

Abstract
Growth factors, such as the transforming growth factor beta (TGFβ), play an important role in promoting metastasis of prostate cancer, thus understanding how TGFβ could induce prostate cancer cell migration may enable us to develop targeted strategies for treatment of advanced metastatic prostate cancer. To more clearly define the mechanism(s) involved in prostate cancer cell migration, we undertook a series of studies utilizing non‐malignant prostate epithelial cells RWPE1 and prostate cancer DU145 and PC3 cells. Our studies show that increased cell migration was observed in prostate cancer cells, which was mediated through epithelial‐to‐mesenchymal transition (EMT). Importantly, addition of Mg2+, but not Ca2+, increased cell migration. Furthermore, TRPM7 expression, which functions as an Mg2+ influx channel, was also increased in prostate cancer cells. Inhibition of TRPM7 currents by 2‐APB, significantly blocked cell migration in both DU145 and PC3 cells. Addition of growth factor TGFβ showed a further increase in cell migration, which was again blocked by the addition of 2‐APB. Importantly, TGFβ addition also significantly increased TRPM7 expression and function, and silencing of TRPM7 negated TGFβ‐induced cell migration along with a decrease in EMT markers showing loss of cell adhesion. Furthermore, resveratrol, which decreases prostate cancer cell migration, inhibited TRPM7 expression and function including TGFβ‐induced cell migration and activation of TRPM7 function. Together, these results suggest that Mg2+ influx via TRPM7 promotes cell migration by inducing EMT in prostate cancer cells and resveratrol negatively modulates TRPM7 function thereby inhibiting prostate cancer metastasis.
Keywords: cell migration, EMT, magnesium, prostate cancer, TRPM7
1. INTRODUCTION
Prostate cancer (PCa) is the most common male malignancy and the second most prevalent cause of cancer‐related death accounting for more than 30 000 deaths per year in the US. Although prostate cancer can be treated when diagnosed at an early stage, patients with advanced or metastatic disease have an average 5‐year survival rate of less than 35%.1, 2 Divalent cations such as calcium (Ca2+) and magnesium (Mg2+) have been shown to play a fundamental role in many cellular processes including cell proliferation, survival and cancer metastasis. Importantly, appropriate intracellular Mg2+ levels are essential for adequate function of nucleic acid metabolism, protein synthesis, and energy production.3, 4 Interestingly, it has been recently proposed that alterations in Mg2+ homeostasis affects many cellular functions that are critical for tumor growth and invasion, such as proliferation, migration, and angiogenesis.5, 6, 7, 8 In addition, factors that increase channel activity to promote Mg2+ entry in prostate cancer cells are also not well established. Among many ion channels present in mammalian cells, the transient receptor potential channels (TRPs) function as nonselective cation channels that are mainly permeable to Ca2+, Mg2+, Na+, and K+. The TRP family is divided into three subfamilies: canonical (TRPC), vanilloid (TRPV), and melastatin type (TRPM). Interestingly, the eight TRPM family members differ significantly from other TRP channels in terms of domain structure, cation selectivity, and activation mechanisms. Furthermore, abnormal activation of TRPM channels has a profound influence on various pathologic processes.
Of the eight TRPM members, subtype six and seven have been shown to conduct Mg2+ at negative membrane potentials.9 As compared to TRPM6, TRPM7 channels are widely expressed ion channels and support multiple cellular and physiological functions, including cellular Mg2+ homeostasis, cell viability and growth, neuronal cell death, synaptic transmission, cell adhesion, intestinal pacemaking and growth/proliferation of human carcinoma cells.10, 11 We have previously shown that TRPM7 is associated with cell proliferation and survival of prostate cancer cells.5, 12 Similarly, studies have also indicated that TRPM7 plays a key role in prostate cancer cells.13, 14, 15 Other studies have shown that TRPM7 is partially associated with epidermal growth factor (EGF)‐induced epithelial to mesenchymal transition (EMT) in breast cancer cells16; however its role in EMT in prostate cancer is not well defined. EMT is a process characterized by repression of E‐cadherin expression, production of the type‐III intermediate filament protein vimentin, and increases in cell migration, invasion, and initiation of metastasis. It is well established that during EMT, cells transform from epithelial to mesenchymal phenotype that leads to the loss of cell‐cell adhesion to promote metastatic potential. Expression of EMT markers (manifested by reduced E‐cadherin and induced N‐cadherin and vimentin expression) is a feature of many cancer subtypes with poor clinical prognoses, and there is evidence that EMT mediates the carcinogenesis of prostate cancer cells.17, 18, 19
Growth factors perform a vital role in the growth of normal, hyperplastic, and malignant prostatic epithelium. Several growth factors such as epidermal growth factor (EGF) family, the fibroblast growth factor (FGF) family, the insulin‐like growth factor (IGF) family, the transforming growth factor‐beta (TGFβ) family, and the vascular endothelial growth factor (VEGF) family have shown to play a critical role in prostate cancer development.20 Interestingly, TGFβ is found in a variety of tissues and is important for angiogenesis, promotes extracellular matrix formation, and suppresses the immune response.21 Since TGFβ promotes angiogenesis, it can bring nutrients and oxygen to tumor cells and also function as an immune suppressor thereby protecting cancer cells from the host immune system. Additionally, it can enhance the invasiveness and metastatic ability of malignant cancer cells. TGFβ has an interesting role in cancer since it can act as either stimulator or inhibitor of cellular growth, however the role is dependent on cell type and the state of cellular differentiation. In prostate cancer, TGFβ levels have been shown to be increased and are associated with tumor development and progression,22, 23 but the mechanism as to how TGFβ promotes metastatic ability is not clear. The purpose of the current study was to determine the mechanism of how growth factors, mainly TGFβ, promotes cell migration and metastasis of prostate cancer cells. Our data suggests that TGFβ promotes cancer cell migration by enhancing EMT. Furthermore, we showed that Mg2+ influx, which was mediated via TRPM7 channels is essential for EMT‐induced cancer cell migration. Moreover, TGFβ enhances TRPM7 expression and function and inhibition of TRPM7 activity or suppression of TRPM7 expression inhibits cell migration by blocking EMT.
2. MATERIALS AND METHODS
2.1. Cell culture reagents and transfection
Control human prostate cell line RWPE1 (CRL 11609), human prostate cancer cell line DU145 and PC3 cells were obtained from the American Type Culture Collection (Manassas, VA). RWPE1 cells were cultured in Keratinocyte Serum Free Medium (include bovine pituitary extract and EGF 1‐53) and DU145 and PC3 cells were cultured in MEM medium along with 10% FBS as suggested by ATCC. Cells were maintained at 37°C with 95% humidified air and 5% CO2 and were passaged as needed. Culture medium was changed twice weekly and cells were maintained in complete media, until reaching 90% confluence. For transfection experiments, shRNA plasmid that targets the coding sequence of human TRPM7 were obtained from Origene. Cells were transfected with individual shRNA (50 nM) or control plasmid (50 nM) using Lipofectamine 2000 in Opti‐MEM medium as per supplier's instructions (Invitrogen, Carlsbad, CA) and assayed after 48 h. All other reagents used were of molecular biology grade obtained from Sigma chemicals (Sigma, St. Louis, MO) unless mentioned otherwise.
2.2. Cell migration assays
For wound healing assays, cells were grown on a 12‐well plate until 95% confluence in the regular medium. Prior to the experiment cells were serum starved for 3 h along with the addition of 1 µg/mL of mitomycin C to inhibit cell proliferation. All cells were then incubated in their respective media with 10% FBS and the monolayer was scratched using a 200 µL pipette tip. Finally, images were taken using an Olympus CKX41 microscope with QCapture ×64 software (Surrey, Canada) immediately after the scratch, marked as 0 h and after 24 h. For transwell migration assays, 25 000 DU145 cells were suspended in serum free media along with treatments indicated in figures and placed in transwell inserts with 8 µM pore membranes (Corning, New York, NY). In all experiments, lower wells contained MEM + 10% FBS and cells were allowed to migrate for 6 h. For pre‐treatment experiments, cells were pretreated with shTRPM7 for 48 h and/or TGFβ for 24 h. Cells on the inside of the transwell membrane were removed with a cotton swab and cells on the lower side of membrane were fixed with methanol and stained with hematoxylin. Four random fields at 10× were counted indicating total cells migrated.
2.3. Magnesium/Calcium imaging
Cells were incubated with 2 µM Mag‐Fura 2‐AM (Invitrogen) or Fura‐2 (Molecular Probes, Eugene, OR for 45 min, washed twice with SES (Standard External Solution, includes: 10 mM HEPES, 120 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM glucose, pH 7.4) buffer. For fluorescence measurements, the fluorescence intensity of Fura‐2‐loaded control cells was monitored with a CCD camera‐based imaging system (Compix) mounted on an Olympus XL70 inverted microscope equipped with an Olympus 40× (1.3 NA) objective. A monochromator dual wavelength enabled alternative excitation at 340 and 380 nm, whereas the emission fluorescence was monitored at 510 nm with an Orca Imaging camera (Hamamatsu, Japan). The images of multiple cells collected at each excitation wavelength were processed using the C imaging, PCI software (Compix Inc., Cranbery, PA). Fluorescence traces shown represent [Mg2+]i values that are averages from at least 30 to 40 cells and are a representative of results obtained in at least 3‐4 individual experiments. [Mg2+]i or [Ca2+]i in individual cells were estimated from the 340/380‐nm ratio according to the Grynkiewicz equation formula, using the indicator's dissociation constant for Mg2+(1.5 mM) or Ca2+ (0.22 µM).24 The bar diagram shown in the figures represents the [Mg2+]i or [Ca2+]i in concentrations.
2.4. Electrophysiology
For patch clamp experiments, coverslips with cells were transferred to the recording chamber and perfused with an external Ringer's solution of the following composition (mM): NaCl, 145; CsCl, 5; MgCl2, 1; CaCl2, 1; Hepes, 10; Glucose, 10; pH 7.3 (NaOH). Whole cell currents were recorded using an Axopatch 200B (Axon Instruments, Inc., San Jose, CA). The patch pipette had resistances between 3 and 5 MΩ after filling with the standard intracellular solution that contained the following (mM): cesium methane sulfonate, 150; NaCl, 8; Hepes, 10; EDTA, 10; pH 7.2 (CsOH). To record SOCE currents, 5 mM MgCl2 was add to the intracellular solution, which was to block the TRPM currents. Osmolarity for all solutions was adjusted with D‐mannitol to 305 ± 5 mmol/kg. With a holding potential 0 mV, voltage ramps ranging from −100 mV to +100 mV and 100 ms duration were delivered at 2s intervals after whole cell configuration was formed. Currents were recorded at 2 kHz and digitized at 5‐8 kHz. pClamp 10.1 software was used for data acquisition and analysis. Basal leak currents were subtracted from the final currents and average currents are shown. All experiments were carried out at room temperature.
2.5. Membrane preparations and Western blot analyses
Cells were harvested and stored at −80°C. Crude lysates were prepared from RWPE1 and DU145 cells as described previously in Singh et al.25 Protein concentrations were determined, using the Bradford reagent (Bio‐Rad, Hercules, CA), and 25‐50 μg of proteins were resolved on 3‐8% SDS‐Tris‐acetate gels, transferred to PVDF membranes and probed with respective antibodies. TRPM7 (Epitomics, Burlingame, CA), TRPM6 (Epitomics), E‐cadherin (Santa Cruz, Dallas, TX), N‐cadherin (Santa Cruz), and Vimentin (Cell Signaling, Danvers, MA) and β‐actin (Santa Cruz) antibodies were used at 1:1000 dilution to probe for respective proteins. Peroxidase conjugated respective secondary antibodies were used to label the proteins. The proteins were detected by enhanced chemiluminescence detection kit (SuperSignal West Pico; Pierce). Densitometric analysis was performed using image J analysis and results were corrected for protein loading by normalization for β‐actin expression as described in ref.25, 26, 27, 28
2.6. Statistics
Data analysis was performed using Origin 7.0 (OriginLab) and Graphpad prism 6.0. Statistical comparisons were made using Student's t‐test or one‐way ANOVA. Experimental values are expressed as means ± SEM or SD. Differences in the mean values were significant at P‐value <0.05*, <0.01**, or <0.001*** respectively.
3. RESULTS
3.1. Magnesium levels via TRPM7 promotes EMT induced cell migration in prostate cancer cells
We first examined the ability of prostate cells to migrate by utilizing human non‐malignant prostate epithelial cells RWPE1 and human metastatic prostate cancer DU145 or PC3 using the wound‐healing assay for cellular migration. As shown in Figure 1A, control non‐cancerous RWPE1 cells showed only a modest increase in cell migration as compared with DU145 cells. Similar results were also observed using the metastatic prostate cancer PC3 cells, which also showed a significant increase in cell migration (cumulative data presented in supplemental Figure S2). Importantly, a key feature for prostate cancer metastasis is the initiation of EMT, thus we looked at various EMT markers. Epithelial non‐cancerous cells, such as RWPE1, are shown to express high levels of E‐cadherin, whereas mesenchymal cells, such as DU145 and PC3, express N‐cadherin and vimentin. E‐cadherin is required for the formation of stable adherens junctions and thus the maintenance of an epithelial phenotype, and its loss is considered a fundamental event in EMT. As expected, expression of E‐cadherin was present in control RWPE1 cells and although a modest cell migration was observed in RWPE1 cells, there was no increase in either vimentin or N‐cadherin (Figure 1B). In contrast, the E‐cadherin level was substantially lower in prostate cancer DU145 cells when compared with control RWPE1 cells, and a subsequent higher expression of vimentin and N‐cadherin was observed in prostate cancer DU145 and PC3 cells (Figure 1B). These results suggest that prostate cancer DU145 and PC3 cells express higher mesenchymal markers and has increased cellular migration.
Figure 1.
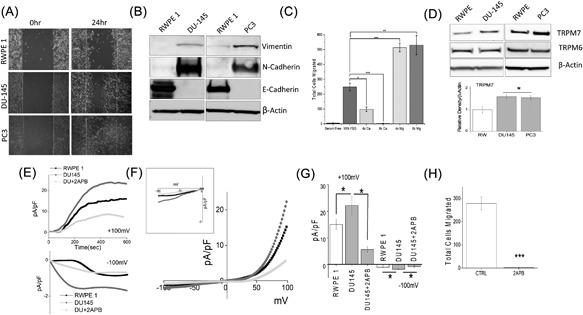
(A) Respective images showing the wound‐healing assay for cellular migration in human prostate epithelial cell RWPE1 and human prostate cancer cell line DU145 and PC3. Images were taken after the wound scratch (0 h) and after 24 h. The pictures are representative of 4 separate experiments performed in duplicate. (B) Western blots showing the expression of Vimentin, N‐cadherin, E‐cadherin, and loading control β‐actin in RWPE1, DU145, and PC3 cells. (C) DU145 cells were migrated using transwell inserts with altered Ca2+ and Mg2+ concentrations in both the upper and lower wells and cells were allowed to migrate for 6 h. 4× and 8× Ca2+ (1.6 mM and 3.2 mM) and Mg2+ (1.6 mM and 3.2 mM) concentrations were made by the addition of CaCl2 or MgCl2 to RPMI + 10% FBS. Four random fields at 10× were counted from three separate experiments indicating total cells migrated in each conditions ± SEM * indicate significance (*P < 0.05, ***P < 0.001). (D) Western blots showing the expression of TRPM6 and TRPM7 and loading control β‐actin in RWPE1, DU145, and PC3 cells. Corresponding densitometric reading of the TRPM7 protein is shown as a bar diagram and each bar is the mean ± SEM of three separate experiments. * indicates significance (P < 0.05). (E) In normal SES (1 mM Ca2+, 1 mM Mg2+) bath solution, whole cell recording showing outward/inward currents at +100 mV/‐100 mV in RWPE1 and DU145 cells. IV curves under various conditions as labelled are shown in (F) (Insert indicate the magnification of inward IV curves) and quantitation (8‐10 recordings) of current intensity at ±100 mV is shown in (G). * indicate significance (P < 0.05). (H) DU145 cells were placed in transwell inserts with RPMI medium with and without 50 μM 2‐APB. Nonmigrating cells were removed from upper membrane and migrated cells were fixed and stained with hematoxylin. Four random fields at 10× were counted from four separate experiments indicating total cells migrated ± SEM. *** indicates significance (P < 0.001)
Growing evidence shows that cancer cell hallmarks are strongly regulated by ion channels including Ca2+, 29, 30, 31 Mg2+,32, 33, 34 and K+,35, 36, 37 Na+ channels.38, 39 Thus, we further investigated the effect of extracellular Ca2+ and Mg2+ on the migration of prostate cancer cells DU145. Importantly, the ratio of Ca2+ to Mg2+ was important for the ability of DU145 cells to migrate using a transwell migration assay (Figure 1C). Stimulation of cells (using FBS) lead to an increase in the migration of DU145 cells (5.33 ± 0.72 in SFM, 251.30 ± 20.27 in 10%FBS); however, increasing Ca2+ concentrations resulted in a gradual reduction in the migration of DU145 cells with complete inhibition of cell migration observed at 8x Ca2+ concentration (Figure 1C). In contrast, increasing the Mg2+ concentration resulted in a significant increase in cell migration, where both 4× (513.67 ± 28.35) and 8× (530.00 ± 64.76) concentration showed similar migration capacity (Figure 1C). Together these results suggest that ion channels involved in Mg2+ homeostasis/entry could promote prostate cancer cell migration. Thus, we next evaluated the expression of TRPM (mainly 6, and 7) channels that are known to modulate Mg2+ homeostasis/entry. Interestingly, TRPM7 but not TRPM6 levels, were significantly higher (Figure 1D) in DU145 and PC3 cancer cells as compared with normal RWPE1 cells (Figure 1D). To further characterize the ion channel(s) involved in cell migration, electrophysiological recordings were performed. As shown in Figures 1E and 1F, a strong outward rectifying non‐selective current was detected in both control and DU145 cells, which has similar current characteristics to what has been previously reported for TRPM6/7 channels.5, 12 More importantly, the TRPM6/7 channel activity was upregulated in cancer cells, when compared with control cells (at +100 mV, 15.01 ± 1.89 pA/pF in RWPE, 22.14 ± 3.40 pA/pF in DU145) (Figure 1E‐G). To further characterize these currents on the basis of their pharmacological properties, we treated the cells with 50 μM 2‐APB, which is previously shown to potentiate TRPM6 currents, whereas TRPM7 currents are inhibited.40, 41 Addition of 2‐APB strongly inhibited currents in DU145 cells, suggesting that TRPM7 rather than TRPM6 underlies this current in cancer cells. (Figure 1E‐G). Importantly, treatment of 2‐APB also significantly inhibited migration in DU145 cancer cells (251.30 ± 20.27 in control, whereas 9.78 ± 2.52 was observed in the presence of 2‐APB) (Figure 1H). As 2‐APB also effect SOCE currents, which may confound the results, we performed SOCE current recordings in these cells. Under similar condition, within 10‐20 min, no recordable SOCE currents was observed in these cells (Supplemental Figure S1A). Collectively, these results demonstrate that TRPM7 may play an important role in inducing EMT in prostate cancer cells.
3.2. TGFβ induces TRPM7 expression and enhances EMT in prostate cancer cells
TGFβ is known to induce tumor cell invasion by activating EMT in metastatic cancer thereby enhancing invasiveness and metastasis.29, 42 Thus, we further evaluated the role of TRPM7 in TGFβ‐induced EMT in prostate cancer cells using both transwell and wound healing assays. Importantly, increase in migration of DU145 and PC3 cells were observed in cells treated with TGFβ, but was attenuated when cells were pretreated with 2‐APB (Figure 2A, quantification provided in supplemental Figures S2B and S2C). Transwell migration assays also showed similar results where addition of 2‐APB eliminated the increase in TGFβ‐induced migration of DU145 cells (209.00 ± 13.19 in control, vs 263.80 ± 9.30 in TGFβ, 126.00 ± 19.29 in 2‐APB) (Figure 2C). To further establish that increase in cell migration is due to EMT, expression of proteins that are associated with EMT were assessed. Again, E‐cadherin levels were decreased along with a subsequent increase in both N‐cadherin and vimentin levels in cells treated with TGFβ (Figure 2B, quantification provided in supplemental Figures S1B and S1C). Interestingly, the expression of TRPM7 also increased in a time dependent manner upon treatment with TGFβ, which mimicked increased expression of EMT marker vimentin (Figure 2D, quantification provided in supplemental Figure S1D). Notably, cells treated with TGFβ showed a significant increase in TRPM7 currents without altering the current‐voltage (I‐V) relationship (at +100 mV, 22.14 ± 3.40 pA/pF in control, 31.06 ± 3.84 pA/pF in TGFβ). In contrast, addition of 50μM 2‐APB to DU145 cells significantly abolished this TGFβ‐mediated increase (at +100 mV, 8.09 ± 1.15 pA/pF) (Figures 2E and 2F). Overall, the data presented thus far strongly suggests a correlation between TRPM7 and TGFβ‐mediated increase in EMT in prostate cancer cells.
Figure 2.
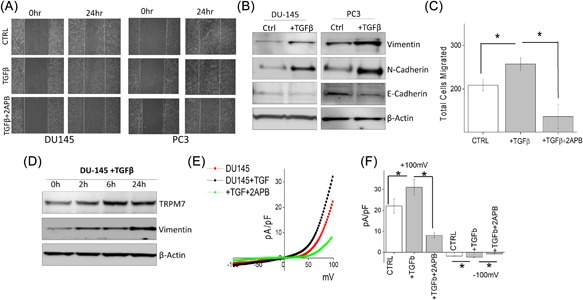
(A) Images showing the wound‐healing assay for cellular migration in human prostate cancer cell line DU145 and PC3 pretreated for 24 h with 5 ng/mL of TGFβ or TGFβ and 50 μM 2‐APB. Images were taken immediately after the wound scratch (0 h) and after 24 h. The pictures are representative of four separate experiments. (B) Western blotting showing the expression of Vimentin, N‐cadherin, E‐cadherin, and loading control β‐actin in DU145 and PC3 cells. (C) DU145 cells were treated with 5 ng/mL TGFβ for 24 h before being trypsinized, re‐suspended in serum free media with or without 50 μM 2‐APB, and placed in transwell inserts for 6 h. Four random fields at 10× were counted from four separate experiments indicating total cells migrated ± SEM. * indicates significance (P < 0.05). (D) DU145 cells were treated with TGFβ for various times as included in the figure, lysed proteins were subjected to SDS‐PAGE followed by Western blotting with respective antibodies. (E) IV curves of TRPM7 current under various conditions (control, TGFβ or TGFβ and 2‐APB.) and quantitation (8‐10 recordings) of current intensity at ±100 mV is shown in (F). * indicates significance (P < 0.05)
3.3. Loss of TRPM7 expression inhibits TGFβ induced increase in EMT
To date, specific pharmacological TRPM7 blockers are not available, 2‐APB is a non‐specific inhibitor that also inhibit other calcium channels, thus we used genetic methods to inhibit TRPM7 expression and directly explored the role of TRPM7 channel in EMT. DU145 cells transfected with control shRNA (mock) showed no change in TRPM7 expression. However, silencing of TRPM7 (using shTRPM7) resulted in a significant decrease in TRPM7 expression (Figure 3A). Importantly, decreased EMT markers vimentin and N‐cadherin was also observed in TRPM7 silenced cells when compared with control mock transfected cells (Figure 3A). Furthermore, TRPM7 silenced cells treated with TGFβ failed to restore vimentin and N‐cadherin expression when compared with mock treated cells (Figure 3A). Moreover, at a functional level, TGFβ showed an increase in TRPM7 currents, but the effect of TGFβ on TRPM7 currents was significantly abolished by the transfection of shTRMP7(at +100 mV, 22.14 ± 3.40 pA/pF in control, 31.14 ± 3.84 pA/pF in TGFβ, 6.80 ± 1.86 pA/pF in TGFβ + shM7) (Figure 3B‐D). TRPM7 silencing failed to affect basal Ca2+ levels; whereas, a significant decrease in basal Mg2+ levels were observed in DU145 cells (Figure 3E). Interestingly, elevating extracellular Mg2+ induced an increase in intracellular MagFura‐2 fluorescence, which was abolished by TRMP7 silencing indicating that Mg2+ entry is mainly regulated by TRPM7 channels (Figures 3F and 3G). To further establish that cell migration is also affected upon TRPM7 silencing, wound healing assays were performed under these conditions. Consistent with the results shown above, cells pretreated with TGFβ showed an increase in cell migration when compared with control untreated, whereas silencing of TRPM7 attenuated TGFβ‐induced cell migration (Figure 3H, quantification provided in supplemental Figure S2D). Additionally, results of transwell migration assays also indicate that TGFβ promotes cell migration of DU145 cells (202.00 ± 12.20 in control, 263.80 ± 9.33 in TGFβ treated cells), whereas silencing of TRPM7 significantly reduces the ability of DU145 cells to migrate and attenuate TGFβ‐induced cell migration (Figure 3I). Similar results were also observed with PC3 cells, where knockdown of TRPM7 significantly decreased cell migration (data not shown). These results indicate that loss of TRPM7 will reverse the EMT status and inhibit cell migration, which is consistent with a recently published report.13 Taken together, these results, show that TRPM7 not only regulates EMT, but also modulates TGFβ‐induced enhancement of EMT.
Figure 3.
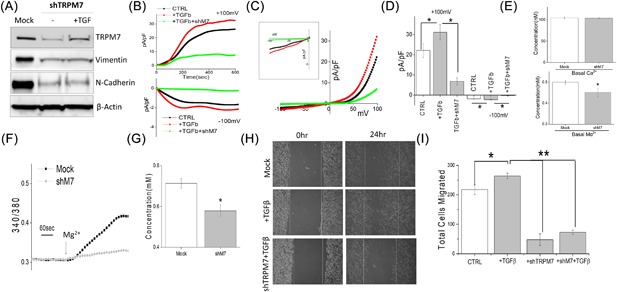
(A) Western blot showing the expression of TRPM7, Vimentin, N‐cadherin, and β‐actin as loading control in DU145 cells under various conditions. Mock indicates cells treated with control shRNA, whereas the other set of cells were treated with shTRPM7 with and without TGFβ. Whole cell recordings showing outward/inward currents at +100 mV/‐100 mV under various conditions in DU145 cells are shown in (B). Respectively IV curves are shown in (C) (insert indicate the magnification of inward IV curves) and quantitation (5‐8 recordings) of current intensity at ±100 mV is shown in (D). * indicate significance (P < 0.05). (E) Quantification of basal intracellular calcium or magnesium concentration under normal SES solution in mock and TRPM7 knockdown DU145 cells. (F) Mg2+ imaging was performed using mag‐fura in mock and TRPM7 knockdown DU145 cells. Bath application of Mg2+ induces Mg2+ influx and analog plots of the fluorescence ratio (340/380) from an average of 30‐50 cells are shown. (G) Quantification (mean ± SD) of Mg2+ concentration. * indicate significance (P < 0.05). (H) Respective images showing the wound‐healing assay for cellular migration in DU145 pretreated for 24 h with 5 ng/mL TGFβ in mock (control shRNA) and TRPM7 knockdown DU145 cells (shTRPM7) pretreated with 24 h with TGFβ. Images were taken after the wound scratch (0 h) and after 24 h, respectively. The pictures are representative of four separate experiments. (I) Transwell migration of mock or shTRPM7 treated cells after TGFβ stimulation with and migrated for 6 h. Four random fields at 10× were counted from four experiments indicating total cells migrated ± SEM. *** indicates significance (P < 0.001)
3.4. RSV inhibits EMT via blocking TRPM7 channel activity in prostate cancer cells
Resveratrol (RSV), a natural polyphenol found in wide variety of plants and fruits, has been suggested to induce cell cycle arrest and activate apoptosis‐mediated cell death in cancer cells. Importantly, the anti‐carcinogenic effect of RSV in prostate cancer has been well established and studies have noticed that RSV inhibits the proliferation of androgen dependent and independent prostate cancer cell lines along with inhibiting EMT.43, 44, 45, 46 To assess whether TRPM7 channels are critically involved in RSV‐mediated inhibition on EMT, analysis of cell migration was performed. In DU145 and PC3 cells, pretreatment with RSV led to an inhibition of cell migration as observed using wound healing assays when compared to untreated cells (Figure 4A, quantification provided in supplemental Figures S2E and S2F). Moreover, transwell migration assays also showed that RSV treatment of DU145 cells resulted in a significant reduction in total number of cells migrated (268.00 ± 10.70 in control vs 123.90 ± 9.78 in RSV) (Figure 4C). Expression of EMT markers vimentin and N‐cadherin was also significantly decreased in RSV treated cells with a complete loss of vimentin and N‐cadherin within 24 h of RSV treatment (Figure 4B). Importantly, in both DU145 and PC3 cells, TRPM7 expression levels were also noticeably downregulated by RSV treatment, which is correlated to the levels of EMT markers (Figure 4B, quantification provided in supplemental Figure S1E). Correspondingly, TRPM7 currents were also significantly inhibited by the addition of RSV (at +100 mV, 22.14 ± 3.40 pA/pF in control, 10.44 ± 2.42 pA/pF in RSV treated cells), (Figures 4D and 4E), suggesting that RSV treatment inhibits TRPM7 expression and function that correspond to the decrease in EMT and decrease in the migration of prostate cancer cells.
Figure 4.
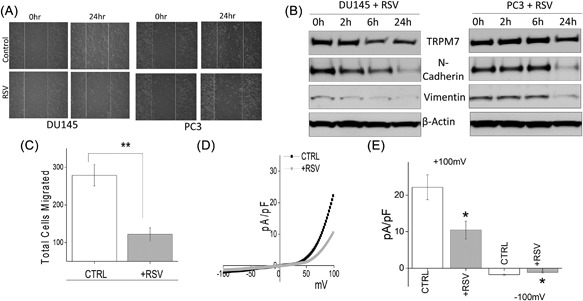
(A) Wound healing assay images for cell migration in human prostate cancer DU145 and PC3 cell lines pretreated for 24 h with 50 μM RSV. (B) Western blot showing the expression of vimentin, N‐cadherin, TRPM7, and loading control β‐actin in DU145 and PC3 cells with RSV treatment under different time points. (C) DU145 cells were migrated using transwell inserts and serum free RPMI ± 50 μM RSV. Four random fields at 10× were counted from seven separate experiments indicating total cells migrated ± SEM. ** indicate significance (P < 0.01). (D) IV curves of TRPM7 current under various conditions (control or RSV pretreatment) and quantitation (5‐10 recordings) of current intensity at ±100 mV is shown in (E). * indicate significance (P < 0.05)
3.5. RSV inhibits EMT enhanced by TGFβ by inhibiting TRPM7
Evidence has shown that RSV suppresses cancer metastasis through inhibiting TGFβ induced EMT.47, 48, 49 To further delineate the role for TRPM7 in modulating RSV‐mediated inhibition of EMT, we assessed migration of DU145 and PC3 cells with and without TGFβ treatment. Similar to the data presented in Figure 4, pretreatment of cells with RSV significantly abolished TGFβ induced increase of EMT markers with a significant decrease in both vimentin and N‐cadherin levels (Figure 5A, quantification provided in supplemental Figure S1F). Importantly, similar effects were also observed in TRPM7 expression levels showing that RSV inhibits TGFβ‐induced increases in TRPM7 expression (Figure 5A). Consistent with these results, TGFβ‐induced increase in TRPM7 currents was also abolished by the addition of RSV without altering the channel properties (at +100 mV, 22.14 ± 3.40 pA/pF in control, 31.06 ± 3.84 pA/pF in TGFβ, 11.4 ± 2.06 pA/pF in TGFβ + RSV) (Figures 5B and 5C). Finally, wound healing assays showed that RSV treatment attenuated the TGFβ‐induced increase in the migration of DU145 cells (Figure 5D, quantification provided in supplemental Figures S2G and S2H). Transwell migration assays further confirmed that the addition of RSV abolishes TGFβ‐mediated increase in cell migration (209.00 ± 13.19 in control, 263.80 ± 9.30 in TGFβ, 143.70 ± 5.35 in TGFβ + RSV) (Figure 5E). Together these results suggest that RSV inhibit TRPM7 function and expression that could contribute to the inhibition of EMT in prostate cancer cells.
Figure 5.
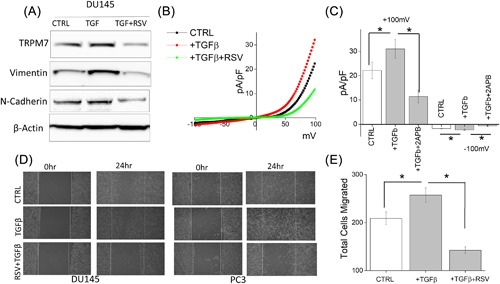
(A) Western blot showing the expression of TRPM7, Vimentin, N‐cadherin, and β‐actin under various conditions in DU145 and PC3 cells. (B) IV curves of TRPM7 current under various conditions and quantitation (5‐10 recordings) of current intensity at ±100 mV is shown in (C). * indicate significance (P < 0.05). (D) Images showing the wound‐healing assay for DU145 and PC3 pretreated for 24 h with 5 ng/mL TGFβ and 50 μM RSV and 10 μM TGFβ. Images were taken after the wound scratch (0 h) and after 24. The pictures are representative of four separate experiments. (E) Cells were treated with TGFβ for 24 h before being trypsinized, re‐suspended in serum free media with or without 50 μM RSV, and placed in transwell inserts for 6 h. Four random fields at 10× were counted from four separate experiments indicating total cells migrated ± SEM. * indicate significance (P < 0.05)
4. DISCUSSION
This study delineates the role of TRPM7 in EMT by using a combination of live cell assays, biochemical and electrophysiological approaches including protein expression and channel activity. We have demonstrated that Mg2+ homeostasis is essential for EMT‐induced cell migration in prostate cancer cells. Moreover, our data shows that TRPM7 regulates constitutive cation entry in prostate cancer cells and is critical for Mg2+ homeostasis and influx. Although both TRPM6 and TRPM7 have been shown to modulate Mg2+ homeostasis and thus could contribute to cell migration and EMT, inhibition of channel activity using 2‐APB points towards TRPM7 as being the major channel. TRPM7 has been shown to be inhibited by the addition of 2‐APB, whereas TRPM6 is shown to be stimulated by the addition of 2‐APB. 2‐APB has also been shown to inhibit SOCE currents, however under these conditions no SOCE currents were observed suggesting that the currents observed are again mainly through TRPM7 channels. In addition, genetic studies using silencing method further established that TRPM7 is the major Mg2+ homeostasis channel in prostate cancer cells that could inhibit cell migration by inhibiting EMT. Although our studies did not fully identify the mechanism as to how TRPM7 modulates EMT, the ion channel signaling is a mechanism well suited to the rapid translation of signals from the tumor microenvironment into cellular responses.50, 51, 52 TRPM7 channel is a Mg2+ channel fused with a functional kinase domain that belongs to the α‐kinase family and could also phosphorylate key proteins involved in cell migration. In addition, TRPM7 has also been shown to modulate epigenetic changes that could also modulate the expression of key proteins required for EMT; however, additional experiments are needed to fully explore these mechanism.
Our studies show that an increase in TRPM7 function and expression in cancer cells along with EMT markers is essential for cell migration. TGFβ enhanced TRPM7 function and expression facilitate cellular migration and was essential for the expression of essential EMT markers, providing further evidence for the importance of TRPM7 in EMT‐induced migration of prostate cancer cells. A direct role for TRPM7 in EMT was demonstrated by silencing TRPM7 that resulted in decreased cellular migration and EMT markers. Resveratrol has been shown to inhibit prostate cancer metastasis, however the exact mechanism or the ion channels involved have yet to be determined. Our results show that RSV significantly inhibit EMT in prostate cancer cells and as expected, both TRPM7 expression and function was significantly decreased upon RSV treatment. More importantly, the suppression of TRPM7 is correlated with the suppression of EMT markers. Based on our results, we hypothesize that treatment of prostate cancer cells with RSV will lead to an inhibition of TRPM7 channel function/expression, thereby inhibiting EMT and cell migration.
TRPM7 is an ion channel that is permeable to both Mg2+ and Ca2+ and thus could be involved in both divalent cation homeostasis. Ca2+ signaling pathways have been shown to be important for cancer cell migration.16, 46 Previous reports suggested that TRPM7 is a regulator of EMT and probably dependent on Ca2+ signaling.13, 16 Here, we show evidence that the effect of TRPM7 on EMT is tightly related to its magnesium‐transporting ability. This is in accordance with previous reports that TRPM7 does not alter the global cytosolic Ca2+ response in EGF‐induced EMT in breast cancer cells and that TRPM7 mainly regulates Mg2+ homeostasis rather than Ca2+ in pancreatic cancer cells.8, 16 Constitutive activation of TRPM7 is still unclear and TRPM7 currents should be inhibited in the physiological cytosolic Mg2+ levels8, 53; however, increases in TRPM7 activity upon the addition of growth factors provides an important clue towards these mechanisms. In addition, several pieces of evidence indicate that Mg2+ homeostasis is altered in cancer and elevated bound Mg2+ in cancer cells may buffer free Mg2+ leading to constitutive TRPM7 activation.5, 54 However, further studies on free intracellular Mg2+ measurements in cancer conditions are still needed to better understand how TRPM7 is constitutively active. Clinical evidence also indicates the role of Mg2+ in sustaining cell proliferation and the possible inhibitory effect of hypomagnesaemia on tumor growth corroborating the idea that reduced serum Mg2+ might improve the response to cancer treatment.6, 55, 56 Low Mg2+ availability also inhibits cell‐cycle progression leading to a G0/G1 arrest through the up‐regulation of p27, p21, and p16.57 In addition, proliferating cells were shown to contain more Mg2+ than resting ones,58 again supporting the overall role of TRPM7 in cancer progression. Experimental evidence had also showed that TRPM7‐mediated Mg2+ influx could influence signaling along the entire PI3K (phosphoinositide 3‐kinase)/Akt/mTOR protein translation cascade.59, 60, 61 Importantly, proteins inhibiting PI3K pathway efficiently suppress EMT,62, 63, 64 suggesting that this pathway is crucial for reprogramming of tumor cells. Furthermore, this also might be a fundamental pathway that transduces Mg2+ availability into cell EMT behavior. Together, these findings clearly outline the activation of TRPM7 is essential for EMT. We further show that the effect of RSV‐mediated inhibition of EMT is via the decrease in TRPM7 channel activity. Furthermore, increases in these early signaling events that are regulated by TRPM7 and are important for EMT induction may offer a novel approach for the prevention of prostate cancer metastasis.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Figure S1. (A) In normal SES (1 mM Ca2+, 1 mM Mg2+) bath solution, whole cell recording showing inward SOCE currents under −100 mV in DU145 and PC3 cells.
Figure S2. (A) Bar diagram showing quantitation of wound‐healing assay in RWPE, DU145 and PC3 cells. (n = 4).
ACKNOWLEDGMENTS
This work was funded by grant support from the National Institutes of Health (R01DE017102; R01DE022765; R21DE024300; P20GM113123) awarded to B.B.S. The funders (NIH) had no further role in study design, data analysis, and/or interpretation of the data.
Sun Y, Schaar A, Sukumaran P, Dhasarathy A, Singh BB. TGFβ‐induced epithelial‐to‐mesenchymal transition in prostate cancer cells is mediated via TRPM7 expression. Molecular Carcinogenesis. 2018;57:752–761. https://doi.org/10.1002/mc.22797
Present address of Yuyang Sun UT Health Science Center San Antonio TX.
REFERENCES
- 1. Center MM, Jemal A, Lortet‐Tieulent J, et al. International variation in prostate cancer incidence and mortality rates. Eur Urol 2012; 61:1079–1092. [DOI] [PubMed] [Google Scholar]
- 2. Neupane S, Bray F, Auvinen A. National economic and development indicators and international variation in prostate cancer incidence and mortality: an ecological analysis. World J Urol. 2017; 35:851–858. [DOI] [PubMed] [Google Scholar]
- 3. Hazelton B, Mitchell B, Tupper J. Calcium, magnesium, and growth control in the WI‐38 human fibroblast cell. J Cell Biol. 1979; 83:487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anghileri LJ. Magnesium, calcium and cancer. Magnes Res. 2009; 22:247–255. [DOI] [PubMed] [Google Scholar]
- 5. Sun Y, Selvaraj S, Varma A, Derry S, Sahmoun AE, Singh BB. Increase in serum Ca2+/Mg2+ ratio promote proliferation of prostate cancer cells by activating TRPM7 channel. J Biol Chem. 2012; 288:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wolf FI, Trapani V. Magnesium and its transporters in cancer: a novel paradigm in tumour development. Clin Sci. 2012; 123:417–427. [DOI] [PubMed] [Google Scholar]
- 7. Trapani V, Arduini D, Cittadini A, Wolf FI. From magnesium to magnesium transporters in cancer: tRPM7, a novel signature in tumour development. Magnes Res. 2013; 26:149–155. [DOI] [PubMed] [Google Scholar]
- 8. Rybarczyk P, Vanlaeys A, Brassart B, et al. The transient receptor potential melastatin 7 channel regulates pancreatic cancer cell invasion through the Hsp90alpha/uPA/MMP2 pathway. Neoplasia. 2017; 19:288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nadler MJ, Hermosura MC, Inabe K, et al. LTRPC7 is a Mg.ATP‐regulated divalent cation channel required for cell viability. Nature. 2001; 411:590–595. [DOI] [PubMed] [Google Scholar]
- 10. Park HS, Hong C, Kim BJ, So I. The pathophysiologic roles of TRPM7 channel. Korean J Physiol Pharmacol. 2014; 18:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fleig A, Chubanov V. Trpm7. Handb Exp Pharmacol. 2014; 222:521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun Y, Sukumaran P, Varma A, Derry S, Sahmoun AE, Singh BB. Cholesterol‐induced activation of TRPM7 regulates cell proliferation, migration, and viability of human prostate cells. Biochim Biophys Acta. 2014; 1843:1839–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen L, Cao R, Wang G, et al. Downregulation of TRPM7 suppressed migration and invasion by regulating epithelial‐mesenchymal transition in prostate cancer cells. Med Oncol. 2017; 34:127. [DOI] [PubMed] [Google Scholar]
- 14. Luo Y, Wu JY, Lu MH, Shi Z, Na N, Di JM. Carvacrol alleviates prostate cancer cell proliferation, migration, and invasion through regulation of PI3K/Akt and MAPK signaling pathways. Oxid Med Cell Longev. 2016; 2016:1469693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin CM, Ma JM, Zhang L, et al. Inhibition of transient receptor potential melastain 7 enhances apoptosis induced by TRAIL in PC‐3 cells. Asian Pac J Cancer Prev. 2015; 16:4469–4475. [DOI] [PubMed] [Google Scholar]
- 16. Davis FM, Azimi I, Faville RA, et al. Induction of epithelial‐mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene. 2014; 33:2307–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abdelrahman AE, Arafa SA, Ahmed RA. Prognostic value of twist‐1, E‐cadherin and EZH2 in prostate cancer: an immunohistochemical study. Turk Patoloji Derg 2017; 1:198–210. [DOI] [PubMed] [Google Scholar]
- 18. Lawrence MG, Veveris‐Lowe TL, Whitbread AK, Nicol DL, Clements JA. Epithelial‐mesenchymal transition in prostate cancer and the potential role of kallikrein serine proteases. Cells Tissues Organs. 2007; 185:111–115. [DOI] [PubMed] [Google Scholar]
- 19. Klarmann GJ, Hurt EM, Mathews LA, et al. Invasive prostate cancer cells are tumor initiating cells that have a stem cell‐like genomic signature. Clin Exp Metastasis. 2009; 26:433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hennenberg M, Schreiber A, Ciotkowska A, et al. Cooperative effects of EGF, FGF, and TGF‐beta1 in prostate stromal cells are different from responses to single growth factors. Life Sci. 2015; 123:18–24. [DOI] [PubMed] [Google Scholar]
- 21. Taylor AW. Review of the activation of TGF‐beta in immunity. J Leukoc Biol. 2009; 85:29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reis ST, Pontes‐Junior J, Antunes AA, et al. Tgf‐beta1 expression as a biomarker of poor prognosis in prostate cancer. Clinics. 2011; 66:1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu CT, Chang YH, Lin WY, Chen WC, Chen MF. TGF beta1 expression correlates with survival and tumor aggressiveness of prostate cancer. Ann Surg Oncol. 2015; 22:S1587–S1593. [DOI] [PubMed] [Google Scholar]
- 24. Trapani V, Schweigel‐Rontgen M, Cittadini A, Wolf FI. Intracellular magnesium detection by fluorescent indicators. Methods Enzymol. 2012; 505:421–444. [DOI] [PubMed] [Google Scholar]
- 25. Singh BB, Lockwich TP, Bandyopadhyay BC, et al. VAMP2‐dependent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist‐stimulated Ca2+ influx. Mol Cell. 2004; 15:635–646. [DOI] [PubMed] [Google Scholar]
- 26. Pani B, Cornatzer E, Cornatzer W, et al. Up‐regulation of transient receptor potential canonical 1 (TRPC1) following sarco(endo)plasmic reticulum Ca2+ ATPase 2 gene silencing promotes cell survival: a potential role for TRPC1 in Darier's disease. Mol Biol Cell. 2006; 17:4446–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pani B, Ong HL, Brazer SC, et al. Activation of TRPC1 by STIM1 in ER‐PM microdomains involves release of the channel from its scaffold caveolin‐1. Proc Natl Acad Sci U S A. 2009; 106:20087–20092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Selvaraj S, Watt JA, Singh BB. TRPC1 inhibits apoptotic cell degeneration induced by dopaminergic neurotoxin MPTP/MPP(+). Cell Calcium. 2009; 46:209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schaar A, Sukumaran P, Sun Y, Dhasarathy A, Singh BB. TRPC1‐STIM1 activation modulates transforming growth factor beta‐induced epithelial‐to‐mesenchymal transition. Oncotarget. 2016; 7:80554–80567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stewart TA, Yapa KT, Monteith GR. Altered calcium signaling in cancer cells. Biochim Biophys Acta. 1848; 2015:2502–2511. [DOI] [PubMed] [Google Scholar]
- 31. Mignen O, Constantin B, Potier‐Cartereau M, et al. Constitutive calcium entry and cancer: updated views and insights. Eur Biophys J. 2017; 46:395–413. [DOI] [PubMed] [Google Scholar]
- 32. Chen Y, Yu Y, Sun S, et al. Bradykinin promotes migration and invasion of hepatocellular carcinoma cells through TRPM7 and MMP2. Exp Cell Res. 2016; 349:68–76. [DOI] [PubMed] [Google Scholar]
- 33. Shen F, Cai WS, Li JL, Feng Z, Cao J, Xu B. The association between serum levels of selenium, copper, and magnesium with thyroid cancer: a meta‐analysis. Biol Trace Elem Res. 2015; 167:225–235. [DOI] [PubMed] [Google Scholar]
- 34. Castiglioni S, Maier JA. Magnesium and cancer: a dangerous liason. Magnes Res. 2011; 24:S92–100. [DOI] [PubMed] [Google Scholar]
- 35. Pardo LA, Stuhmer W. The roles of K(+) channels in cancer. Nat Rev Cancer. 2014; 14:39–48. [DOI] [PubMed] [Google Scholar]
- 36. Huang X, Jan LY. Targeting potassium channels in cancer. J Cell Biol. 2014; 206:151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang L, Zou W, Zhou SS, Chen DD. Potassium channels and proliferation and migration of breast cancer cells. Sheng Li Xue Bao. 2009; 61:15–20. [PubMed] [Google Scholar]
- 38. Luiz AP, Wood JN. Sodium channels in pain and cancer: new therapeutic opportunities. Adv Pharmacol. 2016; 75:153–178. [DOI] [PubMed] [Google Scholar]
- 39. Roger S, Potier M, Vandier C, Besson P, Le Guennec JY. Voltage‐gated sodium channels: new targets in cancer therapy? Curr Pharm Des. 2006; 12:3681–3695. [DOI] [PubMed] [Google Scholar]
- 40. Mishra R, Rao V, Ta R, Shobeiri N, Hill CE. Mg2+− and MgATP‐inhibited and Ca2+/calmodulin‐sensitive TRPM7‐like current in hepatoma and hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2009; 297:G687–G694. [DOI] [PubMed] [Google Scholar]
- 41. Li M, Du J, Jiang J, et al. Molecular determinants of Mg2+ and Ca2+ permeability and pH sensitivity in TRPM6 and TRPM7. J Biol Chem. 2007; 282:25817–25830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ha B, Ko H, Kim B, et al. Regulation of crosstalk between epithelial to mesenchymal transition molecules and MMP‐9 mediates the antimetastatic activity of anethole in DU145 prostate cancer cells. J Nat Prod. 2014; 77:63–69. [DOI] [PubMed] [Google Scholar]
- 43. Xu J, Liu D, Niu H, et al. Resveratrol reverses Doxorubicin resistance by inhibiting epithelial‐mesenchymal transition (EMT) through modulating PTEN/Akt signaling pathway in gastric cancer. J Exp Clin Cancer Res. 2017; 36:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sun Y, Wang H, Liu M, Lin F, Hua J. Resveratrol abrogates the effects of hypoxia on cell proliferation, invasion and EMT in osteosarcoma cells through downregulation of the HIF‐1alpha protein. Mol Med Rep. 2015; 11:1975–1981. [DOI] [PubMed] [Google Scholar]
- 45. Salagierski M. Resveratrol in prostate diseases. Cent European J Urol. 2013; 66:150–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Selvaraj S, Sun Y, Sukumaran P, Singh BB. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol Carcinog. 2016; 55:818–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang H, Zhang H, Tang L, et al. Resveratrol inhibits TGF‐beta1‐induced epithelial‐to‐mesenchymal transition and suppresses lung cancer invasion and metastasis. Toxicology. 2013; 303:139–146. [DOI] [PubMed] [Google Scholar]
- 48. Ji Q, Liu X, Han Z, et al. Resveratrol suppresses epithelial‐to‐mesenchymal transition in colorectal cancer through TGF‐beta1/Smads signaling pathway mediated Snail/E‐cadherin expression. BMC Cancer. 2015; 15:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen CL, Chen YH, Tai MC, Liang CM, Lu DW, Chen JT. Resveratrol inhibits transforming growth factor‐beta2‐induced epithelial‐to‐mesenchymal transition in human retinal pigment epithelial cells by suppressing the Smad pathway. Drug Des Devel Ther. 2017; 11:163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Davis FM, Parsonage MT, Cabot PJ, et al. Assessment of gene expression of intracellular calcium channels, pumps and exchangers with epidermal growth factor‐induced epithelial‐mesenchymal transition in a breast cancer cell line. Cancer Cell Int. 2013; 13:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shapovalov G, Skryma R, Prevarskaya N. Calcium channels and prostate cancer. Recent Pat Anticancer Drug Discov. 2013; 8:18–26. [DOI] [PubMed] [Google Scholar]
- 52. Bates E. Ion channels in development and cancer. Annu Rev Cell Dev Biol. 2015; 31:231–247. [DOI] [PubMed] [Google Scholar]
- 53. Kozak JA, Cahalan MD. MIC channels are inhibited by internal divalent cations but not ATP. Biophys J. 2003; 84:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chandra S, Parker DJ, Barth RF, Pannullo SC. Quantitative imaging of magnesium distribution at single‐cell resolution in brain tumors and infiltrating tumor cells with secondary ion mass spectrometry (SIMS). J. Neurooncol. 2016; 127:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wolf FI, Maier JA, Nasulewicz A, et al. Magnesium and neoplasia: from carcinogenesis to tumor growth and progression or treatment. Arch Biochem Biophys. 2007; 458:24–32. [DOI] [PubMed] [Google Scholar]
- 56. Vincenzi B, Santini D, Tonini G. Biological interaction between anti‐epidermal growth factor receptor agent cetuximab and magnesium. Expert Opin Pharmacother. 2008; 9:1267–1269. [DOI] [PubMed] [Google Scholar]
- 57. Killilea DW, Ames BN. Magnesium deficiency accelerates cellular senescence in cultured human fibroblasts. Proc Natl Acad Sci U S A. 2008; 105:5768–5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rubin H. Central role for magnesium in coordinate control of metabolism and growth in animal cells. Proc Natl Acad Sci U S A. 1975; 72:3551–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sahni J, Scharenberg AM. TRPM7 ion channels are required for sustained phosphoinositide 3‐kinase signaling in lymphocytes. Cell Metab. 2008; 8:84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fang L, Zhan S, Huang C, et al. TRPM7 channel regulates PDGF‐BB‐induced proliferation of hepatic stellate cells via PI3K and ERK pathways. Toxicol Appl Pharmacol. 2013; 272:713–725. [DOI] [PubMed] [Google Scholar]
- 61. Chen WL, Turlova E, Sun CL, et al. Xyloketal B suppresses glioblastoma cell proliferation and migration in vitro through inhibiting TRPM7‐regulated PI3K/Akt and MEK/ERK signaling pathways. Marine Drugs. 2015; 13:2505–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rumman M, Jung KH, Fang Z, et al. HS‐173, a novel PI3K inhibitor suppresses EMT and metastasis in pancreatic cancer. Oncotarget. 2016; 7:78029–78047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Niederst MJ, Benes CH. EMT twists the road to PI3K. Cancer Discov. 2014; 4:149–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Salt MB, Bandyopadhyay S, McCormick F. Epithelial‐to‐mesenchymal transition rewires the molecular path to PI3K‐dependent proliferation. Cancer Discov. 2014; 4:186–199. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Figure S1. (A) In normal SES (1 mM Ca2+, 1 mM Mg2+) bath solution, whole cell recording showing inward SOCE currents under −100 mV in DU145 and PC3 cells.
Figure S2. (A) Bar diagram showing quantitation of wound‐healing assay in RWPE, DU145 and PC3 cells. (n = 4).