

Summary
Development and standardization of fibrinolysis methods have progressed more slowly than coagulation testing and routine high‐throughput screening tests for fibrinolysis are still lacking. In laboratory research, a variety of approaches are available and are applied to understand the regulation of fibrinolysis and its contribution to the hemostatic balance. Fibrinolysis in normal blood is slow to develop. For practical purposes plasminogen activators can be added to clotting plasma, or euglobulin prepared to reduce endogenous inhibitors, but results are complicated by these manipulations. Observational studies to identify a ‘fibrinolysis deficit’ have concluded that excess fibrinolysis inhibitors, plasminogen activator inhibitor 1 (PAI‐1) or thrombin‐activatable fibrinolysis inhibitor (TAFI), zymogen or active enzyme, may be associated with an increased risk of thrombosis. However, results are not always consistent and problems of adequate standardization are evident with these inhibitors and also for measurement of fibrin degradation products (D‐dimer). Few methods are available to investigate fibrinolysis under flow, or in whole blood, but viscoelastic methods (VMs) such as ROTEM and TEG do permit the contribution of cells, and importantly platelets, to be explored. VMs are used to diagnose clinical hyperfibrinolysis, which is associated with high mortality. There is a debate on the usefulness of VMs as a point‐of‐care test method, particularly in trauma. Despite the difficulties of many fibrinolysis methods, research on the fibrinolysis system, taking in wider interactions with hemostasis proteins, is progressing so that in future we may have more complete models and better diagnostic methods and therapeutics.
Keywords: carboxypeptidase B2, euglobulin clot lysis time, fibrin clot lysis time, plasminogen activator inhibitor 1, plasminogen activators, thromboelastometry
Background to fibrinolysis
The significance of fibrinolysis in the hemostatic balance was identified early 1, 2. However, methods available for investigating fibrinolysis have lagged behind methods used to study coagulation, which are widespread and standardized for routine testing purposes (activated partial thromboplastin time (APTT), international normalized ratio (INR) or prothrombin time (PT), for example). Under normal circumstances, fibrinolysis takes many hours or days to develop in healthy blood after clotting 3, which is a major obstacle in assessing the ‘global fibrinolysis capacity’. This is in clear contrast to coagulation reactions, which are conveniently monitored for seconds or minutes. In order to make some assessment of fibrinolysis it is necessary to remove some inhibitors or add plasminogen activators, which obviously hinders complete understanding of the in vivo situation. So, for example, plasma‐based systems, where clotting and lysis may be easily followed turbidimetrically, have tissue plasminogen activator (tPA) added to speed up lysis. Alternatively, euglobulin may be prepared from plasma, which reduces the concentration of fibrinolytic inhibitors. More details on these methods will be given below. Fibrinolysis techniques are often technically more difficult and time consuming and not so amenable to automation. It is also the case that congenital deficiencies of fibrinolytic components are not widespread in humans, like hemophilia A and B for example, which has driven research and therapeutic development in coagulation. All these factors contribute to the lower profile of fibrinolysis and potentially some underestimation of the significance of fibrinolysis in hemostasis. Some key proteins involved in fibrinolysis are summarized in Table 1.
Table 1.
The balance between thrombosis and bleeding is maintained by coagulation and fibrinolysis factors. Fibrinolysis is regulated by many proteins, molecules and cells that enhance or dampen plasminogen activation and fibrin degradation. The main players in modulating fibrinolysis from studies over many years are shown. Further details may be found in 4, 5
| Profibrinolytic | Antifibrinolytic | ||
|---|---|---|---|
| Enzymes | Specific inhibitors | Other proteins or biomolecules | Cells** |
| tPA | Alpha‐2‐antiplasmin | Fibrinogen | Platelets |
| uPA | PAI‐1 | HRG§ | Erythrocytes |
| Exogenous PAs | Alpha‐2‐macroglobulin* | Lipoprotein (a) | Neutrophils (NETs) |
| Substrate | TAFIa† | Phospholipids | |
| Plasminogen | FXIII‡ | Polyphosphate¶ | |
*Under some circumstances plasmin may be inhibited by this broad specificity inhibitor. †TAFI zymogen is activated by thrombin in complex with thrombomodulin, or alternatively by plasmin. TAFIa reduces plasminogen and plasmin binding by cleaving C‐terminal lysines in fibrin. ‡FXIII is a transglutaminase that stabilizes fibrin by crosslinking fibrin chains and alpha‐2‐antiplasmin to fibrin. §Histidine‐rich glycoprotein and lipoprotein (a) also act to block lysine binding sites on plasminogen. ¶Polyphosphate (PP) may have anti‐ or profibrinolytic effects on enzymes and fibrin structure. **Cells interfere with fibrin breakdown by physical means, including clot retraction by platelets and fibrin structure changes by erythrocytes. Platelets release inhibitors, including PAI‐1, FXIII and PP, and neutrophils release nuclear DNA and histones, as neutrophil extracellular traps.
Review organization
Earlier reviews highlighting the molecular components and mechanisms and regulation of fibrinolysis are available and these aspects will not be covered here 4, 5. The purpose of this review is to consider methods that are available to quantitatively assess fibrinolysis, which includes diagnostic methods, activity measurement for therapeutics, standardization and other aspects of research. Problems and drawbacks will be highlighted. The review is grouped by technique and according to the complexity of the matrix, with distinctions for diagnostic and other types of quantitative measurements. Animal work or genetic studies are not covered.
Why measure fibrinolysis
Although fibrinolysis is slow to develop under normal circumstances, enhanced fibrinolysis or hyperfibrinolysis may develop and lead to life‐threatening blood loss. Congenital deficiencies of fibrinolysis inhibitors are not common but do occur and can lead to bleeding 6. Accelerated fibrinolysis has been known for a long time in patients who have suffered shock (or adrenaline administration) or major trauma and after surgery, especially involving extracorporeal surgery, and lung, liver and prostate surgery 7, 8, 9. Increased fibrinolysis is also seen in cirrhosis 10, renal failure, menorrhagia and obstetric complications 11, and some malignancies, particularly in some leukemia patients 12. Increased fibrinolysis possibly linked to clot instability is also noted in hemophilia patients (e.g. reviewed, 6). In some of these examples, administration of antifibrinolytic lysine analogues, epsilon‐aminocaproic acid (EACA), tranexamic acid (TXA) or aprotinin may be beneficial and indicates an imbalance between clotting and fibrinolysis that can be rectified by suppressing fibrin breakdown.
Conversely, impaired fibrinolysis or hypofibrinolysis, which may be hereditary or of environmental origin, may be linked to the development of thrombosis and has been associated with atherosclerosis and obesity, diabetes and hyperlipidemia. This fibrinolysis deficit may contribute to thrombotic incidents, both venous and arterial. Researchers have looked at global assays for fibrinolysis capacity or fibrinolysis potential (see 13 for recent review) and for biomarkers that would indicate reduced fibrinolysis, such as elevated alpha‐2‐antiplasmin (α2AP, plasmin inhibitor), plasminogen activator inhibitor 1 (PAI‐1) and thrombin‐activatable fibrinolysis inhibitor (TAFI, carboxypeptidase U, CBP2 gene product) or changes in active tPA levels.
Another goal of studying fibrinolysis is to improve our understanding of the system and develop realistic models and computer simulations. To achieve this, as a first step it is necessary to accumulate knowledge of molar concentrations, binding constants and kinetic parameters of enzyme‐substrate and enzyme‐inhibitor reactions, to understand the basic enzymology (e.g. 14). More complete models require cellular effects, inclusion of the complexities of fibrin interactions and breakdown and the influence of flow. Progress on modelling fibrinolysis has lagged behind coagulation modelling (e.g. 15, 16). Fibrinolysis reactions take place in a heterogeneous environment where critical reactions take place at the liquid–solid interface. However, some progress is evident 17, and it should be possible in future to increase model complexity in line with additional knowledge and improved computing power.
Plasminogen activators and plasmin in purified systems
In purified systems, chromogenic and fluorogenic substrate assays provide a convenient way to measure enzyme activity and a simple way to titrate and quantify enzyme inhibitors. However, kinetic studies in purified systems overlook important regulatory interactions. For example, very high inhibition rate constants (of the order of 107 mol L−1 s−1) are often quoted for α2AP and PAI‐1, but these are modulated by fibrinogen and fibrin in vivo 18. Solution studies are definitely easier to perform than studies in a fibrin matrix and are useful for plasminogen activators such as urokinase (uPA) or therapeutic streptokinase (but note, streptokinase variants from other streptococcus strains do interact with fibrinogen and fibrin 19). Experience has shown that caution is needed when using tPA with soluble stimulators such as cyanogen bromide fragments of fibrinogen 20, compared with fibrin 21, as the kinetics of binding and plasminogen activation are different. It should also be noted that quoted values for K m and k cat from many tPA studies over the years are inconsistent because they will be highly dependent on the composition of the system (concentration of stimulator for example) so are not comparable.
A special group of amidolytic substrates are used to perform active site titrations on many serine proteases, including hemostasis enzymes. Common examples are p‐nitrophenyl p‐guanidinobenzoate (NPGB) and 4‐methylumbelliferyl p‐guanidinobenzoate hydrocholoride (MUGB). The requirement for an active site titrant is good Michaelis Complex formation (fast binding and low K m) with the target enzyme, followed by slow turnover (low k cat). The reaction time‐course is seen as a burst of release of chromophore or fluorophore, followed by steady state turnover. The magnitude of the burst can be converted to a molar concentration from a standard curve of released product and this is equivalent to the concentration of active sites of the enzyme. We have used MUGB to estimate the conversion factor for several WHO International Standards (IS), including plasmin (the 4th IS for plasmin,13/206, contains 5.3 IU mL−1, corresponding to approximately 1.5 μmol L−1 plasmin 22), thrombin (1st WHO IS for alpha‐thrombin, 89/588, contained 100 IU mL−1 or 1.1 μmol L−1 thrombin) and uPA (the WHO 2nd IS for high‐molecular‐weight urokinase, 11/184, is estimated to contain 3200 IU mL−1 uPA, corresponding to 380 nmol L−1 active enzyme 23). From the uPA results it can also be estimated that 7 IU mL−1 of PAI‐1 in the WHO IS for PAI‐1 activity, 92/654, corresponds to 0.83 nmol L−1 active PAI‐1. Interestingly, the 3rd IS for tPA, 98/714, contains 20 μg mL−1 tPA per ampoule, and although there is no suitable active site titrant for tPA, we can calculate that the anti‐tPA activity in 92/654 corresponds to 0.83 nmol L−1 inhibitor, in agreement with the anti‐uPA activity. It should be emphasized that these figures are not endorsed by WHO but are requested by users of IS as conversion factors from IU to μg or molar equivalents, which are helpful in fundamental enzymology studies.
Fibrin matrix in purified systems
Early methods of quantifying plasminogen activators, such as the fibrin plate were reported to be time consuming and unreliable but microtiter plate‐based methods solved many problems 24. Variations include internal and external lysis, in which plasminogen activator is either mixed with fibrinogen, plasminogen and thrombin to form a clot that is subsequently lysed evenly throughout, or where activator is added to the top of a preformed clot. The internal, mixed arrangement is suggested to reflect normal hemostasis, whereas superficially added activator is said to more closely reflect the situation during thrombolytic therapy. A combination method that includes fibrin and chromogenic substrate allows rates of plasminogen activation to be monitored in the presence of fibrin, or by running parallel plates to simultaneously measure plasmin generation and fibrinolysis 25, 26. A driver behind the development of this method was to permit calculation of rates of plasmin generation in pmol L−1 s−1, and enable specific activities of plasminogen activators, tPA, uPA and streptokinase to be compared. WHO assigned IU are arbitrary and cannot be directly compared. Other available methods for measuring the activity of plasminogen activators include the European Pharmacopoeia method for Alteplase (tPA) 27 in which clot lysis time is determined by release of trapped bubbles from the fibrin network in tubes, or the related method of timing the passage of a glass or steel ball through the clot 28. A new and interesting method for measuring thrombolytic drugs in plate format is the Halo method, which is microtiter plate‐based, allowing high throughput, but has the advantage of using whole blood 29.
Euglobulin, plasma, whole blood
The basic method of euglobulin preparation involves dilution and acidification of chilled plasma to form a precipitate, which is collected and re‐dissolved in higher pH buffer containing calcium. Clotting occurs rapidly and clot lysis takes place over several hours. Results have been reported as lysis time or time to half lysis 30, area under the curve or rate of lysis 31, for example. The euglobulin fraction is reported to have a greater than 90% reduction in α2AP, but there is also significant depletion of PAI‐1 and TAFI 31. The euglobulin lysis time is strongly influenced by PAI‐1 (and free tPA) concentrations, so is sensitive to the precise method of euglobulin preparation.
Plasma clot lysis
Table 2 shows a selection of methods used to measure plasma clot lysis. The simplest turbidimetric methods report time to 50% lysis, but later developments were aimed at generating a global figure for patient fibrinolytic capacity. The introduction of fluorogenic substrates has developed from thrombin generation assays (TGAs) 32, but fibrinolysis is explored in the presence of two substrates sensitive to thrombin or plasmin, added to separate wells run in parallel 33, 34 or in the same well 35. These TGA‐like assay systems have been explored to identify what factors regulate thrombin and plasmin generation and probe the relationships between clotting and lysis. In particular, like TGA, they have the advantage of permitting calculation of molar concentrations of thrombin and plasmin developed during clotting and lysis phases. However, for all plasma clot lysis assays, the requirement to add a large excess of tPA makes these methods insensitive to the influence of intrinsic tPA and subtle variations in the concentration of PAI‐1. By contrast, euglobulin methods require no tPA to be added but are less sensitive to the effects of TAFI on clot lysis due to significant losses during euglobulin preparation 31.
Table 2.
Methods for assessing fibrinolytic potential in plasma
| Name | Clotting | Lysis [tPA] ng mL−1 | Readout | Analysis | References |
|---|---|---|---|---|---|
| Plasma clot lysis | Thr = 0.04 IU mL−1 * | 400 | Turbidity | Time to 50% lysis read from absorbance profile† | 92 |
| OHPP | Thr = 0.2 IU mL−1 | 700 | Turbidity | Absorbances summed over 30 min from clot lysis curves and used to calculate the overall hemostatic potential in plasma (OHPP) | 93 |
| OFP | Thr = 0.04 IU mL−1 | 300 | Turbidity | Summed absorbances over 40 min in parallel reactions of clotting and lysis curve (OHP, +tPA) and overall coagulation potential (OCP no tPA). Overall fibrinolysis potential, OFP = (OCP‐OHP)/OCP | 94 |
|
Thr = 0.04 IU mL−1
TF = 0.1 pmol L−1 |
330 | Turbidity | As above, with addition of platelet reagent containing tissue factor (TF) | 95 | |
| OHI | TF = 2.1 pmol L−1 | 135 | Turbidity | Recombinant TF and phospholipids (PL) in reaction mixture. One single reaction for clotting and lysis profiles; parameters taken from first derivative and used to calculate overall hemostasis index (OHI) | 96 |
| TF = 2.1 pmol L−1 | 135 | Turbidity | As above, PL replaced by washed, frozen–thawed platelets | 97 | |
| CloFAL | TF = 5 pmol L−1 | 450 | Turbidity | Clot formation and lysis (CloFAL) gives a coagulation index from AUC over 30‐min reaction. Fibrinolysis index calculated using times to maximum absorbance and to end of first phase of lysis, indicated by inflection in profile from faster to slower lysis rate | 98, 99 |
| STP | TF = 5 pmol L−1 | 450 | Fluorescence | Simultaneous thrombin and plasmin generation (STP) with two fluorescent substrates, in parallel wells. First derivative of fluorescence reads used to calculate time of lag and max, Vmax rates and AUC | 34 |
| TF = 1 pmol L−1 | 215 | Fluorescence | As STP, with 4 μmol L−1 PL. First derivative of fluorescence curves allows calculation of peak (nmol L−1) or endogenous potential from AUC (nmol L−1 min) for thrombin and plasmin | 33 | |
| NHA | TF = 0.28 pmol L−1 | 390 | Fluorescence | Novel hemostasis assay (NHA), thrombin and plasmin fluorescent substrates with non‐overlapping spectra in one well. First derivatives used to determine values for lag and peak times, peak nmol L−1 levels of plasmin and thrombin and AUC | 35 |
Thr, thrombin; tPA, tissue plasminogen activator; TF, tissue factor; PL, phospholipid; AUC, area under the curve. *CaCl2 is added to all systems. PL is also carefully controlled in some methods as it does influence clot lysis times 100. †The simplest methods use thrombin and CaCl2 to initiate clotting, add tPA for lysis, use turbidimetry to assess time to 50% lysis, and work equally well with plasma or fibrinogen substrates 92. Later methods have added other readouts such as lag times, time to maximum clotting, maximum clot absorbance, maximum rates of clotting and lysis and AUC, for example, which can be extracted from turbidimetric time‐courses.
Data and curve analysis
Variations in the way data are analyzed are also summarized in Table 2. The publications cited may report analysis in some detail, but unless bespoke software is readily available, the methods are difficult to transfer. We have developed freely available open source software for download or for use online in a browser that may be used for clot lysis and zymogen activation kinetics analysis 36. Analysis is thus better standardized and can improve transparency and reproducibility 37. In line with much common practice, lysis is expressed as time to a chosen % of lysis to avoid problems of misinterpretation of rates of lysis when raw data (as opposed to normalized data) are used. This problem is illustrated in Fig. 1 and has been highlighted previously 38.
Figure 1.
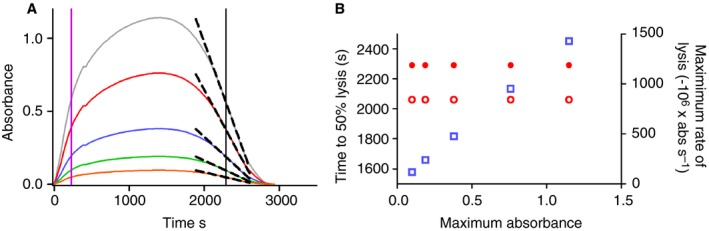
The effect of clot absorbance on lysis rate. In panel A, absorbance values of a clot lysis curve have been scaled to give a hypothetical set of clotting and lysis profiles. The time to clotting and lysis is the same for all curves and the time to 50% clotting and lysis is shown by the magenta and black lines, respectively. The maximum rate of lysis around the 50% lysis point is shown as the dashed line for each curve. Panel B shows the relationship between maximum absorbance and apparent clot lysis rate (open squares), using raw absorbances, or time to 50% lysis from reaction start time (closed circles), or time between 50% clotting and 50% lysis (open circles). The absolute rate of lysis depends on the absorbance range and should not necessarily be interpreted as being influenced by fibrin structure (which also affects absorbance). The time to 50% lysis, however selected, is independent of absolute absorbances. Normalized curves may also be used to avoid this artefact 38. [Color figure can be viewed at http://wileyonlinelibrary.com]
Other methods
It is possible to use turbidimetric methods that include some blood cells to understand their potential role, but there are limits before the fibrin clots become too turbid (e.g. see 4). Measurement of fibrin formation and lysis by turbidimetric methods in microtiter plates has the advantage of high throughput compared with more cumbersome methods that follow release of radiolabeled 39 or fluorescently labeled fibrin fragments, which require separation. However, fluorescently labeled fibrin can be used to investigate flow effects in a closed‐loop system with plasma or blood 40, 41. Clot lysis may be followed by confocal microscopy methods and using Ibidi microslides, so long as there is a lysis front that can be used to calculate a lysis rate (e.g. 42). Rijken and colleagues have addressed the difficulties of using whole blood by developing a method of estimating fibrinolytic capacity, which relies on measurement of fibrin degradation products (FDP) by ELISA in clotted‐lysing blood 43. Global fibrinolytic capacity (GFC) is calculated after correcting for background FDP in parallel blood samples incubated with plasmin inhibitor, aprotinin.
Individual protein components
Insights into the functioning of the fibrinolytic system may be gained from protein levels during health and disease or therapy. Deficiencies of fibrinolysis inhibitors in human populations are rare but when found often lead to increased bleeding risk (e.g. 6), or high circulating PAI‐1 and TAFI may indicate a ‘fibrinolysis deficit’. It is not possible to review conclusions from the many large‐scale population studies that include fibrinolysis proteins, but a flavor of results, problems and conflicts is presented in example reviews 13, 44. There are many studies involving PAI‐1 (antigen and activity) and TAFI (zymogen and activated enzyme), covering circulating levels, polymorphisms, influence on venous and arterial thrombosis, in men and women, young and old, first and later or recurring events. Generally speaking, plasminogen deficiency appears to be rare but does not cause thrombosis and is rarely included as a biomarker in observational studies, and tPA is less often a focus of studies than inhibitors. Markers of ongoing fibrinolysis such as D‐dimer and plasmin‐α2AP (PAP) complexes are commonly used. Interest in PAI‐1 and TAFI as drug targets to modulate hemostasis has focused attention on the need for reliable assays 45, 46, 47.
PAI‐1 and tPA
The association of elevated PAI‐1 with cardiovascular disease, metabolic syndrome, diabetes, obesity and senescence, and as a prognostic marker for several cancers, has long been investigated (e.g. 48). Evidence for an influence on venous thrombosis may be weaker and mixed. These studies or methods will not be discussed in detail but general problems that affect interpretation of results will be approached. Methods are available to measure PAI‐1 antigen (including free active, inactive and complexed PAI‐1) or activity. There is poor harmonization between commercial methods for PAI‐1 antigen or activity or tPA antigen 49, 50. Poor agreement was also found in a more recent study organized by the Fibrinolysis Subcommittee of the SSC, which included five plasma pool samples, with a range of PAI‐1 levels, measured in 12 laboratories, using seven different methods (in‐house or commercial kits). For any given sample, the reported PAI‐1 antigen level varied 4–6‐fold between different methods (report by C.L. and P. Declerck, ISTH/SSC Fibrinolysis Subcommittee minutes, 2011).
Harmonization could be improved by using a common standard but one obstacle to harmonization is the reporting of results in ng mL−1. Existing methods report results in ng mL−1, now the expected unit, but the origin of kit protein standards is variable, so each ‘ng’ is different. It is undesirable to establish a standard with a ‘consensus ng’, taken as a mean of values from a collaborative study. As a consequence of these observations it is clear that the absolute amounts of PAI‐1 in ng mL−1 between different studies are not comparable unless they used exactly the same kit or method.
An alternative approach to avoid using antibody methods associated with different local calibrators has been explored: isotope dilution mass spectrometry (IDMS). PAI‐1 contains several unique peptide sequences that can be liberated by trypsin digestion. One or two chosen peptides can be purified and quantified (using liquid chromatography and mass spectrometry) after spiking digests with synthetic 13C‐labelled peptides of known concentration to act as an internal calibrator. After accounting for losses and calculating recovery, the concentration of peptides is used to determine the molar concentration of PAI‐1 in the starting plasma. This approach has been demonstrated to work in principle but some technical challenges remain before a plasma sample with an agreed ng mL−1 concentration of PAI‐1 can be proposed as a common standard. Aside from these testing issues, other pre‐test problems may also affect PAI‐1 measurements. These include temporal variations, both diurnal and seasonal 51, and blood sample collection and processing, as platelets will release PAI‐1 if activated during venepuncture 52.
TAFI
It is not uncommon to find expressions such as ‘inconsistent’ or ‘counter‐intuitive’ to summarize results from studies involving TAFI (e.g. see 13, 44), and there have been many studies on TAFI links with arterial and venous thrombosis, responses in myocardial infarction and stroke, and in inflammation, infection and sepsis. Details of the biochemistry of TAFI are well understood 53. Several issues are known to have caused difficulties during assay method development, such as the Thr325Ile polymorphism, which affects some assays, and TAFI activation and stability 54, 55. Furthermore, although TAFI activation is regulated by thrombin‐thrombomodulin, plasmin also appears to be involved in activation and inactivation 56. The thermal instability of TAFI is an established mechanism of inactivation and has to be taken into account in functional methods that rely on activity measurements. A variety of ELISA (e.g. 57) and functional methods are available 58 for quantifying TAFI in plasma. Some recent studies have focused on circulating levels of TAFIa as a more important biomarker than zymogen 59, using highly sensitive activity assays (the proportion of TAFIa vs. TAFI proenzyme is very small) 60, 61 or ELISA methods 62.
D‐dimer
D‐dimer, as precisely defined, is an observed late stage FDP formed as a result of FXIII crosslinking of adjacent D domains in fibrin polymers 63. Thus circulating D‐dimer is a measure of fibrin breakdown and ongoing fibrinolysis as distinct from fibrinogenolysis. Early studies were restricted to detecting FDPs in patient serum but progress accelerated with the arrival of monoclonal antibody technology 64 that could generate antibodies that were insensitive to fibrinogen. There is currently a plethora of different commercial D‐dimer assay methods and kits using different platforms to detect clinically significant levels of circulating D‐dimer, including many point‐of‐care (POC) devices. It has long been known that antibodies to ‘D‐dimer’ react with high‐molecular‐weight FDPs and the spectrum of fragments differs between disease types. Antibodies used to measure FDPs also have different specificity for high and low‐molecular‐weight fragments and these factors make harmonization of D‐dimer test results particularly challenging 65, 66. It should be noted that the clinical application of D‐dimer testing is restricted to exclusion of thrombosis because D‐dimer test methods tend to have poor specificity, with better sensitivity, making false‐positive results quite likely. Low specificity may be a particular problem for POC devices, highlighting the need to restrict the application of such testing (e.g. 67).
Negative predictive value (NPV) and positive predictive value (PPV) are useful concepts to help understand how D‐dimer testing is used. High NPVs give confidence when excluding patients from further testing for thrombosis, and are further improved by algorithms such as the Wells score that limit D‐dimer screening to populations with low pre‐test probability of a thrombosis. In this way, more confidence is gained that a negative result is truly negative. A simple app is available to explore the relationship between test sensitivity, specificity and prevalence that calculates PPV and NPV 68. The results produced by D‐dimer testing, either as ‘D‐dimer’ or fibrinogen equivalent units (FEU), depend on manufacturer calibrators and need to be interpreted with caution as they bear little relationship to real ng mL−1, as discussed for PAI‐1 above. Cut‐off values for ‘normal’ stated in tests kits need to be validated locally. However, there are indications that harmonization may be improved by appropriate common standards. Several collaborative studies so far suggest this may be best achieved by having a pool of D‐dimer plasma with a heterogeneous mix of fibrin breakdown products 66, 69, 70. In the future it may also be possible to find a way to incorporate some traceable calibration of such a standard back to a real gravimetric concentration of ng mL−1 D‐dimer fragment using IDMS.
Besides technical and standardization difficulties associated with assays for fibrinolytic proteins, there are other, more general, factors that need to be considered when evaluating these kinds of studies. The goal of many studies seems to be the identification of a statistically significant difference between groups. However, one should also consider whether the size of any difference is likely to be physiologically significant given the common wide variations seen in many hemostasis proteins in normal populations (e.g. 71). Caution is needed where P values are close to the chosen level of significance (often P < 0.05) as this may be very weak evidence of any real effect. There is increasing concern around the low bar that is set to determine statistical significance across diverse areas of scientific study, and debate around how P values should be interpreted 72, 73. To address these problems some workers have advocated more stringent tests of significance (e.g. P < 0.005 or P < 0.001), or suggested P values close to 0.05 are merely an indication that further investigations are needed. Some scientists and journals argue for abandoning the notion of statistical significance. Another proposal is to estimate the prior probability of a real effect before any experiment is run to better interpret final P values and properly assess the risk of false‐positive results (see 74 for discussion and a link to an online app to calculate false‐positive risk). Other relevant concerns are publication bias, dealing adequately with confounders, and repeated post hoc subset analysis until an effect is found.
Viscoelastic methods
As a research tool, viscoelastic methods (VMs) provide the opportunity to study clotting and fibrinolysis in whole blood and hence investigate the contribution of platelets and other blood cells. In a clinical setting these methods also have a vital advantage of speed and can act as POC tests during surgery associated with major blood loss, including cardiothoracic surgery, liver transplant and traumatic bleeding. Here the goal of VM testing is to optimize transfusion regimes of red cells or platelets, or application or clotting factors, fibrinogen, fresh frozen plasma or cryopreciptate, and antifibrinolytics such as TXA.
Three commercial platforms exist to monitor viscoelastic changes in blood: thromboelastograpy (TEG), thromboelastometry (ROTEM) and Sonoclot. Most published work uses ROTEM or TEG. All approaches detect changes in viscosity through movement of a pin through blood in a cup after coagulation has been triggered. Beyond this common principle, the mechanics of the systems differ, as do the reagents supplied and the readouts. Precise details can be found in manufacturer literature and are reviewed elsewhere 75, 76. Considering the ROTEM delta machine for example, differential testing on one patient is accomplished on a single unit, using four channels, and available tests are Intem (intrinsic coagulation with ellagic acid), Extem (tissue factor triggers extrinsic activation), Fibtem (cytochalasin D is added to extem to elimate the platelet contribution) and Aptem (aprotinin is added to extem to stop fibrinolysis). A modification of Intem is available with the addition of heparinase to detect heparin in the blood sample. The Natem test adds only CaCl2 to generate low thrombin concentrations and takes longer to run than the other tests. All results are displayed as a set of clotting and lysis curves called Temograms and up to 32 parameters are derived describing clotting and lysis kinetics and clot properties. Some published reference ranges are available for some of these parameters on healthy individuals and trauma patients (for example 77, 78). Similar, but not identical, sets of tests exist for TEG, but with differences in reagents (kaolin in place of ellagic acid to study the intrinsic coagulation pathway being one example). Significantly for fibrinolysis, the reduction in clot firmness generated by the ROTEM as lysis index at 30 min LI(30) is calculated in a different way from LY30 in TEG, so the results are not interchangeable. Different definitions of hyperfibrinolysis as detected by VMs used in various studies have been reviewed and summarized elsewhere 79, and efforts are underway to standardize how diagnosis is performed, and to identify the best parameters to guide therapeutic interventions. For example, research is being carried out by the European TACTIC consortium to develop data‐driven algorithms for the management of hemorrhage 80.
Interest in VMs in research and diagnostic settings has been increasing in recent years. Trauma‐induced coagulopathy has long been studied but the importance of fibrinolysis has only been addressed more recently (e.g. 81). VMs are attractive as a means of diagnosing ongoing fibrinolysis in trauma as results may be generated quickly, but there is disagreement over what benefits may be gained from these diagnostic procedures at the cost of time taken. Concerns have been raised about the sensitivity of VMs for identifying fibrinolysis in all patients. Raza and colleagues 82 compared results from ROTEM with other fibrinolysis assay methods and found the majority of patients with ongoing fibrinolysis, associated with increased PAP complexes, D‐dimer, raised tPA, prothrombin fragents 1 + 2 and TAFIa, were not identified by ROTEM. Severe hyperfibrinolysis was picked up, defined as maximum lysis (ML) of > 15% at 60 min after the start of clot formation, and was associated with very high PAP > 1500 μg L−1. The severe group accounted for 5% of patients in this study and had high mortality (40% with a mean time to death of < 1 day). A moderate fibrinolysis group, not identified by ROTEM (ML < 15%), but with raised fibrinolysis markers, had a 12% mortality rate, higher than the normal fibrinolysis group, and required more transfusions and medical support. This moderate group was highlighted as the group that were most likely to benefit from antifibrinolytic therapy. Interestingly, this report also identified a small group of patients (eight out of 288) who showed apparent hyperfibrinolysis as ML > 15%, but without raised fibrinolysis markers. This group was not explored in detail. Assay methods available for circulating markers of fibrinolysis used in this study are too slow to be POC tests that could guide trauma therapy, but this study 82 suggests VMs do not give a full picture of fibrinolysis activation in trauma patients. Current European guidelines advise earliest possible use of antifibrinolytics to avoid delays due to diagnostic procedures. Earliest possible infusion of TXA is advisable following results from several trials which show that TXA given later than 3 h after trauma increases mortality 79, 83.
A number of studies using TEG have defined hyperfibrinolysis as LY30 > 3%, meaning lysis > 3%, 30 min after maximum amplitude (e.g. reviewed 79). However, another area of interest has been fibrinolysis shutdown, or hypofibrinolysis, in trauma patients, which may also be associated with high mortality 84, and which potentially could be exacerbated by antifibrinolytic drugs. Moore and co‐workers have developed a diagnostic approach to identify patients with fibrinolysis shutdown, normal fibrinolysis and hyperfibrinolysis, based on TEG LY30 values of 0–0.08%<0.09–2.9%<3%, respectively 84. However, these rather precise readouts are complicated by platelet‐induced clot retraction, which may be interpreted as a false‐positive result for hyperfibrinolysis. To differentiate between hyperfibrinolysis and clot retraction these workers have suggested parallel blood samples be run, including one with added TXA to block fibrinolysis and identify the contribution to the LY30 value from platelet contraction alone 85. The work cited above 82 suggests some caution is needed in relying exclusively on VMs to make therapeutic choices on transfusion strategies or antifibrinolytic therapy, at least in trauma. The conclusions from many studies and systematic reviews seem to be that more research is needed before there is sufficient confidence in VMs for routine diagnostic testing, although there are promising signals of benefit 86, 87, 88. Collaborative trials are needed to explore positive and negative predicative values from VM testing and calculate the costs and benefits of testing in reducing transfusion or blood component requirements and ultimately saving lives.
In the trauma situation, testing must be balanced against protocols for rapid TXA administration. Trials will be complicated by the fact that VM diagnosis cannot be used on patients who have received antifibrinolytics because fibrinolysis is blocked. There are also questions around the effects of alcohol on VM parameters, possibly acting on platelets, which is particularly important given that the majority of trauma patients arriving in emergency departments in some countries have measurable blood alcohol levels 89. However, some groups are generally enthusiastic about the routine use of VM testing 90, and VMs certainly will continue to be a research tool that can be used to investigate the kinetics of coagulation and fibrinolysis in whole blood.
Conclusions
By comparison with common diagnostic coagulation tests (APTT, PT, INR, etc.), fibrinolysis methods are more cumbersome and poorly standardized. However, improvements are being made. It is desirable to increase the application of SI units including traceable gravimetric and molar concentrations, for inhibitors and enzymes, and find reaction rate and binding constants that are realistic for a blood matrix. These parameters form the basis of quantitative reaction schemes and computer simulations of fibrinolysis. Biophysical methods such as IDMS will help with this endeavor and facilitate the harmonization of results from different methods and studies. Global tests such as euglobulin clot lysis time or plasma clot lysis have drawbacks and cannot provide a complete picture of fibrinolysis. Fluorescent substrate methods outlined in Table 2 offer the chance to express results as molar concentrations, but have so far not seen wide uptake. The relationship to TGA is clear, but even though TGA methods have been around for many years they remain largely a research tool and are difficult to standardize. The questions surrounding VMs when looking at fibrinolysis in routine testing situations, and particularly in the challenging arena of trauma, have been touched upon. Nevertheless, VMs are a valuable approach to studying the effects of cells in whole blood on fibrinolysis. Fibrinolysis studies lag behind coagulation studies in terms of methods and data covering transport‐physics aspects such as flow and pressure differences across blood clots 91. Despite all the difficulties highlighted above, which need to be realistically addressed, no doubt progress in method development will continue and contribute to better models to improve our understanding of the regulation of fibrinolysis in health and disease.
Disclosure of Conflict of Interests
The author states that he has no conflict of interest.
Acknowledgements
This work was funded, in part, by a grant from UK Department of Health's Policy Research Programme, Grant Number 044/0069. The views expressed in the publication are those of the author and not necessarily those of the NHS, the NIHR, the Department of Health, ‘arms’ length bodies or other government departments.
Longstaff C. Measuring fibrinolysis: from research to routine diagnostic assays. J Thromb Haemost 2018; 16: 652–62.
Manuscript handled by: P. H. Reitsma
Final decision: P. H. Reitsma, 4 January 2018
References
- 1. Astrup T. The biological significance of fibrinolysis. Lancet 1956; 271: 565–8. [DOI] [PubMed] [Google Scholar]
- 2. Macfarlane RG, Biggs R. Fibrinolysis; its mechanism and significance. Blood 1948; 3: 1167–87. [PubMed] [Google Scholar]
- 3. Fearnley GR, Lackner R. The fibrinolytic activity of normal blood. Br J Haematol 1955; 1: 189–98. [DOI] [PubMed] [Google Scholar]
- 4. Longstaff C, Kolev K. Basic mechanisms and regulation of fibrinolysis. J Thromb Haemost 2015; 13(Suppl 1): S98–105. [DOI] [PubMed] [Google Scholar]
- 5. Rijken DC, Lijnen HR. New insights into the molecular mechanisms of the fibrinolytic system. J Thromb Haemost 2009; 7: 4–13. [DOI] [PubMed] [Google Scholar]
- 6. Kolev K, Longstaff C. Bleeding related to disturbed fibrinolysis. Br J Haematol 2016; 175: 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Borowiecki B, Sharp AA. Trauma and fibrinolysis. J Trauma 1969; 9: 522–36. [PubMed] [Google Scholar]
- 8. Innes D, Sevitt S. Coagulation and fibrinolysis in injured patients. J Clin Pathol 1964; 17: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pellman CM, Ridlon HC, Lang Phillips L. Manifestation and management of hypofibrinogenemia and fibrinolysis in patients with carcinoma of the prostate. J Urol 1966; 96: 375–9. [DOI] [PubMed] [Google Scholar]
- 10. Booth NA, Anderson JA, Bennett B. Plasminogen activators in alcoholic cirrhosis: demonstration of increased tissue type and urokinase type activator. J Clin Pathol 1984; 37: 772–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mackinnon S, Walker ID, Davidson JF, Walker JJ. Plasma fibrinolysis during and after normal childbirth. Br J Haematol 1987; 65: 339–42. [DOI] [PubMed] [Google Scholar]
- 12. Bell WR. The fibrinolytic system in neoplasia. Semin Thromb Hemost 1996; 22: 459–78. [DOI] [PubMed] [Google Scholar]
- 13. Lisman T. Decreased plasma fibrinolytic potential as a risk for venous and arterial thrombosis. Semin Thromb Hemost 2017; 43: 178–84. [DOI] [PubMed] [Google Scholar]
- 14. Horrevoets AJG, Pannekoek H, Nesheim ME. A steady‐state template model that describes the kinetics of fibrin‐stimulated. J Biol Chem 1997; 272: 2183–91. [DOI] [PubMed] [Google Scholar]
- 15. Brummel‐Ziedins K. Models for thrombin generation and risk of disease. J Thromb Haemost 2013; 11(Suppl 1): 212–23. [DOI] [PubMed] [Google Scholar]
- 16. Diamond SL. Systems biology of coagulation. J Thromb Haemost 2013; 11(Suppl 1): 224–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bannish BE, Chernysh IN, Keener JP, Fogelson AL, Weisel JW. Molecular and physical mechanisms of fibrinolysis and thrombolysis from mathematical modeling and experiments. Sci Rep 2017; 7: 6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thelwell C, Longstaff C. The regulation by fibrinogen and fibrin of tissue plasminogen activator kinetics and inhibition by plasminogen activator inhibitor 1. J Thromb Haemost 2007; 5: 804–11. [DOI] [PubMed] [Google Scholar]
- 19. Huish S, Thelwell C, Longstaff C. Activity regulation by fibrinogen and fibrin of streptokinase from streptococcus pyogenes. PLoS ONE 2017; 12: e0170936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Verheijen JH, Mullaart E, Chang GT, Kluft C, Wijngaards G. A simple, sensitive spectrophotometric assay for extrinsic (tissue‐type) plasminogen activator applicable to measurements in plasma. Thromb Haemost 1982; 48: 266–9. [PubMed] [Google Scholar]
- 21. Sands D, Whitton C, Merton R, Longstaff C. A collaborative study to establish the 3rd international standard for tissue plasminogen activator. Thromb Haemost 2002; 88: 294–7. [PubMed] [Google Scholar]
- 22. Thelwell C, Longstaff C, Rigsby P; Subcommittee on Fibrinolysis . An International Collaborative Study to establish the World Health Organization 4th International Standard for Plasmin (13/206): communication from the SSC of the ISTH. J Thromb Haemost 2016; 14: 215–8. [DOI] [PubMed] [Google Scholar]
- 23. Longstaff C, Thelwell C, Rigsby P; SSC Fibirinolysis Subcommittee . An International Collaborative Study to establish the WHO 2nd International Standard for High Molecular Weight Urokinase: communication from SSC of the ISTH. J Thromb Haemost 2014; 12: 415–7. [DOI] [PubMed] [Google Scholar]
- 24. Beebe DP, Aronson DL. An automated fibrinolytic assay performed in microtiter plates. Thromb Res 1987; 47: 123–8. [DOI] [PubMed] [Google Scholar]
- 25. Longstaff C, Whitton CM. A proposed reference method for plasminogen activators that enables calculation of enzyme activities in SI units. J Thromb Haemost 2004; 2: 1416–21. [DOI] [PubMed] [Google Scholar]
- 26. Silva MM, Thelwell C, Williams SC, Longstaff C. Regulation of fibrinolysis by C‐terminal lysines operates through plasminogen and plasmin but not tissue plasminogen activator (tPA). J Thromb Haemost 2012; 10: 2354–60. [DOI] [PubMed] [Google Scholar]
- 27. European Commission . Alteplase for injection. Monograph, European Pharmacopoeia: 2013; 2007: 1170 corrected 8.0.
- 28. Wohner N, Sotonyi P, Machovich R, Szabo L, Tenekedjiev K, Silva MM, Longstaff C, Kolev K. Lytic resistance of fibrin containing red blood cells. Arterioscler Thromb Vasc Biol 2011; 31: 2306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bonnard T, Law LS, Tennant Z, Hagemeyer CE. Development and validation of a high throughput whole blood thrombolysis plate assay. Sci Rep 2017; 7: 2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Urano T, Sakakibara K, Rydzewski A, Urano S, Takada Y, Takada A. Relationships between euglobulin clot lysis time and the plasma levels of tissue plasminogen activator and plasminogen activator inhibitor 1. Thromb Haemost 1990; 63: 82–6. [PubMed] [Google Scholar]
- 31. Smith AA, Jacobson LJ, Miller BI, Hathaway WE, Manco‐Johnson MJ. A new euglobulin clot lysis assay for global fibrinolysis. Thromb Res 2003; 112: 329–37. [DOI] [PubMed] [Google Scholar]
- 32. Hemker HC, Giesen PL, Ramjee M, Wagenvoord R, Beguin S. The thrombogram: monitoring thrombin generation in platelet‐rich plasma. Thromb Haemost 2000; 83: 589–91. [PubMed] [Google Scholar]
- 33. Matsumoto T, Nogami K, Shima M. Simultaneous measurement of thrombin and plasmin generation to assess the interplay between coagulation and fibrinolysis. Thromb Haemost 2013; 110: 761–8. [DOI] [PubMed] [Google Scholar]
- 34. Simpson ML, Goldenberg NA, Jacobson LJ, Bombardier CG, Hathaway WE, Manco‐Johnson MJ. Simultaneous thrombin and plasmin generation capacities in normal and abnormal states of coagulation and fibrinolysis in children and adults. Thromb Res 2011; 127: 317–23. [DOI] [PubMed] [Google Scholar]
- 35. van Geffen M, Loof A, Lap P, Boezeman J, Laros‐van Gorkom BA, Brons P, Verbruggen B, van Kraaij M, van Heerde WL. A novel hemostasis assay for the simultaneous measurement of coagulation and fibrinolysis. Hematology 2011; 16: 327–36. [DOI] [PubMed] [Google Scholar]
- 36. Longstaff C, for the Subcommittee on Fibrinolysis . Development of Shiny app tools to simplify and standardize the analysis of hemostasis assay data: communication from the SSC of the ISTH. J Thromb Haemost 2017; 15: 1044–6. [DOI] [PubMed] [Google Scholar]
- 37. Rosendaal FR, Reitsma PH. Reproducibility. J Thromb Haemost 2017; 15: 833. [DOI] [PubMed] [Google Scholar]
- 38. Weisel JW. Structure of fibrin: impact on clot stability. J Thromb Haemost 2007; 5(Suppl 1): 116–24. [DOI] [PubMed] [Google Scholar]
- 39. Matsuo O, Rijken DC, Collen D. Thrombolysis by human tissue plasminogen activator and urokinase in rabbits with experimental pulmonary embolus. Nature 1981; 291: 590–1. [DOI] [PubMed] [Google Scholar]
- 40. Mutch NJ, Koikkalainen JS, Fraser SR, Duthie KM, Griffin M, Mitchell J, Watson HG, Booth NA. Model thrombi formed under flow reveal the role of factor XIII‐mediated cross‐linking in resistance to fibrinolysis. J Thromb Haemost 2010; 8: 2017–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Robbie LA, Young SP, Bennett B, Booth NA. Thrombi formed in a chandler loop mimic human arterial thrombi in structure and PAI‐1 content and distribution. Thromb Haemost 1997; 77: 510–5. [PubMed] [Google Scholar]
- 42. Varju I, Longstaff C, Szabo L, Farkas AZ, Varga‐Szabo VJ, Tanka‐Salamon A, Machovich R, Kolev K. DNA, histones and neutrophil extracellular traps exert anti‐fibrinolytic effects in a plasma environment. Thromb Haemost 2015; 113: 1289–98. [DOI] [PubMed] [Google Scholar]
- 43. Rijken DC, Kock EL, Guimarães AHC, Talens S, Murad SD, Janssen HLA, Leebeek FWG. Evidence for an enhanced fibrinolytic capacity in cirrhosis as measured with two different global fibrinolysis tests. J Thromb Haemost 2012; 10: 2116–22. [DOI] [PubMed] [Google Scholar]
- 44. Meltzer ME, Doggen CJ, de Groot PG, Rosendaal FR, Lisman T. The impact of the fibrinolytic system on the risk of venous and arterial thrombosis. Semin Thromb Hemost 2009; 35: 468–77. [DOI] [PubMed] [Google Scholar]
- 45. Hendrickx ML, Zatloukalova M, Hassanzadeh‐Ghassabeh G, Muyldermans S, Gils A, Declerck PJ. Identification of a novel, nanobody‐induced, mechanism of TAFI inactivation and its in vivo application. J Thromb Haemost 2014; 12: 229–36. [DOI] [PubMed] [Google Scholar]
- 46. Wyseure T, Rubio M, Denorme F, Martinez de Lizarrondo S, Peeters M, Gils A, De Meyer SF, Vivien D, Declerck PJ. Innovative thrombolytic strategy using a heterodimer diabody against TAFI and PAI‐1 in mouse models of thrombosis and stroke. Blood 2014; 125: 1325–32. [DOI] [PubMed] [Google Scholar]
- 47. Willemse JL, Heylen E, Nesheim ME, Hendriks DF. Carboxypeptidase U (TAFIa): a new drug target for fibrinolytic therapy? J Thromb Haemost 2009; 7: 1962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Song C, Burgess S, Eicher JD, O'Donnell CJ, Johnson AD, Huang J, Sabater‐Lleal M, Asselbergs FW, Tregouet D, Shin S‐Y, Ding J, Baumert J, Oudot‐Mellakh T, Folkersen L, Smith NL, Williams SM, Ikram MA, Kleber ME, Becker DM, Truong V, et al Causal effect of plasminogen activator inhibitor type 1 on coronary heart disease. J Am Heart Assoc 2017; 6: e004918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Declerck PJ, Moreau H, Jespersen J, Gram J, Kluft C. Multicenter evaluation of commercially available methods for the immunological determination of plasminogen activator inhibitor‐1 (PAI‐1). Thromb Haemost 1993; 70: 858–63. [PubMed] [Google Scholar]
- 50. Gram J, Declerck PJ, Sidelmann J, Jespersen J, Kluft C. Multicentre evaluation of commercial kit methods: plasminogen activator inhibitor activity. Thromb Haemost 1993; 70: 852–7. [PubMed] [Google Scholar]
- 51. Rudnicka AR, Rumley A, Lowe GD, Strachan DP. Diurnal, seasonal, and blood‐processing patterns in levels of circulating fibrinogen, fibrin D‐dimer, C‐reactive protein, tissue plasminogen activator, and von Willebrand factor in a 45‐year‐old population. Circulation 2007; 115: 996–1003. [DOI] [PubMed] [Google Scholar]
- 52. Macy EM, Meilahn EN, Declerck PJ, Tracy RP. Sample preparation for plasma measurement of plasminogen activator inhibitor‐1 antigen in large population studies. Arch Pathol Lab Med 1993; 117: 67–70. [PubMed] [Google Scholar]
- 53. Declerck P. TAFI structure and function. J Thromb Haemost 2012; 10: e27. [Google Scholar]
- 54. Schneider M, Boffa M, Stewart R, Rahman M, Koschinsky M, Nesheim M. Two naturally occurring variants of TAFI (Thr‐325 and Ile‐325) differ substantially with respect to thermal stability and antifibrinolytic activity of the enzyme. J Biol Chem 2002; 277: 1021–30. [DOI] [PubMed] [Google Scholar]
- 55. Frère C, Morange PE, Saut N, Tregouet DA, Grosley M, Beltran J, Juhan‐Vague I, Alessi M‐C. Quantification of thrombin activatable fibrinolysis inhibitor (TAFI) gene polymorphism effects on plasma levels of TAFI measured with assays insensitive to isoform‐dependent artefact. Thromb Haemost 2005; 94: 373–9. [DOI] [PubMed] [Google Scholar]
- 56. Marx PF, Dawson PE, Bouma BN, Meijers JC. Plasmin‐mediated activation and inactivation of thrombin‐activatable fibrinolysis inhibitor. Biochemistry 2002; 41: 6688–96. [DOI] [PubMed] [Google Scholar]
- 57. Gils A, Alessi MC, Brouwers E, Peeters M, Marx P, Leurs J, Bouma B, Hendriks D, Juhan‐Vague I, Declerck PJ. Development of a genotype 325‐specific proCPU/TAFI ELISA. Arterioscler Thromb Vasc Biol 2003; 23: 1122–7. [DOI] [PubMed] [Google Scholar]
- 58. Willemse JL, Hendriks DF. Measurement of procarboxypeptidase U (TAFI) in human plasma: a laboratory challenge. Clin Chem 2006; 52: 30–6. [DOI] [PubMed] [Google Scholar]
- 59. Tregouet DA, Schnabel R, Alessi MC, Godefroy T, Declerck PJ, Nicaud V, Munzel T, Bickel C, Rupprecht HJ, Lubos E, Zeller T, Juhan‐Vague I, Blankenberg S, Tiret L, Morange P‐E. Activated thrombin activatable fibrinolysis inhibitor levels are associated with the risk of cardiovascular death in patients with coronary artery disease: the Athero Gene study. J Thromb Haemost 2009; 7: 49–57. [DOI] [PubMed] [Google Scholar]
- 60. Neill EK, Stewart RJ, Schneider MM, Nesheim ME. A functional assay for measuring activated thrombin‐activatable fibrinolysis inhibitor in plasma. Anal Biochem 2004; 330: 332–41. [DOI] [PubMed] [Google Scholar]
- 61. Heylen E, Van Goethem S, Augustyns K, Hendriks D. Measurement of carboxypeptidase U (active thrombin‐activatable fibrinolysis inhibitor) in plasma: challenges overcome by a novel selective assay. Anal Biochem 2010; 403: 114–6. [DOI] [PubMed] [Google Scholar]
- 62. Ceresa E, Brouwers E, Peeters M, Jern C, Declerck PJ, Gils A. Development of ELISAs measuring the extent of TAFI activation. Arterioscler Thromb Vasc Biol 2006; 26: 423–8. [DOI] [PubMed] [Google Scholar]
- 63. Walker JB, Nesheim ME. The molecular weights, mass distribution, chain composition, and structure of soluble fibrin degradation products released from a fibrin clot perfused with plasmin. J Biol Chem 1999; 274: 5201–12. [DOI] [PubMed] [Google Scholar]
- 64. Elms MJ, Bunce IH, Bundesen PG, Rylatt DB, Webber AJ, Masci PP, Whitaker AN. Measurement of crosslinked fibrin degradation products – an immunoassay using monoclonal antibodies. Thromb Haemost 1983; 50: 591–4. [PubMed] [Google Scholar]
- 65. Gaffney PJ, Edgell T, Creighton‐Kempsford LJ, Wheeler S, Tarelli E. Fibrin degradation product (FnDP) assays: analysis of standardization issues and target antigens in plasma. Br J Haematol 1995; 90: 187–94. [DOI] [PubMed] [Google Scholar]
- 66. Dempfle CE, Zips S, Ergul H, Heene DL. The Fibrin Assay Comparison Trial (FACT): evaluation of 23 quantitative D‐dimer assays as basis for the development of D‐dimer calibrators. FACT study group. Thromb Haemost 2001; 85: 671–8. [PubMed] [Google Scholar]
- 67. Dempfle CE, Korte W, Schwab M, Zerback R, Huisman MV; CARDIM Study Group . Sensitivity and specificity of a quantitative point of care D‐dimer assay using heparinized whole blood, in patients with clinically suspected deep vein thrombosis. Thromb Haemost 2006; 96: 79–83. [DOI] [PubMed] [Google Scholar]
- 68. Longstaff C. Shiny app for screening test exploration. RStudio Shiny Apps, 2017, Shiny app to explore test sensitivity, specificity and prevalence. https://drclongstaff.shinyapps.io/ScreeningCL, last accessed 22 December 2017.
- 69. Jennings I, Woods TA, Kitchen DP, Kitchen S, Walker ID. Laboratory D‐dimer measurement: improved agreement between methods through calibration. Thromb Haemost 2007; 98: 1127–35. [DOI] [PubMed] [Google Scholar]
- 70. Meijer P, Haverkate F, Kluft C, de Moerloose P, Verbruggen B, Spannagl M. A model for the harmonisation of test results of different quantitative D‐dimer methods. Thromb Haemost 2006; 95: 567–72. [DOI] [PubMed] [Google Scholar]
- 71. Chetaille P, Alessi MC, Kouassi D, Morange PE, Juhan‐Vague I. Plasma TAFI antigen variations in healthy subjects. Thromb Haemost 2000; 83: 902–5. [PubMed] [Google Scholar]
- 72. Nuzzo R. Scientific method: statistical errors. Nature 2014; 506: 150–2. [DOI] [PubMed] [Google Scholar]
- 73. Chawla DS. Big names in statistics want to shake up much‐maligned P value. Nature 2017; 548: 16–7. [DOI] [PubMed] [Google Scholar]
- 74. Colquhoun D. The reproducibility of research and the misinterpretation of p‐values. R Soc Open Sci 2017; 4: 171085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ganter MT, Hofer CK. Coagulation monitoring: current techniques and clinical use of viscoelastic point‐of‐care coagulation devices. Anesth Analg 2008; 106: 1366–75. [DOI] [PubMed] [Google Scholar]
- 76. Whiting D, Dinardo JA. TEG and ROTEM: technology and clinical applications. Am J Hematol 2014; 89: 228–32. [DOI] [PubMed] [Google Scholar]
- 77. Lang T, Bauters A, Braun SL, Potzsch B, von Pape KW, Kolde HJ, Lakner M. Multi‐centre investigation on reference ranges for ROTEM thromboelastometry. Blood Coagul Fibrinolysis 2005; 16: 301–10. [DOI] [PubMed] [Google Scholar]
- 78. Levrat A, Gros A, Rugeri L, Inaba K, Floccard B, Negrier C, David JS. Evaluation of rotation thrombelastography for the diagnosis of hyperfibrinolysis in trauma patients. Br J Anaesth 2008; 100: 792–7. [DOI] [PubMed] [Google Scholar]
- 79. Gall LS, Brohi K, Davenport RA. Diagnosis and treatment of hyperfibrinolysis in trauma (A European Perspective). Semin Thromb Hemost 2017; 43: 224–34. [DOI] [PubMed] [Google Scholar]
- 80. Balvers K, van Dieren S, Baksaas‐Aasen K, Gaarder C, Brohi K, Eaglestone S, Stanworth S, Johansson PI, Ostrowski SR, Stensballe J, Maegele M, Goslings JC, Juffermans NP. Combined effect of therapeutic strategies for bleeding injury on early survival, transfusion needs and correction of coagulopathy. Br J Surg 2017; 104: 222–9. [DOI] [PubMed] [Google Scholar]
- 81. Schochl H, Voelckel W, Maegele M, Solomon C. Trauma‐associated hyperfibrinolysis. Hamostaseologie 2012; 32: 22–7. [DOI] [PubMed] [Google Scholar]
- 82. Raza I, Davenport R, Rourke C, Platton S, Manson J, Spoors C, Khan S, De'Ath HD, Allard S, Hart DP, Pasi KJ, Hunt BJ, Stanworth S, MacCallum PK, Brohi K. The incidence and magnitude of fibrinolytic activation in trauma patients. J Thromb Haemost 2013; 11: 307–14. [DOI] [PubMed] [Google Scholar]
- 83. Roberts I. Tranexamic acid in trauma: how should we use it? J Thromb Haemost 2015; 13(Suppl 1): S195–9. [DOI] [PubMed] [Google Scholar]
- 84. Moore HB, Moore EE, Liras IN, Gonzalez E, Harvin JA, Holcomb JB, Sauaia A, Cotton BA. Acute fibrinolysis shutdown after injury occurs frequently and increases mortality: a multicenter evaluation of 2,540 severely injured patients. J Am Coll Surg 2016; 222: 347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chapman MP, Moore EE, Moore HB, Gonzalez E, Gamboni F, Chandler JG, Mitra S, Ghasabyan A, Chin TL, Sauaia A, Banerjee A, Silliman CC. Overwhelming tPA release, not PAI‐1 degradation, is responsible for hyperfibrinolysis in severely injured trauma patients. J Trauma Acute Care Surg 2016; 80: 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hunt H, Stanworth S, Curry N, Woolley T, Cooper C, Ukoumunne O, Zhelev Z, Hyde C. Thromboelastography (TEG) and rotational thromboelastometry (ROTEM) for trauma induced coagulopathy in adult trauma patients with bleeding. Cochrane Database Syst Rev 2015; 2: CD010438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wikkelso A, Wetterslev J, Moller AM, Afshari A. Thromboelastography (TEG) or thromboelastometry (ROTEM) to monitor haemostatic treatment versus usual care in adults or children with bleeding. Cochrane Database Syst Rev 2016; CD007871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rugeri L, Levrat A, David JS, Delecroix E, Floccard B, Gros A, Allaouchiche B, Negrier C. Diagnosis of early coagulation abnormalities in trauma patients by rotation thrombelastography. J Thromb Haemost 2007; 5: 289–95. [DOI] [PubMed] [Google Scholar]
- 89. Howard BM, Kornblith LZ, Redick BJ, Vilardi RF, Balhotra KS, Crane JM, Forde MR, Nelson MF, Callcut RA, Cohen MJ. The effects of alcohol on coagulation in trauma patients: interpreting thrombelastography with caution. J Trauma Acute Care Surg 2014; 77: 865–71; discussion 71–2. [DOI] [PubMed] [Google Scholar]
- 90. Spahn DR. TEG®‐ or ROTEM®‐based individualized goal‐directed coagulation algorithms: don't wait – act now! Crit Care 2014; 18: 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Brass LF, Diamond SL. Transport physics and biorheology in the setting of hemostasis and thrombosis. J Thromb Haemost 2016; 14: 906–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bajzar L, Fredenburgh JC, Nesheim M. The activated protein C‐mediated enhancement of tissue‐type plasminogen activator‐induced fibrinolysis in a cell‐free system. J Biol Chem 1990; 265: 16948–54. [PubMed] [Google Scholar]
- 93. He S, Bremme K, Blomback M. A laboratory method for determination of overall haemostatic potential in plasma. I. Method design and preliminary results. Thromb Res 1999; 96: 145–56. [DOI] [PubMed] [Google Scholar]
- 94. He S, Antovic A, Blomback M. A simple and rapid laboratory method for determination of haemostasis potential in plasma. II. Modifications for use in routine laboratories and research work. Thromb Res 2001; 103: 355–61. [DOI] [PubMed] [Google Scholar]
- 95. Antovic A, Blombäck M, Sten‐Linder M, Petrini P, Holmström M, He S. Identifying hypocoagulable states with a modified global assay of overall haemostasis potential in plasma. Blood Coagul Fibrinolysis 2005; 16: 585–96. [DOI] [PubMed] [Google Scholar]
- 96. He S, Zhu K, Skeppholm M, Vedin J, Svensson J, Egberg N, Blombäck M, Wallen H. A global assay of haemostasis which uses recombinant tissue factor and tissue‐type plasminogen activator to measure the rate of fibrin formation and fibrin degradation in plasma. Thromb Haemost 2007; 98: 871–82. [PubMed] [Google Scholar]
- 97. He S, Wallèn HK, Bark N, Blombäck M. In vitro studies using a global hemostasis assay to examine the anticoagulation effects in plasma by the direct thrombin inhibitors: dabigatran and argatroban. J Thromb Thrombolysis 2013; 35: 131–9. [DOI] [PubMed] [Google Scholar]
- 98. Goldenberg NA, Hathaway WE, Jacobson L, Manco‐Johnson MJ. A new global assay of coagulation and fibrinolysis. Thromb Res 2005; 116: 345–56. [DOI] [PubMed] [Google Scholar]
- 99. Goldenberg NA, Hathaway WE, Jacobson L, McFarland K, Manco‐Johnson MJ. Influence of factor VIII on overall coagulability and fibrinolytic potential of haemophilic plasma as measured by global assay: monitoring in haemophilia A. Haemophilia 2006; 12: 605–14. [DOI] [PubMed] [Google Scholar]
- 100. von dem Borne PA, Mosnier LO, Tans G, Meijers JC, Bouma BN. Factor XI activation by meizothrombin: stimulation by phospholipid vesicles containing both phosphatidylserine and phosphatidylethanolamine. Thromb Haemost 1997; 78: 834–9. [PubMed] [Google Scholar]