

Abstract
Halogen‐ and chalcogen‐based σ‐hole interactions have recently received increased interest in non‐covalent organocatalysis. However, the closely related pnictogen bonds have been neglected. In this study, we introduce conceptually simple, neutral, and monodentate pnictogen‐bonding catalysts. Solution and in silico binding studies, together with high catalytic activity in chloride abstraction reactions, yield compelling evidence for operational pnictogen bonds. The depth of the σ holes is easily varied with different substituents. Comparison with homologous halogen‐ and chalcogen‐bonding catalysts shows an increase in activity from main group VII to V and from row 3 to 5 in the periodic table. Pnictogen bonds from antimony thus emerged as by far the best among the elements covered, a finding that provides most intriguing perspectives for future applications in catalysis and beyond.
Keywords: anion binding, catalysis, chalcogen bonds, halogen bonds, pnictogen bonds
Integrating conceptually new non‐covalent interactions into functional systems is of fundamental importance.1, 2, 3, 4 It enables the creation of novel, unprecedented architectures and holds promise to access new properties. Well‐known interactions such as dispersion forces,3 ion pairing,4 hydrogen bonding,5 and cation–π interactions6 play a key role in the function of organocatalysts. In an effort to expand the interaction toolbox available to chemists, lesser known interactions such as anion–π interactions, the unorthodox counterpart of cation–π interactions, have been introduced recently to organocatalysis.7 Another family of unorthodox interactions are the so called σ‐hole interactions (Figure 1).8 While their exact origin is still under debate, it is clear that they originate from an anisotropic distribution of electron density around an organo main group atom. It is believed that chiefly electrostatic, but also charge transfer and other forces give rise to σ‐hole interactions.8 Aware of the current situation but without better alternatives, the term “σ‐hole interactions” is thus used with all due reservation and for convenience only.
Figure 1.
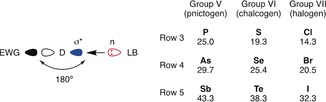
Pnictogen, chalcogen, and halogen bonds between the σ* orbital of a donor (D, with electron‐withdrawing groups (EWGs) to deepen the σ hole) and a lone pair n of a bound Lewis base (LB) afford bond angles around 180° (left) and are predicted to increase in strength with the polarizability of the donor in main groups V–VII (right; computational values based on MP2 calculations in a.u.).13
Arguably the best studied σ‐hole interaction is the halogen bond,9 which has found elegant applications in organocatalysis (Figure 2 a, green).2 Possibly owing to higher steric demands, non‐covalent chalcogen bonding in solution has only recently received increased attention,10 and was applied to non‐covalent catalysis only last year (Figure 2 a, red).11, 12 σ‐Hole interactions not only increase with heavier atoms, but it is also predicted that upon going from right to left in the periodic table, the atomic polarizability, and thus the donor ability, increases (Figure 1).13 Pnictogen bonds have been heavily investigated computationally,8e, 10e, 14 also as potential catalysts.15 Not named as such, pnictogen bonds in the solid and solution state of trisubstituted pnictogens have been observed with strongly electron‐withdrawing cyano or halogen substituents for phosphorus,16 arsenic,17 antimony,18, 19 and bismuth.19 Moreover, pnictogen–π interactions have been used to build supramolecular self‐assemblies.20 To the best of our knowledge, pnictogen bonds have not been used in catalysis thus far.
Figure 2.
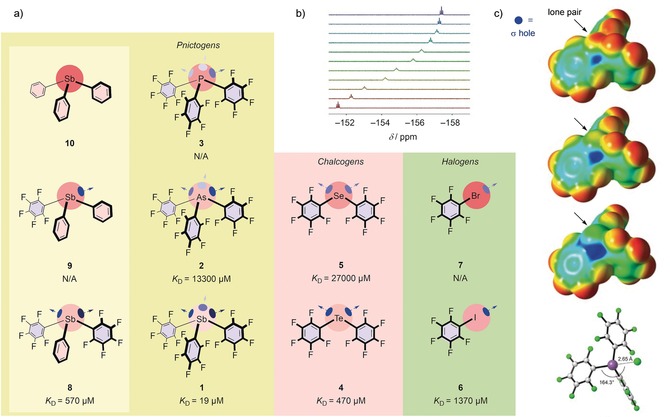
a) Structures of catalyst candidates 1–10 explored in this study, with dissociation constants of chloride complexes in THF. b) 19F NMR spectra of antimony donor 1 in the presence of increasing concentrations of TBACl (maroon to ultramarine). c) Molecular electrostatic potential surfaces (MEPs) of catalysts 3, 2, and 1 (from top to bottom; M06‐2X/6–311G**/aug‐cc‐pVTZ‐pp; isosurface: 0.001 a.u. (0.627 kcal mol−1); red: −0.01327 a.u. (−8.3 kcal mol−1), blue: +0.04527 a.u. (+28.4 kcal mol−1)), and minimized structure of the chloride complex of 1, with relevant bond lengths and angles. C gray, Cl− green, F green, Sb violet.
For a comparative assessment of the promise of pnictogen, chalcogen, and halogen bonds in catalysis, a robust catalyst platform was needed to minimize structural changes between different catalysts that is synthetically accessible for Br, I, Se, Te, P, As, and Sb. Neutral and monovalent designs were desirable to assure comparability and rule out other interactions such as ion pairing. Owing to their excellent electron‐withdrawing capability, stability, and accessibility, pentafluorophenyl substituents were selected for potential catalysts 1–10 (Figure 2). If not commercially available, they were synthesized by known procedures (see the Supporting Information, Schemes S1 and S2).21
The dissociation constants (K D values) of the chloride complexes of catalysts 1–9 were determined by 19F NMR spectroscopic titration with TBACl in THF (Figure 2 a, b; Table 1).24 For a neutral monodentate binder, tris(pentafluorophenyl)stibane 1 showed with K D=19±7 μm an exceptionally high activity, especially considering that it might be underestimated.25 Chloride binding via pnictogen bonds to 1 was over one order of magnitude stronger than chalcogen bonding to bis(pentafluorophenyl)tellurium 4 (K D=470±70 μm), which in turn was three times stronger than the known halogen bonding to iodopentafluorobenzene 6 (K D=1370±30 μm).22, 23 This trend correlates with the increasing molecular polarizability going from main group VII to V in row 5 in the periodic table (Table 1).13
Table 1.
Characteristics of σ‐hole catalysts.
| Entry | Catalyst[a] | Element[b] | K D | E int | η [%] | k cat/k uncat | ΔE a [kJ mol−1] | |||
|---|---|---|---|---|---|---|---|---|---|---|
| [μm][c] | [kcal mol−1][d] | 15 [e] | 18 [f] | 15 [g] | 18 [h] | 15 [i] | 18 [j] | |||
| 1 | 1 | Sb | 19±7 | −51.8 | 53 | 91 | 4090 | 99 | −20.3 | −11.2 |
| 2 | 8 | Sb | 570±70 | −44.4 | 40 | 30 | 209 | 14 | −13.0 | −6.5 |
| 3 | 9 | Sb | n.d.[k] | −37.2 | 3 | 11 | – | – | – | – |
| 4 | 10 | Sb | n.d.[l] | −22.6 | 3 | 8 | – | – | – | – |
| 5 | 4 | Te | 470±70 | −39.7 | 47 | 48 | 52 | 39 | −9.6 | −8.9 |
| 6 | 6 | I | 1370±30 | −27.8 | 47 | 15 | 50 | 5 | −9.5 | −4.1 |
| 7 | 2 | As | 13300±800 | −40.8 | 3 | 7 | – | – | – | – |
| 8 | 5 | Se | 27000±4000 | −28.5 | 6 | 6 | – | – | – | – |
| 9 | 7 | Br | n.d.[k] | −17.7 | 4 | 6 | – | – | – | – |
| 10 | 3 | P | n.d.[k] | −30.0 | 3 | n.d. | – | – | – | – |
[a] Catalyst candidates; structures shown in Figure 2. [b] Central atom of the catalyst, engaged in σ‐hole interactions. [c] Dissociation constant determined by 19F NMR titration with TBACl in THF. [d] Chloride binding energy in the gas phase calculated at the M06‐2X/6–311G**/aug‐cc‐pVTZ‐pp level of theory. [e] Yield of product 15 determined after 4–6 h by 1H NMR integration (see Scheme 1). [f] Yield of 18, determined after 55 h. [g] Rate enhancement for the formation of 15, compared to k uncat=110 mm −1 h−1. [h] Rate enhancement for the formation of 18, compared to k uncat=168 mm −1 h−1. [i] Change in the activation energy for the formation of 15, compared to k uncat=110 mm −1 h−1, from k cat/k uncat. [j] Change in the activation energy for the formation of 18, compared to k uncat=168 mm −1 h−1, from k cat/k uncat. [k] No saturation was observed; see Figure S7. [l] Not determined owing to the lack of fluorine substituents.
Going one row up in the periodic table, a sharp drop in binding ability was observed for all catalysts. However, similar trends were measured. Namely, tris(pentafluorophenyl)arsine 2 was the strongest binder in this group, with K D=13.3±0.8 mm, followed by bis(pentafluorophenyl)selenium 5 with K D=27.0±0.8 mm. For bromopentafluorobenzene 7, no significant shift was observed in the 19F NMR spectra in the presence of up to 15 mm of TBACl (Figure S7). An identical lack of responsiveness was found for tris(pentafluorophenyl)phosphine 3 in main group V but row 3, which illustrates the supremacy of heavier atoms in σ‐hole interactions that is due to their increased polarizability.
To clarify the influence of the substituents on the outstanding binding ability of stibane 1, we successively substituted pentafluorophenyl by phenyl groups in catalysts 8–10 (Figure 2 a and Scheme S1). With one pentafluorophenyl group exchanged in 8, binding dropped from K D=19±7 μm for the perfluorinated antimony donor 1 to K D=570±70 μm, which is comparable to the strength of the perfluorinated tellurium donor 4. With two pentafluorophenyl groups exchanged in stibane 9, binding was not detectable, and partial decomposition occurred at higher TBACl concentrations (Figure S7 e,f ). This decreasing anion binding with decreasing fluorination provided corroborative evidence for σ‐hole binding because the stronger the electron‐withdrawing substituents are, the more potent the binders become.
To probe the exact nature of the σ‐hole binding, chloride binding energies and molecular electrostatic potential (MEP) energy surfaces were calculated in the gas phase for catalysts 1–10 (Figures 2 c and S13). The trends for chloride binding correlated well with the experimental findings (Table 1). The perfluorinated antimony donor 1 was confirmed as the most potent, with E int=−51.8 kcal mol−1. Going right or up one element in the periodic table amounted to a loss of roughly 10 kcal mol−1 in binding strength (Table S7).
The MEP surfaces provide interesting insight into the geometrical constraints that apply to pnictogen‐bonded systems. As can be seen for the surface of 1, in the tetrahedral geometry, only one of the three potential σ holes is truly accessible (Figure 2 c). This is due to the highly asymmetric organization of the three pentafluorophenyl rings, which applies also to catalysts 2 and 3 (Figures 2, S12, and S13) and to crystal structures.24 Interestingly, the computed bond angle of Cl−⋅⋅⋅Pn‐C is with 164.3° significantly smaller in catalyst 1 than in catalysts 2 (170.6°) and 3 (173.4°; Table S7). The ideal angle for σ‐hole interactions would be 180° (Figure 1), but it is expected to be somewhat distorted for chalcogens and pnictogens to minimize lone‐pair repulsion. It appears that larger atoms enable more freedom of movement for the chloride in order to avoid the lone pair, while still fully profiting from the σ hole.
Having secured convincing evidence that σ holes account for strong binding, both experimentally and theoretically, we set out to test whether or not this could be translated into potent catalytic activity in chloride‐binding catalysis. As a starting point, we selected the Reissert‐type substitution of isoquinoline 11 (Schemes 1 and S3), which is known to be catalyzed by numerous, conceptually different anion‐binding catalysts.26 They accelerate the reaction by stabilizing the rate‐limiting transition state 1 (TS1), that is, the elimination of chloride following the addition of Troc chloride 12. Passing through TS2, the resulting cationic intermediate then readily reacts with nucleophiles 13 and 14 to afford products 15 and 16, respectively. After an initial screen of solvents and conditions (Table S1), we were pleased to find that with 5 mol % of catalyst 1 and nucleophile 13, the product 15 was obtained in 51 % yield within 30 min at −100 °C, in THF as the solvent. Without catalyst, only a slow background reaction of ≤2 % was observed.
Scheme 1.
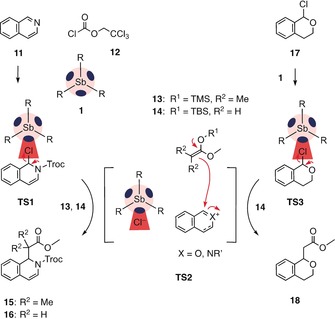
Reactions tested with the potential σ‐hole catalysts 1–10, with the proposed mechanism for antimony catalyst 1. Substrate 11 (25 mm) was reacted with 12 (27 mm), nucleophiles 13 or 14 (38 mm), and the catalyst (20 mol %) in dry THF at −100 °C; substrate 17 (167 mm) was reacted with 14 (250 mm) and the catalyst (20 mol %) in dry THF at −78 °C, together with 1,4‐bis(trimethylsilyl)benzene (6 mm) as an internal standard. See the Supporting Information for details.
Increasing the catalyst loading or reaction time did not lead to significant improvements (Tables S1 and S2). The catalytic activity was almost completely suppressed by the addition of 1.1 equiv of TBACl per catalyst, providing excellent evidence for a chloride‐binding mechanism (Figure S9). In the presence of only 1.0 equiv of TBACl, the yield dropped only moderately to 40 %, indicating that already trace amounts of antimony catalyst 1 are sufficient to drive the reaction forward.
To evaluate the relative strength of pnictogen compared to chalcogen and halogen bonding, catalysts 2–10 were tested under the same conditions as catalyst 1. In line with the chloride binding studies, the weak donors 2, 3, 5, and 7 with elements of row 4 and above were essentially inactive (Table 1, entries 7–10). On the other hand, iodopentafluorobenzene 6 accelerated the reaction by a factor of 50 over the uncatalyzed reaction (entry 6). The tellurium catalyst 4 with k cat/k uncat=52 (entry 5) was only marginally faster than 6. However, the pnictogen‐bonding catalyst 1 was clearly superior, with k cat/k uncat=4090 (entry 1). Exchange of one pentafluorophenyl group for a phenyl group in antimony catalyst 8 reduced the activity to k cat/k uncat=209, but the catalyst was still more active than fully fluorinated chalcogen‐ or halogen‐bonding analogues (entry 2). Further weakening of the electron‐withdrawing nature of the substituents in stibanes 9 and 10 rendered them inactive, as expected for a σ‐hole‐driven process (entries 3 and 4).
Next, we tackled the more challenging transformation of 1‐chloroisochromane 17 to ester 18, a reaction that proceeds through chloride abstraction in TS3 and successive attack of silyl enol ether 14. This reaction is of particular interest because of the significance of the oxonium intermediate in TS2 (X=O) in carbohydrate chemistry. Addition of 20 mol % of antimony catalyst 1 furnished the product 18 in an excellent yield of 91 % after 55 h at −78 °C in THF (Table 1, entry 1 and Scheme S4). This is remarkable for a neutral monodentate catalyst as only multidentate and/or charged hydrogen‐,27 halogen‐,28 chalcogen‐bonding,12 and coulombic29 catalysts had proved to be proficient thus far. Without catalyst, product 18 was formed in only 3 % yield, even after 90 h reaction time. Tellurium catalyst 4 still gave 18 in 48 % yield (Table 1, entry 5) whereas the yield dropped to 15 % for iodopentafluorobenzene 6 (entry 6). The less electron‐deficient antimony catalyst 8 with one phenyl group gave a reduced yield of 30 % compared to catalyst 1 (entry 2). With two and three phenyl groups in catalysts 9 and 10, the yield dropped further to 11 % and 8 %, respectively (entries 3 and 4). As for the Reissert‐type substitution of isoquinolines, catalysts based on row 4 elements 2, 5, and 7 showed only negligible activity (entries 7–9). Kinetic analysis revealed the strongest rate increase for catalyst 1, with k cat/k uncat=99 (entry 1), which gradually decreased from tellurium to iodine catalysts 4 and 6 with k cat/k uncat=39 and 5, respectively (entries 5 and 6). Again, a drop in activity was observed upon exchanging a pentafluorophenyl group with a phenyl group in catalyst 8 (k cat/k uncat=14; Table 1, entry 2).
For the two reactions tested, catalyst 1 was most active, with a gradual decrease in activity upon either reducing the electron‐withdrawing capability of the substituents or going towards chalcogen and halogen bonding. In contrast to more sophisticated systems,2, 11, 12 catalysts based on elements of row 4 and above failed to yield significant activity within this series of conceptually simple, neutral, and monodentate catalysts. This direct comparison ignores that pnictogen variants contain three activating substituents, whereas chalcogen ones contain only two and halogen variants only one. Halogen donor 6 with one activating pentafluorophenyl moiety could also be compared with pnictogen donor 8 with one activating pentafluorophenyl compensated by one inactivating phenyl substituent plus one extra pentafluorophenyl activator. In this comparison, however, pnictogen donors (8: E int=−44.4 kcal mol−1, K D=570±70 μm) still emerge as superior with regard to anion binding in experiment and particularly in theory (6: E int=−27.8 kcal mol−1, K D=1370±30 μm), and are clearly the better catalysts (Table 1). Moreover, with the electron configuration of the elements as the ultimate origin of all chemical properties, the inability of halogens to accommodate three pentafluorophenyl activators can be viewed as an intrinsic disadvantage compared to pnictogens and thus merits full consideration in any systematic comparison.
In conclusion, we have shown that the theoretically predicted increase in anion binding from halogen to chalcogen and ultimately to pnictogen bonding can be experimentally observed by titration experiments with chloride. The observed binding trends are directly reflected in increased catalytic activity for C−Cl bond activation. The tetrahedral geometry of trisubstituted pnictogens enables efficient access to the σ hole. Therefore, the pnictogen family represents an ideal compromise, upon going from right to left in the periodic table, between increased steric repulsion on the one hand and beneficial deeper σ holes resulting from increased polarizability on the other hand. Preliminary results with group IV elements, namely germanium and tin, confirmed theoretically and experimentally that tetrel bonds are insufficiently accessible for anion binding, at least in the pentafluorophenyl series (not shown). These findings demonstrate that there is no reason to ignore pnictogen bonds. On the contrary, they suggest that pnictogens in general and antimony in particular will remain the most powerful donors for operational σ‐hole interactions by far, and will hopefully stimulate their integration into functional systems for catalysis and beyond. With configurational stability and the σ holes not exposed on the surface as with halogens and chalcogens but embedded within a structurable cavity, chiral pnictogen‐bond donors appear particularly promising with regard to asymmetric catalysis.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Professor Antonio Frontera (Universitat de les Illes Balears) for assistance, the NMR and MS platforms for services, and the University of Geneva, the Swiss National Centre of Competence in Research (NCCR) Chemical Biology, the NCCR Molecular Systems Engineering, and the Swiss NSF for financial support.
S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders, S. Matile, Angew. Chem. Int. Ed. 2018, 57, 5408.
Contributor Information
Sebastian Benz, http://www.unige.ch/sciences/chiorg/matile/.
Prof. Stefan Matile, Email: stefan.matile@unige.ch.
References
- 1. Zhao Y., Cotelle Y., Sakai N., Matile S., J. Am. Chem. Soc. 2016, 138, 4270–4277. [DOI] [PubMed] [Google Scholar]
- 2. Bulfield D., Huber S. M., Chem. Eur. J. 2016, 22, 14434–14450. [DOI] [PubMed] [Google Scholar]
- 3. Wagner J. P., Schreiner P. R., Angew. Chem. Int. Ed. 2015, 54, 12274–12296; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12446–12471. [Google Scholar]
- 4.
- 4a. Lacour J., Moraleda D., Chem. Commun. 2009, 7073–7089; [DOI] [PubMed] [Google Scholar]
- 4b. Mahlau M., List B., Angew. Chem. Int. Ed. 2013, 52, 518–533; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 540–556; [Google Scholar]
- 4c. Brak K., Jacobsen E. N., Angew. Chem. Int. Ed. 2013, 52, 534–561; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 558–588. [Google Scholar]
- 5.
- 5a. Hof K., Lippert M., Schreiner P. R. in Science of Synthesis (Eds.: B. List, K. Maruoka), Georg Thieme, Stuttgart, 2012, pp. 297–412; [Google Scholar]
- 5b. Uraguchi D., Ooi T. in Science of Synthesis (Eds.: B. List, K. Maruoka), Georg Thieme, Stuttgart, 2012, pp. 413–435; [Google Scholar]
- 5c. Fang X., Wang C.-J., Chem. Commun. 2015, 51, 1185–1197. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Kennedy C. R., Lin S., Jacobsen E. N., Angew. Chem. Int. Ed. 2016, 55, 12596–12624; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12784–12814; [Google Scholar]
- 6b. Bräuer T. M., Zhang Q., Tiefenbacher K., Angew. Chem. Int. Ed. 2016, 55, 7698–7701; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7829–7832. [Google Scholar]
- 7.
- 7a. Zhao Y., Benz S., Sakai N., Matile S., Chem. Sci. 2015, 6, 6219–6223; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Cotelle Y., Lebrun V., Sakai N., Ward T. R., Matile S., ACS Cent. Sci. 2016, 2, 388–393; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Zhang X., Liu L., López-Andarias J., Wang C., Sakai N., Matile S., Helv. Chim. Acta 2018, 101, e1700288. [Google Scholar]
- 8.
- 8a. Thirman J., Engelage E., Huber S., Head-Gordon M., Phys. Chem. Chem. Phys. 2018, 20, 905–915; [DOI] [PubMed] [Google Scholar]
- 8b. Huber S. M., Jimenez-Izal E., Ugalde J. M., Infante I., Chem. Commun. 2012, 48, 7708–7771; [DOI] [PubMed] [Google Scholar]
- 8c. Pascoe D. J., Ling K. B., Cockroft S. L., J. Am. Chem. Soc. 2017, 139, 15160–15167; [DOI] [PubMed] [Google Scholar]
- 8d. Angarov V., Kozuch S., New J. Chem. 2018, 42, 1413–1422; [Google Scholar]
- 8e. Guan L., Mo Y., J. Phys. Chem. A 2014, 118, 8911–8921. [DOI] [PubMed] [Google Scholar]
- 9. Cavallo G., Metrangolo P., Milani R., Pilati T., Priimagi A., Resnati G., Terraneo G., Chem. Rev. 2016, 116, 2478–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Ho P. C., Szydlowski P., Sinclair P. J., Elder W., Kübel J., Gendy C., Lee L. M., Jenkins H., Britten J. F., Morim D. R., Vargas-Baca I., Nat. Commun. 2016, 7, 11299; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Lim J. Y. C., Marques I., Thompson A. L., Christensen K. E., Felix V., Beer P. D., J. Am. Chem. Soc. 2017, 139, 3122–3133; [DOI] [PubMed] [Google Scholar]
- 10c. Zhao H., Gabbaï F. P., Nat. Chem. 2010, 2, 984–990; [DOI] [PubMed] [Google Scholar]
- 10d. Garrett G. E., Gibson G. L., Straus R. N., Seferos D. S., Taylor M. S., J. Am. Chem. Soc. 2015, 137, 4126–4133; [DOI] [PubMed] [Google Scholar]
- 10e. Scheiner S., Chem. Eur. J. 2016, 22, 18850–18858; [DOI] [PubMed] [Google Scholar]
- 10f. Ho H.-A., Najari A., Leclerc M., Acc. Chem. Res. 2008, 41, 168–178; [DOI] [PubMed] [Google Scholar]
- 10g. Verolet Q., Dal Molin M., Colom A., Roux A., Guénée L., Sakai N., Matile S., Helv. Chim. Acta 2017, 100, e1600328; [Google Scholar]
- 10h. Biot N., Bonifazi D., Chem. Eur. J. 2018, https://doi.org/10.1002/chem.201705428. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Benz S., Mareda J., Besnard C., Sakai N., Matile S., Chem. Sci. 2017, 8, 8164–8169; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Wonner P., Vogel L., Düser M., Gomes L., Kniep F., Mallick B., Werz D. B., Huber S. M., Angew. Chem. Int. Ed. 2017, 56, 12009–12012; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12172–12176; [Google Scholar]
- 11c. Benz S., Lopez-Andarias J., Mareda J., Sakai N., Matile S., Angew. Chem. Int. Ed. 2017, 56, 812–815; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 830–833. [Google Scholar]
- 12. Wonner P., Vogel L., Kniep F., Huber S. M., Chem. Eur. J. 2017, 23, 16972–16975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bauzá A., Mooibroek T. J., Frontera A., ChemPhysChem 2015, 16, 2496–2517. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Murray J. S., Lane P., Politzer P., Int. J. Quantum Chem. 2007, 107, 2286–2292; [Google Scholar]
- 14b. George J., Deringer V. L., Dronskowski R., J. Phys. Chem. A 2014, 118, 3193–3200. [DOI] [PubMed] [Google Scholar]
- 15. Schmauck J., Breugst M., Org. Biomol. Chem. 2017, 15, 8037–8045. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Wilkie C. A., Parry R. W., Inorg. Chem. 1980, 19, 1499–1502; [Google Scholar]
- 16b. Dillon K. B., Platt A. W. G., Schmidpeter A., Sheldrick W. S., Zwaschka F., Z. Anorg. Allg. Chem. 1982, 488, 7–26; [Google Scholar]
- 16c. Deng R. M. K., Dillon K. B., Sheldrick W. S., J. Chem. Soc. Dalton Trans. 1990, 551–554; [Google Scholar]
- 16d. Ali R., Dillon K. B., J. Chem. Soc. Dalton Trans. 1990, 2593–2596; [Google Scholar]
- 16e. Müller G., Jörg B., Jetter S., Z. Naturforsch. B 2001, 56, 1163; [Google Scholar]
- 16f. Marchenko A. P., Koidan G. N., Hurieva A. N., Rozhenko A. B., Kostyuk A. N., Heteroat. Chem. 2016, 27, 12–22. [Google Scholar]
- 17. Emerson K., Britton D., Acta Crystallogr. 1963, 16, 113–118. [Google Scholar]
- 18. Christianson A. M., Gabbaï F. P., Organometallics 2017, 36, 3013–3015. [Google Scholar]
- 19. Arlt S., Harloff J., Schulz A., Stoffers A., Villinger A., Chem. Eur. J. 2016, 22, 16012–16016. [DOI] [PubMed] [Google Scholar]
- 20. Watt M. M., Collins M. S., Johnson D. W., Acc. Chem. Res. 2013, 46, 955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Kemmitt R. D. W., Nichols D. I., Peacock R. D., J. Chem. Soc. A 1968, 2149–2152; [Google Scholar]
- 21b. Tyrra W. E., J. Fluorine Chem. 2001, 112, 149–152. [Google Scholar]
- 22. Vargas Jentzsch A., Emery D., Mareda J., Metrangolo P., Resnati G., Matile S., Angew. Chem. Int. Ed. 2011, 50, 11675–11678; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 11879–11882. [Google Scholar]
- 23. Dimitrijević E., Kvak O., Taylor M. S., Chem. Commun. 2010, 46, 9025–9027. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Rheingold A. L., Staley D. L., Fountain M. E., J. Organomet. Chem. 1989, 365, 123–135; [Google Scholar]
- 24b. Cosio C. M., Karipides A., Acta Crystallogr. Sect. C 1989, 45, 1743–1745; [DOI] [PubMed] [Google Scholar]
- 24c. Mahalakshmi H. I., Jain V. K. I., Tiekink E. R. T., Z. Kristallogr. 2003, 218, 71–72. [Google Scholar]
- 25. Thordarson P., Chem. Soc. Rev. 2011, 40, 1305–1323. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Schafer A. G., Wieting J. M., Fisher T. J., Mattson A. E., Angew. Chem. Int. Ed. 2013, 52, 11321–11324; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11531–11534; [Google Scholar]
- 26b. Zurro M., Asmus S., Bamberger J., Beckendorf S., García Mancheño O., Chem. Eur. J. 2016, 22, 3785–3793; [DOI] [PubMed] [Google Scholar]
- 26c. Kaneko S., Kumatabara Y., Shimizu S., Maruoka K., Shirakawa S., Chem. Commun. 2017, 53, 119–122; [DOI] [PubMed] [Google Scholar]
- 26d. Shirakawa S., Liu S., Kaneko S., Kumatabara Y., Fukuda A., Omagari Y., Maruoka K., Angew. Chem. Int. Ed. 2015, 54, 15767–15770; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15993–15996; [Google Scholar]
- 26e. Wieting J. M., Fisher T. J., Schafer A. G., Visco M. D., Gallucci J. C., Mattson A. E., Eur. J. Org. Chem. 2015, 525–533; [Google Scholar]
- 26f. Taylor M. S., Tokunaga N., Jacobsen E. N., Angew. Chem. Int. Ed. 2005, 44, 6700–6704; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 6858–6862. [Google Scholar]
- 27. Reisman S. E., Doyle A. G., Jacobsen E. N., J. Am. Chem. Soc. 2008, 130, 7198–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Kniep F., Jungbauer S. H., Zhang Q., Walter S. M., Schindler S., Schnapperelle I., Herdtweck E., Huber S. M., Angew. Chem. Int. Ed. 2013, 52, 7028–7032; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7166–7170; [Google Scholar]
- 28b. Jungbauer S. H., Huber S. M., J. Am. Chem. Soc. 2015, 137, 12110–12120. [DOI] [PubMed] [Google Scholar]
- 29. Berkessel A., Das S., Pekel D., Neudörfl J.-M., Angew. Chem. Int. Ed. 2014, 53, 11660–11664; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11846–11850. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary