

Abstract
There is increasing evidence for a sudden and unprecedented rise in the incidence of multiple sclerosis (MS) in Westernized countries over the past decades, emphasizing the role of environmental factors. Among many candidates, rapid changes in dietary habits seem to play a role in the pathogenesis of MS. Here, we summarize and discuss the available evidence for the role of dietary nutrients, such as table salt, fatty acids, and flavonoids, in the development and pathogenesis of MS. We also discuss new and emerging risk factors accompanying Western lifestyle, such as shift work, sleep, and circadian disruption.
Keywords: diet, environmental risk factors, FOXP3+ Treg cells, multiple sclerosis, Western lifestyle, TH17 cells
Introduction
Multiple sclerosis (MS) is a chronic demyelinating disease of the central nervous system (CNS) that afflicts at least 2.3 million individuals worldwide and is among the most common causes of neurological disability in young adults.1 The pathophysiology of MS is characterized by chronic inflammation, blood–brain barrier (BBB) breakdown, and immune cell infiltration into the CNS, leading to the destruction of the protective myelin sheath of neurons, gliosis, and axonal loss. Generally, MS can be divided into relapsing form of the disease (relapsing–remitting multiple sclerosis (RRMS)) and progressive forms of the disease (primary progressive multiple sclerosis or secondary progressive multiple sclerosis (SPMS)).2
The precise cause of MS still remains unknown. Genetic, environmental, and immunological factors have been implicated in the etiology of this complex and heterogeneous disease. The rise in the incidence and prevalence of MS in the world in the past decades paralleled the rapid socioeconomic development, urbanization, and westernization, which was marked by radical change in dietary and lifestyle habits. The industrial revolution and the contemporary age in Western countries gave rise to the fast‐food industry and widespread consumption of excessive salt, refined vegetable oils, and sugars and also led to reduced physical activity, exposure to artificial light at atypical biological times, and insufficient and poor‐quality sleep. The influence of other environmental factors, such as Epstein–Barr infection, vitamin D levels, smoking, obesity, and geographical location, has already been extensively reviewed.3, 4, 5 Here, we mainly focus on how Western diet and sleep‐circadian disruption may contribute to our understanding of MS etiology.
MS and immune cells
MS is characterized by lesions or so‐called plaques of demyelination and consequent neurodegeneration, which is associated with inflammation mediated by autoreactive myelin‐specific T and B cells, macrophages, and activated microglia.6 It is still not known whether the disease is triggered by peripheral activation of the autoreactive lymphocytes or by intrinsic events in the brain, which is then followed by infiltration of immune cells from peripheral tissues across the BBB into the CNS. The former mechanism is employed to induce experimental autoimmune encephalomyelitis (EAE), the most common model to study MS in rodents. Initially, based on findings in EAE, MS was thought to be mainly a T helper (TH) 1 cell–mediated disease. However, later studies in mice highlighted an important pathogenic role for CD4+ cells that secrete interleukin (IL)‐17 driven by IL‐23 (later referred to as TH17 cells).7 Soon after their discovery, TH17 cells became a main culprit in EAE/MS. TH17 cells secrete proinflammatory cytokines IL‐17A, IL‐17F, IL‐21, IL‐22, and granulocyte–macrophage colony‐stimulating factor, promoting autoimmune neuroinflammation.8 Human TH17 cells are capable of inducing a functional breakdown of the BBB, expressing high levels of granzyme B, and directly damaging the neurons in brains of MS patients.9 Tzartos et al. revealed a significant increase in the number of IL‐17–producing T cells in active lesions in the brains of MS patients.10 Similarly, higher frequencies of TH17 cells were detected in the cerebrospinal fluid of patients with RRMS during relapse and in patients with a first demyelinating event. Furthermore, TH17 cells from these patients showed a highly pathogenic phenotype, with higher expression of costimulatory molecules and higher resistance to suppression.11
Further support for a pivotal role for TH17 cells in MS comes from clinical studies, in which effectiveness of immunoablative chemotherapy and autologous hematopoietic stem cell transplantation in patients with aggressive MS was due to decreased TH17 responses.12 Unlike EAE lesions, the frequencies of IL‐17+ CD8+ cells were almost comparable to the number of IL‐17+ CD4+ T cells in acute MS lesions.10 Moreover, expanded clones of CD8+ T but not CD4+ T cells from MS brain lesions were found in the cerebrospinal fluid and blood, highlighting an important role of CD8+ T cells in MS pathogenesis.13
Suppression of autoreactive cells by regulatory T (Treg) cells may contribute to the protection and recovery from both MS and EAE. Transfer of in vitro–induced Treg cells ameliorates EAE,14 while depletion of Treg cells in vivo by anti‐CD25 antibody worsens disease outcome.15 Initially, no significant alterations in frequencies of Treg cells were found in the peripheral blood of MS. Yet, altered functional state and plasticity of Treg cells and their reduced capacity for suppression may play a role (reviewed in Refs. 16 and 17). Furthermore, decreased forkhead box P3 (FOXP3) and cytotoxic T lymphocyte–associated antigen 4 (CTLA‐4) expression is noted in many MS patients.18, 19
B cells seem to be another important player in MS pathology, as B cell depletion is a successful therapeutic strategy.20, 21, 22, 23, 24, 25 B cells, as well as plasmablasts and plasma cells, are found in lesions and cerebrospinal fluid in most patients with MS, and their numbers correlate with lesion activity.26, 27, 28 Despite the rising evidence in MS, the pathological role of CD8+ T cells and B cells has been far less well studied in experimental animal models compared with CD4+ T cells.
Besides T and B lymphocytes, monocyte‐derived macrophages and activated resident microglia are also abundantly present in active MS lesions.29, 30, 31 In vivo positron emission tomography imaging suggests that macrophage and microglial activity correlates with disability scores in individuals with MS.32 However, recent studies indicate that macrophages and microglia are highly heterogeneous cells displaying spatial‐ and temporal‐dependent identities in the CNS of MS patients.31 For instance, while uptake of myelin by phagocytes is well known to promote demyelination, it also drives phagocytes toward a less inflammatory, neuroprotective phenotype.33, 34 This suggests that phagocytes play roles in both disease‐promoting and disease‐controlling processes in MS. Other immune cells that may be involved in the pathogenesis of MS include innate‐like lymphocytes, such as γδ T cells, natural killer cells, innate lymphoid cells, and mucosal‐associated invariant T cells.35, 36, 37, 38, 39, 40, 41, 42 Investigation of some of these cell subsets in both MS patients and EAE models yielded contradictory observations, and their role in the disease has not been clearly defined.43
Genetic and environmental risk factors
MS has a complex etiology that involves both permissive genetic backgrounds and environmental factors. Of note, genetic predisposition can only explain a fraction of the increased susceptibility risk.44, 45 Genetic studies identified roughly about 200 genetic risk loci involved in MS susceptibility, mostly genes involved in the innate and adaptive immune responses.46, 47, 48 A recent study on genetic and epigenetic fine mapping of causal variants in several autoimmune diseases revealed the cell types most likely to contribute to MS by showing that causal variants of MS are enriched in cis‐regulatory elements mostly in immune cells, particularly Treg cells, TH cells, and B cells.49
Classical twin studies that assess the contribution of genes and environment to a disease yielded conflicting results. Some studies revealed a higher concordance rate between identical (monozygotic) compared with nonidentical (dizygotic) pairs (approximately 25–30% in monozygotic versus 5% in dizygotic twins).50, 51 In contrast, other studies showed equivalent concordance rates.52, 53
Epidemiological studies of families with twins50 and adoptees54 and of migrant individuals who moved between high‐ and low‐risk regions demonstrated that the environment plays an even bigger role in MS than genetic factors. Studies on migrants revealed that individuals moving from an area with high to low MS incidence show a decreased MS risk, while migrants moving in the opposite direction usually maintain the low risk of MS.55, 56 Interestingly, the risk of developing MS in migrants is dependent on the age at migration,57, 58, 59 suggesting that there is a critical time window during childhood or adolescence in determining the risk of developing MS.58
Dietary factors
Diet and obesity as well as intestinal microbiota are well‐known players in MS. For instance, an increased body mass index is positively associated with an increased risk of developing MS in both children and adults.60, 61, 62
The importance of the intestinal microbiota in EAE has been recognized in pioneering work in germ‐free mice and mice after antibiotic treatment. Berer et al. showed that germ‐free mice are protected against spontaneous EAE,63 and treatment with broad‐spectrum antibiotics dramatically reduced EAE clinical symptoms.64 Interestingly, EAE‐resistant germ‐free mice became susceptible after colonization with segmented filamentous bacterium,65 which is known to promote intestinal TH17 responses.66 Studies in MS patients revealed that individuals with MS possess a distinct fecal microbiota composition.67, 68, 69, 70 Indeed, the intestinal flora comprises thousands of different bacterial species; thus, dissecting the role of specific subsets or even single bacteria strain in MS pathogenesis is currently being investigated in further detail. For instance, Lactobacillus reuteri can produce indoles from dietary tryptophan, which is in turn metabolized into ligands for the aryl‐hydrocarbon receptor (AHR), which directly affects astrocytes and restricts CNS inflammation in mice.71 Moreover, serum levels of indoles and its metabolites are decreased in patients with MS.71 However, a more recent study suggests a differential mechanism of regulation: AHR agonistic activity is increased during relapse episodes of MS and decreased during remission.72 When discussing the role of nutrients in MS, we should carefully take into account not only a direct immunomodulatory role of diet but also its associated effects on microbiota composition and the development of obesity.
Salt and MS
The mean global salt intake in 2010 was estimated to be around 10 g per person a day, corresponding to 4 g/day of sodium, which is twice the reference level recommended by the World Health Organization.73 The salt consumption varies considerably among countries, ranging from 1.5 g of sodium per day in East African countries to 3.9 g of sodium per day in high‐income countries.73 Recently, many studies have demonstrated that a high intake of sodium chloride can be an important factor potentially influencing autoimmunity in both humans and rodents.74, 75, 76, 77, 78, 79, 80, 81, 82 In patients with RRMS in Argentina, increased sodium intake was associated with enhanced disease exacerbation and new lesion development.76 On average, the chance of developing a new lesion on magnetic resonance imaging scans was increased 2.8‐fold in individuals whose sodium intake was above recommended levels (2 g/day) and 3.4‐fold higher in individuals who consumed 4.8 g of sodium a day or more. These findings were also accompanied by a higher exacerbation rate in patients with medium and high sodium intake compared with the normal‐intake group.76 Interestingly, there is a positive correlation between the amount of salt consumed and IL‐17 plasma levels in healthy individuals.83 However, a recent large European–Canadian trial study in MS patients treated with interferon‐β1b did not find any association between salt intake and MS activity or progression in this cohort.84 A possible explanation of these divergent results is the different treatments and baseline characteristics of the MS patients included in the studies, as well as the divergent approaches to assess sodium intake in these retrospective analyses. Several studies have shown that high sodium diets could exacerbate EAE.74, 75, 77, 79 When analyzing CD4+ T cells, this effect was accompanied by increased numbers of TH17 cells and a higher infiltration of TH17 cells into the CNS.74, 75 In agreement with these findings, TH17 cells generated under salt‐enriched conditions show a highly pathogenic phenotype in vitro.74, 75 This increase in TH17 responses under high salt conditions was shown to be mediated by p38 mitogen‐activated protein kinase (MAPK), nuclear factor of activated T cells (NFAT5), and serum glucocorticoid kinase‐1 (SGK1).74, 75 Consistently, a T cell–specific knockout of Sgk1 in mice ameliorated EAE disease severity that was seen on a high salt diet.75 Of note, high salt concentrations also alter the regulatory arm of adaptive immunity. Human and mouse Treg cells in high salt environments lose their suppressive function while gaining a TH1‐like phenotype.85
In addition to its effects on T cells, a high salt diet was also associated with altered macrophage responses in EAE.77 Enhanced infiltration and activation of macrophages in the CNS was accompanied by increased levels of the myeloid‐attracting chemokines CCL2 and CXCL10. Both in vitro and in vivo high salt concentrations promoted a proinflammatory M1 phenotype of macrophages, with upregulation of iNOS and IL‐1β. Mechanistically, macrophages pretreated with NaCl showed activation of members of the MAPK pathway, such as ERK1/2, JNK, p38, and p38‐dependent NFAT5, leading to increased production of nitric oxide and tumor necrosis factor‐α.77, 86 Moreover, adoptive transfer of salt‐treated bone marrow–derived macrophages (BMMs) aggravated ongoing EAE compared with control BMMs.77 Dietary salt could also affect anti‐inflammatory macrophage responses. It was recently demonstrated that high salt levels suppress alternative macrophage activation to an M2 or M(IL‐4+IL‐13+) phenotype, promoting tissue inflammation and attenuating wound healing.87
The adoptive and innate responses to high salt seem to be tightly interconnected. Another recent study demonstrated that increased TH17 responses under high sodium diets might also depend on inflammasome activation in macrophages, most likely through the enhanced production of the TH17‐promoting cytokine IL‐1β. Heightened TH17 responses in an inflammation model were significantly abrogated in caspase‐1–deficient mice.88
Although translational evidence of these findings is still scarce, Yi et al. demonstrated in a long‐term longitudinal study that there is a strong positive correlation between serum monocytes and dietary salt intake in healthy donors. Interestingly, high salt intake was associated with increased levels of IL‐6 and IL‐23 and decreased levels of IL‐10, thus creating a milieu that promotes TH17 differentiation.83 Furthermore, 1 week of high salt diet in healthy volunteers resulted in expansion of nonclassical CD14+ CD16+ monocytes and monocyte–platelet aggregates with enhanced reactive oxygen species production by monocytes.89
Besides its effects on immune cells, a high salt challenge can directly affect BBB permeability by suppressing the expression of tight junction proteins on endothelial cells.90 Taken together, these studies demonstrate that a high sodium environment could influence and shape the balance between pro‐ and anti‐inflammatory immune cells, which are involved in the pathogenesis of MS, indicating that high salt intake could represent an environmental risk factor for disease.
Involvement of fatty acids in CNS autoimmunity: saturated versus unsaturated fatty acids
Another hallmark of a typical Western diet is increased consumption of fat (especially saturated fatty acids (SFA), ω‐6 polyunsaturated fatty acids (PUFAs), and trans‐fatty acids) accompanied by an overall decrease in ω‐3 PUFA intake.
Epidemiological and animal studies indicate that the consumption of excessive fats is closely associated with an increased MS incidence and more severe EAE.91, 92, 93, 94, 95, 96, 97 An increasing amount of literature shows that fat‐induced alterations in T cell lineages and the activation status of macrophages and microglia exacerbate neuroinflammation in MS and the EAE model.98, 99, 100, 101, 102, 103 Interestingly, excessive fat consumption is also a key factor inducing obesity, which increases the risk of developing MS.60, 61, 62 Obesity‐induced inflammation may partially underlie changes in the inflammatory status of immune cell subsets in MS.97 Interestingly, a plethora of studies indicate that the fatty acid saturation status and carbon chain length largely define whether fatty acids promote or suppress inflammation. Below, we elaborate on how these structural differences among fatty acids may influence MS pathogenesis.
Fatty acids can be subdivided into unsaturated and saturated fats. Generally, excessive intake of SFAs is suggested to be a causative factor in MS.104 However, to date, there is still controversy regarding the detrimental impact that SFA intake has on MS disease initiation and progression.95 Similarly, the precise underlying pathogenic molecular mechanisms remain largely unknown. Recently, increased consumption of saturated medium‐ and long‐chain fatty acids was found to worsen autoimmunity in the EAE model.98 The exacerbated EAE severity was paralleled by increased TH17 cell frequencies in the CNS and elevated TH1 and TH17 cell numbers in the spleen. In line with the latter, exogenous SFAs are reported to enrich the binding of RORγt at the IL‐17 and IL‐23R loci, thereby inducing the polarization of “pathogenic” TH17 cells.99 Similarly, an increase in systemic endotoxin levels after SFA consumption105 may partially account for exacerbated neuroinflammation after SFA intake. Endotoxins can promote TH1 and TH17 polarization,106, 107 as well as the inflammatory activation of peripheral and CNS‐resident innate immune cells.108, 109 Of interest, SFAs, such as palmitate (C16:0) and laurate (C12:0), can also directly activate TLR‐mediated proinflammatory signaling pathways in innate immune cells.100 Finally, while feeding dietary monounsaturated fats reduces low‐density lipoprotein (LDL) levels, SFAs are reported to elevate circulating LDL.110 Elevated levels of LDL may stimulate inflammatory innate and adaptive immune responses.111, 112 In contrast to SFAs, high intake of PUFAs is associated with a lower risk of MS.113 In particular, the PUFA α‐linolenic acid is inversely associated with MS risk, while the long‐chain fatty acids, such as eicosapentaenoic acid and docosahexaenoic acid, are not.113 Interestingly, a recent case–control study in Australia showed that high intake of PUFA from fish but not plants was associated with a reduced risk of first clinical attack of CNS demyelination.114 This study suggests that the dietary source of PUFAs is crucial for its beneficial impact on CNS demyelination. Of note, not all PUFA species harbor anti‐inflammatory properties. Omega‐3 PUFAs are known to have anti‐inflammatory properties, repressing NLRP3 inflammasome activation in macrophages,101 reducing TH17 cell differentiation,102 activating peroxisome proliferator–activated receptors,115 and inhibiting the migratory activity of leukocytes.116 By using the fat‐1 mouse model, in which ω‐3 PUFAs are endogenously formed from ω‐6 PUFAs, ω‐3 PUFAs were found to boost remyelination.117 Some of the anti‐inflammatory effects of ω‐3 PUFAs may be explained by the fact that they act as precursors of anti‐inflammatory resolvins and protectins.118, 119 Unlike ω‐3 PUFAs, ω‐6 PUFAs have been suggested to be proinflammatory and may stimulate TH17 cell activation,120 likely by acting as precursors of inflammatory lipid mediators, such as eicosanoids.120 Interestingly, ω‐3 PUFAs are also reported to inhibit the formation of ω‐6 PUFA–derived proinflammatory eicosanoids,121 which may be another way in which ω‐3 PUFAs are beneficial in MS. However, a Cochrane review of randomized trials of dietary interventions for MS did not observe a significant effect of ω‐6 PUFAs on disease progression and relapse rate.122 Moreover, oral feeding of ω‐6 PUFAs attenuated the disease course of acute and chronic EAE, which was paralleled by an increased production of prostaglandin E2 (PGE2) and transforming growth factor beta 1 (TGF‐β1).123 It is worth mentioning that trans‐fatty acids, unsaturated fatty acids that contain at least one double bond in the trans‐configuration, promote the inflammatory activation of macrophages and TH17 polarization.124 Collectively, these studies indicate that the SFA/UFA balance is important for MS disease initiation and progression, but more research is needed to unravel the culprit fatty acid species involved (Fig. 1).
Figure 1.
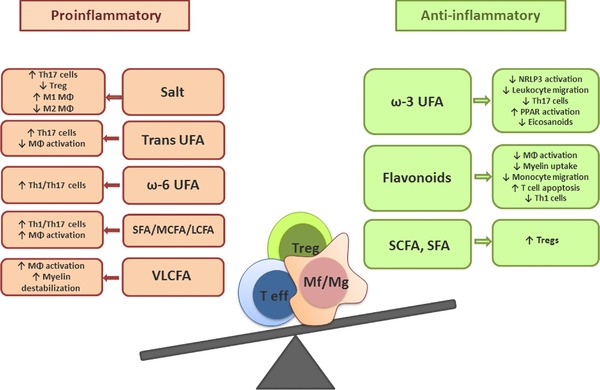
The impact of dietary fats, salt, and flavonoids on the immunopathogenesis of MS. Dietary factors are increasingly acknowledged to affect immune cell function in MS. For example, diets rich in salt and diverse fatty acids species, such as trans‐unsaturated fatty acids (UFAs), saturated fatty acids (SFAs), ω‐6 unsaturated fatty acids (ω‐6 UFAs), medium‐chain fatty acids (MCFAs), long‐chain fatty acids (LCFAs), or very‐long‐chain fatty acids (VLCFAs), induce TH1/TH17 cells and the inflammatory activation of phagocytes, such as macrophages and microglia. A diet rich in these nutritional factors may promote MS disease progression. In contrast, saturated short‐chain fatty acids (SCFA), ω‐3 unsaturated fatty acids (ω‐3 UFAs), and flavonoids may beneficially affect immune cell function in MS. Upon dietary consumption or after intestinal formation, SCFAs are reported to increase Treg cell differentiation and proliferation. ω‐3 UFAs may reduce inflammasome activation, the formation of inflammatory eicosanoids, TH17 cell numbers, and leukocyte migration into the CNS. In addition, ω‐3 UFAs can activate the anti‐inflammatory peroxisome proliferator–activated receptor. Plant‐derived flavonoids reduce phagocyte activation and migration, as well as the uptake of myelin by these cells. Furthermore, they may promote T cell apoptosis and the differentiation and proliferation of TH1 cells.
Long‐ versus short‐chain (saturated) fatty acids
Increasing evidence indicates that the length of fatty acids also determines how they affect inflammation in the context of autoimmunity. Fatty acids can be classified into short‐ (SCFA, <10 carbon atoms), medium‐ (MCFA, 10–14 carbon atoms), long‐ (LCFA, 16–20 carbon atoms), and very‐long‐chain fatty acids (VLCFA, >20 carbon atoms). SCFAs were recently found to ameliorate EAE, as well as other autoimmune diseases, such as type 1 diabetes, by expanding lamina propria–derived Treg cells through suppression of the JNK1 and p38 pathway.98, 125 In line with these studies, SFCAs also increase the number of colonic Treg cells in an experimental model of inflammatory bowel disease.126, 127 In addition to controlling JNK1 and p38 signaling, SCFA may also function as histone deacetylase inhibitors, thereby potentially controlling the acetylation of Treg‐associated genes.128
The main source of SCFA in humans is a fiber‐rich diet (i.e., consisting of complex polysaccharides) that is fermented by gut bacteria: diet and the gut microbiota are closely interlinked and determine the level of SCFAs available.129 It is believed that the amount of plant‐derived fiber in the diet and hence the concentration of fermented SCFAs, both of which dramatically reduced into the Western diet, are inversely correlated with metabolic and cardiovascular disease. The resulting lower diversity of bacterial taxa due to high levels of fats and carbohydrates in the Western diet further contributes to this vicious circle.
Propionic acid (PA), a three‐carbon saturated SCFA for which much of the data are available from immune‐ and metabolic‐related research, is also readily available in crustaceans and Swiss cheese. PA had also been extensively used as food preservative in the past (e.g., in bread and pastries) and thus is approved by the European (European Food Safety Authority; E280 and E281) and American (Food and Drug Administration) food agencies as a food additive.130, 131 While, in recent decades, there has been a decline in the use of PA and its salts as food additives, there has been a contemporaneous rise in the incidence of several autoimmune diseases, including MS, as mentioned above. However, it has not been tested whether there is a causal link between these trends.
MS patients show reduced levels of species belonging to Clostridia XIVa and IV clusters, which are known to produce anti‐inflammatory SCFAs, such as propionate and butyrate.69 This argues for a dysfunctional microbiome being a possible culprit for MS disease initiation or progression. SCFAs, MCFAs, and LCFAs, the most abundant components of a Western diet, worsen EAE severity by enhancing TH1 and TH17 differentiation and proliferation and possibly by decreasing intestinal SCFA levels.98 In line with this finding, LCFA also induced the polarization of human naive T cells toward TH1 and TH17 fates in vitro.98 In addition to its effects on T cells, MCFA may also affect macrophage physiology through the activation of G protein‐coupled receptor 84 (GPR84).132 The expression of GPR84 is elevated in the CNS of EAE animals, and GPR84 promotes the inflammatory activation of macrophages.133, 134 VLFCAs can be derived from both dietary and endogenous origin through elongation of LCFAs. In X‐linked adrenoleukodystrophy (X‐ALD), elevated levels of VLCFAs are well known to promote neuroinflammation and demyelination.135 Classically, structural changes in myelin lipid components were suggested to trigger destabilization of the myelin sheath in X‐ALD patients, leading to demyelination and eventually neuroinflammation.103 However, accumulation of VLCFAs in membrane domains associated with signal transduction pathways may also directly trigger the inflammatory activation of macrophages and microglia.136, 137, 138 In MS, SPMS, and RRMS, patients with longer disease duration show elevated levels of VLCFAs compared with controls and RRMS patients with shorter disease duration. However, another study demonstrated no changes in serum levels of VLCFAs in MS patients.139 More research is warranted to define the impact that dietary VLCFAs or VLCFAs formed upon dietary intake of LCFAs have on MS pathogenesis. In summary, these findings indicate that SCFAs, either dietary or formed by the microbiota, may have therapeutic value for MS by inducing Treg cells. In contrast, diets rich in particular MCFA, LCFA, and VLCFA species may exert a detrimental effect on MS disease progression.
Flavonoids
Despite the detrimental effects of a Western diet on MS, it also contains factors that may counteract these disease‐promoting constituents. An example of a class of beneficial factors is flavonoids, which are present in a wide range in plant‐derived food products. Flavonoids are present in a Western diet, although in limited amounts. Flavonoids are a large class of plant components with immunomodulatory and antioxidative properties.140 Some flavonoids have been shown to modulate disease mechanisms that are involved in the pathology of MS and may thus have a beneficial influence on the progression of the disease. In a Western diet, flavonoids are present in food products, such as cereals, chocolate, beans, wine, beer, tea, and coffee. The most abundant flavonoids in these dietary products are quercetin, resveratrol, catechin, epicatechin, and kaemperfol.141
Some of the flavonoids present in a Western diet are described to have immunomodulatory, neuroprotective, and repair‐promoting properties. Quercetin and epigallocatechin gallate (EGCG) reduce the production of proinflammatory cytokines by macrophages and microglia.142, 143, 144, 145 Moreover, flavonoids, such as quercetin, suppress the capacity of monocytes to cross the BBB and limit the uptake of myelin by macrophages.146, 147 Interestingly, quercetin was shown to suppress proinflammatory cytokine production by monocytes derived from MS patients.148 Moreover, quercetin, EGCG, and resveratrol reduce disease severity in the EAE model.147, 149, 150 In EAE, flavonoids mainly exert their beneficial effects through their anti‐inflammatory properties. While quercetin modulates neuroinflammation predominantly by interfering with TH1 differentiation, resveratrol ameliorates EAE by promoting the integrity of the BBB and the induction of T cell apoptosis.149, 151, 152 Various signaling pathways may underlie these functional changes. Flavonoids inhibit STAT signaling through the induction of SOCS3, and they inhibit RhoA GTPases, thus impairing cytoskeletal changes.147, 153 While the flavonoids present in a Western diet may have some capacity to modulate disease mechanisms in MS, flavonoids that are less abundant may have a higher potential to modulate CNS inflammation. Here, the flavonoid subclass of flavones may be particularly effective. Flavones, such as hesperidin, apigenin, and luteolin, are present in herbs, fruits, and vegetables, like parsley, oregano, celery, citrus fruit, artichoke, and lettuce, of which the intake is typically low in a Western diet and high in a Mediterranean diet.141 These flavones effectively suppress disease progression in the EAE model.154, 157 Interestingly, it was recently demonstrated that flavonoids, such as scutellarin, promote CNS repair, as demonstrated in the cuprizone model, which is commonly used to study de‐ and remyelination.155, 156 This indicates that flavonoids may be useful not only to target the inflammatory response in MS but also to promote repair of damage within the CNS.
Most of the data that suggest beneficial effects of flavonoids for MS are based on cell culture and animal models. Thus, their efficacy in MS patients has not yet been studied. Therefore, caution has to be taken when extrapolating these results to the human situation. When EGCG was tested in a clinical phase I/II study in MS patients, no neuroprotective effects were found. The dose tested induced hepatotoxicity, which led to a premature termination of the study, which questions the potential of flavonoids to be used as a complementary treatment in MS.157 Since flavonoids could differ in their mechanism of action, testing long‐term intake of physiological doses of flavonoids may still be a potential strategy to slow chronic inflammatory and neurodegenerative diseases like MS. However, this has first to be explored in larger studies in detail.
Shift work, sleep, and circadian disruption in MS
The Industrial Revolution and urbanized living not only changed eating habits but also altered life behaviors in ways that were never possible before. The advent of artificial light made it possible for individuals to be exposed to light at unnatural biological times, leading to sleep and circadian misalignment. As a consequence of a Western “24/7” society, there is an increase in numbers of workers employed in shift work, which is estimated to be between 15% and 25% in industrialized countries. Shift work is work on a schedule outside of the traditional working hours from 9 am to 5 pm for 5 days a week. In Europe, it is estimated that only 24% of work force follows the classical working schedule.158 Shift work can be permanent or rotating, with or without night shifts. Importantly, an increasing amount of epidemiological studies have shown a strong evidence that shift work is associated with severe health conditions, including cardiovascular diseases, obesity, metabolic syndrome, type 2 diabetes, certain cancers, and gastrointestinal disorders and can bear a potential risk to reproduction.159 Research in the past decade also revealed the increased risk of autoimmune diseases, such as psoriasis,160 rheumatoid arthritis,161 systemic lupus erythematosus,162 and thyroid disorders163 linked to shift work. Recently, two large case–control studies in Sweden uncovered an association between shift work and increased risk of MS.164, 165 Hedström et al. showed that there is a twofold greater risk of developing MS for individuals who worked night shifts for at least 3 years as teens before age 20 compared with those who only worked day shifts, which was possibly due to a circadian rhythm and sleep disruption. Another Scandinavian study, from Denmark, confirmed the link between night shift work during the critical period between 15 and 19 years and the risk of developing MS. However, no association was found between the duration of shift work in years and increased risk for MS.166
Although the biological mechanisms underlying increased risk of MS among shift workers are not fully understood, possible mechanisms have been proposed. The direct consequences of shift work are circadian disruption and sleep restriction, since most shift workers are not able to adapt their biological rhythms to these shifted times of activity, sleep, and food intake. Disruption of the circadian rhythms primarily induced by light exposure at night, as experienced by shift workers, may alter the circadian expression of clock genes and their epigenetic status. Clock gene expression is regulated by several interconnected feedback loops. The main autoregulatory feedback loop is regulated by positive (the heterodimers brain and muscle aryl hydrocarbon receptor nuclear translocator (ARNT)‐like protein 1 (also known as BMAL1)/circadian locomotor output cycles kaput (CLOCK) and BMAL1/neuronal PAS (Per–Arnt–Sim) domain protein 2 (NPAS2)) and negative components (period 1–3 (PER1–3) and cryptochrome 1 and 2 (CRY1–2)).167 A pilot study of women working night shifts in Denmark compared to day‐working women (i.e., never worked the night shift) found promoter hypomethylation of CLOCK and hypermethylation of CRY2.168 The degree of promoter hypomethylation usually results in increased gene expression, and hypermethylation is consistent with decreased gene expression, which was observed in another study of the expression of main circadian genes among female nurses working a rotating shift schedule compared with nurses on a day‐only shift. This study identified impairment of clock gene expression, showing the upregulation of positive regulators, such as BMAL1, CLOCK, and NPAS2, and downregulation of negative components, such as PER3, CRY1, and CRY2, in peripheral blood mononuclear cells.169 Interestingly, CRY2, a gene involved in regulation of the circadian clock, is hypermethylated and expressed at lower levels in MS–affected brains than in controls.170 However, whether this epigenetic change associated with MS may be caused by circadian disruption through exposure to light at night is not fully understood.
There is evidence of a circadian disruption of corticosterone and leptin levels, as well as rhythmic disruption in heart rate and blood pressure in EAE mice.171, 172 Moreover, a recent study showed a time‐of‐day circadian variation of EAE induction, which is dependent on a functional circadian clock in T cells.173 Daytime immunization induced a steep and more severe EAE, with increased demyelination and increased number of TH17 cells compared with night‐time immunization. This difference was no longer seen in mice lacking CLOCK in T cells. Furthermore, the circadian clock in lymphocytes was found to control the cyclic variation in cell numbers in lymph nodes throughout the day. CC‐chemokine receptor 7, one of the main lymph node homing receptors for lymphocytes, is increased during the day just before the peak of lymph node cellularity, while the sphingosine‐1‐phosphate receptor, which controls egress of lymphocytes from lymph nodes, is increased during the night when the cell number in lymph nodes is low.173
Another possible mechanism behind the negative health consequences of shift work is sleep loss, which might be associated with disturbed melatonin secretion and shortened sleep. Approximately 60% of patients with MS report sleep disturbances and poor sleep quality.174, 175 Sleep disorders, such as circadian rhythm sleep disorder, obstructive sleep apnea, restless legs syndrome, and periodic limb movement disorder, are more frequent in MS patients relative to healthy control subjects, especially in those who experience severe fatigue.176, 177 Interestingly, in a mouse model of MS, sleep fragmentation before and after EAE induction led to a worse disease outcome by promoting leukocyte infiltration across the BBB and impairing immune regulation, but termination of sleep fragmentation improved EAE scores in these mice compared with mice that had normal sleep pattern.178 Light exposure at night and sleep deprivation suppress circulating levels of melatonin,179, 180 a hormone released by the pineal gland during the dark phase that plays a critical role in the regulation of circadian and seasonal rhythms.181 A role for melatonin in MS has been suggested by several clinical observations. Both melatonin levels in saliva and levels of the melatonin metabolite 6‐sulfatoxymelatonin in urine were significantly lower in MS patients compared with controls, suggesting deregulated production of this hormone in individuals with MS.182, 183, 184, 185 A recent study by Farez et al. showed that melatonin levels are inversely correlated with MS relapses in a seasonal pattern: Higher melatonin levels in autumn and winter corresponded to lower number of relapses during this time of a year. Using melatonin as a treatment in rodent models of MS yielded conflicting results. Some studies showed detrimental effects of melatonin,186, 187 while most demonstrated beneficial outcomes in EAE.188, 189, 190, 191, 192 The latter studies reported several mechanisms. Kang et al. proposed that the melatonin effects in rat EAE were mediated by reducing inflammatory cell infiltration into the CNS by suppressing the expression of the intracellular adhesion molecule 1 (ICAM‐1).189 Farez et al. demonstrated that melatonin can directly bind to T cells via melatonin receptor type 1A and interfere with TH17 and IL‐10–producing Tr1 cell differentiation, thus boosting levels of Tr1 cells while suppressing the generation of TH17 cells.191 However, so far, only one case report of a female MS patient was published in which melatonin was used as the only treatment for MS for 4 years, and the disability status improved by two points from Expanded Disability Status Scale (EDSS) 8 to EDSS 6.193 Overall, most published reports on sleep and circadian disruption in MS patients are descriptive in nature and cannot causally link the disruption to the pathogenesis. More research is needed to gain a deeper understanding of the effects of shift work, sleep, and circadian disruption and their underlying mechanisms in MS (Fig. 2).
Figure 2.
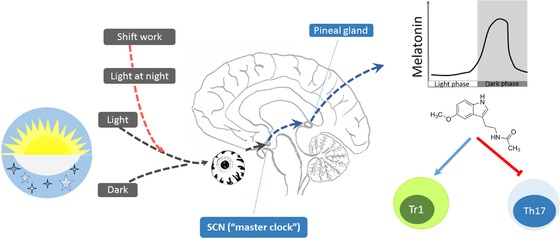
Shift work and circadian disruption: role of disrupted melatonin rhythm in immune regulation. Light stimulation of the retina signals to the circadian clock, located in the suprachiasmatic nucleus in the hypothalamus. This information about the length of day and night is passed on to the pineal gland, where melatonin is produced. Melatonin, which is secreted in the dark with a robust circadian rhythm, has its highest expression levels at night and lowest during the day, regulating the sleep–wake cycle and the balance between immunosuppressive Tr1 cells and proinflammatory TH17 cells. Artificial lighting during the night as experienced by shift workers disrupts this circadian melatonin rhythm.
Concluding remarks
MS remains one of the most challenging neurologic diseases to understand and treat. Several new and emerging risk factors associated with a Westernized lifestyle may improve our risk assessment for MS. Here, we summarized the currently available evidence for the role of dietary factors, such as salt, fatty acids, flavonoids, and sleep and circadian disruption, in the immunopathology of MS. Although more research is needed on the mechanisms underlying the impact of lifestyle on MS, studying the combined lifestyle factors simultaneously would help to enhance our understanding of the multifactorial impact on MS etiology. Furthermore, unbiased, controlled, and prospective randomized trials including large numbers of diverse patients are needed before personalized lifestyle recommendations can be given. The advancement of our knowledge of molecular mechanisms that trigger and drive MS may lead to the development of individualized dietary and lifestyle interventions that may control the development, progression, and clinical course of MS.
Competing interests
The authors declare no competing interests.
Acknowledgments
M.K. was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (640116), by an SALK grant from the government of Flanders, Belgium, and by an Odysseus grant from the Research Foundation Flanders, Belgium (FWO). J.H. and J.B. are supported by the FWO and the Charcot Foundation Belgium. R.A.L. holds an endowed professorship supported by Novartis Pharma.
References
- 1. Browne, P. , Chandraratna D., Angood C., et al 2014. Atlas of Multiple Sclerosis 2013: a growing global problem with widespread inequity. Neurology 83: 1022–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nylander, A. & Hafler D.A.. 2012. Multiple sclerosis. J. Clin. Invest. 122: 1180–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olsson, T. , Barcellos L.F. & Alfredsson L.. 2017. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 13: 25–36. [DOI] [PubMed] [Google Scholar]
- 4. Hewer, S. , Lucas R., van der Mei I., et al 2013. Vitamin D and multiple sclerosis. J. Clin. Neurosci. 20: 634–641. [DOI] [PubMed] [Google Scholar]
- 5. Belbasis, L. , Bellou V., Evangelou E., et al 2015. Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta‐analyses. Lancet Neurol. 14: 263–273. [DOI] [PubMed] [Google Scholar]
- 6. Sospedra, M. & Martin R.. 2005. Immunology of multiple sclerosis. Annu. Rev. Immunol. 23: 683–747. [DOI] [PubMed] [Google Scholar]
- 7. Langrish, C.L. , Chen Y., Blumenschein W.M., et al 2005. IL‐23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201: 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Petermann, F. & Korn T.. 2011. Cytokines and effector T cell subsets causing autoimmune CNS disease. FEBS Lett. 585: 3747–3757. [DOI] [PubMed] [Google Scholar]
- 9. Kebir, H. , Kreymborg K., Ifergan I., et al 2007. Human TH17 lymphocytes promote blood–brain barrier disruption and central nervous system inflammation. Nat. Med. 13: 1173–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tzartos, J.S. , Friese M.A., Craner M.J., et al 2008. Interleukin‐17 production in central nervous system‐infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 172: 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brucklacher‐Waldert, V. , Stuerner K., Kolster M., et al 2009. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 132: 3329–3341. [DOI] [PubMed] [Google Scholar]
- 12. Darlington, P.J. , Touil T., Doucet J.S., et al 2013. Diminished Th17 (not Th1) responses underlie multiple sclerosis disease abrogation after hematopoietic stem cell transplantation. Ann. Neurol. 73: 341–354. [DOI] [PubMed] [Google Scholar]
- 13. Skulina, C. , Schmidt S., Dornmair K., et al 2004. Multiple sclerosis: brain‐infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proc. Natl. Acad. Sci. USA 101: 2428–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kohm, A.P. , Carpentier P.A., Anger H.A., et al 2002. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen‐specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J. Immunol. 169: 4712–4716. [DOI] [PubMed] [Google Scholar]
- 15. Kohm, A.P. , McMahon J.S., Podojil J.R., et al 2006. Cutting edge: anti‐CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells. J. Immunol. 176: 3301–3305. [DOI] [PubMed] [Google Scholar]
- 16. Kleinewietfeld, M. & Hafler D.A.. 2014. Regulatory T cells in autoimmune neuroinflammation. Immunol. Rev. 259: 231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kleinewietfeld, M. & Hafler D.A.. 2013. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin. Immunol. 25: 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sellebjerg, F. , Krakauer M., Khademi M., et al 2012. FOXP3, CBLB and ITCH gene expression and cytotoxic T lymphocyte antigen 4 expression on CD4(+) CD25(high) T cells in multiple sclerosis. Clin. Exp. Immunol. 170: 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Venken, K. , Hellings N., Thewissen M., et al 2008. Compromised CD4+ CD25(high) regulatory T‐cell function in patients with relapsing–remitting multiple sclerosis is correlated with a reduced frequency of FOXP3‐positive cells and reduced FOXP3 expression at the single‐cell level. Immunology 123: 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kappos, L. , Li D., Calabresi P.A., et al 2011. Ocrelizumab in relapsing–remitting multiple sclerosis: a phase 2, randomised, placebo‐controlled, multicentre trial. Lancet 378: 1779–1787. [DOI] [PubMed] [Google Scholar]
- 21. CAMMS223 Trial Investigators , Coles A.J., Composton D.A., et al 2008. Alemtuzumab vs. interferon beta‐1a in early multiple sclerosis. N. Engl. J. Med. 359: 1786–1801. [DOI] [PubMed] [Google Scholar]
- 22. Coles, A.J. , Fox E., Vladic A., et al 2012. Alemtuzumab more effective than interferon β‐1a at 5‐year follow‐up of CAMMS223 clinical trial. Neurology 78: 1069–1078. [DOI] [PubMed] [Google Scholar]
- 23. Hawker, K. , O'Connor P., Freedman M.S., et al 2009. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double‐blind placebo‐controlled multicenter trial. Ann. Neurol. 66: 460–471. [DOI] [PubMed] [Google Scholar]
- 24. Hauser, S.L. , Comi G.C. & Hartung H.‐P.. 2015. Efficacy and safety of ocrelizumab in relapsing multiple sclerosis—results of the interferon‐beta‐1a‐controlled, double blind, phase III OPERA I and II studies. Mult. Scler. 21(S11): 61–62. [Google Scholar]
- 25. Montalban, X. , Hemmer B., Rammohan K., et al; ORATORIO Clinical Investigators. 2015. Efficacy and safety of ocrelizumab in primary progressive multiple sclerosis—results of the placebo‐controlled, double‐blind, phase III ORATORIO study. ECTRIMS Congress, Barcelona, abstract 228.
- 26. Magliozzi, R. , Howell O., Vora A., et al 2007. Meningeal B‐cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 130: 1089–1104. [DOI] [PubMed] [Google Scholar]
- 27. Cepok, S. , Rosche B., Grummel V., et al 2005. Short‐lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain 128: 1667–1676. [DOI] [PubMed] [Google Scholar]
- 28. Kuenz, B. , Lutterotti A., Ehling R., et al 2008. Cerebrospinal fluid B cells correlate with early brain inflammation in multiple sclerosis. PLoS One 3: e2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Howell, O.W. , Rundle J.L., Garg A., et al 2010. Activated microglia mediate axoglial disruption that contributes to axonal injury in multiple sclerosis. J. Neuropathol. Exp. Neurol. 69: 1017–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trebst, C. , Sorensen T.L., Kivisakk P., et al 2001. CCR1+/CCR5+ mononuclear phagocytes accumulate in the central nervous system of patients with multiple sclerosis. Am. J. Pathol. 159: 1701–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bogie, J.F. , Stinissen P. & Hendriks J.J.. 2014. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol. 128: 191–213. [DOI] [PubMed] [Google Scholar]
- 32. Politis, M. , Giannetti P., Su P., et al 2012. Increased PK11195 PET binding in the cortex of patients with MS correlates with disability. Neurology 79: 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bogie, J.F. , Jorissen W., Mailleux J., et al 2013. Myelin alters the inflammatory phenotype of macrophages by activating PPARs. Acta Neuropathol. Commun. 1: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bogie, J.F. , Timmermans S., Huynh‐Thu V.A., et al 2012. Myelin‐derived lipids modulate macrophage activity by liver X receptor activation. PLoS One 7: e44998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shimonkevitz, R. , Colburn C., Burnham J.A., et al 1993. Clonal expansions of activated gamma/delta T cells in recent‐onset multiple sclerosis. Proc. Natl. Acad. Sci. USA 90: 923–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schirmer, L. , Rothhammer V., Hemmer B., et al 2013. Enriched CD161high CCR6+ γδ T cells in the cerebrospinal fluid of patients with multiple sclerosis. JAMA Neurol. 70: 345–351. [DOI] [PubMed] [Google Scholar]
- 37. Plantone, D. , Marti A., Frisullo G., et al 2013. Circulating CD56dim NK cells expressing perforin are increased in progressive multiple sclerosis. J. Neuroimmunol. 265: 124–127. [DOI] [PubMed] [Google Scholar]
- 38. Rodriguez‐Martin, E. , Picon C., Costa‐Frossard L., et al 2015. Natural killer cell subsets in cerebrospinal fluid of patients with multiple sclerosis. Clin. Exp. Immunol. 180: 243–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gross, C.C. , Schulte‐Mecklenbeck A., Hanning U., et al 2017. Distinct pattern of lesion distribution in multiple sclerosis is associated with different circulating T‐helper and helper‐like innate lymphoid cell subsets. Mult. Scler. 23: 1025–1030. [DOI] [PubMed] [Google Scholar]
- 40. Abrahamsson, S.V. , Angelini D.F., Dubinsky A.N., et al 2013. Non‐myeloablative autologous haematopoietic stem cell transplantation expands regulatory cells and depletes IL‐17 producing mucosal‐associated invariant T cells in multiple sclerosis. Brain 136: 2888–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Illes, Z. , Shimamura M., Newcombe J., et al 2004. Accumulation of Valpha7.2–Jalpha33 invariant T cells in human autoimmune inflammatory lesions in the nervous system. Int. Immunol. 16: 223–230. [DOI] [PubMed] [Google Scholar]
- 42. Miyazaki, Y. , Miyake S., Chiba A., et al 2011. Mucosal‐associated invariant T cells regulate Th1 response in multiple sclerosis. Int. Immunol. 23: 529–535. [DOI] [PubMed] [Google Scholar]
- 43. Bianchini, E. , De Biasi S., Simone A.M., et al 2017. Invariant natural killer T cells and mucosal‐associated invariant T cells in multiple sclerosis. Immunol. Lett. 183: 1–7. [DOI] [PubMed] [Google Scholar]
- 44. International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2 , Sawcer S., et al 2011. Genetic risk and a primary role for cell‐mediated immune mechanisms in multiple sclerosis. Nature 476: 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taylor, B.V. 2011. The major cause of multiple sclerosis is environmental: genetics has a minor role—yes. Mult. Scler. 17: 1171–1173. [DOI] [PubMed] [Google Scholar]
- 46. Moutsianas, L. , Jostins L., Beecham A.H., et al 2015. Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat. Genet. 47: 1107–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. International Multiple Sclerosis Genetics Consortium (IMSGC) , Beecham A.H., Patsopoulos N.A., et al 2013. Analysis of immune‐related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 45: 1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Patsopoulos, N.A. , International Multiple Sclerosis Genetics Consortium . 2016. 200 loci complete the genetic puzzle of multiple sclerosis. In American Society of Human Genetics 2016 Annual Meeting, Vancouver, BC, Canada.
- 49. Farh, K.K. , Marson A., Zhu J., et al 2015. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518: 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Willer, C.J. , Dyment D.A., Risch N.J., et al 2003. Twin concordance and sibling recurrence rates in multiple sclerosis. Proc. Natl. Acad. Sci. USA 100: 12877–12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mumford, C.J. , Wood N.W., Kellar‐Wood H., et al 1994. The British Isles survey of multiple sclerosis in twins. Neurology 44: 11–15. [DOI] [PubMed] [Google Scholar]
- 52. French Research Group on Multiple Sclerosis . 1992. Multiple sclerosis in 54 twinships: concordance rate is independent of zygosity. French Research Group on Multiple Sclerosis. Ann. Neurol. 32: 724–727. [DOI] [PubMed] [Google Scholar]
- 53. Ristori, G. , Cannoni S., Stazi M.A., et al 2006. Multiple sclerosis in twins from continental Italy and Sardinia: a nationwide study. Ann. Neurol. 59: 27–34. [DOI] [PubMed] [Google Scholar]
- 54. Ebers, G.C. , Sadovnick A.D. & Risch N.J.. 1995. A genetic basis for familial aggregation in multiple sclerosis. Canadian Collaborative Study Group. Nature 377: 150–151. [DOI] [PubMed] [Google Scholar]
- 55. Berg‐Hansen, P. , Moen S.M., Sandvik L., et al 2015. Prevalence of multiple sclerosis among immigrants in Norway. Mult. Scler. 21: 695–702. [DOI] [PubMed] [Google Scholar]
- 56. Kurtzke, J.F. , Beebe G.W. & J.E. Norman, Jr . 1985. Epidemiology of multiple sclerosis in US veterans: III. Migration and the risk of MS. Neurology 35: 672–678. [DOI] [PubMed] [Google Scholar]
- 57. McLeod, J.G. , Hammond S.R. & Kurtzke J.F.. 2011. Migration and multiple sclerosis in immigrants to Australia from United Kingdom and Ireland: a reassessment. I. Risk of MS by age at immigration. J. Neurol. 258: 1140–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dean, G. & Kurtzke J.F.. 1971. On the risk of multiple sclerosis according to age at immigration to South Africa. Br. Med. J. 3: 725–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Orton, S.M. , Ramagopalan S.V., Brocklebank D., et al 2010. Effect of immigration on multiple sclerosis sex ratio in Canada: the Canadian Collaborative Study. J. Neurol. Neurosurg. Psychiatry 81: 31–36. [DOI] [PubMed] [Google Scholar]
- 60. Mokry, L.E. , Ross S., Timpson N.J., et al 2016. Obesity and multiple sclerosis: a Mendelian randomization study. PLoS Med. 13: e1002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Langer‐Gould, A. , Brara S.M., Beaber B.E., et al 2013. Childhood obesity and risk of pediatric multiple sclerosis and clinically isolated syndrome. Neurology 80: 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hedström, A.K. , Olsson T. & Alfredsson L.. 2012. High body mass index before age 20 is associated with increased risk for multiple sclerosis in both men and women. Mult. Scler. 18: 1334–1336. [DOI] [PubMed] [Google Scholar]
- 63. Berer, K. , Mues M., Koutrolos M., et al 2011. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479: 538–541. [DOI] [PubMed] [Google Scholar]
- 64. Ochoa‐Reparaz, J. , Mielcarz D.W., Ditrio L.E., et al 2009. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J. Immunol. 183: 6041–6050. [DOI] [PubMed] [Google Scholar]
- 65. Lee, Y.K. , Menezes J.S., Umesaki Y., et al 2011. Proinflammatory T‐cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 108(Suppl. 1): 4615–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ivanov, I.I. , Atarashi K., Manel N., et al 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jangi, S. , Gandhi R., Cox L.M., et al 2016. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 7: 12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tremlett, H. , Fadrosh D.W., Faruqi A.A., et al 2016. Gut microbiota composition and relapse risk in pediatric MS: a pilot study. J. Neurol. Sci. 363: 153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Miyake, S. , Kim S., Suda W., et al 2015. Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLoS One 10: e0137429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cosorich, I. , Dalla‐Costa G., Sorini C., et al 2017. High frequency of intestinal TH17 cells correlates with microbiota alterations and disease activity in multiple sclerosis. Sci. Adv. 3: e1700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rothhammer, V. , Mascanfroni I.D., Bunse L., et al 2016. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 22: 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rothhammer, V. , Borucki D.M., Garcia Sanchez M.I., et al 2017. Dynamic regulation of serum aryl hydrocarbon receptor agonists in MS. Neurol. Neuroimmunol. Neuroinflamm. 4: e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mozaffarian, D. , Fahimi S., Singh G.M., et al 2014. Global sodium consumption and death from cardiovascular causes. N. Engl. J. Med. 371: 624–634. [DOI] [PubMed] [Google Scholar]
- 74. Kleinewietfeld, M. , Manzel A., Titze J., et al 2013. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496: 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wu, C. , Yosef N., Thalhamer T., et al 2013. Induction of pathogenic TH17 cells by inducible salt‐sensing kinase SGK1. Nature 496: 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Farez, M.F. , Fiol M.P., Gaitan M.I., et al 2015. Sodium intake is associated with increased disease activity in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 86: 26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hucke, S. , Eschborn M., Liebmann M., et al 2016. Sodium chloride promotes pro‐inflammatory macrophage polarization thereby aggravating CNS autoimmunity. J. Autoimmun. 67: 90–101. [DOI] [PubMed] [Google Scholar]
- 78. Jorg, S. , Kissel J., Manzel A., et al 2016. High salt drives TH17 responses in experimental autoimmune encephalomyelitis without impacting myeloid dendritic cells. Exp. Neurol. 279: 212–222. [DOI] [PubMed] [Google Scholar]
- 79. Krementsov, D.N. , Case L.K., Hickey W.F., et al 2015. Exacerbation of autoimmune neuroinflammation by dietary sodium is genetically controlled and sex specific. FASEB J. 29: 3446–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Salgado, E. , Bes‐Rastrollo M., de Irala J., et al 2015. High sodium intake is associated with self‐reported rheumatoid arthritis: a cross sectional and case control analysis within the SUN cohort. Medicine 94: e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wu, H. , Huang X., Qiu H., et al 2016. High salt promotes autoimmunity by TET2‐induced DNA demethylation and driving the differentiation of Tfh cells. Sci. Rep. 6: 28065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yang, X. , Yao G., Chen W., et al 2015. Exacerbation of lupus nephritis by high sodium chloride related to activation of SGK1 pathway. Int. Immunopharmacol. 29: 568–573. [DOI] [PubMed] [Google Scholar]
- 83. Yi, B. , Titze J., Rykova M., et al 2015. Effects of dietary salt levels on monocytic cells and immune responses in healthy human subjects: a longitudinal study. Transl. Res. 166: 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fitzgerald, K.C. , Munger K.L., Hartung H.P., et al 2017. Sodium intake and multiple sclerosis activity and progression in BENEFIT. Ann. Neurol. 82: 20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hernandez, A.L. , Kitz A., Wu C., et al 2015. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J. Clin. Invest. 125: 4212–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jantsch, J. , Schatz V., Friedrich D., et al 2015. Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage‐driven host defense. Cell Metab. 21: 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Binger, K.J. , Gebhardt M., Heinig M., et al 2015. High salt reduces the activation of IL‐4‐ and IL‐13‐stimulated macrophages. J. Clin. Invest. 125: 4223–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ip, W.K. & Medzhitov R.. 2015. Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat. Commun. 6: 6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhou, X. , Zhang L., Ji W.J., et al 2013. Variation in dietary salt intake induces coordinated dynamics of monocyte subsets and monocyte–platelet aggregates in humans: implications in end organ inflammation. PLoS One 8: e60332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhang, T. , Fang S., Wan C., et al 2015. Excess salt exacerbates blood–brain barrier disruption via a p38/MAPK/SGK1‐dependent pathway in permanent cerebral ischemia. Sci. Rep. 5: 16548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Timmermans, S. , Bogie J.F., Vanmierlo T., et al 2014. High fat diet exacerbates neuroinflammation in an animal model of multiple sclerosis by activation of the renin angiotensin system. J. Neuroimmune Pharmacol. 9: 209–217. [DOI] [PubMed] [Google Scholar]
- 92. Swank, R.L. , Lerstad O., Strom A., et al 1952. Multiple sclerosis in rural Norway its geographic and occupational incidence in relation to nutrition. N. Engl. J. Med. 246: 722–728. [DOI] [PubMed] [Google Scholar]
- 93. Alter, M. , Yamoor M. & Harshe M.. 1974. Multiple sclerosis and nutrition. Arch. Neurol. 31: 267–272. [DOI] [PubMed] [Google Scholar]
- 94. Lauer, K. 1997. Diet and multiple sclerosis. Neurology 49: S55–S61. [DOI] [PubMed] [Google Scholar]
- 95. Zhang, S.M. , Willett W.C., Hernan M.A., et al 2000. Dietary fat in relation to risk of multiple sclerosis among two large cohorts of women. Am. J. Epidemiol. 152: 1056–1064. [DOI] [PubMed] [Google Scholar]
- 96. Schwarz, S. & Leweling H.. 2005. Multiple sclerosis and nutrition. Mult. Scler. 11: 24–32. [DOI] [PubMed] [Google Scholar]
- 97. Winer, S. , Paltser G., Chan Y., et al 2009. Obesity predisposes to Th17 bias. Eur. J. Immunol. 39: 2629–2635. [DOI] [PubMed] [Google Scholar]
- 98. Haghikia, A. , Jorg S., Duscha A., et al 2015. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity 43: 817–829. [DOI] [PubMed] [Google Scholar]
- 99. Wang, C. , Yosef N., Gaublomme J., et al 2015. CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell 163: 1413–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Huang, S. , Rutkowsky J.M., Snodgrass R.G., et al 2012. Saturated fatty acids activate TLR‐mediated proinflammatory signaling pathways. J. Lipid Res. 53: 2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yan, Y. , Jiang W., Spinetti T., et al 2013. Omega‐3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity 38: 1154–1163. [DOI] [PubMed] [Google Scholar]
- 102. Allen, M.J. , Fan Y.Y., Monk J.M., et al 2014. n‐3 PUFAs reduce T‐helper 17 cell differentiation by decreasing responsiveness to interleukin‐6 in isolated mouse splenic CD4(+) T cells. J. Nutr. 144: 1306–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Singh, I. & Pujol A.. 2010. Pathomechanisms underlying X‐adrenoleukodystrophy: a three‐hit hypothesis. Brain Pathol. 20: 838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Swank, R.L. & Goodwin J.W.. 2003. How saturated fats may be a causative factor in multiple sclerosis and other diseases. Nutrition 19: 478. [DOI] [PubMed] [Google Scholar]
- 105. Mani, V. , Hollis J.H. & Gabler N.K.. 2013. Dietary oil composition differentially modulates intestinal endotoxin transport and postprandial endotoxemia. Nutr. Metab. 10: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Davila, E. & Kolls J.. 2010. A “Toll” for Th17 cell expansion. J. Leukoc. Biol. 88: 5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. McAleer, J.P. & Vella A.T.. 2008. Understanding how lipopolysaccharide impacts CD4 T‐cell immunity. Crit. Rev. Immunol. 28: 281–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Qin, L. , Wu X., Block M.L., et al 2007. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55: 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Rivest, S. 2009. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 9: 429–439. [DOI] [PubMed] [Google Scholar]
- 110. Siri‐Tarino, P.W. , Sun Q., Hu F.B., et al 2010. Saturated fatty acids and risk of coronary heart disease: modulation by replacement nutrients. Curr. Atheroscler. Rep. 12: 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Tall, A.R. & Yvan‐Charvet L.. 2015. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 15: 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Lim, H. , Kim Y.U., Sun H., et al 2014. Proatherogenic conditions promote autoimmune T helper 17 cell responses in vivo . Immunity 40: 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bjornevik, K. , Chitnis T., Ascherio A., et al 2017. Polyunsaturated fatty acids and the risk of multiple sclerosis. Mult. Scler. 23: 1830–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hoare, S. , Lithander F., van der Mei I., et al 2016. Higher intake of omega‐3 polyunsaturated fatty acids is associated with a decreased risk of a first clinical diagnosis of central nervous system demyelination: results from the Ausimmune Study. Mult. Scler. 22: 884–892. [DOI] [PubMed] [Google Scholar]
- 115. Schmitz, G. & Ecker J.. 2008. The opposing effects of n‐3 and n‐6 fatty acids. Prog. Lipid Res. 47: 147–155. [DOI] [PubMed] [Google Scholar]
- 116. Ferrante, A. , Goh D., Harvey D.P., et al 1994. Neutrophil migration inhibitory properties of polyunsaturated fatty acids. The role of fatty acid structure, metabolism, and possible second messenger systems. J. Clin. Invest. 93: 1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Siegert, E. , Paul F., Rothe M., et al 2017. The effect of omega‐3 fatty acids on central nervous system remyelination in fat‐1 mice. BMC Neurosci. 18: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kohli, P. & Levy B.D.. 2009. Resolvins and protectins: mediating solutions to inflammation. Br. J. Pharmacol. 158: 960–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Basil, M.C. & Levy B.D.. 2016. Specialized pro‐resolving mediators: endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 16: 51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sakata, D. , Yao C. & Narumiya S.. 2010. Prostaglandin E2, an immunoactivator. J. Pharmacol. Sci. 112: 1–5. [DOI] [PubMed] [Google Scholar]
- 121. Calviello, G. , Su H.M., Weylandt K.H., et al 2013. Experimental evidence of omega‐3 polyunsaturated fatty acid modulation of inflammatory cytokines and bioactive lipid mediators: their potential role in inflammatory, neurodegenerative, and neoplastic diseases. Biomed. Res. Int. 2013: 743171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Farinotti, M. , Vacchi L., Simi S., et al 2012. Dietary interventions for multiple sclerosis. Cochrane Database Syst. Rev. 12: CD004192. [DOI] [PubMed] [Google Scholar]
- 123. Harbige, L.S. , Layward L., Morris‐Downes M.M., et al 2000. The protective effects of omega‐6 fatty acids in experimental autoimmune encephalomyelitis (EAE) in relation to transforming growth factor‐beta 1 (TGF‐beta1) up‐regulation and increased prostaglandin E2 (PGE2) production. Clin. Exp. Immunol. 122: 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Okada, Y. , Tsuzuki Y., Ueda T., et al 2013. Trans fatty acids in diets act as a precipitating factor for gut inflammation? J. Gastroenterol. Hepatol. 28(Suppl. 4): 29–32. [DOI] [PubMed] [Google Scholar]
- 125. Marino, E. , Richards J.L., McLeod K.H., et al 2017. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat. Immunol. 18: 552–562. [DOI] [PubMed] [Google Scholar]
- 126. Smith, P.M. , Howitt M.R., Panikov N., et al 2013. The microbial metabolites, short‐chain fatty acids, regulate colonic Treg cell homeostasis. Science 341: 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Furusawa, Y. , Obata Y., Fukuda S., et al 2013. Commensal microbe‐derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504: 446–450. [DOI] [PubMed] [Google Scholar]
- 128. Wang, L. , de Zoeten E.F., Greene M.I., et al 2009. Immunomodulatory effects of deacetylase inhibitors: therapeutic targeting of FOXP3+ regulatory T cells. Nat. Rev. Drug Discov. 8: 969–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Sonnenburg, J.L. & Backhed F.. 2016. Diet–microbiota interactions as moderators of human metabolism. Nature 535: 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. U.S. Food & Drug Administration . 2016. CFR—Code of Federal Regulations Title 21. September 21, 2016. Accessed April 21, 2017. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=184.1081.
- 131. EFSA Panel on Food Additives and Nutrient Sources added to Food (ANS) . 2016. Safety of the extension of use of sodium propionate (E 281) as a food additive. EFSA J. 14: 4546–4559. [Google Scholar]
- 132. Alvarez‐Curto, E. & Milligan G.. 2016. Metabolism meets immunity: the role of free fatty acid receptors in the immune system. Biochem. Pharmacol. 114: 3–13. [DOI] [PubMed] [Google Scholar]
- 133. Bouchard, C. , Page J., Bedard A., et al 2007. G protein‐coupled receptor 84, a microglia‐associated protein expressed in neuroinflammatory conditions. Glia 55: 790–800. [DOI] [PubMed] [Google Scholar]
- 134. Nicol, L.S. , Dawes J.M., La Russa F., et al 2015. The role of G‐protein receptor 84 in experimental neuropathic pain. J. Neurosci. 35: 8959–8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Brites, P. , Mooyer P.A., El Mrabet L., et al 2009. Plasmalogens participate in very‐long‐chain fatty acid‐induced pathology. Brain 132: 482–492. [DOI] [PubMed] [Google Scholar]
- 136. Paintlia, A.S. , Gilg A.G., Khan M., et al 2003. Correlation of very long chain fatty acid accumulation and inflammatory disease progression in childhood X‐ALD: implications for potential therapies. Neurobiol. Dis. 14: 425–439. [DOI] [PubMed] [Google Scholar]
- 137. Yanagisawa, N. , Shimada K., Miyazaki T., et al 2008. Enhanced production of nitric oxide, reactive oxygen species, and pro‐inflammatory cytokines in very long chain saturated fatty acid‐accumulated macrophages. Lipids Health Dis. 7: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Weber, F.D. , Wiesinger C., Forss‐Petter S., et al 2014. X‐linked adrenoleukodystrophy: very long‐chain fatty acid metabolism is severely impaired in monocytes but not in lymphocytes. Hum. Mol. Genet. 23: 2542–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wilkins, A. , Ingram G., Brown A., et al 2009. Very long chain fatty acid levels in patients diagnosed with multiple sclerosis. Mult. Scler. 15: 1525–1527. [DOI] [PubMed] [Google Scholar]
- 140. Kumar, S. & Pandey A.K.. 2013. Chemistry and biological activities of flavonoids: an overview. Sci. World J. 2013: 162750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Rothwell, J.A. , Perez‐Jimenez J., Neveu V., et al 2013. Phenol‐Explorer 3.0: a major update of the Phenol‐Explorer database to incorporate data on the effects of food processing on polyphenol content. Database 2013: bat070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Cheng‐Chung Wei, J. , Huang H.C., Chen W.J., et al 2016. Epigallocatechin gallate attenuates amyloid β‐induced inflammation and neurotoxicity in EOC 13.31 microglia. Eur. J. Pharmacol. 770: 16–24. [DOI] [PubMed] [Google Scholar]
- 143. Endale, M. , Park S.C., Kim S., et al 2013. Quercetin disrupts tyrosine‐phosphorylated phosphatidylinositol 3‐kinase and myeloid differentiation factor‐88 association, and inhibits MAPK/AP‐1 and IKK/NF‐κB‐induced inflammatory mediators production in RAW 264.7 cells. Immunobiology 218: 1452–1467. [DOI] [PubMed] [Google Scholar]
- 144. Manjeet, K.R. & Ghosh B.. 1999. Quercetin inhibits LPS‐induced nitric oxide and tumor necrosis factor‐alpha production in murine macrophages. Int. J. Immunopharmacol. 21: 435–443. [DOI] [PubMed] [Google Scholar]
- 145. Sun, G.Y. , Chen Z., Jasmer K.J., et al 2015. Quercetin attenuates inflammatory responses in BV‐2 microglial cells: role of MAPKs on the Nrf2 pathway and induction of heme oxygenase‐1. PLoS One 10: e0141509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Hendriks, J.J. , de Vries H.E., van der Pol S.M., et al 2003. Flavonoids inhibit myelin phagocytosis by macrophages; a structure–activity relationship study. Biochem. Pharmacol. 65: 877–885. [DOI] [PubMed] [Google Scholar]
- 147. Hendriks, J.J. , Alblas J., van der Pol S.M., et al 2004. Flavonoids influence monocytic GTPase activity and are protective in experimental allergic encephalitis. J. Exp. Med. 200: 1667–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Sternberg, Z. , Chadha K., Lieberman A., et al 2008. Quercetin and interferon‐beta modulate immune response(s) in peripheral blood mononuclear cells isolated from multiple sclerosis patients. J. Neuroimmunol. 205: 142–147. [DOI] [PubMed] [Google Scholar]
- 149. Singh, N.P. , Hegde V.L., Hofseth L.J., et al 2007. Resveratrol (trans‐3,5,4′‐trihydroxystilbene) ameliorates experimental allergic encephalomyelitis, primarily via induction of apoptosis in T cells involving activation of aryl hydrocarbon receptor and estrogen receptor. Mol. Pharmacol. 72: 1508–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Aktas, O. , Prozorovski T., Smorodchenko A., et al 2004. Green tea epigallocatechin‐3‐gallate mediates T cellular NF‐kappa B inhibition and exerts neuroprotection in autoimmune encephalomyelitis. J. Immunol. 173: 5794–5800. [DOI] [PubMed] [Google Scholar]
- 151. Muthian, G. & Bright J.J.. 2004. Quercetin, a flavonoid phytoestrogen, ameliorates experimental allergic encephalomyelitis by blocking IL‐12 signaling through JAK–STAT pathway in T lymphocyte. J. Clin. Immunol. 24: 542–552. [DOI] [PubMed] [Google Scholar]
- 152. Wang, D. , Li S.P., Fu J.S., et al 2016. Resveratrol defends blood–brain barrier integrity in experimental autoimmune encephalomyelitis mice. J. Neurophysiol. 116: 2173–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Zhang, Y. , Li X., Ciric B., et al 2015. Therapeutic effect of baicalin on experimental autoimmune encephalomyelitis is mediated by SOCS3 regulatory pathway. Sci. Rep. 5: 17407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Ginwala, R. , McTish E., Raman C., et al 2016. Apigenin, a natural flavonoid, attenuates EAE severity through the modulation of dendritic cell and other immune cell functions. J. Neuroimmune Pharmacol. 11: 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Liang, M. , Chen Y., Zhang L., et al 2015. Epimedium flavonoids ameliorate neuropathological changes and increases IGF‐1 expression in C57BL/6 mice exposed to cuprizone. Neurochem. Res. 40: 492–500. [DOI] [PubMed] [Google Scholar]
- 156. Wang, W.W. , Lu L., Bao T.H., et al 2016. Scutellarin alleviates behavioral deficits in a mouse model of multiple sclerosis, possibly through protecting neural stem cells. J. Mol. Neurosci. 58: 210–220. [DOI] [PubMed] [Google Scholar]
- 157. Lovera, J. , Ramos A., Devier D., et al 2015. Polyphenon E, non‐futile at neuroprotection in multiple sclerosis but unpredictably hepatotoxic: phase I single group and phase II randomized placebo‐controlled studies. J. Neurol. Sci. 358: 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Costa, G. , Akerstedt T., Nachreiner F., et al 2004. Flexible working hours, health, and well‐being in Europe: some considerations from a SALTSA project. Chronobiol. Int. 21: 831–844. [DOI] [PubMed] [Google Scholar]
- 159. Costa, G. 2010. Shift work and health: current problems and preventive actions. Saf. Health Work 1: 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Li, W.Q. , Qureshi A.A., Schernhammer E.S., et al 2013. Rotating night‐shift work and risk of psoriasis in US women. J. Invest. Dermatol. 133: 565–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Puttonen, S. , Oksanen T., Vahtera J., et al 2010. Is shift work a risk factor for rheumatoid arthritis? The Finnish Public Sector study. Ann. Rheum. Dis. 69: 779–780. [DOI] [PubMed] [Google Scholar]
- 162. Cooper, G.S. , Parks C.G., Treadwell E.L., et al 2004. Occupational risk factors for the development of systemic lupus erythematosus. J. Rheumatol. 31: 1928–1933. [PubMed] [Google Scholar]
- 163. Magrini, A. , Pietroiusti A., Coppeta L., et al 2006. Shift work and autoimmune thyroid disorders. Int. J. Immunopathol. Pharmacol. 19: 31–36. [PubMed] [Google Scholar]
- 164. Hedström, A.K. , Akerstedt T., Hillert J., et al 2011. Shift work at young age is associated with increased risk for multiple sclerosis. Ann. Neurol. 70: 733–741. [DOI] [PubMed] [Google Scholar]
- 165. Hedström, A.K. , Akerstedt T., Olsson T., et al 2015. Shift work influences multiple sclerosis risk. Mult. Scler. 21: 1195–1199. [DOI] [PubMed] [Google Scholar]
- 166. Gustavsen, S. , Sondergaard H.B., Oturai D.B., et al 2016. Shift work at young age is associated with increased risk of multiple sclerosis in a Danish population. Mult. Scler. Relat. Disord. 9: 104–109. [DOI] [PubMed] [Google Scholar]
- 167. Lowrey, P.L. & Takahashi J.S.. 2011. Genetics of circadian rhythms in Mammalian model organisms. Adv. Genet. 74: 175–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Zhu, Y. , Stevens R.G., Hoffman A.E., et al 2011. Epigenetic impact of long‐term shiftwork: pilot evidence from circadian genes and whole‐genome methylation analysis. Chronobiol. Int. 28: 852–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Bracci, M. , Manzella N., Copertaro A., et al 2014. Rotating‐shift nurses after a day off: peripheral clock gene expression, urinary melatonin, and serum 17‐β‐estradiol levels. Scand. J. Work Environ. Health 40: 295–304. [DOI] [PubMed] [Google Scholar]
- 170. Huynh, J.L. , Garg P., Thin T.H., et al 2014. Epigenome‐wide differences in pathology‐free regions of multiple sclerosis‐affected brains. Nat. Neurosci. 17: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Buenafe, A.C. , Zwickey H., Moes N., et al 2008. A telemetric study of physiologic changes in mice with induced autoimmune encephalomyelitis. Lab. Anim. 37: 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Buenafe, A.C. 2012. Diurnal rhythms are altered in a mouse model of multiple sclerosis. J. Neuroimmunol. 243: 12–17. [DOI] [PubMed] [Google Scholar]
- 173. Druzd, D. , Matveeva O., Ince L., et al 2017. Lymphocyte circadian clocks control lymph node trafficking and adaptive immune responses. Immunity 46: 120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Bamer, A.M. , Johnson K.L., Amtmann D., et al 2008. Prevalence of sleep problems in individuals with multiple sclerosis. Mult. Scler. 14: 1127–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Boe Lunde, H.M. , Aae T.F., Indrevag W., et al 2012. Poor sleep in patients with multiple sclerosis. PLoS One 7: e49996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Tachibana, N. , Howard R.S., Hirsch N.P., et al 1994. Sleep problems in multiple sclerosis. Eur. Neurol. 34: 320–323. [DOI] [PubMed] [Google Scholar]
- 177. Najafi, M.R. , Toghianifar N., Etemadifar M., et al 2013. Circadian rhythm sleep disorders in patients with multiple sclerosis and its association with fatigue: a case–control study. J. Res. Med. Sci. 18: S71–S73. [PMC free article] [PubMed] [Google Scholar]
- 178. He, J. , Hsuchou H., He Y., et al 2014. Leukocyte infiltration across the blood–spinal cord barrier is modulated by sleep fragmentation in mice with experimental autoimmune encephalomyelitis. Fluids Barriers CNS 11: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Figueiro, M.G. , Bierman A. & Rea M.S.. 2013. A train of blue light pulses delivered through closed eyelids suppresses melatonin and phase shifts the human circadian system. Nat. Sci. Sleep 5: 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Trinder, J. , Armstrong S.M., O'Brien C., et al 1996. Inhibition of melatonin secretion onset by low levels of illumination. J. Sleep Res. 5: 77–82. [DOI] [PubMed] [Google Scholar]
- 181. Reiter, R.J. 1993. The melatonin rhythm: both a clock and a calendar. Experientia 49: 654–664. [DOI] [PubMed] [Google Scholar]
- 182. Ghorbani, A. , Salari M., Shaygannejad V., et al 2013. The role of melatonin in the pathogenesis of multiple sclerosis: a case–control study. Int. J. Prev. Med. 4: S180–S184. [PMC free article] [PubMed] [Google Scholar]
- 183. Melamud, L. , Golan D., Luboshitzky R., et al 2012. Melatonin dysregulation, sleep disturbances and fatigue in multiple sclerosis. J. Neurol. Sci. 314: 37–40. [DOI] [PubMed] [Google Scholar]
- 184. Gholipour, T. , Ghazizadeh T., Babapour S., et al 2015. Decreased urinary level of melatonin as a marker of disease severity in patients with multiple sclerosis. Iran J. Allergy Asthma Immunol. 14: 91–97. [PubMed] [Google Scholar]
- 185. Damasceno, A. , Moraes A.S., Farias A., et al 2015. Disruption of melatonin circadian rhythm production is related to multiple sclerosis severity: a preliminary study. J. Neurol. Sci. 353: 166–168. [DOI] [PubMed] [Google Scholar]
- 186. Constantinescu, C.S. , Hilliard B., Ventura E., et al 1997. Luzindole, a melatonin receptor antagonist, suppresses experimental autoimmune encephalomyelitis. Pathobiology 65: 190–194. [DOI] [PubMed] [Google Scholar]
- 187. Ghareghani, M. , Dokoohaki S., Ghanbari A., et al 2017. Melatonin exacerbates acute experimental autoimmune encephalomyelitis by enhancing the serum levels of lactate: a potential biomarker of multiple sclerosis progression. Clin. Exp. Pharmacol. Physiol. 44: 52–61. [DOI] [PubMed] [Google Scholar]
- 188. Alvarez‐Sanchez, N. , Cruz‐Chamorro I., Lopez‐Gonzalez A., et al 2015. Melatonin controls experimental autoimmune encephalomyelitis by altering the T effector/regulatory balance. Brain Behav. Immun. 50: 101–114. [DOI] [PubMed] [Google Scholar]
- 189. Kang, J.C. , Ahn M., Kim Y.S., et al 2001. Melatonin ameliorates autoimmune encephalomyelitis through suppression of intercellular adhesion molecule‐1. J. Vet. Sci. 2: 85–89. [PubMed] [Google Scholar]
- 190. Chen, S.J. , Huang S.H., Chen J.W., et al 2016. Melatonin enhances interleukin‐10 expression and suppresses chemotaxis to inhibit inflammation in situ and reduce the severity of experimental autoimmune encephalomyelitis. Int. Immunopharmacol. 31: 169–177. [DOI] [PubMed] [Google Scholar]
- 191. Farez, M.F. , Mascanfroni I.D., Mendez‐Huergo S.P., et al 2015. Melatonin contributes to the seasonality of multiple sclerosis relapses. Cell 162: 1338–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Wen, J. , Ariyannur P.S., Ribeiro R., et al 2016. Efficacy of N‐acetylserotonin and melatonin in the EAE model of multiple sclerosis. J. Neuroimmune Pharmacol. 11: 763–773. [DOI] [PubMed] [Google Scholar]
- 193. Lopez‐Gonzalez, A. , Alvarez‐Sanchez N., Lardone P.J., et al 2015. Melatonin treatment improves primary progressive multiple sclerosis: a case report. J. Pineal Res. 58: 173–177. [DOI] [PubMed] [Google Scholar]