

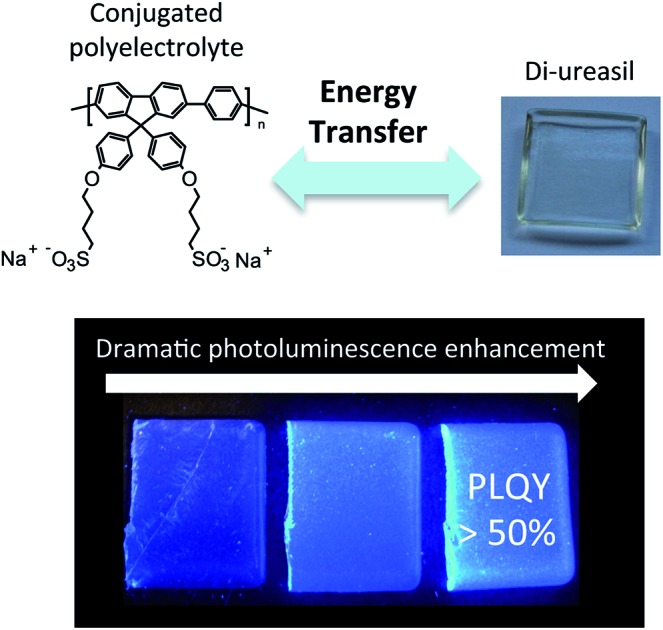
 Energy transfer between a hybrid di-ureasil host and conjugated polymer dopants results in a dramatic enhancement in the photoluminescence quantum yield due to exciton isolation at long-lived trap sites.
Energy transfer between a hybrid di-ureasil host and conjugated polymer dopants results in a dramatic enhancement in the photoluminescence quantum yield due to exciton isolation at long-lived trap sites.
Abstract
Poly(fluorene) conjugated polyelectrolyte (CPE)-di-ureasil organic–inorganic composites have been prepared using a versatile sol–gel processing method, which enables selective localisation of the CPE within the di-ureasil matrix. Introduction of the CPE during the sol–gel reaction leads to a homogeneous distribution of the CPE throughout the di-ureasil, whereas a post-synthesis solvent permeation route leads to the formation of a confined layer of the CPE at the di-ureasil surface. The CPE and the di-ureasil both function as photoactive components, contributing directly to, and enhancing the optical properties of their composite material. The bright blue photoluminescence exhibited by CPE-di-ureasils is reminiscent of the parent CPE; however the distinct contribution of the di-ureasil to the steady-state emission profile is also apparent. This is accompanied by a dramatic increase in the photoluminescence quantum yield to >50%, which is a direct consequence of the synergy between the two components. Picosecond time-correlated single photon counting measurements reveal that the di-ureasil effectively isolates the CPE chains, leading to emissive trap sites which have a high radiative probability. Moreover, intimate mixing of the CPE and the di-ureasil, coupled with their strong spectral overlap, results in efficient excitation energy transfer from the di-ureasil to these emissive traps. Given the simple, solution-based fabrication method and the structural tunability of the two components, this approach presents an efficient route to highly desirable CPE-hybrid materials whose optoelectronic properties may be enhanced and tailored for a targeted application.
Introduction
Solution-processable conjugated polymers (CP) continue to dominate the field of low-cost, organic device technologies such as optical sensors,1–3 polymer light-emitting diodes,4–6 photovoltaic devices7,8 and field effect transistors.9,10 Such devices typically require a multi-layer architecture to meet the optoelectronic requirements.11,12 Device performance thus depends critically on both the interfacial contact between active layers and the nanoscale morphology of individual layers, which in turn is intrinsically linked to the optoelectronic properties.13–15 Although multi-layer devices can be fabricated using inexpensive solution-phase coating or printing techniques,16 this is non-trivial, and obtaining the fine balance between the polymer morphology in solution and the wettability of the deposited films can be challenging to achieve.
Conjugated polymer organic–inorganic hybrid and composite materials present an elegant approach to indirectly manipulate the conformation and orientation of the polymer within the active layer17–20 as well as improving environmental stability.21 The confinement of a conjugated polymer within a nanoporous inorganic structure has been shown to enhance electroluminescence.22 Moreover, non-covalent interactions (e.g. hydrogen and ionic bonding, π–π stacking, hydrophobic) can also be exploited and coordinated to yield desirable morphologies at the organic–inorganic phase interface.23 However, in each of these examples, the inorganic component has served primarily to isolate or aggregate the polymer chains and makes no contribution to the functional properties of the material. This represents a missed opportunity, since one of the inherent curiosities of organic–inorganic hybrid materials is the potential for emergent or enhanced properties deriving from the interaction between the two components.24
To address this limitation, we have designed a novel series of CP-materials that utilise a family of organic–inorganic hybrid polymers known as the di-ureasils as the host. Di-ureasils are comprised of a siliceous skeleton that is chemically-grafted to poly(ethylene oxide) (PEO)/poly(propylene oxide) (PPO) chains through two urea [NHC( O)HN] cross-linkages (Fig. 1a).25,26 We chose to use a di-ureasil as our host material for several reasons. Firstly, an organic–inorganic hybrid should show improved chemical compatibility with the CP compared with a purely inorganic matrix, thereby minimising the risk of phase separation. Secondly, the di-ureasil structure presents multiple chemical groups for physical interaction with the CP via hydrogen or electrostatic bonding. Thirdly, the di-ureasils themselves are intrinsically photoluminescent and have been frequently employed in optical applications, e.g. as light-emitting materials27 and in luminescent solar concentrators.28 As such, they should function as active hosts and contribute to the optoelectronic properties of the resultant material. Further, the fact that the di-ureasil emissive sites are relatively localised within thermally accessible trap states provides the intriguing possibility of enhancing yields in electroluminescent devices based on these materials through thermal activation.29 This is similar to the thermally activated delayed fluorescence (TADF) approach currently being exploited in the development of the next generation of high efficiency organic light emitting diodes.30 Finally, as the di-ureasils combine the inherent flexibility of the PEO/PPO component with the thermal and mechanical stability of the organosilica, they are prime candidates as materials for flexible display technologies.31
Fig. 1. (a) Chemical structures of the non-hydrolysed di-ureasil precursor (d-UPTES) and the CPEs investigated. (b) Schematic representation of the two synthetic routes used to prepare CPE-di-ureasil composites. In the Direct Insertion method, the d-UPTES precursor is mixed with a solution of the CPE prior to initiation of the sol–gel reaction. Hydrolysis and condensation of the silica network proceeds with the CPE in situ, such that it becomes trapped within the di-ureasil matrix. In the solvent Permeation method, a pre-formed di-ureasil sample is immersed in a solution of the CPE. The di-ureasil absorbs the solution, leading to uptake of the CPE. Upon drying the di-ureasil contracts to its initial size.

Herein, we report the synthesis, structural characterisation and optical performance of CPE-di-ureasil materials incorporating the water-soluble poly(fluorene) conjugated polyelectrolytes (CPEs), PBS-PFP32,33 and HTMA-PFP34,35,36 (Fig. 1a). Two distinct synthetic procedures have been developed to incorporate the CPE, which enable us to probe the influence of the local host environment on the photophysical properties of the CP. We demonstrate that these CPE-di-ureasil composites exhibit a dramatic enhancement of their photoluminescence in the solid state, with photoluminescence quantum yields (PLQY) reaching > 50%. We show using picosecond time-resolved emission studies that the observed photoluminescence enhancement results from electronic communication between the CPE and the di-ureasil and potential mechanisms for the energy/charge transfer are discussed. To the best of our knowledge, this is the first demonstration of enhanced PLQYs in a CPE-composite material due to a synergistic interaction between the CPE and an active host. Given the simple, solution-based fabrication method and the structural tunability of the two components, this approach presents a versatile and efficient route to highly desirable CPE-hybrid materials whose optoelectronic properties may be enhanced and tailored for a targeted end-application.
Results and discussion
Synthesis of CPE-di-ureasil composites
The synthesis of the dU-(600) di-ureasil has been reported in detail elsewhere.25,26,37 In brief, 3-isocyanatoproplytriethoxysilane (ICPTES) is first reacted with the commercial polyetheramine Jeffamine ED-600 to form the intermediate di-ureapropyltriethoxysilane (d-UPTES) (Fig. 1a). Acid-catalysed hydrolysis and condensation of the siliceous framework subsequently yields the di-ureasil d-U(600). PBS-PFP and HTMA-PFP were incorporated into the di-ureasil using two different approaches, as illustrated in Fig. 1b. Detailed sample compositions are shown in Table 1. In the Direct Insertion (DI) method, a fixed volume of CPE stock solution is mixed with the d-UPTES precursor solution prior to condensation of the silica network. The samples in this series are designated as DI-PBS-x and DI-HTMA-x, for PBS-PFP and HTMA-PFP di-ureasils, respectively, where x represents the concentration (in × 10–3 wt%) of the CPE within the di-ureasil.
Table 1. Composition of CPE-di-ureasils prepared in this study. [CPE] is the concentration of the stock CPE solution (in mM repeat units (r.u.)), that is either added to the d-UPTES intermediate (DI method) or in which the di-ureasil is immersed (SP method). The weight percent (wt%) of CPE incorporated was estimated from [CPE] and the resultant mass of the dry CPE-di-ureasil for the DI method and from the solvent uptake, [CPE] and the mass of the dry CPE-di-ureasil for the SP method.
| Method | Polymer | Sample | [CPE] (mM r.u.) | CPE wt% |
| Direct Insertion (DI) | PBS-PFP | DI-PBS-0.0 | 0 | 0 |
| DI-PBS-1.0 | 0.2 | 1 × 10–3 | ||
| DI-PBS-2.0 | 0.4 | 2 × 10–3 | ||
| DI-PBS-4.0 | 0.8 | 4 × 10–3 | ||
| DI-PBS-8.0 | 1.6 | 8 × 10–3 | ||
| Direct Insertion (DI) | HTMA-PFP | DI-HTMA-0.0 | 0 | 0 |
| DI-HTMA-1.0 | 0.2 | 1 × 10–3 | ||
| DI-HTMA-2.0 | 0.4 | 2 × 10–3 | ||
| DI-HTMA-4.0 | 0.8 | 4 × 10–3 | ||
| DI-HTMA-8.0 | 1.6 | 8 × 10–3 | ||
| Solvent Permeation (SP) | PBS-PFP | SP-PBS-0.0 | 0 | 0 |
| SP-PBS-0.7 | 0.01 | 0.7 × 10–3 | ||
| SP-PBS-2.0 | 0.03 | 2.0 × 10–3 | ||
| SP-PBS-3.4 | 0.05 | 3.4 × 10–3 | ||
| SP-PBS-7.0 | 0.1 | 7.0 × 10–3 |
In the Solvent Permeation (SP) method, a preformed dU-600 di-ureasil sample is immersed in a solution (of fixed volume and concentration) of the CPE. Gradual swelling of the di-ureasil is observed with increasing immersion time due to permeation of the CPE-solvent system into the cross-linked hybrid network. We focus our discussion here on the SP-PBS-x series, but comparable results were also obtained for SP-HTMA, indicating that the nature of the ionic side chain has little influence on this mode of synthesis. Notably, no variation in the solvent percentage uptake trends was observed for different batches of the parent di-ureasil, suggesting that the synthetic procedure leads to highly reproducible samples (see Fig. S3†). At longer immersion times (>1000 min), the percentage solvent uptake slows and eventually plateaus at ∼180% for SP-PBS-0, which corresponds to the solvent equilibrium point.38 Comparable swelling trends were previously reported for di-ureasil membranes prepared from the related Jeffamine ED-2000 in ethylene carbonate/propylene carbonate mixtures.39 Although longer immersion times led to increased gel swelling, the samples displayed a higher tendency towards cracking upon drying, which is due to a reduction in the degree of cross-linking in the swollen state.40,41 Thus, an immersion time of 5 hours, corresponding to a percentage uptake of ∼70%, was found to be the best compromise between maintaining the mechanical strength of the di-ureasil, and simultaneously enabling sufficient CPE incorporation. We note that the percentage uptake is greater for the CPE solution when compared to the pure solvent (Fig. S3†); however, this increase is not strongly dependent on the concentration of the CPE solution (within the range examined).
Both methods yield pristine elastomeric monoliths, which exhibit intense blue photoluminescence under UV illumination (Fig. 2). Notably, once incorporated, the CPE is physically-retained within the di-ureasil network. Following immersion of CPE-di-ureasils in an appropriate solvent system for up to 24 h, no detectable CPE photoluminescence was observed from the immersion solution (Fig. S4†). The distribution of the CPEs within the di-ureasil was investigated using fluorescence confocal microscopy. From Fig. 2 we observe that for the DI method, the CPE forms isolated domains of ca. 2 μm diameter, which are homogeneously distributed within the di-ureasil matrix. Each domain is anticipated to comprise of multiple CPE chains. In contrast, for materials prepared by the SP method, a dense CPE layer of ∼12–14 μm in thickness is observed at the surface of the sample. Comparison of the intensity depth profiles (Fig. 2c and d) reveals that for the DI method, the PL emission originates at depths >4 μm, whereas for the SP method the emissive layer is confined to the surface, which accounts for the observed differences in the absolute emission intensities for these samples (Fig. 2a and b). These observations are consistent with the synthetic conditions: for DI, the di-ureasil network condenses with the CPE in situ, whereas for SP the CPE is incorporated via diffusion through the swollen di-ureasil network. For SP, however, the permeation distance is determined by the immersion time, the solubility parameters of the di-ureasil, CPE and the solvent, and the degree of cross-linking. As such, under the experimental conditions employed here, the CPE is unable to penetrate much further than the surface of the di-ureasil.
Fig. 2. Confocal microscopy images (λex = 405 nm) from 20 and 4 μm beyond the surface of (a) DI-PBS-1.0 and (b) SP-PBS-0.7, respectively. (c) Lateral cross-section of DI-PBS-1.0 reconstructed from confocal microscopy images (λex = 405 nm) showing fluorescence throughout the sample depth investigated, 68 μm. (d) Lateral cross-section of SP-PBS-0.7 reconstructed from confocal microscopy images (λex = 405 nm) showing fluorescence from a 12–14 μm surface layer. Sample depth investigated, 32 μm. (e) Photograph of DI-PBS-0.0, DI-PBS-1.0, DI-PBS-2.0 and DI-PBS-4.0 under UV-illumination (λex = 366 nm).

Bulk and local structural characterisation
The bulk structural features of CPE-di-ureasils were examined by powder X-ray diffraction (XRD), solid-state magic angle spinning nuclear resonance spectroscopy (MAS-NMR) and Fourier transform infrared (FTIR) spectroscopy. All samples exhibit XRD patterns that are typical of di-ureasils, characterised by two broad overlapping peaks, centred at 16.6° and 22.5°, respectively (Fig. S5†). The more intense peak at 22.5° is associated with the presence of coherent diffracting regions of the siliceous network,27,37,42,43 whose structural unit distance, d, was determined to be 4.0 ± 0.1 Å (from Bragg's law). The weak second order peak is observed at 43–46°. The overlapping peak at 16.6° is attributed to the in-plane ordering of additional intra-siloxane domains, as previously observed for di-ureasil and di-urethane hybrids prepared via carboxylic acid solvolysis.43 The coherence length, L, over which the structural unit survives (i.e. the crystallite size) was calculated as 21 ± 2 Å (from the Scherrer equation),44 which is comparable to the diameter of siliceous domains reported for self-assembled alkyl-siloxane hybrids.45 The structural unit distances are somewhat larger than the average bond distances for Si–O, O–O and Si–Si (1.62, 2.65, and 3.12 Å, respectively), reflecting a local order that is developed over distances beyond the first few nearest neighbour shells. However, the XRD patterns are essentially independent of both the charge and concentration of the CPE and the preparation method, suggesting that sub-nanometre organisation of the siliceous network is unaffected by CPE incorporation.
The corresponding 29Si MAS-NMR spectra exhibit broad signals characteristic of T1 (R′Si(OSi)-(OR)2, ∼49 ppm), T2 (R′Si(OSi)2(OR), ∼58 ppm) and T3 (R′Si(OSi)3, ∼66 ppm) organosiloxane units (Fig. S6†).27,42,43 The absence of a T0 signal confirms that no unreacted precursor remains. The degree of condensation, C, of the siliceous network, was found to be between 73–81%, which is consistent with values previously reported for di-ureasils doped with lanthanide ions (Table S1†).27 Notably, the degree of condensation is not significantly affected by either the method of incorporation, the charge on the CPE or its concentration. The 13C CP/MAS NMR spectra agree with those previously reported for di-ureasils of this type (Fig. S7†)43 further confirming that the integrity of the network structure is maintained upon CPE incorporation.
FTIR spectroscopy has been shown to provide valuable insight into the influence of the preparation method and/or doping with lanthanide ions/complexes on the di-ureasil structure.27,42 The amide I region (1610–1770 cm–1) in particular, can provide diagnostic information about the specificity and degree of hydrogen bonding interactions associated with C O stretching frequencies in different local environments.26 The amide I band of CPE-di-ureasils is moderately influenced by the type and concentration of CPE incorporated, most notably at the higher concentrations (4 × 10–3 wt%); however significant differences are observed for the two preparation methods (Fig. 3 and Table S2†). For the DI samples, Gaussian deconvolution reveals the presence of two components centred at 1708 (Peak 1) and 1663 cm–1 (Peak 2), attributed to νC O stretches located within disordered hydrogen-bonded POE-urea associations of increasing strength, and a third component at 1635 cm–1 (Peak 3) due to self-organised, hydrogen-bonded urea–urea interactions.26,27,42 The absence of a peak at 1750 cm–1 further confirms that all urea groups undergo hydrogen-bonding interactions.26,27 A slightly decreased contribution from Peak 3 is observed for DI-PBS-4.0 when compared to DI-HTMA-4.0, suggesting the formation of a less ordered host network upon incorporation of the anionic PBS-PFP. The SP-samples also exhibit a three-component amide I band; however their relative contributions differ considerably. A decrease in Peak 1 is accompanied by a concomitant increase in the relative contribution of Peak 2 (10%). This suggests that the incorporation of CPE via Solvent Permeation disrupts and reorders the disordered hydrogen-bonded POE/urea associations, which is consistent with swelling and contraction of the di-ureasil network. We note that the hydrogen-bonded urea–urea interactions (Peak 3) appear to be unaffected by this process.
Fig. 3. Results of curve-fitting preformed in the ‘amide I’ region of the samples prepared via both the Direct Insertion and Solvent Permeation methods: (i) DI-PBS-0.0, (ii) SP-PBS-0.0, (iii) DI-PBS-4.0 and (iv) SP-PBS-3.4. The numbers identify the peaks described in the text.

Thermal- and photostability
The thermograms for the undoped samples (e.g. DI-PBS-0.0) are in excellent agreement with that previously reported for the d-U(600) di-ureasil,46,47 with the onset of sample decomposition observed at ∼339 °C (Fig. 4a). Incorporation of PBS-PFP or HTMA-PFPvia either method yields a material with good thermal stability with the onset of thermal decomposition occurring between 337–352 °C. This thermal stability considerably surpasses the moderate operating temperatures (∼65–140 °C) present in organic electronic devices.48 The photostability of the CPE-di-ureasils was also investigated. Fig. 4b shows the integrated emission intensity of DI-PBS-4.0, SP-PBS-3.4, DI-HTMA-4.0 and a pure PBS-PFP film as a function of irradiation time upon excitation at 370 nm. Irradiation of the PBS-PFP film over time results in the growth of a green emission band centred at 560 nm that is characteristic of the formation of fluorenone defects on poly(fluorene) chains.49 This band eventually becomes more intense than the initial blue-poly(fluorene) emission. Thus to examine purely the poly(fluorene) contribution to the emission, the integrated intensity for all samples was measured in the range 385–490 nm. The integrated intensity of the PBS-PFP film dropped gradually to 40% of the initial value in the first hour, and continued to decrease to 25% at the end of the irradiation period (2 h). This is in contrast to DI-BPS-4.0, SP-PBS-3.4 and DI-HTMA-4.0, whose emission shows improved stability to extended irradiation, retaining >80% of the initial intensity at the end of the study.
Fig. 4. (a) TGA thermograms of DI-PBS-0.0 (blue line), DI-PBS-4.0 (red line), SP-PBS-3.4 (blue line) and DI-HTMA-4.0 (green line). TGA measurements were performed in air at a heating rate of 10 °C min–1. (b) Integrated emission intensity of a pure PBS-PFP thin film on glass (open blue circles), DI-PBS-4.0 (open red squares), SP-PBS-3.4 (open black triangles) and DI-HTMA-4.0 (open green diamonds) under irradiation at 370 nm. The black lines serve only to guide the eye.

Steady-state photoluminescence studies
Undoped samples prepared by both the DI and SP methods exhibit a broad emission band centred between 380 and 450 nm, which is characteristic of di-ureasils (Fig. 5a). This emission has previously been assigned to radiative electron–hole recombination mediated by donor–acceptor pairs occurring at distinct localised trap states: (i) oxygen defects in the siliceous nanodomains (purple-blue component centred at ∼2.78–3.28 eV) and (ii) photo-induced proton transfer between NH2+ and N– defects localised at the urea linkages (blue component centred at ∼2.53–3.13 eV).37,50 The emission maximum is strongly dependent on the excitation energy, red-shifting to longer wavelengths as λex increases. A similar trend is observed in the corresponding excitation spectrum (Fig. S8†).
Fig. 5. PL (λex = 320, 330, 340, 350, 360, 370 and 380 nm) of (a) DI-PBS-0.0 and (b) DI-PBS-1.0. (c) Excitation spectra (λem = 400, 420, 440, 460 and 480 nm) of DI-PBS-1.0. (d) Photoluminescence quantum yield (λex = 370 nm) for SP-PBS-x (closed squares), DI-PBS-x (closed circles) and DI-HTMA-x (open triangles). The solid lines serve solely to guide the eye.

In contrast, DI-PBS samples exhibit the characteristic, poly(fluorene) emission profile, featuring a structured band between 400–500 nm, as shown in Fig. 5b for DI-PBS-1.0. A small blue-shift (∼2 nm) in the emission maximum is observed upon transferring the CPEs from a good solvent to the solid-state demonstrating that isolation of individual polymer chains is improved upon incorporation into the di-ureasil.32,51 This suggests that rather than undergoing phase separation, as could be anticipated when blending two chemically-dissimilar polymers,52 ion–dipole interactions between the CPE and the di-ureasil network effectively break-up polymer chain aggregates. Furthermore, a distinct contribution from the di-ureasil to the emission spectrum can also be discerned. Excitation between 320–350 nm, which is semi-selective for di-ureasil excitation, results in significant broadening of the high-energy edge of the emission band. Similarly, as λex is increased between 320–380 nm, gradual broadening of the lower energy region of the emission spectrum is also observed. The corresponding excitation spectra reveal similar emission wavelength dependence (Fig. 5c). As the PBS-PFP concentration is increased, the extent of broadening at the high energy, blue-edge is decreased in favour of the lower energy region (see Fig. S9†). SP-PBS-di-ureasils show a similar trend in the PL emission and excitation spectra, with spectral broadening occurring at both edges of the band, although to a somewhat reduced extent (see Fig. S10†).
We note that for the DI-HTMA-x series, the emergence of poly(fluorene)-like emission is observed only upon incorporation of a critical concentration of the CPE. For example, while DI-HTMA-1.0 exhibits the characteristic emission profile of the undoped di-ureasil (see Fig. S11a and b†), the emission spectrum of DI-HTMA-4.0 resembles that of the CPE in solution, with moderate contribution from the di-ureasil in the form of excitation wavelength dependent emission and band-broadening These subtle differences provide the first indication for a weaker interaction between the CPE and the di-ureasil for the HTMA-PFP system (Fig. S11c and d†).
The corresponding photoluminescence quantum yields (λex = 370) as a function of wt% of CPE incorporated are shown in Fig. 5d.53 Undoped di-ureasils prepared by both methods exhibit a PLQY of 2.3–6.8%, which is in good agreement with previously reported values for di-ureasils synthesised via hydrochloric acid catalysis.27 Remarkably, a dramatic increase in PLQY as a function of increasing CPE wt% is observed, reaching 56% and 47% for DI-PBS-4.0 and DI-HTMA-4.0, respectively. A significant increase in PLQY is also observed for the SP-PBS series, reaching 31% for SP-PBS-0.7. This increase in the photoluminescence yield cannot arise simply from the summed contribution of the individual components. In the solvent systems used to prepare the CPE-di-ureasils, PBS-PFP (25% (v/v) 1,4-dioxane/water) and HTMA-PFP (25% (v/v) MeCN/water) exhibit a maximum PLQY of 23.5% and 28.7%, respectively. Similarly, pure thin films of these CPEs typically display low PL quantum yields (PLQY ∼ 7.5% for HTMA-PFP54). If these values are simply combined with the typical PLQY for di-ureasils (4–10%),43 the obtained quantum yield would be substantially lower than the values determined here. An alternative pathway must operate in CPE-di-ureasils to yield this dramatic photoluminescence enhancement.
Picosecond (ps) time-resolved emission studies
Picosecond time-correlated single photon counting (TC-SPC) emission lifetime measurements were performed to gain further insight into the mechanism responsible for the observed PL enhancement. Individually, the CPEs and di-ureasil relax over sufficiently different timescales to enable distinct contributions from each component to the emission lifetime to be identified. Experimental considerations restrict us to excitation at 365 nm, which means that selective excitation is unattainable due to strong overlap of the absorption bands of the two components (Fig. S12†). However, PLE data mapping suggests that variation of the monitoring wavelength should provide more insight (Fig. 6). As expected, the undoped DI-PBS-0 sample exhibits two distinct weak emission contributions, located at 370–420 nm (λex = 320–350 nm) and 425–500 nm (λex = 350–370 nm). In contrast, DI-PBS-1.0 emits strongly between 400–420 nm, with a systematic increase in the intensity as the excitation wavelength is increased to 370 nm (the absorbance maximum of the CPE). A weaker emission between 470–500 nm is also observed for λex = 350–370 nm. Based on these observations, monitoring wavelengths of 420 nm and 500 nm should enable us to primarily observe emission from the CPE and the di-ureasil, respectively.
Fig. 6. Excitation-emission data maps for (a) DI-PBS-0.0 and (b) DI-PBS-1.0, where λex = 320–375 nm (1 nm intervals) and λem = 380–650 nm (1 nm intervals). (c) Emission lifetimes and (d) corresponding pre-exponential αi values as determined from the fitting of tri-exponential functions to the fluorescence decays of the sample DI-PBS-1.0 as a function of emission wavelength (λem = 400, 410, 420, 430, 440, 450, 460, 470, 480, 490 and 500 nm) (λex = 365 nm). (e) Emission lifetimes and (f) corresponding pre-exponential αi values as determined from the fitting of tri-exponential functions to the fluorescence decays for the DI-PBS-x series at the di-ureasil emission wavelength (λex = 365 nm, λem = 500 nm).

In solution, poly(fluorene-phenylene) polyelectrolytes often exhibit complex decay curves, which can be resolved into three exponential components: τCPE1 ∼ 10–60 ps, assigned to fast intra-/inter-chain energy migration and/or conformational relaxation along the polymer backbone,55,56 τCPE2 ∼ 400 ps, attributed to radiative relaxation of aggregated polymer clusters,57,58 and τCPE3 ∼ 800 ps, due to radiative decay of isolated polymer chains.56,57 The relative contribution of τCPE2 and τCPE3 to the total decay profile is thus dependent on the extent of polymer aggregation. Di-ureasils, also exhibit a tri-exponential decay curve in the solid-state at room temperature, with τDU1 < 1 ns, τDU2 ∼ 3.0–3.5 ns and τDU3 ∼ 9.0–9.8 ns.42
The emission decay curves were recorded for DI-PBS-1.0 using monitoring wavelengths at 10 nm intervals in the 400–500 nm spectral window (Fig. S13 and Table S3†). The decay curves are best fit with three discrete exponential components, whose lifetime (τi) and pre-exponential contribution (αi) exhibit significant wavelength dependence (Fig. 6c and d). Between λem = 400–480 nm, a short component of 50–200 ps (τ1), an intermediate component of 400–440 ps (τ2) and a long component of 0.7–1.5 ns (τ3) are identified. Each contribution becomes longer-lived as the monitoring wavelength is increased.
We now consider the origin of each component. τ1 is reminiscent of τCPE1 and is assigned to fast intra-/inter-chain energy migration. However, it is longer-lived, indicating that incorporation of the CPE into the di-ureasil decreases the rate of these relaxation processes. While τ1 increases up to λem = 460 nm, it subsequently decreases and disappears altogether at λem = 490 nm. The intermediate lifetime, τ2, is assigned to contributions from both τCPE2 and τDU1, with the CPE providing the major contribution at λem < 450 nm, switching to the di-ureasil at longer wavelengths. The pre-exponential coefficients support this assignment (Fig. 6d): τ1 appears as a decay time at the onset of the emission band and as a rise time at the end (demonstrated by the switch from a positive to negative α1 value). Simultaneously, the pre-exponential coefficient (α2) associated with τ2 increases in magnitude with analogous wavelength dependence, suggesting interconversion between the species is responsible for these decay pathways. A very long-lived component, τ4 ∼ 8 ns, emerges at emission wavelengths >480 nm, coinciding with the disappearance of τ1. The emission wavelength dependence and lifetimes of τ3 and τ4 are typical of τDU2 and τDU3, respectively. These observations support our assumption that at 420 nm the emission arises largely from the CPE, whereas at 500 nm the di-ureasil contribution predominates.
The PL decays for the DI-PBS-x series were also measured to probe the effect of the CPE concentration on the relaxation dynamics (Fig. S14†). Global analysis of the decay curves reveals that at 420 nm, τ1, τ2 and τ3 and their corresponding pre-exponential coefficients remain effectively constant as the CPE concentration is varied from 1–8 × 10–3 wt% (see Table 2). At the highest concentration, however, α1 decreases significantly and is accompanied by an increase in α2 and α3 (see Table 2). At 500 nm, while τ2 and τ3 remain constant, τ4 decreases significantly from 5.5 to 2.2 ns as the CPE wt% increases (Fig. 6e). This is accompanied by a simultaneous decrease in α2 and increase in α3 (Fig. 6f).
Table 2. Decay times (τi) and pre-exponential coefficients (αi) resulting from Global analysis of the photoluminescence decays (λex = 365 nm) of DI-PBS-x (λem = 420 nm).
| Sample | τ 1 (ns) | τ 2 (ns) | τ 3 (ns) | α 1 | α 2 | α 3 |
| DI-PBS-0.0 | 0.453 | 2.459 | 9.890 | 0.744 | 0.208 | 0.047 |
| DI-PBS-1.0 | 0.008 | 0.342 | 0.679 | 0.688 | 0.262 | 0.050 |
| DI-PBS-2.0 | 0.015 | 0.343 | 0.634 | 0.607 | 0.308 | 0.085 |
| DI-PBS-4.0 | 0.008 | 0.324 | 0.592 | 0.617 | 0.285 | 0.098 |
| DI-PBS-8.0 | 0.035 | 0.372 | 0.635 | 0.283 | 0.576 | 0.141 |
A similar pattern was observed for the DI-HTMA-x series (see Fig. S15 and Table S4†). We note that DI-HTMA-1.0 exhibits di-ureasil-like decay times at both 420 nm and 500 nm, which corroborates the earlier evidence from steady-state PL studies. Finally, the PL decay curves for the SP-PBS-x series were measured to probe the method of incorporation (see Fig. S16†). The trends in τi and αi are similar to those observed for the DI-PBS-x series at both emission wavelengths, but occur to a reduced extent, suggesting decreased electronic interaction between the CPE and the di-ureasil (see Table S5†). Moreover, at 420 nm, the emission decays are longer and are dominated by τ2 (∼500 ps), suggesting that aggregation of CPE chains is increased for the SP-PBS-x series (see Table S5†). These observations are consistent with the results from confocal microscopy, which show the CPE is confined to a surface layer.
Mechanism for photoluminescence enhancement?
The steady-state PL spectra clearly show that the emission originates primarily from the CPE. However, the di-ureasil is an “active” host, which also contributes to the emission properties of the hybrid sample. The significant spectral overlap indicates the potential for energy transfer (via either Dexter or Förster mechanisms) between the CPE and the di-ureasil and vice versa (Fig. 7). The time-resolved PL measurements confirm this hypothesis. As the detection wavelength is increased, the emission decay profile transitions from CPE-like to more di-ureasil-like (Fig. 6), with the CPE emission effectively switched-off at 460 nm.
Fig. 7. Schematic energy level diagram showing proposed mechanism for energy transfer between PBS-PFP and the di-ureasil host. The solid black lines represent the tabulated energies for the emissive states taken from ref. 59 for PBS-PFP and ref. 60 for the di-ureasil. The shaded bands correspond to the full-width half-maxima (FWHM) values of the corresponding emission spectra.

Fig. 7 presents a schematic diagram for the proposed energy transfer mechanism in DI-PBS. Energy transfer and charge carrier trapping both rely on electronic coupling of the donor and the acceptor, the extent of which will depend on both the spatial distribution of the CPE within the di-ureasil matrix and the energy of the electronic states involved. As noted earlier, the DI method results in the formation of CPE domains that are well-distributed throughout the di-ureasil matrix. However, despite displaying these structural characteristics of a composite material, the dramatic increase in the PLQY indicates a synergistic interaction more typical of a hybrid material occurs.24 The extended nature of the electronic states in conjugated polymers means that excitons are free to migrate along an individual backbone or to hop between chains.18 Both PBS-PFP and HTMA-PFP have relatively short chain lengths, corresponding to ∼8–9 repeat units. Assuming a conjugation length of ∼3–4 for a fluorene-phenylene copolymer,34 this implies that exciton migration along the polymer backbone should not be a significant prerequisite for energy transfer to the di-ureasil.
The strong spectral overlap means that it is challenging to unequivocally assign the direction of energy transfer upon excitation at 370 nm. However, detailed analysis of the emission lifetime data allows some conclusions to be drawn. The fastest decay component, τ1, becomes a rise time at 440 nm, and since α2 simultaneously increases, it is reasonable to propose that interconversion occurs between these two species. This response is characteristic of exciton energy transfer (EET) from a high to a lower energy chromophore.61 We therefore propose that τ1 corresponds to a combination of EET from the CPE to the di-ureasil and on-chain exciton migration. The lifetime and contribution of the longer-lived components (τ2, τ3, τ4) also depend on wavelength, which suggests that the absorbing and emitting species are chemically distinct.62 The intermediate lifetime, τ2, is similar to both τCPE2 (polymer clusters) and τDU1 and based on the steady state PL spectra, it seems reasonable that the CPE contribution dominates at λem < 450 nm, and switches to the di-ureasil at longer wavelengths. Notably, at 420 nm, the corresponding α2 is effectively independent of the CPE concentration, with the exception of the highest wt% studied. Moreover, at this wavelength, τ3 is comparable to the reported lifetime for radiative relaxation of isolated CPE chains (τCPE3)56,57 and does not fluctuate with the CPE wt%. These trends suggest that the di-ureasil matrix is extremely effective at inhibiting aggregation of the CPE. Between 440–500 nm, τ3 becomes longer-lived (1.3–1.6 ns) and is more reminiscent of the reported di-ureasil lifetime, τDU2.42 Remarkably, at λem = 500 nm, τ4 decreases significantly as the CPE wt% increases, with a concomitant rise in α3. This trend indicates that the longest-lived di-ureasil component undergoes energy transfer to isolated CPE chains rather than CPE clusters, which is supported by the constant α2 value at 420 nm. The fact that the di-ureasil can energy transfer to the CPE is fully consistent with the description of localised excited states in the inorganic moiety.29
The CPE emission profile dominates the steady-state photoluminescence spectra of the CPE-di-ureasils and must provide the major contribution to the PLQY. One of the key processes that controls the PLQY of emissive conjugated polymers is the quenching of excitons at non-emissive defect sites.63 Aggregation can increase the rate of exciton migration, leading to a decrease in the PLQY due to trapping at these sites. This should be exacerbated with increasing CPE concentration, which is in direct contrast to the trend observed here. The lifetime data indicate that the di-ureasil matrix is in fact very efficient at inhibiting CPE aggregation. Moreover, the FTIR data reveal a high degree of hydrogen-bonding is present in our system, and indeed this may be attributed in part to interactions between the di-ureasil and the polar terminal groups on the side chains of both CPEs. Particularly at higher concentrations, DI-PBS-x samples possess a less ordered di-ureasil matrix, suggesting greater interaction between CPE and host when compared with their DI-HTMA counterparts. Matrix-inhibited aggregation has recently been demonstrated as an effective mechanism to isolate CPE chains in poly(vinyl alcohol) (PVA) films.54 A significant increase in the PLQY was observed for water-soluble poly(fluorenephenylene) CPEs (including HTMA-PFP and PBS-PFP) upon their incorporation into the rigid, hydrogen-bonded PVA matrix, which was attributed to a combined reduction in chain flexibility and increase in chain isolation upon incorporation.52 Di-ureasils have also been shown to effectively inhibit the aggregation of molecular organic dyes (e.g. rhodamine B).64
In the CPE-di-ureasil system, however, the lifetime data indicate an energy transfer process also contributes to the enhanced PLQY. We propose that the CPE is the primary light-absorbing component and that after photogeneration, excitons migrate to lower energy subunits. Given the relatively short conjugation length of the CPEs used, it is reasonable to suggest that EET to the di-ureasil will be as, if not more, important than on-chain exciton migration. The individual lifetime components of the pure di-ureasil hybrid at room temperature have yet to be formally assigned.42 However, at 14 K, time-resolved emission spectra revealed the predominance of the “purple” component from silica defects at 1–5 ms, with the emergence of the lower-energy “blue” component associated with the NH centres at longer times (>10 ms).37 Interestingly, an abrupt decrease in the emission lifetime from the milli- to nanosecond timescale is observed at temperatures above 220 K.25 It is therefore tempting to assign τ4 at 500 nm to this emission component. The decrease in τ4 with increasing CPE concentration, in conjunction with the increase in α3, would suggest that energy transfer additionally occurs from the NH-centred trap states to the CPE. At first glance, analysis of the relative energy levels of the individual components indicates that this would require an uphill jump in energy. However, strong electronic coupling results in excitation sharing across a large number of sub-units (in both the CPE and the di-ureasil), meaning that there are a significant number of electronic energy levels in each exciton manifold.65 As such, a band structure description, rather than discrete energy levels, is more appropriate for this system. In this case, at room temperature (kBT ∼ 0.03 eV), there is sufficient band overlap to facilitate forward and thermally-activated reverse energy transfer between all components, provided that the long-range Coulomb transfer integrals are sufficient.65 This leads us to propose that EET occurs to an isolated CPE chain (as indicated by the lifetime data), which has a high radiative probability, giving rise to a long emission lifetime (τ3) and correspondingly high PLQY.
Conclusions
CPE-di-ureasil organic–inorganic composites have been successfully synthesised using mild, solution-processing conditions, with no phase separation observed. Direct Insertion leads to a homogeneous distribution of isolated CPE domains throughout the bulk sample, whereas Solvent Permeation results in the formation of a confined layer of the CPE at the surface. The structural integrity of the di-ureasil is retained upon incorporation of the CPE at all wt% concentrations investigated. However, the Solvent Permeation method does lead to disruption and reordering of the disordered hydrogen-bonded POE/urea associations present within the di-ureasil network.
The CPE and the di-ureasil both contribute to the photoluminescence properties of the material, which leads to a dramatic enhancement in the PLQY. This is partly due to effective disentanglement and isolation of individual CPE chains through weak physical interactions with the di-ureasil matrix, which are more favourable for PBS-PFP than HTMA-PFP. However, steady-state and ps-time-resolved emission studies also indicate that cooperative electronic interactions between the constituent building blocks are important. While the emission profile is predominantly characteristic of the poly(fluorene) CPE, the excitation energy dependence and red- and blue-edge band broadening are indicative of the di-ureasil contribution. The emission decay dynamics are complex but clearly demonstrate that the di-ureasil effectively isolates CPE chains, yielding emissive sites that have a high radiative probability. Moreover, intimate mixing between the two components and their high spectral overlap, lead to efficient energy transfer from both the siliceous and urea domains of the di-ureasil to these emissive centres, which results in the observed increase in the PLQY for the hybrid system.
CPE-di-ureasils and related hybrid materials offer a wealth of potential applications from hybrid photovoltaics, to optical sensors and luminescent solar concentrators. We note, in particular, that the electronic coupling between the CPE and the ureasil may be important for both TADF and light-harvesting in hybrid solar cells. This could be invaluable for extending the spectral response in perovskite or other photovoltaic devices. The power of the approach presented here lies both in its simplicity and versatility. Through judicious selection of the constituents, it is possible to tailor the optical properties (e.g. emission colour, energy transfer, PLQY) for a targeted application. Moreover, a huge variety of Jeffamine™ precursors66 are commercially-available, which also makes tuning of the mechanical properties possible (e.g. strength, flexibility, porosity). Controlled placement of the CPE is facilitated through the synthetic procedure used. Indeed, Solvent Permeation may offer a convenient route for the introduction of one or more lumophore layers within a confined region of an active layer. This approach has the potential to reduce the complexity of multi-layer device architectures and may yield improved device performance.
Supplementary Material
Acknowledgments
The authors thank Prof. Rute A. S. Ferreira for thoughtful insight into the discussion of the results presented. This work was supported in part by the Science Foundation Ireland under Grant No. 12/IP/1608. NWF thanks the Irish Research Council for a Government of Ireland postgraduate studentship. The authors acknowledge financial support from the European Commission under the Seventh Framework Programme by means of the grant agreement for the Integrated Infrastructure Initiative No. 262348 European Soft Matter Infrastructure (ESMI). Support from COST action MP1202 (HINT – Hybrid Interfaces) is also gratefully acknowledged. Solid-state NMR spectra were obtained at the EPSRC UK National Solid-state NMR Service at Durham. HDB thanks the Coimbra Chemistry Centre for support from the Fundação para a Ciência e a Tecnologia (FCT), Portuguese Agency for Scientific Research, through the project PEst-OE/QUI/UI0313/2014. This work was partially developed in the scope of the project CICECO-Aveiro Institute of Materials (Ref. FCT UID/CTM/50011/2013), financed by Portuguese funds through the FCT/MEC and when applicable co-financed by FEDER under the PT2020 Partnership Agreement.
Footnotes
†Electronic supplementary information (ESI) available: Synthesis and experimental methods, solvent permeation data, structural characterisation data (powder X-ray diffractograms, 29Si and 13C solid-state MAS NMR spectra), supporting PL and excitation spectra, ps-TCSPC decay curves and fitting data, and FTIR fitting data. See DOI: 10.1039/c5sc02409a
References
- Kim H. N., Guo Z., Zhu W., Yoon J., Tian H. Chem. Soc. Rev. 2011;40:79–93. doi: 10.1039/c0cs00058b. [DOI] [PubMed] [Google Scholar]
- Rochat S., Swager T. M. Angew. Chem., Int. Ed. 2014;53:9792–9796. doi: 10.1002/anie.201404439. [DOI] [PubMed] [Google Scholar]
- Wu C., Chiu D. T. Angew. Chem., Int. Ed. 2013;52:3086–3109. doi: 10.1002/anie.201205133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan C., Zhang K., Guan X., Zhong C., Xie H., Huang F., Chen J., Peng J., Cao Y. Chem. Sci. 2013;4:1298–1307. [Google Scholar]
- Ying L., Ho C.-L., Wu H., Cao Y., Wong W.-Y. Adv. Mater. 2014;26:2459–2473. doi: 10.1002/adma.201304784. [DOI] [PubMed] [Google Scholar]
- Zalar P., Henson Z. B., Welch G. C., Bazan G. C., Nguyen T.-Q. Angew. Chem., Int. Ed. 2012;51:7495–7498. doi: 10.1002/anie.201202570. [DOI] [PubMed] [Google Scholar]
- Huo L., Liu T., Sun X., Cai Y., Heeger A. J., Sun Y. Adv. Mater. 2015;27:2938–2944. doi: 10.1002/adma.201500647. [DOI] [PubMed] [Google Scholar]
- Wu J.-S., Cheng S.-W., Cheng Y.-J., Hsu C.-S. Chem. Soc. Rev. 2015;44:1113–1154. doi: 10.1039/c4cs00250d. [DOI] [PubMed] [Google Scholar]
- Liu C., Jang J., Xu Y., Kim H.-J., Khim D., Park W.-T., Noh Y.-Y., Kim J.-J. Adv. Funct. Mater. 2015;25:758–767. [Google Scholar]
- Nketia-Yawson B., Lee H.-S., Seo D., Yoon Y., Park W.-T., Kwak K., Son H. J., Kim B., Noh Y.-Y. Adv. Mater. 2015;27:3045–3052. doi: 10.1002/adma.201500233. [DOI] [PubMed] [Google Scholar]
- Park J., Lee C., Jung J., Kang H., Kim K.-H., Ma B., Kim B. J. Adv. Funct. Mater. 2014;24:7588–7596. [Google Scholar]
- Trattnig R., Pevzner L., Jäger M., Schlesinger R., Nardi M. V., Ligorio G., Christodoulou C., Koch N., Baumgarten M., Müllen K., List E. J. W. Adv. Funct. Mater. 2013;23:4897–4905. [Google Scholar]
- Hamadani B. H., Jung S., Haney P. M., Richter L. J., Zhitenev N. B. Nano Lett. 2010;10:1611–1617. doi: 10.1021/nl9040516. [DOI] [PubMed] [Google Scholar]
- Li G., Shrotriya V., Huang J., Yao Y., Moriarty T., Emery K., Yang Y. Nat. Mater. 2005;4:864–868. [Google Scholar]
- (a) Scherf U., List E. J. W. Adv. Mater. 2002;14:477–487. [Google Scholar]; (b) Monkman A., Rothe C., King S., Dias F. Adv. Polym. Sci. 2008;212:187–225. [Google Scholar]; (c) Henson Z. B., Müllen K., Bazan G. C. Nat. Chem. 2012;4:699–704. doi: 10.1038/nchem.1422. [DOI] [PubMed] [Google Scholar]
- Zheng H., Zheng Y., Liu N., Ai N., Wang Q., Wu S., Zhou J., Hu D., Yu S., Han S., Xu W., Luo C., Meng Y., Jiang Z., Chen Y., Li D., Huang F., Wang J., Peng J., Cao Y. Nat. Commun. 2013;4:1971. doi: 10.1038/ncomms2971. [DOI] [PubMed] [Google Scholar]
- Evans R. C., Marr P. C. Chem. Commun. 2012;48:3742–3744. doi: 10.1039/c2cc18022g. [DOI] [PubMed] [Google Scholar]
- Nguyen T. Q., Wu J., Doan V. V., Schwartz B. J., Tolbert S. H. Science. 2000;288:652–656. doi: 10.1126/science.288.5466.652. [DOI] [PubMed] [Google Scholar]
- Nguyen T. Q., Wu J., Tolbert S. H., Schwartz B. J. Adv. Mater. 2001;13:609–611. [Google Scholar]
- Wu J., Gross A. F., Tolbert S. H. J. Phys. Chem. B. 1999;103:2374–2384. [Google Scholar]
- Álvaro M., Corma A., Ferrer B., Galletero M. S., García H., Peris E. Chem. Mater. 2004;16:2142–2147. [Google Scholar]
- Aharon E., Kalina M., Frey G. L. J. Am. Chem. Soc. 2006;128:15968–15969. doi: 10.1021/ja066290m. [DOI] [PubMed] [Google Scholar]
- Evans R. C. J. Mater. Chem. C. 2013;1:4190–4200. [Google Scholar]
- (a) Sanchez C., Lebeau B., Chaput F., Boilot J.-P. Adv. Mater. 2003;15:1969–1994. [Google Scholar]; (b) Sanchez C., Julián B., Belleville P., Popall M. J. Mater. Chem. 2005;15:3559–3592. [Google Scholar]; (c) Nicole L., Laberty-Robert C., Rozes L., Sanchez C. Nanoscale. 2014;6:6267–6292. doi: 10.1039/c4nr01788a. [DOI] [PubMed] [Google Scholar]
- Carlos L. D., Ferreira R. A. S., Bermudez V. de Zea, Ribeiro S. J. L. Adv. Funct. Mater. 2001;11:111–115. [Google Scholar]
- de Zea Bermudez V., Carlos L. D., Alcácer L. Chem. Mater. 1999;11:569–580. [Google Scholar]
- Lima P. P., Ferreira R. A. S., Júnior S. A., Malta O. L., Carlos L. D. J. Photochem. Photobiol., A. 2009;201:214–221. [Google Scholar]
- Correia S. F. H., Lima P. P., André P. S., Ferreira R. A. S., Carlos L. D. Sol. Energy Mater. Sol. Cells. 2015;138:51–57. [Google Scholar]
- Ferreira R. A. S., Ferreira A. L., Carlos L. D. Eur. Phys. J. B. 2006;50:371–378. [Google Scholar]
- Uoyama H., Goushi K., Shizu K., Nomura H., Adachi C. Nature. 2012;492:234–238. doi: 10.1038/nature11687. [DOI] [PubMed] [Google Scholar]
- Sanchez C., Belleville P., Popall M., Nicole L. Chem. Soc. Rev. 2011;40:696–753. doi: 10.1039/c0cs00136h. [DOI] [PubMed] [Google Scholar]
- Burrows H. D., Lobo V. M. M., Pina J., Ramos M. L., de Melo J. S., Valente A. J. M., Tapia M. J., Pradhan S., Scherf U. Macromolecules. 2004;37:7425–7427. [Google Scholar]
- Evans R. C., Macedo A. G., Pradhan S., Scherf U., Carlos L. D., Burrows H. D. Adv. Mater. 2010;22:3032–3037. doi: 10.1002/adma.200904377. [DOI] [PubMed] [Google Scholar]
- Pinto S. M., Burrows H. D., Pereira M. M., Fonseca S. M., Dias F. B., Mallavia R., Tapia M. J. J. Phys. Chem. B. 2009;113:16093–16100. doi: 10.1021/jp904959h. [DOI] [PubMed] [Google Scholar]
- Yang R., Wu H., Cao Y., Bazan G. C. J. Am. Chem. Soc. 2006;128:14422–14423. doi: 10.1021/ja063723c. [DOI] [PubMed] [Google Scholar]
- HTMA-PFP is also referred to as PFP-NR3 in the literature. See ref. 33 and 56
- Carlos L. D., de Zea Bermudez V., Ferreira R. A. S., Marques L., Assunção M. Chem. Mater. 1999;11:581–588. [Google Scholar]
- Wu Z. G., Bouklas N., Huang R. Int. J. Solids Struct. 2013;50:578–587. [Google Scholar]
- Saikia D., Wu W.-H., Pan Y.-C., Liao C.-C., Chen C.-F., Fey G. T. K., Kao H. M. Electrochim. Acta. 2009;54:7156–7166. [Google Scholar]
- Ding Y., Li Y., Qin M., Cao Y., Wang W. Langmuir. 2013;29:13299–13306. doi: 10.1021/la4029639. [DOI] [PubMed] [Google Scholar]
- Plunkett K. N., Kraft M. L., Yu Q., Moore J. S. Macromolecules. 2003;36:3960–3966. [Google Scholar]
- Fu L., Ferreira R. A. S., Fernandes M., Nunes S. C., de Zea Bermudez V., Hungerford G., Rocha J., Carlos L. D. Opt. Mater. 2008;30:1058–1064. [Google Scholar]
- Fu L., Ferreira R. A. S., Silva N. J. O., Carlos L. D., de Zea Bermudez V., Rocha J. Chem. Mater. 2004;16:1507–1516. [Google Scholar]
- Scherrer equation: L = 0.94λ/(A cos θ), where A (in radians) is the full-width-half-maximum of the Bragg peak. See Guinier A., X-ray Diffraction in Crystals, Imperfect Crystals and Amorphous Bodies, Dover, New York, 1994. [Google Scholar]
- Dahmouche K., Carlos L. D., de Zea Bermudez V., Ferreira R. A. S., Santilli C. V., Craievich A. F. J. Mater. Chem. 2001;11:3249–3257. [Google Scholar]
- Mesquita M. E., Nobre S. S., Fernandes M., Ferreira R. A. S., Santos S. C. G., Rodrigues M. O., Carlos L. D., de Zea Bermudez V. J. Photochem. Photobiol., A. 2009;205:156–160. [Google Scholar]
- Fernandes M., de Zea Bermudez V., Ferreira R. A. S., Carlos L. D., Martins N. V. J. Lumin. 2008;128:205–212. [Google Scholar]
- (a) Sachs-Quintana I. T., Heumüller T., Mateker W. R., Orozco D. E., Cheacharoen R., Sweetnam S., Brabec C. J., McGehee M. D. Adv. Funct. Mater. 2014;24:3978–3985. [Google Scholar]; (b) Loy D. E., Koene B. E., Thompson M. Adv. Funct. Mater. 2002;12:245–249. [Google Scholar]
- (a) Kuik M., Wetzelaer G. A. H., Laddé J. G., Nicolai H. T., Wildeman J., Sweelssen J., Blom P. W. M. Adv. Funct. Mater. 2011;21:4502–4509. [Google Scholar]; (b) Chan K. L., Sims M., Pascu S. I., Ariu M., Holmes A. B., Bradley D. D. C. Adv. Funct. Mater. 2009;19:2147–2154. [Google Scholar]; (c) Yang X. H., Jaiser F., Neher D., Lawson P. V., Brédas J., Zojer E., Güntner R., de Freitas P. S., Forster M., Scherf U. Adv. Funct. Mater. 2004;14:1097–1104. [Google Scholar]
- Carlos L. D., Ferreira R. A. S., Pereira R. N., Assunção M., de Zea Bermudez V. J. Phys. Chem. B. 2004;108:14924–14932. [Google Scholar]
- Burrows H. D., Tapia M. J., Fonseca S. M., Valente A. J., Lobo V. M., Justino L. L., Qiu S., Pradhan S., Scherf U., Chattopadhyay N., Knaapila M., Garamus V. M. ACS Appl. Mater. Interfaces. 2009;1:864–874. doi: 10.1021/am800267n. [DOI] [PubMed] [Google Scholar]
- Geoghegan M., Krausch G. Prog. Polym. Sci. 2003;28:261. [Google Scholar]
- We note that for optically-thick samples, waveguiding of the emitted light and/or reabsorption of the emitted photons may cause the measured PLQY to deviate from its true value. However, correction of the PLQY to account for re-absorption/re-emission processes in these samples using the method described by Ahn et al. results in a negligible change in the PLQY for the CPE-di-ureasil samples reported here. Further details may be found in the ESI. T.-S. Ahn, R. O. Al-Kayasi, A. M. Müller, K. M. Wentz, C. J. Bardeen, Rev. Sci. Instrum. 78, 2007, 086105
- Al-Attar H. A., Monkman A. P. Adv. Funct. Mater. 2012;22:3824–3832. [Google Scholar]
- Costa T., Marques A. T., Seixas de Melo J. S., Thomas A. W., Garner L. E., Scherf U., Bazan G. C., Burrows H. D. J. Phys. Chem. B. 2014;118:613–623. doi: 10.1021/jp409577y. [DOI] [PubMed] [Google Scholar]
- Davies M. L., Douglas P., Burrows H. D., Miguel M. da G., Douglas A. J. Phys. Chem. B. 2011;115:6885–6892. doi: 10.1021/jp202446a. [DOI] [PubMed] [Google Scholar]
- Attar H. A. A., Monkman A. P. Adv. Funct. Mater. 2008;18:2498–2509. [Google Scholar]
- Marques A. T., Burrows H. D., Seixas de Melo J. S., Valente A. J., Justino L. L., Scherf U., Fron E., Rocha S., Hofkens J., Snedden E. W., Monkman A. P. J. Phys. Chem. B. 2012;116:7548–7559. doi: 10.1021/jp3000703. [DOI] [PubMed] [Google Scholar]
- Fonseca S. M., Pina J., Arnaut L. G., Seixas de Melo J., Burrows H. D., Chattopadhyay N., Alcácer L., Charas A., Morgado J., Monkman A. P., Asawapirom U., Scherf U., Edge R., Navaratnam S. J. Phys. Chem. B. 2006;110:8278–8283. doi: 10.1021/jp060251f. [DOI] [PubMed] [Google Scholar]
- Nobre S. S., Lima P. P., Mafra L., Ferreira R. A. S., Freire R. O., Fu L. S., Pischel U., de Zea Bermudez V., Malta O. L., Carlos L. D. J. Phys. Chem. C. 2007;111:3275–3284. [Google Scholar]
- Snedden E. W., Cury L. A., Bourdakos K. N., Monkman A. P. Chem. Phys. Lett. 2010;490:76–79. [Google Scholar]
- Strickler S. J., Berg R. A. J. Chem. Phys. 1962;37:814. [Google Scholar]
- Hintschich S. I., Rothe C., Sinha S., Monkman A. P., Scandiucci de Freitas P., Scherf U. J. Chem. Phys. 2003;119:12017–12022. [Google Scholar]
- Fernández J., García-Revilla S., Carlos L. D., Pecoraro E., Arriandiaga M. A., Balda R. Laser Photonics Rev. 2014;8:L32–L36. [Google Scholar]
- Scholes G. D., Rumbles G. Nat. Mater. 2006;5:683–696. doi: 10.1038/nmat1710. [DOI] [PubMed] [Google Scholar]
- Huntsman International LLC, Products, http://www.huntsman.com/performance_products/a/Products, accessed May 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.