

Abstract
Chromatin function depends on a dense network of interactions between nucleosomes and a wide range of proteins. A detailed description of these protein–nucleosome interactions is required to reach a full molecular understanding of chromatin function in both genetics and epigenetics. Herein, we show that the structure, dynamics, and interactions of nucleosomes can be interrogated in a residue‐specific manner by using state‐of‐the‐art solid‐state NMR spectroscopy. Using sedimented nucleosomes, high‐resolution spectra were obtained for both flexible histone tails and the non‐mobile histone core. Through co‐sedimentation of a nucleosome‐binding peptide, we demonstrate that protein‐binding sites on the nucleosome surface can be determined. We believe that this approach holds great promise as it is generally applicable, extendable to include the structure and dynamics of the bound proteins, and scalable to interactions of proteins with higher‐order chromatin structures, including isolated and cellular chromatin.
Keywords: magic-angle spinning, NMR spectroscopy, nucleosomes, protein–protein interactions, proton detection
Eukaryotic genomes are present in the cell as a complex of DNA and histone proteins, called chromatin. The fundamental building block of chromatin, the nucleosome, is an assembly of a histone octamer protein core around which approximately 147 base pairs (bp) of DNA are wrapped1 (Figure 1 A). Nucleosomes compact and protect DNA and, importantly, serve as the binding platform for a multitude of proteins that regulate gene transcription, DNA repair, and replication.2 Recently, the structures of various nucleosome–protein complexes have been solved, offering new and exciting insight into chromatin function.3 The wide range of interaction modes in terms of affinities, binding sites, and contribution of dynamics calls for a wide range of techniques to describe the molecular basis of these interactions. The combination of state‐of‐the‐art solid‐state NMR (ssNMR) spectroscopy and sedimentation has developed into an attractive method for the detailed characterization of soluble biomolecular systems.4 Sedimentation has been widely used to study the folding and compaction of nucleosomes and nucleosomal arrays.5 Jaroniec and co‐workers were the first to realize the great potential of ssNMR spectroscopy in the context of nucleosomes through their study on histone tails in nucleosomal arrays.6 Herein, we introduce the use of nucleosome sedimentation, ultra‐fast magic angle spinning (MAS), and 1H‐detected ssNMR spectroscopy to characterize the structure and dynamics of nucleosomes and their protein complexes. High‐quality ssNMR spectra were obtained for both flexible and rigid parts of the nucleosome, allowing near‐complete and residue‐specific assignment. As proof‐of‐principle, we exploited the sensitivity of the amide backbone chemical shifts to map the binding surface of the N‐terminal segment of the LANA protein on the nucleosome surface.7
Figure 1.
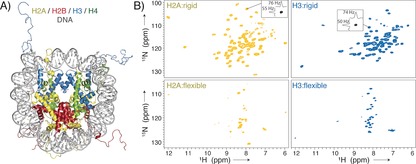
Nucleosome sedimentation yields high‐quality protein “fingerprint” NMR spectra covering both the globular core and flexible tails of the histones. A) The nucleosome is formed by an octamer of four core histones, H2A, H2B, H3, and H4, and binds ca. 147 bp of DNA around its surface (crystal structure from PDB ID 1KX515). B) 1H‐detected 2D NH spectra of sedimented nucleosomes with either 2H/15N/13C‐labeled H2A or H3, using either dipolar‐coupling‐based (top) or scalar‐coupling‐based (bottom) magnetization transfer. Slices along the 1H/15N dimension in the dipolar spectra are shown to highlight typical linewidths.
We reconstituted milligram amounts of mononucleosomes from recombinantly expressed core histones (H2A, H2B, H3, and H4) and a high‐affinity DNA template8 according to established methods (see the Supporting Information for a detailed description and Figure S1).9 The modular nature of the nucleosome (molecular weight ca. 210 kDa) afforded selective isotope labeling of one histone protein per sample (here H2A or H3, each ca. 14 kDa), thereby alleviating signal overlap. Isotope‐labeled histones were labeled uniformly with 15N/13C isotopes in combination with fractional deuteration.10 Typically, ca. 2 mg of the nucleosomes were sedimented directly into a 1.3 mm ssNMR rotor by ultracentrifugation. Proton 1D spectra established that the sediment is highly hydrated (Figure S1).
Sedimented nucleosomes with either labeled H2A or H3 give excellent 2D 1H‐15N correlation spectra in terms of sensitivity, resolution, and signal dispersion (Figure 1). The spectral quality suggests that the nucleosomes in the sediment are well‐folded and homogenously arranged on a microscopic scale. Spectra were recorded using either dipolar‐ or scalar‐coupling‐based magnetization transfer to identify and characterize rigid and dynamic parts in the histones, respectively.11 As expected for folded nucleosomes, the dipolar‐coupling‐based spectra exhibit high chemical shift dispersion in line with the folded core of the histones (Figure 1 B, top). The scalar‐coupling‐based spectra show fewer resonances, which are clustered around δ(1H)=8.2 ppm, characteristic for dynamic and unstructured histone tails (Figure 1 B, bottom). For both H2A and H3, the observed peak pattern is highly similar to that in the solution‐state spectra of nucleosomes,12 allowing the assignment of nearly all observed resonances (Figure S2). For H3, all resonances correspond to the N‐terminal tail (residues 3–35), indicating that the H3 tail of sedimented nucleosomes is unstructured and highly flexible, as in solution13 and in Mg2+‐precipitated nucleosomal arrays.6 For H2A, part of the N‐terminal (residues 3–8), and C‐terminal tails (residues 117–123) are highly dynamic, as judged from their observation in the J‐based ssNMR spectrum. Chemical shift changes to isolated H2A/H2B dimer or H3 peptide, where DNA is absent, suggest that the N‐terminal tails of both H2A and H3 are (transiently) bound to nucleosomal DNA (Figure S2), in line with previous NMR studies.13, 14
The high quality of the spectra, in particular for the core region of the nucleosome, enabled the per‐residue characterization of histone structure, dynamics, and interactions. We performed a de novo sequential resonance assignment of H2A within the sedimented nucleosomes, aided by the assignments of H2A in the H2A–H2B dimer in solution (unpublished data).
Using 3D 1H‐detected CANH/CA(CO)NH and CONH/CO(CA)NH experiments,16 we assigned 93 % of the amide backbone resonances between residues 18–112, which corresponds to nearly all resonances in the dipolar‐based 1H‐15N spectrum of H2A (Figure 2 A, B and Figure S3). Notably, the significant resolution and sensitivity gains afforded by 1H compared to 13C detection were instrumental herein (Figure S4). Based on the assigned H, N, Cα, and C′ chemical shifts, the H2A secondary structure propensities were analyzed using TALOS.17 The characteristic three‐helix histone fold is clearly present, as well as the two β‐strands that mediate interactions with H2B in the H2A–H2B dimer (Figure 2 C). Importantly, the C‐terminal β‐strand, which forms a “docking” sheet with H4 in the nucleosome crystal structure,18 is also observed. This region of H2A, the docking sequence (residues 97–116), binds to H3–H4 and thus stabilizes the histone octamer. In our dipolar ssNMR spectra, resonances for several stretches in this region, including parts of the 310 helix observed in the crystal, are missing, suggesting increased mobility. In addition, this region has, compared to the histone‐fold region, slightly decreased order parameters, as predicted from the chemical shifts (Figure 2 C).19
Figure 2.
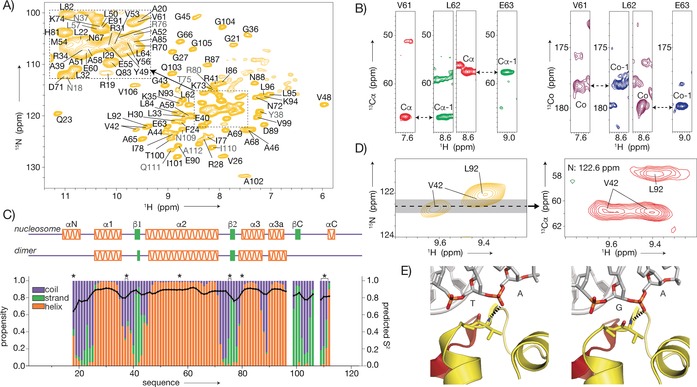
Resonance assignment and secondary structure of H2A in sedimented nucleosomes. A) 2D NH spectrum with (tentative) assignments indicated in (gray) black. Side chain resonances in light colors. B) Representative strips illustrating the sequential backbone assignment based on 3D CANH and CA(CO)NH (red and green) or CONH and CO(CA)NH (magenta and blue) spectra. C) Secondary structure propensities (colored bars) and predicted S2 values (black line) based on assigned backbone chemical shifts. Secondary structure in the nucleosome crystal structure (PDB ID 2PYO18) and isolated H2A–H2B dimer (unpublished results) shown at the top. Asterisks indicate tentative assignments. D, E) Peak doubling of V42 observed in the 2D NH (left) and 3D CANH (right) spectra (D), correlated to the asymmetric environment in 601 nucleosomes (PDB UD 3LZ020). Dashed lines indicate hydrogen bonds (E).
Interestingly, residues 15–17, which are part of the N‐terminal α‐helix in the crystal structure, are absent in both the dipolar and J‐based spectra, and residues 18–20 exhibit low helical propensities and low predicted order parameters, suggesting that the αN helix is not a stably folded structure (Figure 2 C). Comparison of the chemical shifts of H2A in sedimented nucleosomes to those of the H2A–H2B dimer in solution shows significant changes for regions that interact with either other histones or DNA (Figure S5). As an example of the sensitivity to the chemical environment afforded by 1H detection, we note the peak doubling observed for V42 (Figure 2 D). As the 601 DNA is not palindromic, this residue is hydrogen‐bonded to either a TpA or GpA base pair step in the two copies of H2A in the nucleosome (Figure 2 E).20
We next used the assigned amide backbone resonances as sensitive reporters on nucleosome–protein interactions. We chose to study the interaction with the N‐terminal domain of the viral LANA protein, for which a crystal structure of the complex with the nucleosome is available.7 LANA binds to the acidic patch, a negatively charged region on the nucleosome surface formed by acidic residues from H2A and H2B.1a, 2c A 20 residue peptide derived from the LANA N‐terminus binds tightly (K D=0.16 μm) to nucleosomes, in agreement with previous studies using longer fragments21 (Figure S6 A). The LANA peptide (2 kDa) was added in fivefold molar excess to a dilute solution of H2A‐labeled nucleosomes from the same batch as the sample used for assignment, followed by sedimentation into the rotor. Comparison of the remaining supernatant and pure LANA peptide solution by NMR spectroscopy suggested that the peptide had been successfully co‐sedimented with the nucleosome (Figure S6 B). The dipolar 1H‐15N correlation spectrum of the nucleosome–LANA complex, measured under identical conditions to the spectrum of free nucleosomes, shows perturbations specifically for a subset of resonances (Figure 3 A and Figure S7). Importantly, there is no peak doubling upon binding, indicating that LANA is bound to both H2A copies in the nucleosome whereas only one copy is bound in the crystal structure.7a Transfer of H2A assignments to the LANA‐bound state was aided by making use of 13Cα chemical shifts from a 3D CANH spectrum. The lack of significant changes in the Cα shifts indicated that there is little to no structural change in H2A upon binding and underscores the benefit of 1H detection. Thus an unambiguous comparison of the 15N/1H chemical shifts in the free and LANA‐bound states was possible for the majority of the H2A residues (Figure 3 B). Notably, resonances from the acidic patch residues E55, E63, E90, and E91 show clear chemical shift changes. In addition, a number of residues, with L92 in Figure 3 A given as an example, display significant peak broadening in the LANA‐bound state, either reflecting increased local 1H density or local structural or dynamic disorder as a result of binding. Displaying the observed chemical shift perturbations (CSPs) on the crystal structure reveals a striking co‐localization of the changes with the interaction surface of LANA seen in the crystal (Figure 3 C).7a The highest CSPs are observed for the residues in the center of the acidic patch, which are in contact with the LANA arginine anchor.
Figure 3.
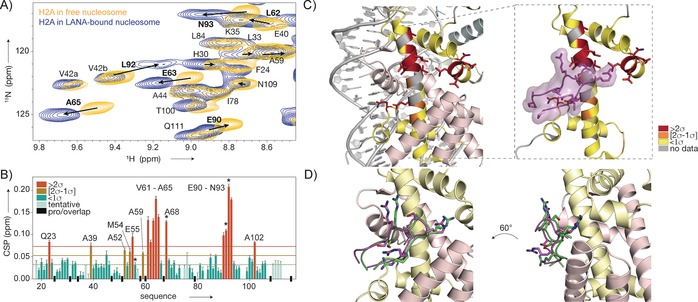
Mapping of the LANA binding site on the nucleosome surface by ssNMR spectroscopy. A) Overlay of the 2D NH spectra of H2A in sedimented nucleosomes (yellow) and nucleosome–LANA complexes (blue). Residues with significant chemical shift changes labeled in bold, peak displacement indicated with arrows. B) Weighted chemical shift perturbations (CSPs) per residue. The 10 % trimmed mean values (dashed line) plus one (orange line) or two (red line) standard deviations (σ) are indicated. Residues with significant peak broadening in the bound state are labeled with asterisks. C) CSPs color‐coded on the structure of H2A. Residues with high CSPs co‐localize with the binding site of LANA (PDB ID 1ZLA). D) NMR data driven model of the LANA–nucleosome complex, showing the best‐scoring water‐refined solution (green). This model corresponds to the crystal structure (magenta) within 1.5 Å backbone RMSD.
We next used our ssNMR data and published mutagenesis data on the LANA–nucleosome interaction7a to generate a molecular model of the complex using HADDOCK.22 The best scoring solution in terms of energetics and agreement with experimental data closely resembles the binding mode observed in the crystal, illustrating the potential of this approach to reveal the architecture of nucleosome–protein complexes (Figure 3 D and Figure S8). We note that for this approach to work well, the structures of the two interacting molecules must be known, and the conformational changes upon binding must be limited.23
In conclusion, we have reported the first high‐resolution solid‐state NMR spectroscopy study on a non‐mobile region of the nucleosome and demonstrated the accurate mapping of binding sites on the nucleosome surface, which was the main goal of this study. High‐quality spectra of histone proteins in sedimented nucleosomes allowed us to obtain near‐complete assignments of histone H2A. Our data indicate that the H2A N‐ and C‐terminal regions, including the αN element and docking domain, have increased flexibility compared to the core, and that its N‐ and C‐terminal tails are highly dynamic, yet DNA‐bound. It is noteworthy that these regions are rich in post‐translational modifications, suggesting that the structural flexibility may be exploited by the enzymes that install, read, or remove these epigenetic marks. In addition, this is the first high‐resolution study of histone structure and nucleosome binding in the crowded, solvated environment of the sediment, thereby mimicking the cellular conditions. Next to this biochemical insight, our approach provides a starting point for the more detailed analysis of the structure and dynamics of nucleosome complexes using the entire toolbox of biomolecular ssNMR spectroscopy. Whereas the solution NMR methyl‐TROSY approach24 is superior in terms of spectral sensitivity and resolution,25 the approach described here enables the observation of all non‐proline residues on the interface and poses fewer labeling requirements. Even though fractional deuteration and considerable measurement times are required, a major advantage is the lack of an intrinsic size limit for ssNMR, making the approach extendable to studies of larger complexes, including larger chromatin substrates such as nucleosomal arrays. We anticipate that in particular for dynamic complexes, for interactions with smaller proteins that cannot easily be captured by cryo‐EM or crystallography, or when plasticity of histones is thought to play a role,26 the use of sedimented nucleosome–protein complexes will be an effective and complementary method in the structural biology of chromatin function.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work received financial support from the Dutch Science Foundation NWO (VIDI grant 723.013.010 to H.v.I., VENI grant 722.016.002 to S.X., NWO‐Groot grant 175.010.2009.002 to M.B., and grant 184.032.207 to the uNMR‐NL National Roadmap Large‐Scale Facility of the Netherlands). We thank Jelmer Eerland for solution‐state assignments of H2A and the Leiden MacBio group members for their support.
S. Xiang, U. B. le Paige, V. Horn, K. Houben, M. Baldus, H. van Ingen, Angew. Chem. Int. Ed. 2018, 57, 4571.
References
- 1.
- 1a. Luger K., Mader A. W., Richmond R. K., Sargent D. F., Richmond T. J., Nature 1997, 389, 251–260; [DOI] [PubMed] [Google Scholar]
- 1b. Kornberg R. D., Lorch Y., Cell 1999, 98, 285–294. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Luger K., Dechassa M. L., Tremethick D. J., Nat. Rev. Mol. Cell Biol. 2012, 13, 436–447; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. McGinty R. K., Tan S., Chem. Rev. 2015, 115, 2255–2273; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. McGinty R. K., Tan S., Curr. Opin. Struct. Biol. 2016, 37, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Speranzini V., Pilotto S., Sixma T. K., Mattevi A., EMBO J. 2016, 35, 376–388; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Liu X., Li M., Xia X., Li X., Chen Z., Nature 2017, 544, 440–445; [DOI] [PubMed] [Google Scholar]
- 3c. Farnung L., Vos S. M., Wigge C., Cramer P., Nature 2017, 550, 539–542; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Kitevski-LeBlanc J., Fradet-Turcotte A., Kukic P., Wilson M. D., Portella G., Yuwen T., Panier S., Duan S., Canny M. D., van Ingen H., Arrowsmith C. H., Rubinstein J. L., Vendruscolo M., Durocher D., Kay L. E., eLife 2017, 6, e23872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Luca S., White J. F., Sohal A. K., Filippov D. V., van Boom J. H., Grisshammer R., Baldus M., Proc. Natl. Acad. Sci. USA 2003, 100, 10706–10711; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Bertini I., Luchinat C., Parigi G., Ravera E., Reif B., Turano P., Proc. Natl. Acad. Sci. USA 2011, 108, 10396–10399; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Gardiennet C., Schutz A. K., Hunkeler A., Kunert B., Terradot L., Bockmann A., Meier B. H., Angew. Chem. Int. Ed. 2012, 51, 7855–7858; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7977–7980; [Google Scholar]
- 4d. Lamley J. M., Iuga D., Öster C., Sass H.-J., Rogowski M., Oss A., Past J., Reinhold A., Grzesiek S., Samoson A., Lewandowski J. R., J. Am. Chem. Soc. 2014, 136, 16800–16806; [DOI] [PubMed] [Google Scholar]
- 4e. Barbet-Massin E., Huang C.-T., Daebel V., Hsu S.-T. D., Reif B., Angew. Chem. Int. Ed. 2015, 54, 4367–4369; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4441–4444; [Google Scholar]
- 4f. Dannatt H. R. W., Felletti M., Jehle S., Wang Y., Emsley L., Dixon N. E., Lesage A., Pintacuda G., Angew. Chem. Int. Ed. 2016, 55, 6638–6641; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6750–6753. [Google Scholar]
- 5.
- 5a. Hansen J. C., Ausio J., Stanik V. H., Van Holde K. E., Biochemistry 1989, 28, 9129–9136; [DOI] [PubMed] [Google Scholar]
- 5b. Shogren-Knaak M., Ishii H., Sun J. M., Pazin M. J., Davie J. R., Peterson C. L., Science 2006, 311, 844–847; [DOI] [PubMed] [Google Scholar]
- 5c. Bertin A., Mangenot S., Renouard M., Durand D., Livolant F., Biophys. J. 2007, 93, 3652–3663; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Korolev N., Allahverdi A., Yang Y., Fan Y., Lyubartsev A. P., Nordenskiold L., Biophys. J. 2010, 99, 1896–1905; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Maeshima K., Rogge R., Tamura S., Joti Y., Hikima T., Szerlong H., Krause C., Herman J., Seidel E., DeLuca J., Ishikawa T., Hansen J. C., EMBO. J. 2016, 35, 1115–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gao M., Nadaud P. S., Bernier M. W., North J. A., Hammel P. C., Poirier M. G., Jaroniec C. P., J. Am. Chem. Soc. 2013, 135, 15278–15281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Barbera A. J., Chodaparambil J. V., Kelley-Clarke B., Joukov V., Walter J. C., Luger K., Kaye K. M., Science 2006, 311, 856–861; [DOI] [PubMed] [Google Scholar]
- 7b. Chodaparambil J. V., Barbera A. J., Lu X., Kaye K. M., Hansen J. C., Luger K., Nat. Struct. Mol. Biol. 2007, 14, 1105–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lowary P. T., Widom J., J. Mol. Biol. 1998, 276, 19–42. [DOI] [PubMed] [Google Scholar]
- 9. Dyer P. N., Edayathumangalam R. S., White C. L., Bao Y., Chakravarthy S., Muthurajan U. M., Luger K., Methods Enzymol. 2004, 375, 23–44. [DOI] [PubMed] [Google Scholar]
- 10. Mance D., Sinnige T., Kaplan M., Narasimhan S., Daniëls M., Houben K., Baldus M., Weingarth M., Angew. Chem. Int. Ed. 2015, 54, 15799–15803; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 16025–16029. [Google Scholar]
- 11.
- 11a. Andronesi O. C., Becker S., Seidel K., Heise H., Young H. S., Baldus M., J. Am. Chem. Soc. 2005, 127, 12965–12974; [DOI] [PubMed] [Google Scholar]
- 11b. Ward M. E., Ritz E., Ahmed M. A. M., Bamm V. V., Harauz G., Brown L. S., Ladizhansky V., J. Biomol. NMR 2015, 63, 375–388. [DOI] [PubMed] [Google Scholar]
- 12. Zhou B. R., Feng H., Ghirlando R., Kato H., Gruschus J., Bai Y., J. Mol. Biol. 2012, 421, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stützer A., Liokatis S., Kiesel A., Schwarzer D., Sprangers R., Soding J., Selenko P., Fischle W., Mol. Cell 2016, 61, 247–259. [DOI] [PubMed] [Google Scholar]
- 14. Kato H., Gruschus J., Ghirlando R., Tjandra N., Bai Y., J. Am. Chem. Soc. 2009, 131, 15104–15105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Davey C. A., Sargent D. F., Luger K., Maeder A. W., Richmond T. J., J. Mol. Biol. 2002, 319, 1097–1113. [DOI] [PubMed] [Google Scholar]
- 16. Zhou D. H., Nieuwkoop A. J., Berthold D. A., Comellas G., Sperling L. J., Tang M., Shah G. J., Brea E. J., Lemkau L. R., Rienstra C. M., J. Biomol. NMR 2012, 54, 291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shen Y., Bax A., J. Biomol. NMR 2013, 56, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clapier C. R., Chakravarthy S., Petosa C., Fernandez-Tornero C., Luger K., Muller C. W., Proteins Struct. Funct. Bioinf. 2008, 71, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Berjanskii M. V., Wishart D. S., J. Am. Chem. Soc. 2005, 127, 14970–14971. [DOI] [PubMed] [Google Scholar]
- 20. Vasudevan D., Chua E. Y., Davey C. A., J. Mol. Biol. 2010, 403, 1–10. [DOI] [PubMed] [Google Scholar]
- 21. Beauchemin C., Moerke N. J., Faloon P., Kaye K. M., J. Biomol. Screening 2014, 19, 947–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Zundert G. C. P., Rodrigues J. P. G. L. M., Trellet M., Schmitz C., Kastritis P. L., Karaca E., Melquiond A. S. J., van Dijk M., de Vries S. J., Bonvin A. M. J. J., J. Mol. Biol. 2016, 428, 720–725. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Bonvin A. M. J. J., Curr. Opin. Struct. Biol. 2006, 16, 194–200; [DOI] [PubMed] [Google Scholar]
- 23b. Lensink M. F., Velankar S., Wodak S. J., Proteins Struct. Funct. Bioinf. 2017, 85, 359–377. [DOI] [PubMed] [Google Scholar]
- 24. Tugarinov V., Hwang P. M., Ollerenshaw J. E., Kay L. E., J. Am. Chem. Soc. 2003, 125, 10420–10428. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Kato H., van Ingen H., Zhou B. R., Feng H., Bustin M., Kay L. E., Bai Y., Proc. Natl. Acad. Sci. USA 2011, 108, 12283–12288; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. van Nuland R., van Schaik F. M., Simonis M., van Heesch S., Cuppen E., Boelens R., Timmers H. M., van Ingen H., Epigenet. Chromatin 2013, 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sinha K. K., Gross J. D., Narlikar G. J., Science 2017, 355, eaaa3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary