000 each) and research support from UCB, Pfizer, and Roche. Dr. McInnes has received consulting fees, speaking fees, and/or honoraria from MedImmune, A2, Eli Lilly and Company, Janssen, Novartis, AbbVie, and UCB (less than $10,000 each). Dr. Kremer has received consulting fees, speaking fees, and/or honoraria from AbbVie, Bristol‐Myers Squibb, Genentech, Eli Lilly and Company, Pfizer, Novartis, and MedImmune (less than $10,000 each) and owns stock or stock options in the Consortium of Rheumatology Researchers of North America (CORRONA). Dr. Miranda has received payment for a clinical study from MedImmune (more than $ 10,000). Dr. Vencovský has received consulting fees, speaking fees, and/or honoraria from AbbVie, Biogen, Bristol‐Myers Squibb, CSL Behring, Eli Lilly and Company, MSD, Novartis, Pfizer, and UCB (less than $10,000 each). Mr. Godwood and Drs. Albulescu, Michaels, Guo, and Close own stock or stock options in AstraZeneca. Dr. Close owns stock or stock options in MedImmune. Dr. Weinblatt has received consulting fees, speaking fees, and/or honoraria from Amgen, AbbVie, Roche Pharmaceuticals, Novartis, AstraZeneca, and MedImmune (less than $10,000 each) and from Pfizer, Eli Lilly and Company, Bristol‐Myers Squibb, and UCB (more than $10,000 each) and research support from Amgen, Crescendo Bioscience, Bristol‐Myers Squibb, and UCB.
References
- 1. Van Onna M, Boonen A. The challenging interplay between rheumatoid arthritis, ageing and comorbidities. BMC Musculoskelet Disord 2016;17:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Listing J, Gerhold K, Zink A. The risk of infections associated with rheumatoid arthritis, with its comorbidity and treatment. Rheumatology (Oxford) 2013;52:53–61. [DOI] [PubMed] [Google Scholar]
- 3. Singh JA, Cameron C, Noorbaloochi S, Cullis T, Tucker M, Christensen R, et al. Risk of serious infection in biological treatment of patients with rheumatoid arthritis: a systematic review and meta‐analysis. Lancet 2015;386:258–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berghen N, Teuwen LA, Westhovens R, Verschueren P. Malignancies and anti‐TNF therapy in rheumatoid arthritis: a single‐center observational cohort study. Clin Rheumatol 2015;34:1687–95. [DOI] [PubMed] [Google Scholar]
- 5. Cornish AL, Campbell IK, McKenzie BS, Chatfield S, Wicks IP. G‐CSF and GM‐CSF as therapeutic targets in rheumatoid arthritis. Nat Rev Rheumatol 2009;5:554–9. [DOI] [PubMed] [Google Scholar]
- 6. Avci AB, Feist E, Burmester GR. Targeting GM‐CSF in rheumatoid arthritis. Clin Exp Rheumatol 2016;34 Suppl 98:39–44. [PubMed] [Google Scholar]
- 7. Greven DE, Cohen ES, Gerlag DM, Campbell J, Woods J, Davis N, et al. Preclinical characterisation of the GM‐CSF receptor as a therapeutic target in rheumatoid arthritis. Ann Rheum Dis 2015;74:1924–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cook AD, Louis C, Robinson MJ, Saleh R, Sleeman MA, Hamilton JA. Granulocyte macrophage colony‐stimulating factor receptor α expression and its targeting in antigen‐induced arthritis and inflammation. Arthritis Res Ther 2016;18:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fleetwood AJ, Cook AD, Hamilton JA. Functions of granulocyte‐macrophage colony‐stimulating factor. Crit Rev Immunol 2005;25:405–28. [DOI] [PubMed] [Google Scholar]
- 10. Hamilton JA. Colony‐stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 2008;8:533–44. [DOI] [PubMed] [Google Scholar]
- 11. Zhan Y, Cheers C. Haemopoiesis in mice genetically lacking granulocyte‐macrophage colony stimulating factor during chronic infection with Mycobacterium avium. Immunol Cell Biol 2000;78:118–23. [DOI] [PubMed] [Google Scholar]
- 12. Calisto Perez C, Leon R, Leon F, Ng SL. Rheumatoid arthritis and anemia: the impact of different anti‐inflammatory therapies on hemoglobin levels: an observational study. Bol Asoc Med P R 2012;104:34–41. [PubMed] [Google Scholar]
- 13. Lazaro E, Morel J. Management of neutropenia in patients with rheumatoid arthritis. Joint Bone Spine 2015;82:235–9. [DOI] [PubMed] [Google Scholar]
- 14. Crotti C, Raimondo MG, Becciolini A, Biggioggero M, Favalli EG. Spotlight on mavrilimumab for the treatment of rheumatoid arthritis: evidence to date. Drug Des Devel Ther 2017;11:211–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen BD, Mueller M, Chou TH. Role of granulocyte/macrophage colony‐stimulating factor in the regulation of murine alveolar macrophage proliferation and differentiation. J Immunol 1988;141:139–44. [PubMed] [Google Scholar]
- 16. Akagawa KS, Kamoshita K, Tokunaga T. Effects of granulocyte‐macrophage colony‐stimulating factor and colony‐stimulating factor‐1 on the proliferation and differentiation of murine alveolar macrophages. J Immunol 1988;141:3383–90. [PubMed] [Google Scholar]
- 17. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM‐CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 2001;15:557–67. [DOI] [PubMed] [Google Scholar]
- 18. Yoshida M, Korfhagen TR, Whitsett JA. Surfactant protein D regulates NF‐κ B and matrix metalloproteinase production in alveolar macrophages via oxidant‐sensitive pathways. J Immunol 2001;166:7514–9. [DOI] [PubMed] [Google Scholar]
- 19. Borie R, Danel C, Debray MP, Taille C, Dombret MC, Aubier M, et al. Pulmonary alveolar proteinosis. Eur Respir Rev 2011;20:98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Minter RR, Cohen ES, Wang B, Liang M, Vainshtein I, Rees G, et al. Protein engineering and preclinical development of a GM‐CSF receptor antibody for the treatment of rheumatoid arthritis. Br J Pharmacol 2013;168:200–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burmester GR, Weinblatt ME, McInnes IB, Porter D, Barbarash O, Vatutin M, et al. Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann Rheum Dis 2013;72:1445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burmester GR, McInnes IB, Kremer J, Miranda P, Korkosz M, Vencovsky J, et al. A randomised phase IIb study of mavrilimumab, a novel GM‐CSF receptor α monoclonal antibody, in the treatment of rheumatoid arthritis. Ann Rheum Dis 2017;76:1020–30. [DOI] [PubMed] [Google Scholar]
- 23. Weinblatt M, McInnes I, Kremer J, Miranda P, Vencovsky J, Godwin D, et al. EARTH EXPLORER 2, a phase IIb exploratory study evaluating efficacy and safety of mavrilimumab, a fully human granulocyte‐macrophage colony‐stimulating factor receptor‐α monoclonal antibody, and the tumor necrosis factor antagonist golimumab in rheumatoid arthritis. Ann Rheum Dis 2016;75:717–8. [Google Scholar]
- 24. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. [DOI] [PubMed] [Google Scholar]
- 25. Prevoo ML, van ‘t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty‐eight–joint counts: development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995;38:44–8. [DOI] [PubMed] [Google Scholar]
- 26. Van Gestel AM, Haagsma CJ, van Riel PL. Validation of rheumatoid arthritis improvement criteria that include simplified joint counts. Arthritis Rheum 1998;41:1845–50. [DOI] [PubMed] [Google Scholar]
- 27. Borg GA. Psychophysical bases of perceived exertion. Med Sci Sports Exerc 1982;14:377–81. [PubMed] [Google Scholar]
- 28. Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum 1995;38:727–35. [DOI] [PubMed] [Google Scholar]
- 29. Aletaha D, Nell VP, Stamm T, Uffmann M, Pflugbeil S, Machold K, et al. Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Res Ther 2005;7:R796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van der Heijde DM, van Riel PL, Gribnau FW, Nuver‐Zwart IH, van de Putte LB. Effects of hydroxychloroquine and sulphasalazine on progression of joint damage in rheumatoid arthritis. Lancet 1989;1:1036–8. [DOI] [PubMed] [Google Scholar]
- 31. Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum 1980;23:137–45. [DOI] [PubMed] [Google Scholar]
- 32. Ware JE Jr, Snow KK, Kosinski M, Gandek B. SF‐36 health survey: manual and interpretation guide. Boston: The Health Institute, New England Medical Center; 1993. [Google Scholar]
- 33. Cella D, Yount S, Sorensen M, Chartash E, Sengupta N, Grober J. Validation of the Functional Assessment of Chronic Illness Therapy Fatigue Scale relative to other instrumentation in patients with rheumatoid arthritis. J Rheumatol 2005;32:811–9. [PubMed] [Google Scholar]
- 34. Achuthan A, Cook AD, Lee MC, Saleh R, Khiew HW, Chang MW, et al. Granulocyte macrophage colony‐stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J Clin Invest 2016;126:3453–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jaguin M, Houlbert N, Fardel O, Lecureur V. Polarization profiles of human M‐CSF‐generated macrophages and comparison of M1‐markers in classically activated macrophages from GM‐CSF and M‐CSF origin. Cell Immunol 2013;281:51–61. [DOI] [PubMed] [Google Scholar]
- 36. Guo X, Wang S, Bay‐Jensen AC, Karsdal MA, Godwood A, Albulescu M, et al. Sustained suppression of peripheral biomarkers by mavrilimumab but not golimumab in anti‐tumor necrosis factor‐inadequate responders: an exploratory analysis in the phase IIb Earth Explorer 2 Clinical Trial [abstract]. Arthritis Rheumatol 2016;68 Suppl 10 URL: http://acrabstracts.org/abstract/sustained-suppression-of-peripheral-biomarkers-by-mavrilimumab-but-not-golimumab-in-anti-tumor-necrosis-factor-inadequate-responders-an-exploratory-analysis-in-the-phase-iib-earth-explorer-2-clinical/. [Google Scholar]
- 37. Brown KK. Rheumatoid lung disease. Proc Am Thorac Soc 2007;4:443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tsuchiya Y, Takayanagi N, Sugiura H, Miyahara Y, Tokunaga D, Kawabata Y, et al. Lung diseases directly associated with rheumatoid arthritis and their relationship to outcome. Eur Respir J 2011;37:1411–7. [DOI] [PubMed] [Google Scholar]
- 39. Kelly C, Iqbal K, Iman‐Gutierrez L, Evans P, Manchegowda K. Lung involvement in inflammatory rheumatic diseases. Best Pract Res Clin Rheumatol 2016;30:870–88. [DOI] [PubMed] [Google Scholar]
- 40. Shaw M, Collins BF, Ho LA, Raghu G. Rheumatoid arthritis‐associated lung disease. Eur Respir Rev 2015;24:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cottin V, Tebib J, Massonnet B, Souquet PJ, Bernard JP. Pulmonary function in patients receiving long‐term low‐dose methotrexate. Chest 1996;109:933–8. [DOI] [PubMed] [Google Scholar]
- 42. Dawson JK, Graham DR, Desmond J, Fewins HE, Lynch MP. Investigation of the chronic pulmonary effects of low‐dose oral methotrexate in patients with rheumatoid arthritis: a prospective study incorporating HRCT scanning and pulmonary function tests. Rheumatology (Oxford) 2002;41:262–7. [DOI] [PubMed] [Google Scholar]
- 43. Antoniu SA. GM‐CSF pathway correction in pulmonary alveolar proteinosis. Expert Opin Biol Ther 2010;10:1357–65. [DOI] [PubMed] [Google Scholar]
- 44. Khan A, Agarwal R. Pulmonary alveolar proteinosis. Respir Care 2011;56:1016–28. [DOI] [PubMed] [Google Scholar]
- 45. Buch MH, Silva‐Fernandez L, Carmona L, Aletaha D, Christensen R, Combe B, et al. Development of EULAR recommendations for the reporting of clinical trial extension studies in rheumatology. Ann Rheum Dis 2015;74:963–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials