

Abstract
Few trials have examined rates of hypersensitivity reactions (HSRs) with intravenous iron formulations used to treat iron deficiency anemia (IDA). This randomized, multicenter, double‐blind clinical trial compared the safety, and efficacy of ferumoxytol versus ferric carboxymaltose (FCM), focusing on rates of HSRs and hypotension as the primary end point. Patients with IDA of any etiology in whom oral iron was unsatisfactory or intolerable received ferumoxytol (n = 997) or FCM (n = 1000) intravenously over ≥15 minutes on days 1 and 8 or 9 for total respective doses of 1.02 g and 1.50 g. Composite incidences of moderate‐to‐severe HSRs, including anaphylaxis, or moderate‐to‐severe hypotension from baseline to week 5 (primary safety end point) were 0.6% and 0.7% in the ferumoxytol and FCM groups, respectively, with ferumoxytol noninferior to FCM. No anaphylaxis was reported in either group. The secondary safety end point of incidences of moderate‐to‐severe HSRs, including anaphylaxis, serious cardiovascular events, and death from baseline to week 5 were 1.3% and 2.0% in the ferumoxytol and FCM groups, respectively (noninferiority test P < .0001). Least‐squares mean changes in hemoglobin at week 5 were 1.4 g/dL and 1.6 g/dL in the ferumoxytol and FCM groups, respectively (noninferiority test P < .0001). Incidence of hypophosphatemia was 0.4% for ferumoxytol and 38.7% for FCM.
1. INTRODUCTION
Iron deficiency anemia (IDA) is the most common form of anemia worldwide. Frequent etiologies include heavy uterine bleeding, pregnancy, inflammatory bowel disease, bariatric surgery, and cancer.1, 2, 3 IDA is a frequent comorbid condition accompanying chronic kidney disease (CKD).3 Whereas fatigue, loss of stamina, and restless leg syndrome are commonly observed, other serious issues (including mortality, impaired cognitive function, increased susceptibility to infection, decreased health‐related quality of life, lower work productivity, and impaired cardiac and sexual function) are also associated with iron deficiency, irrespective of the degree of associated anemia.1, 4, 5
Iron replacement therapy reconstitutes iron stores and increases hemoglobin concentration. Oral iron is inexpensive and convenient when tolerated but confers an increased rate of gastrointestinal perturbation, often leading to poor adherence.5, 6, 7 Intravenous iron formulations may be used in those intolerant of oral iron or for conditions in which oral iron is ineffective or potentially harmful, such as hereditary hemorrhagic telangiectasia and inflammatory bowel disease. Intravenous iron has been shown to raise hemoglobin levels more effectively than oral iron.8 Serious acute hypersensitivity reactions (HSRs) to intravenous iron formulations are rare but do occur and in the past were largely due to high‐molecular‐weight iron dextrans, which are no longer commercially available.9, 10
This study was performed at the request of the US Food and Drug Administration (FDA) and in support of a supplemental new drug application (sNDA) to broaden the indication of ferumoxytol from IDA in patients with CKD only to IDA of all etiologies. Study IDA‐304 (the FIRM study: a phase 3, randomized, multicenter, double‐blind, safety study of Ferumoxytol compared to ferrIc carboxymaltose [FCM] for the treatment of IDA) was designed to formally investigate these concerns by prospectively adjudicating the rates of HSRs and related adverse events (AEs) in 2 approved and available intravenous iron formulations, ferumoxytol and FCM, using dosing regimens indicated for treatment of IDA. Additionally, although previously considered transient and without clinical sequelae, hypophosphatemia is gaining recognition as potentially important.11 Therefore, we included an evaluation of hypophosphatemia in this large, phase 3, randomized, double‐blind study.
2. METHODS
2.1. Study participants
We conducted this phase 3, randomized, multicenter, double‐blind, safety trial (http://www.clinicaltrials.gov identifier: NCT02694978)11 from February 2016 to January 2017 in 2014 patients from 129 study sites in the United States (patients, n = 1661), Latvia (n = 122), Lithuania (n = 111), Canada (n = 35), Hungary (n = 34), and Poland (n = 34). Primary inclusion criteria were age ≥18 years with IDA (hemoglobin <12.0 g/dL for women and <14.0 g/dL for men and transferrin saturation ≤20% or ferritin ≤100 ng/mL within 60 days of dosing), and a history of unsatisfactory oral iron therapy or intolerance, or in whom oral iron was considered medically inappropriate, as documented on a prespecified oral iron history questionnaire. Exclusion criteria included known HSR to any component of ferumoxytol or FCM, history of allergy to intravenous iron, multiple drug allergies, dialysis‐dependent CKD, or hemoglobin ≤7.0 g/dL.
2.2. Study design
This study was approved by institutional review boards or ethics committees in compliance with the Declaration of Helsinki, Good Clinical Practice guidelines, FDA regulations, International Conference on Harmonisation guidelines, and other applicable regulations and policies. All participants gave written informed consent. Data were analyzed by all authors.
We employed FDA‐approved dosing and randomized patients 1:1 via a centralized interactive web response system to receive diluted intravenous infusions of either ferumoxytol (Feraheme®; AMAG Pharmaceuticals, Inc., Waltham, Massachusetts) 510 mg iron over ≥15 minutes on day 1 and a second dose 7 to 8 days later for a total cumulative dose of 1.02 g or FCM (Injectafer®; American Regent, Inc., Shirley, New York) 750 mg iron over ≥15 minutes on day 1 and a second dose 7 to 8 days later for a total cumulative dose of 1.50 g.12, 13 An unblinded test article preparer diluted the study drugs, while participants and other staff at each site were blinded to drug allocation.
2.3. Study assessments
The primary end point was the incidence of moderate‐to‐severe HSRs, including anaphylaxis, or moderate‐to‐severe hypotension. Signs and symptoms potentially representing hypersensitivity included erythema, pruritus, urticaria (hives, welts, wheals), flushing, dyspnea, wheezing, bronchospasm, tachypnea, chest tightness, stridor, rapid‐onset edema (facial, laryngeal, pharyngeal), chest pain, syncope, dizziness, abdominal or back pain, and anxiety. Moderate HSR was defined as requiring minimal medical intervention or corrective treatment; severe HSR was characterized as requiring medical intervention or corrective treatment, with possible hospitalization. We defined anaphylaxis as a sudden, severe, potentially fatal systemic allergic reaction as operationally defined using the Sampson criteria.14 We defined hypotension as a >30% decrease from baseline in systolic blood pressure or a decrease of >20 mm Hg in systolic blood pressure with symptoms (dizziness/lightheadedness, fatigue, syncope, lack of consciousness, blurred vision).
A Clinical Events Committee (CEC) assessed and adjudicated all potential HSRs, moderate‐to‐severe hypotension, and deaths. CEC members were independent of the sponsor and contract research organization, were not participating clinical investigators, and were experienced in evaluating HSRs. The CEC, comprised of expert immunology, renal, or pulmonary physicians, was blinded to treatment assignment and identity. Study sites were instructed to report all potential HSRs even if they were considered unlikely to represent a true HSR. All patients with a suspected HSR had blood drawn 1 to 2 hours after the start of the event and at the week 2 visit for tryptase measurements (to establish a baseline for comparison), utilized by the CEC to assist in adjudicating events.
A secondary safety end point was the incidence of moderate‐to‐severe HSRs, including anaphylaxis, serious cardiovascular events, and death. Efficacy end points were the mean change in hemoglobin and the mean change in hemoglobin per gram of iron administered, both from baseline to week 5. Exploratory end points were the proportion of patients reporting treatment‐emergent AEs (TEAEs), the proportion of patients reporting treatment‐emergent serious AEs (SAEs), incidence of TEAEs, incidence of SAEs, incidence of severe AEs (severity assessed by the investigator), AEs leading to study drug discontinuation, mean change in serum phosphate from baseline to week 2, and the incidence of hypophosphatemia (defined as serum phosphate level <2.0 mg/dL [<0.6 mmol/L], representing grade 3 or “severe” in the Common Terminology Criteria for Adverse Events, version 4.2). Serum phosphate and fractional excretion of phosphate were evaluated in all patients who received study drug. AEs were recorded throughout the study and coded using the Medical Dictionary for Regulatory Activities, version 19.0.
All patients were monitored for 1 hour after dosing and completed a targeted HSR questionnaire. Vital signs were measured within 30 minutes before dosing and approximately 5, 10, and 30 minutes after dosing. Blood and urine samples for hematology, clinical chemistry, iron parameters, and urine tests were collected. Follow‐up visits occurred at weeks 2 and 5.
2.4. Statistical analysis
The safety population was composed of any randomized patient who received any amount of study drug. All safety analyzes were performed on the safety population based on actual treatment received. The intention‐to‐treat (ITT) population comprised any randomized patient with any exposure to ferumoxytol or FCM; all efficacy analyzes were based on the ITT population and randomized treatment assignment.
For the primary and secondary end points, calculations of point estimates and 2‐sided 95% confidence intervals (CIs) for the treatment difference of ferumoxytol minus FCM and the relative risk of ferumoxytol/FCM used normal approximation (Wald); the P value from the noninferiority test was 1‐sided at α = 0.025 using the Wald method. In addition, for the primary end point, confirmatory calculation of the 95% CI for the treatment difference of ferumoxytol minus FCM was also performed without using the large sample, normal approximation (exact method). For analyzes of efficacy end points, treatment differences were assessed using analysis of covariance adjusted for baseline hemoglobin by applying the general linear model procedure in SAS (SAS Institute, Inc., Cary, North Carolina); a 0 was imputed for the change from baseline value missing postbaseline values (a nonresponder imputation). The incidence of hypophosphatemia at week 2 was analyzed using the same method as the primary safety end point and serum phosphate was assessed with the same method as the efficacy end points. Analyzes for all other parameters were descriptive.
Statistical inferences of noninferiority and/or superiority for the primary and secondary safety, efficacy, and some exploratory end points were performed with no adjustments for multiple comparisons. Given a 3.3% AE rate for the primary safety end point in each treatment group, 2000 patients were needed to provide approximately 90% power for the noninferiority test. For primary and secondary safety end point analyzes, noninferiority was defined as when the upper limit of the 95% CI for the treatment difference of ferumoxytol–FCM did not exceed the predefined margins for the primary (2.6%) and secondary (3.6%) safety end points. For the 2 efficacy end point analyzes, noninferiority was defined as when the lower limit of the 95% CI for the treatment difference of ferumoxytol–FCM was greater than the predefined margin of −0.5 g/dL, and superiority when the lower limit of the 95% CI was >0.
3. RESULTS
3.1. Patient disposition and baseline characteristics
Of 2014 patients with IDA who were randomized, 1997 were treated with either ferumoxytol (n = 997) or FCM (n = 1000; Supporting Information Figure S1). Study completion rates between the 2 groups were similar (ferumoxytol, 93.8%; FCM, 94.8%). Of the 114 patients (5.7%) who withdrew from the study, the most common reasons were withdrawal of consent (2.1%), loss to follow‐up (1.6%), and AEs (1.0%).
The mean ± standard deviation (SD) age was 55.2 ± 17.2 years, and most patients were women (76.1%) and white (71.4%; Table 1). The mean baseline hemoglobin concentration was 10.4 g/dL. The most frequent primary causes of IDA, as attributed by the investigators, were gastrointestinal disorders (29.1%), CKD (26.6%), and abnormal uterine bleeding (24.6%).
Table 1.
Baseline patient demographics and disease characteristics (safety population)
| Ferumoxytol (n = 997) | Ferric carboxymaltose (n = 1000) | |
|---|---|---|
| Age, years, mean (SD) | 55.6 (17.3) | 54.8 (17.0) |
| Female, n (%) | 743 (74.5) | 776 (77.6) |
| Race, n (%) | ||
| White | 711 (71.3) | 715 (71.5) |
| Black | 237 (23.8) | 232 (23.2) |
| Asian | 32 (3.2) | 29 (2.9) |
| American Indian/Alaskan Native | 6 (0.6) | 3 (0.3) |
| Other | 8 (0.8) | 15 (1.5) |
| Not reported | 3 (0.3) | 4 (0.4) |
| Weight, kg, mean (SD) | 84.3 (24.9) | 85.8 (26.0) |
| Hemoglobin, g/dL, mean (SD) | 10.4 (1.5) | 10.4 (1.5) |
| Serum phosphate, mg/dL, mean (SD) | 3.7 (0.6)a | 3.7 (0.6)b |
| Transferrin saturation, %, mean (SD) | 13.9 (28.4) | 13.9 (25.0) |
| Serum ferritin, ng/mL, mean (SD) | 54.3 (115.4) | 55.1 (120.2) |
SI units: 1.2 (0.2) mmol/L.
SI units: 1.2 (0.2) mmol/L.
SD, standard deviation.
3.2. Dosing, exposure, and administration time
In the ferumoxytol group, 992 patients (99.5%) received at least 1 dose of ferumoxytol and 946 (94.9%) received 2 doses. The mean ± SD cumulative intravenous iron dose was 994 ± 119 mg. In the FCM group, 994 patients (99.4%) received at least 1 dose of FCM and 946 (94.6%) received 2 doses, with a mean ± SD cumulative intravenous iron dose of 1458 ± 179 mg. Withdrawal of consent, lost to follow‐up, and AEs were the most common reasons for patients in either group to not receive the second dose. The mean administration time for each dose and each treatment group was approximately 27 minutes.
3.3. Primary and secondary safety end points
The composite incidences of moderate‐to‐severe HSRs, including anaphylaxis, and moderate‐to‐severe hypotension from baseline to week 5 (primary safety end point) were 0.6% and 0.7% in the ferumoxytol and FCM groups, respectively (Table 2). Ferumoxytol was noninferior to FCM (noninferiority test P < .0001), with a risk ratio of 0.9 (95% CI, 0.3‐2.5).
Table 2.
Primary and secondary safety end points (composites and their components)
| Treatment group, n (%) | Treatment difference (95% CI) | Relative risk (95% CI) | Noninferiority P | ||
|---|---|---|---|---|---|
| Ferumoxytol (n = 997) | Ferric carboxymaltose (n = 1000) | ||||
| Primary end point—composite incidence of the following: | 6 (0.6) | 7 (0.7) | −0.1 (–0.8 to 0.6) | 0.9 (0.3–2.5) | .0001a |
| Moderate hypersensitivity reaction | 3 (0.3) | 6 (0.6) | |||
| Severe hypersensitivity reaction | 1 (0.1) | 0 (0.0) | |||
| Anaphylaxis | 0 (0.0) | 0 (0.0) | |||
| Moderate hypotension | 2 (0.2) | 1 (0.1) | |||
| Severe hypotension | 0 (0.0) | 0 (0.0) | |||
| Secondary end point—composite incidence of the following: | 13 (1.3) | 20 (2.0) | −0.7 (–1.8 to 0.4) | 0.7 (0.3–1.3) | .0001b |
| Moderate hypersensitivity reaction | 3 (0.3) | 6 (0.6) | |||
| Severe hypersensitivity reaction | 1 (0.1) | 0 (0.0) | |||
| Anaphylaxis | 0 (0.0) | 0 (0.0) | |||
| Serious cardiovascular eventc | 6 (0.6) | 13 (1.3) | |||
| Death | 4 (0.4)d | 2 (0.2)e | |||
From noninferiority test using a large sample assumption (Wald) with margin of 2.64% at α = 0.025 level for the rate difference. Exact 95% CI for treatment difference, −0.91% to +0.70%.
From noninferiority test using a large sample assumption (Wald) with margin of 3.6% at α = 0.025 level for the rate difference.
Time of onset ranged from within the same day of dosing to up to 4 weeks postdose; events included atrial fibrillation and hypertensive emergency/mild heart failure, among others.
Due to respiratory failure, completed suicide/intentional overdose, acute pancreatitis, and cardiorespiratory arrest. Deaths occurred within 16 to 29 days following the last dose.
Due to mixed hepatocellular cholangiocarcinoma and cardiac failure. Deaths occurred within 29 to 33 days following the last dose, with one occurring after study completion.
CI, confidence interval.
The composite incidence of moderate‐to‐severe HSRs, including anaphylaxis, serious cardiovascular events, and death (secondary safety end point), from baseline to week 5 was 1.3% and 2.0% in the ferumoxytol and FCM groups, respectively (Table 2). Ferumoxytol was noninferior to FCM (noninferiority test P < .0001), with a risk ratio of 0.7 (95% CI, 0.3‐1.3). None of the 6 (0.0%) serious cardiovascular events in the ferumoxytol group and 3 of the 13 (23.1%) serious cardiovascular events in the FCM group were considered to be related to treatment. Of note, this study was intended to focus on acute HSRs and AEs that would be expected to occur within minutes to perhaps hours after administration. As with other prior studies evaluating the safety and efficacy of ferumoxytol, patients were followed for approximately 30 days after the second dose of study medication. This follow‐up duration is customary in studies of intravenous iron given the fast response in hemoglobin levels and the interest in especially acute AEs associated with intravenous administration. None of the 6 (0.3%) deaths (4 and 2 in the ferumoxytol and FCM groups, respectively) were considered to be related to treatment or related to HSR as per CEC assessment.
3.4. Adverse events
A total of 1593 TEAEs were reported by 698 of 1997 patients (35.0%; Supporting Information Table S1). There were fewer reported TEAEs (n = 322/997 [32.3%] vs. n = 376/1000 [37.6%]) and TEAEs related to treatment (n = 116/997 [11.6%] vs. n = 167/1000 [16.7%]) in the ferumoxytol group versus the FCM group, respectively.
There were no new safety signals identified in this trial. The most frequent treatment‐related TEAEs were headache (ferumoxytol, 3.4%; FCM, 3.1%), nausea (ferumoxytol, 1.8%; FCM, 3.4%), dizziness (ferumoxytol, 1.5%; FCM, 1.6%), and fatigue (ferumoxytol, 1.5%; FCM, 1.2%). For SAEs, 36 (3.6%) were reported in the ferumoxytol group and 35 (3.5%) in the FCM group; the one SAE considered by the investigator to be related to treatment in the ferumoxytol group was a severe HSR as determined by the CEC. Of the patients with TEAEs that led to permanent discontinuation of study drug, 17 (1.7%) were in the ferumoxytol group and 23 (2.3%) were in the FCM group. TEAEs led to study discontinuation in 10 patients (1.0%) in the ferumoxytol group and 9 (0.9%) in the FCM group.
The majority of reported TEAEs were mild; 37 patients (3.7%) in the ferumoxytol group and 34 (3.4%) in the FCM group had severe TEAEs. Most severe TEAEs were unrelated to treatment, although there were 3 in the ferumoxytol group and 7 in the FCM group; all but 3 events occurring in the FCM group (hypertensive emergency [15 days], severe nausea [31 days], and severe hypophosphatemia [ongoing at end of study]) resolved within 1 to 4 days.
3.5. Hematology, iron, clinical chemistry, and vital signs
Mean baseline values for all hematology parameters were similar between groups. Following intravenous iron administration, increases in hemoglobin, hematocrit, erythrocytes, erythrocyte mean corpuscular volume, erythrocyte mean corpuscular hemoglobin, and erythrocyte mean corpuscular hemoglobin concentration were seen at weeks 2 and 5 in both groups.
All baseline iron parameters were low, as designed. After treatment, iron and transferrin saturation values increased, and total iron binding capacity decreased similarly in both groups. Mean ± SD serum ferritin concentrations peaked at week 2 (567.1 ± 298.6 ng/mL and 864.6 ± 402.9 ng/mL in the ferumoxytol and FCM groups, respectively), decreasing at week 5 but remaining elevated (227.5 ± 193.9 ng/mL and 397.8 ± 301.2 ng/mL, respectively). Similarly, mean ± SD transferrin saturation increased in both groups, with the highest levels at week 2 (32.9 ± 20.9% and 35.2 ± 17.6% in the ferumoxytol and FCM groups, respectively), and decreased somewhat by week 5 (25.9 ± 11.6% and 28.6 ± 11.5%, respectively).
Mean baseline and posttreatment values for all clinical chemistry parameters were similar between the 2 groups, with the exception of serum phosphate. There were no clinically meaningful changes in heart rate, systolic blood pressure, or diastolic blood pressure during the 1‐hour observation period after administration of ferumoxytol or FCM.
3.6. Secondary efficacy end points
Least‐squares mean changes in hemoglobin from baseline to week 5 were 1.4 and 1.6 g/dL in the ferumoxytol and FCM groups, respectively (Figure 1A), indicating that ferumoxytol was noninferior to FCM (noninferiority test P < .0001). Because the FCM dose of iron was about 1.5 times the ferumoxytol dose, the least‐squares mean change in hemoglobin was also examined per gram of iron administered (Figure 1B). Least‐squares mean changes were 1.4 and 1.1 g/dL in the ferumoxytol and FCM groups, respectively (superiority test P < .0001).
Figure 1.

Changes in Hb from baseline. (A) Least‐squares mean changes in Hb from baseline to week 5. (B) Changes in Hb from baseline to week 5 per gram of iron administered (intention‐to‐treat population). Results were from an analysis of covariance model adjusting for BL value. Error bars show the standard error. BL, baseline; CI, confidence interval; Hb, hemoglobin
3.7. Hypophosphatemia
The incidence of hypophosphatemia 2 weeks after treatment, defined as a serum phosphate concentration <2.0 mg/dL (<0.6 mmol/L), was 0.4% in the ferumoxytol group and 38.7% in the FCM group. Serum phosphate concentrations significantly decreased from baseline in the FCM group compared with the ferumoxytol group as early as day 8 (treatment difference, 1.0 mg/dL [0.32 mmol/L]; exploratory P < .0001), decreased further at week 2, and the difference persisted at week 5 (Figure 2A). By week 2, the mean baseline serum phosphate among those who received FCM decreased by 40%. Figure 2B shows the fractional excretion of phosphate in the ferumoxytol and FCM groups, indicating that the sharp decline in serum phosphate seen with FCM was due to increased renal phosphate excretion.
Figure 2.
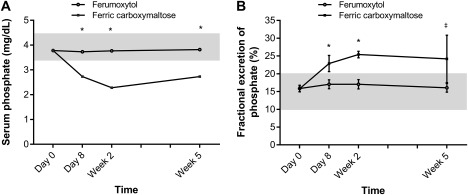
Measures of phosphate. Mean values for (A) serum phosphate and (B) fractional excretion of phosphate by visit. Shading indicates the normal range.15 Error bars show the standard deviation. *P < .0001. † P ≤ .005. ǂ P < .05. To convert serum phosphate to mmol/L, multiply by 0.323
4. DISCUSSION
As noted, subsequent to the filing of the sNDA to obtain the broader indication for treatment of IDA due to any etiology, the FDA issued a Complete Response Letter requesting additional information. This study was performed to support the sNDA for a broader indication of ferumoxytol and at the request of the FDA to perform a randomized controlled trial with ferumoxytol compared to another approved intravenous iron with the primary focus on rates of serious HSRs. The FIRM study was designed in concert with the FDA including a required HSR questionnaire following each administration's observation period and the primary and secondary safety end points. Notable was the fact that dosing would utilize the FDA‐approved regimens for each product. FCM was selected to facilitate a double‐blind study design comparing 2 intravenous irons delivered in 2 administrations.
Although it has been suggested based on possible pathobiologic mechanisms that the rapidity of administration may increase the occurrence of serious HSRs,3 other than so‐called “minor infusion reactions” due to labile or free iron, chiefly noted with higher doses of ferric gluconate and iron sucrose,16 there is no evidence that the total dose of intravenous iron impacts the rate of serious HSRs. The dosing of the 2 intravenous irons studied (1.02 g vs. 1.50 g) reflects the approved dosing regimen, and the difference in dose was not expected to affect the rate of acute HSRs following either of the 2 intravenous iron administrations.
While the primary focus and end points of the study were safety, hematopoietic responses were also assessed. In contrast to HSRs, it was recognized that administering ∼50% more iron could likely affect the secondary end points of efficacy. Therefore, it was prespecified that the change in hemoglobin from baseline would be analyzed as both mean change and mean change per gram of iron administered to assist practitioners in formulating treatment decisions for their patients.
This prospective, randomized, double‐blind trial examined the safety of ferumoxytol (510 mg × 2) compared with FCM (750 mg × 2) in patients with IDA and intolerance of or unresponsiveness to oral iron. Inclusion criteria, including entry hemoglobin and iron indices, were intentionally broad to include a range of different IDA etiologies. While all prior randomized controlled trials of ferumoxytol administered the drug as a rapid injection, this is the first trial in which ferumoxytol was administered as a diluted infusion over ≥15 minutes, and it was well tolerated in this population, consistent with a small, open‐label trial.17
For the primary end point, the incidence of moderate‐to‐severe HSRs, including anaphylaxis, or moderate‐to‐severe hypotension from baseline to week 5, ferumoxytol demonstrated noninferiority to FCM. Ferumoxytol was also noninferior to FCM for the secondary end point of the composite incidence of moderate‐to‐severe HSRs, including anaphylaxis, serious cardiovascular events, or death.
Notably, hypersensitivity to either ferumoxytol or FCM was rare. There were no anaphylactic reactions or severe hypotension and very low incidences of moderate hypotension and moderate‐to‐severe HSRs. The incidence of moderate‐to‐severe HSRs following ferumoxytol treatment was 0.4%, consistent with the 0.2% (n = 3/1726) reported for serious HSRs in a pooled analysis of the 3 pivotal trials in CKD and IDA.18 In other trials that excluded patients with CKD stages 4 and 5, the rate of moderate‐to‐severe HSRs was 2.6% (n = 26/1014).12, 19, 20 Rates of HSRs following FCM treatment in this study were similar to those from other large randomized trials in patients with IDA of any etiology (0.3% to 0.8%) and in patients with nondialysis‐dependent CKD (0.7%).21‐23
No new safety signals were observed, and the types and frequencies of reported AEs were consistent with previously published studies. None of the 6 deaths in this study were considered to be related to the study drugs or HSRs. SAE rates were similar between the 2 formulations.
Before the availability of the FIRM trial, the comparative safety of certain intravenous iron formulations was evaluated in observational studies. While not as rigorous as randomized controlled trials, observational studies, mainly due to their large size, can provide complementary information. Three recent retrospective database analyzes that evaluated the relative risks of HSRs (including anaphylaxis), hypotension, mortality, and cardiovascular morbidity found similar safety profiles among ferumoxytol and other intravenous iron formulations included in the comparisons.10, 24, 25 Of note, all epidemiological studies have inherent weaknesses and/or biases and therefore must be interpreted with caution.
In the FIRM randomized controlled trial, ferumoxytol also demonstrated consistent clinical efficacy, with noninferiority to FCM for the mean change in hemoglobin from baseline to week 5. In absolute terms, the increase in hemoglobin was about 18% higher in the FCM group. However, the FCM dose was nearly 50% higher than that in the ferumoxytol group. The increase in hemoglobin following treatment was consistent with results from prior studies for each agent.18, 19, 20, 21, 23, 26
Hypophosphatemia is a more recently recognized AE that can occur after treatment with certain intravenous iron formulations. Although hypophosphatemia incidence ranged from 41% to 70% in large trials of IDA treated with FCM,22, 27 it was previously thought to be asymptomatic and transient.28 Several cases of severe hypophosphatemia following intravenous iron administration have been reported, some after only a single dose.28, 29, 30, 31, 32 Symptoms of hypophosphatemia include pain, nausea, and asthenia; when severe, hypophosphatemia can result in muscle (including diaphragmatic) weakness, potentially leading to respiratory failure, and rhabdomyolysis, which could lead to acute renal failure, hemolytic anemia, and cardiac dysrhythmias.28 Even mild hypophosphatemia can contribute to osteomalacia over time.28 A retrospective study showed that hypophosphatemia lasted for ≥2 months in 13 of 17 patients treated with a single dose of FCM, suggesting a persistent hypophosphatemic effect.33
The incidence of hypophosphatemia, defined as a serum phosphate level <2.0 mg/dL (<0.6 mmol/L), was 38.7% with FCM. A retrospective study found a similar decrease in phosphate at week 2 with FCM treatment; 64 of 164 (39%)34 and 440 of 1638 (27%) in clinical trials13 had hypophosphatemia (serum phosphate level <2.0 mg/dL [<0.6 mmol/L]). In contrast, in the ferumoxytol group, serum hypophosphatemia was observed in 0.4%.
Strengths of this randomized controlled trial include the large sample size, its double‐blind design, instruction to sites to use a low threshold for consideration of a possible HSR, and rigorous assessment of HSRs by means of a prespecified HSR questionnaire administered to all patients, as well as utilization of the blinded CEC to adjudicate possible HSRs.
Limitations of this study include the relatively short (5 weeks) follow‐up time. However, the overwhelming preponderance of HSRs occur soon after an infusion of intravenous iron, mitigating the need for a longer observation period for the primary end point analysis.3 Efficacy of intravenous iron is usually studied over relatively short time frames, as the maximum hemoglobin increase is achieved within the first few weeks. The follow‐up period of this study would not be sufficient to identify subacute or chronic cardiovascular adverse effects of either intravenous iron formulation, but this trial was designed to capture acute cardiovascular events triggered by hypotension or hypersensitivity.
In addition, the doses of ferumoxytol and FCM differed due to the use of the FDA‐approved doses and schedules. FCM was chosen as a comparator because it is the only other FDA‐approved intravenous iron treatment able to complete a full course of therapy in 2 doses, enabling a fully blinded study. A dose‐response relationship for the occurrence of HSRs with these intravenous iron formulations has not been observed. Both formulations were administered over a period of ≥15 minutes, and a potential HSR would occur within a short time frame after exposure, as a severe delayed reaction is not expected.3 In support of this conclusion, a consensus panel assembled by Kidney Disease: Improving Global Outcomes on intravenous iron safety reported that there is no evidence to support a 30‐minute observation period after the administration of intravenous iron.3 However, while the rapidity of administration has been associated with an increase in the likelihood of HSRs,35, 36 the administered dose of iron has not been associated with HSRs.
In summary, ferumoxytol was noninferior to FCM for both the primary (the incidence of moderate‐to‐severe HSRs, including anaphylaxis, or moderate‐to‐severe hypotension) and secondary (the incidence of moderate‐to‐severe HSRs, including anaphylaxis, serious cardiovascular events, and death) composite safety end points, with equivalent efficacy in raising hemoglobin despite a lower dose. The incidences of TEAEs were similar in both groups, with no new safety signals. Severe hypophosphatemia was seen at higher rates with FCM treatment.
CONFLICT OF INTERESTS
NFA has served on an advisory board and has consulted for AMAG Pharmaceuticals, Inc. ICM has served on an advisory board and has consulted for AMAG Pharmaceuticals, Inc. and has served on an advisory board, has consulted, and is on the speakers' bureau for Vifor Pharma. MA has served on an advisory board, has received funding from AMAG Pharmaceuticals, Inc. and Pharmacosmos and has consulted for AMAG Pharmaceuticals, Inc., Luitpold/American Regent, and Pharmacosmos. GMC has served on an advisory board and has consulted for AMAG Pharmaceuticals, Inc. and Keryx Biopharmaceuticals. WES, KEB, RFK, and JSK are employees of and own stock in AMAG Pharmaceuticals, Inc.
AUTHOR CONTRIBUTIONS
WES, KEB, RFK, and JSK designed the research; collected, analyzed, and interpreted data; and edited and approved the manuscript; NFA, ICM, MA, and GMC analyzed and interpreted data and edited and approved the manuscript.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Information
ACKNOWLEDGMENTS
Jennifer L. Giel, PhD, on behalf of inScience Communications, Springer Healthcare (Philadelphia, PA), provided medical writing support, which was funded by AMAG Pharmaceuticals, Inc.
Adkinson NF, Strauss WE, Macdougall IC, et al. Comparative safety of intravenous ferumoxytol versus ferric carboxymaltose in iron deficiency anemia: A randomized trial. Am J Hematol. 2018;93:683–690. https://doi.org/10.1002/ajh.25060
Funding information AMAG Pharmaceuticals, Inc.
REFERENCES
- 1. Miller JL. Iron deficiency anemia: a common and curable disease. Cold Spring Harb Perspect Med. 2013;3(7):a011866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. World Health Organization (WHO)/Centers for Disease Control and Prevention (CDC) . Worldwide Prevalence of Anaemia 1993–2005. WHO Global Database on Anaemia. Geneva, Switzerland: WHO Press; 2008:1–40. [Google Scholar]
- 3. Macdougall IC, Bircher AJ, Eckardt KU, et al. Iron management in chronic kidney disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) controversies conference. Kidney Int. 2016;89(1):28–39. [DOI] [PubMed] [Google Scholar]
- 4. Shander A, Goodnough LT, Javidroozi M, et al. Iron deficiency anemia—bridging the knowledge and practice gap. Transfus Med Rev. 2014;28(3):156–166. [DOI] [PubMed] [Google Scholar]
- 5. Umbreit J. Iron deficiency: a concise review. Am J Hematol. 2005;78(3):225–231. [DOI] [PubMed] [Google Scholar]
- 6. Macdougall IC, Geisser P. Use of intravenous iron supplementation in chronic kidney disease: an update. Iran J Kidney Dis. 2013;7(1):9–22. [PubMed] [Google Scholar]
- 7. Tolkien Z, Stecher L, Mander AP, Pereira DI, Powell JJ. Ferrous sulfate supplementation causes significant gastrointestinal side‐effects in adults: a systematic review and meta‐analysis. PLoS One. 2015;10(2):e0117383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kidney Disease: Improving Global Outcomes (KDIGO) . KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney Int Suppl. 2012;2(4):280–335. [Google Scholar]
- 9. Macdougall IC, Vernon K. Complement activation‐related pseudo‐allergy: a fresh look at hypersensitivity reactions to intravenous iron. Am J Nephrol. 2017;45(1):60–62. [DOI] [PubMed] [Google Scholar]
- 10. Wang C, Graham DJ, Kane RC, et al. Comparative risk of anaphylactic reactions associated with intravenous iron products. JAMA. 2015;314(19):2062–2068. [DOI] [PubMed] [Google Scholar]
- 11. Adkinson NF, Strauss WE, Bernard K, et al. Comparative safety of intravenous ferumoxytol versus ferric carboxymaltose for the treatment of iron deficiency anemia: rationale and study design of a randomized double‐blind study with a focus on acute hypersensitivity reactions. J Blood Med. 2017;8:155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feraheme [Package Insert]. Waltham, MA: AMAG Pharmaceuticals, Inc; 2015. [Google Scholar]
- 13. Injectafer [Package Insert]. Shirley, NY: American Reagent, Inc; 2013. [Google Scholar]
- 14. Sampson HA, Munoz‐Furlong A, Campbell RL, et al. Second symposium on the definition and management of anaphylaxis: summary report—Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. J Allergy Clin Immunol. 2006;117(2):391–397. [DOI] [PubMed] [Google Scholar]
- 15. Bansal VK. Serum inorganic phosphorus In: Clinical Methods: The History, Physical, and Laboratory Examinations. 3rd ed Boston: Butterworths; 1990:895–899. [PubMed] [Google Scholar]
- 16. Chandler G, Harchowal J, Macdougall IC. Intravenous iron sucrose: establishing a safe dose. Am J Kidney Dis. 2001;38(5):988–991. [DOI] [PubMed] [Google Scholar]
- 17. Auerbach M, Strauss W, Auerbach S, Rineer S, Bahrain H. Safety and efficacy of total dose infusion of 1,020 mg of ferumoxytol administered over 15 min. Am J Hematol. 2013;88(11):944–947. [DOI] [PubMed] [Google Scholar]
- 18. Lu M, Cohen MH, Rieves D, Pazdur R. FDA report: ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. Am J Hematol. 2010;85(5):315–319. [DOI] [PubMed] [Google Scholar]
- 19. Hetzel D, Strauss W, Bernard K, et al. A phase III, randomized, open‐label trial of ferumoxytol compared with iron sucrose for the treatment of iron deficiency anemia in patients with a history of unsatisfactory oral iron therapy. Am J Hematol. 2014;89(6):646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vadhan‐Raj S, Strauss W, Ford D, et al. Efficacy and safety of IV ferumoxytol for adults with iron deficiency anemia previously unresponsive to or unable to tolerate oral iron. Am J Hematol. 2014;89(1):7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Onken JE, Bregman DB, Harrington RA, et al. Ferric carboxymaltose in patients with iron‐deficiency anemia and impaired renal function: the REPAIR‐IDA trial. Nephrol Dial Transplant. 2014;29(4):833–842. [DOI] [PubMed] [Google Scholar]
- 22. Onken JE, Bregman DB, Harrington RA, et al. A multicenter, randomized, active‐controlled study to investigate the efficacy and safety of intravenous ferric carboxymaltose in patients with iron deficiency anemia. Transfusion. 2014;54(2):306–315. [DOI] [PubMed] [Google Scholar]
- 23. Barish CF, Koch T, Butcher A, Morris D, Bregman DB. Safety and efficacy of intravenous ferric carboxymaltose (750 mg) in the treatment of iron deficiency anemia: two randomized, controlled trials. Anemia. 2012;2012:1 2012172104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Airy M, Mandayam S, Mitani AA, et al. Comparative outcomes of predominant facility‐level use of ferumoxytol versus other intravenous iron formulations in incident hemodialysis patients. Nephrol Dial Transplant. 2015;30(12):2068–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wetmore JB, Weinhandl ED, Zhou J, Gilbertson DT. Relative incidence of acute adverse events with ferumoxytol compared to other intravenous iron compounds: a matched cohort study. PLoS One. 2017;12(1):e0171098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qunibi WY, Martinez C, Smith M, et al. A randomized controlled trial comparing intravenous ferric carboxymaltose with oral iron for treatment of iron deficiency anaemia of non‐dialysis‐dependent chronic kidney disease patients. Nephrol Dial Transplant. 2011;26(5):1599–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van Wyck DB, Mangione A, Morrison J, et al. Large‐dose intravenous ferric carboxymaltose injection for iron deficiency anemia in heavy uterine bleeding: a randomized, controlled trial. Transfusion. 2009;49(12):2719–2728. [DOI] [PubMed] [Google Scholar]
- 28. Zoller H, Schaefer B, Glodny B. Iron‐induced hypophosphatemia: an emerging complication. Curr Opin Nephrol Hypertens. 2017;26(4):266–275. [DOI] [PubMed] [Google Scholar]
- 29. Anand G, Schmid C. Severe hypophosphataemia after intravenous iron administration. BMJ Case Rep. 2017;2017:pii: bcr2016219160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blazevic A, Hunze J, Boots JM. Severe hypophosphataemia after intravenous iron administration. Neth J Med. 2014;72(1):49–53. [PubMed] [Google Scholar]
- 31. Mani LY, Nseir G, Venetz JP, Pascual M. Severe hypophosphatemia after intravenous administration of iron carboxymaltose in a stable renal transplant recipient. Transplantation. 2010;90(7):804–805. [DOI] [PubMed] [Google Scholar]
- 32. Sato K, Nohtomi K, Demura H, et al. Saccharated ferric oxide (SFO)‐induced osteomalacia: in vitro inhibition by SFO of bone formation and 1,25‐dihydroxy‐vitamin D production in renal tubules. Bone. 1997;21(1):57–64. [DOI] [PubMed] [Google Scholar]
- 33. Schaefer B, Wurtinger P, Finkenstedt A, et al. Choice of high‐dose intravenous iron preparation determines hypophosphatemia risk. PLoS One. 2016;11(12):e0167146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bager P, Hvas CL, Dahlerup JF. Drug‐specific hypophosphatemia and hypersensitivity reactions following different intravenous iron infusions. Br J Clin Pharmacol. 2017;83(5):1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bircher AJ, Auerbach M. Hypersensitivity from intravenous iron products. Immunol Allergy Clin North Am. 2014;34(3):707–723. x–xi. [DOI] [PubMed] [Google Scholar]
- 36. Rampton D, Folkersen J, Fishbane S, et al. Hypersensitivity reactions to intravenous iron: guidance for risk minimization and management. Haematologica. 2014;99(11):1671–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Information