

Abstract
ELQ-300 is a preclinical antimalarial drug candidate that is active against liver, blood, and transmission stages of Plasmodium falciparum. While ELQ-300 is highly effective when administered in a low multi-dose regimen, poor aqueous solubility and high crystallinity have hindered its clinical development. To overcome its challenging physiochemical properties, a number of bioreversible alkoxycarbonate ester prodrugs of ELQ-300 were synthesized. These bioreversible prodrugs are converted to ELQ-300 by host and parasite esterase action in the liver and bloodstream of the host. One such alkoxycarbonate prodrug, ELQ-331, is curative against Plasmodium yoelii with a single low dose of 3 mg/kg in a murine model of patent malaria infection. ELQ-331 is at least as fully protective as ELQ-300 in a murine malaria prophylaxis model when delivered 24 hours before sporozoite inoculation at an oral dose of 1 mg/kg. Here, we show that ELQ-331 is a promising prodrug of ELQ-300 with improved physiochemical and metabolic properties and excellent potential for clinical formulation.
Keywords: Malaria, Cytochrome bc1, Plasmodium falciparum, Prodrug
Graphical abstract
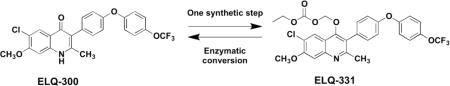
Introduction
Malaria is a potentially fatal parasitic infection that caused 214 million clinical cases and 438,000 deaths in 2015.1 Endemic in 106 countries, most malaria infections occur in tropical and subtropical regions, and the African continent is home to 88% of the world’s cases. The vast majority (99%) of malaria deaths are due to Plasmodium falciparum, the species most common in sub-Saharan Africa.2 Most of the currently available antimalarial drugs require repeat dosing that can be difficult to achieve in areas with limited medical resources.3,4 Interruptions in treatment regimens can lead to parasite recrudescence and drug resistance.5,6
A major global health challenge of today is the threat of widespread resistance to clinically available antimalarials. Since the discovery of chloroquine resistance in the mid twentieth century,7,8 malaria parasites have developed resistance to all antimalarials that have been approved for clinical use.5 Most recently in Southeast Asia, malaria parasites have developed tolerance to artesunate, the main component in some fast-acting artemisinin combination therapeutics (ACTs).9,10 To overcome widespread drug resistance, there is an urgent need for new antimalarials that are curative with a single dose, inexpensive to manufacture, easy to administer, and are active against all life cycle stages of multidrug-resistant strains of malaria, particularly falciparum malaria.11,12
Recently, we reported a new class of antimalarial, the 4(1H)-quinolone-3-diarylethers, based on the structure of the early twentieth century antimalarial endochin.13 These endochin-like quinolones (ELQ) target the cytochrome bc1 complex of Plasmodium parasites.14,15 The Plasmodium cytochrome bc1 complex is also the target of atovaquone, one component of the FDA-approved antimalarial combination drug known as Malarone® (Fig. 1).16 Atovaquone is not feasible for widespread use because it is expensive to manufacture and Malarone® must be taken daily.7,17,18
Figure 1.
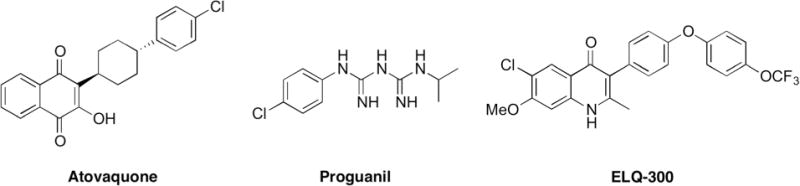
Structures of ELQ-300 and the components of Malarone®, atovaquone and proguanil.
Structural optimization of 4(1H)-quinolone-3-diarylethers led to preclinical drug candidate ELQ-300 (Fig. 1). ELQ-300 targets the highly divergent Qi-site of the cytochrome bc1 complex and it has a low resistance propensity compared to atovaquone, which targets the widely conserved QO-site.19 ELQ-300 and atovaquone together provide strong dual-site inhibition of the P. falciparum cytochrome bc1 complex, and the combination is highly effective against patent malaria in mice.20
ELQ-300 is active against liver and blood stages of P. falciparum, as well as sexual and vector stage parasites.21 Oral absorption of ELQ-300 is limited by its high crystallinity and poor water solubility, preventing the achievement of bloodstream concentrations necessary to deliver single-dose cures, despite being extremely effective when administered in multiple low doses. To mitigate the poor physiochemical properties of ELQ-300, we have employed a prodrug approach.
Prodrugs are chemically modified derivatives of drugs that are inactive until some biological process releases the pharmacologically active drug molecule in vivo. The prodrug approach is used extensively in drug design and development to improve pharmacokinetic, biopharmaceutical, and/or physiochemical qualities of an active drug molecule.22–25 Most recently we reported an ethyl carbonate prodrug of ELQ-300, ELQ-337 (Fig. 2), that shows reduced crystallinity compared to ELQ-300. ELQ-337 increases oral bioavailability of the parent molecule, as indicated by increased bloodstream concentrations of ELQ-300 when administered in prodrug form compared to administration of ELQ-300 alone.26
Figure 2.

Structures of ELQ-300 prodrugs, including carbonate ester prodrug ELQ-337 and novel alkoxycarbonate ester prodrugs ELQ-330, ELQ-331, and ELQ-387.
Here we present a new class of ELQ-300 prodrugs, the alkoxycarbonate esters (Fig. 2). These prodrugs are synthesized in one step and ELQ-300 is liberated as the active drug once host or parasite esterases cleave the promoiety (Fig. 3). Some alkoxycarbonate ester promoieties appear in FDA-approved prodrugs; ELQ-330 utilizes the same promoiety as Tenofovir disoproxil (Viread®), and ELQ-387 uses the same promoiety as Bacampacillin (Spectrobid®, Penglobe®).27, 28
Figure 3.
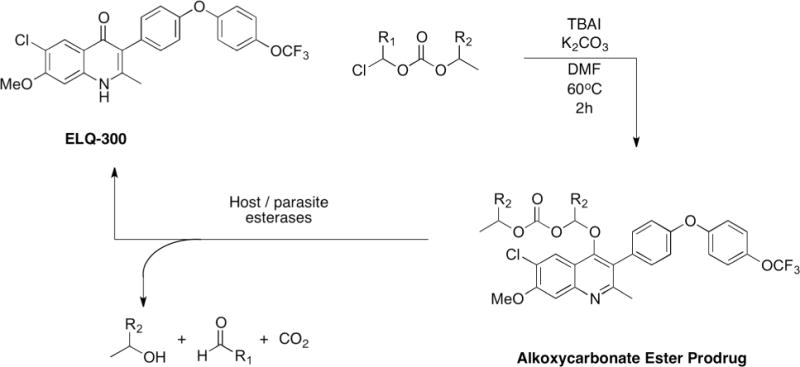
Chemical synthesis and biological cleavage of alkoxycarbonate ester prodrugs of ELQ-300. ELQ-330: R1=H, R2=CH3; ELQ-331: R1=H, R2=H; ELQ-387: R1=CH3, R2=H.
Results and Discussion
Rationale for an alkoxycarbonate ester prodrug
We previously reported the synthesis and antiplasmodial profile of ELQ-337, a carbonate ester prodrug of ELQ-300 (Fig. 2).26 Melting point data suggest that attaching a promoiety to the 4-oxo position of ELQ-300 disrupts the drug’s tendency to form microcrystals with high lattice energy. We hypothesized that alkoxycarbonate ester promoieties would also reduce crystallinity and increase solubility of ELQ-300, facilitating higher bloodstream concentrations necessary to cure patent malaria infection at a single dose.
Chemical synthesis of alkoxycarbonate ester prodrugs
ELQ-330, ELQ-331, and ELQ-387 are synthesized in one step from ELQ-300 using potassium carbonate, tetrabutylammonium iodide (TBAI), and the respective chloromethyl alkoxycarbonate ester in dimethyl formamide (DMF) at 60°C. The conversion is complete overnight in high yield. The alkoxycarbonate ester prodrugs have significantly lower melting points than ELQ-300, indicating a reduction in crystallinity compared to ELQ-300, i.e., ELQ-300 decomposes at ~314°C, while ELQ-330, ELQ-331, and ELQ-387 melt at 99.7°C, 103.5°C, and 135°C, respectively.
In vitro activity of alkoxycarbonate ester prodrugs of ELQ-300
ELQ-331 is equally as effective as ELQ-300 against P. falciparum-infected red blood cells while ELQ-330 and ELQ-387 are less potent than ELQ-300 (Table 1). Since ELQ-300 prodrugs are themselves poor inhibitors of the P. falciparum cytochrome bc1 complex (Table 2),26 the equipotency of ELQ-331 compared to ELQ-300 in vitro indicates that ELQ-331 is likely converted to ELQ-300 via esterase activity in parasite-infected red blood cells. ELQ-330 and ELQ-387 both show diminished antiplasmodial activity in vitro compared to ELQ-300, suggesting that these specific alkoxycarbonate ester promoieties are not as readily cleaved by parasite esterases as in the case of ELQ-331.
Table 1.
In vitro and in vivo comparison of chloroquine, ELQ-300, and alkoxycarbonate ester prodrugs.
| Drug | Melting Point (°C) | In vitro activity vs. P. falciparum IC50 (nM)a | In vivo efficacy (oral) P. yoelii (mg/kg/day) | |||||
|---|---|---|---|---|---|---|---|---|
| D6 | Dd2 | C2B | ED50b | ED90b | NRDc | Single Dose Cured | ||
| CQ | 196-199 | 10 | 140 | 98 | 2.1 | 4.0 | >64 | >64 |
| ELQ-300 | 312-314 | 6.0 | 6.0 | 2.0 | 0.020 | 0.060 | 1.0 | >20e |
| ELQ-330 | 99.7-99.9 | 62 | 38 | 47 | NT | NT | NT | 5.0 |
| ELQ-331 | 102-103 | 6.0 | 6.0 | 4.0 | 0.020 | 0.050 | 1.0 | 3.0 |
| ELQ-387 | 135.0-135.7 | 330 | 140 | 230 | NT | NT | NT | 5.0 |
Seventy-two hour SYBR green assay. P. falciparum strains are D6 (drug sensitive), Dd2 (chloroquine resistant), and Tm90-C2B (Chloroquine and Atovaquone resistant).
ED50 and ED90 values are the doses required to suppress parasitemia by 50% and 90% relative to untreated controls;
NRD = non-recrudescence dose (4-day Peters Test).
Single Dose Cure: Dose required to clear patent infection without recrudescence for 30 days post-infection.
Solubility limit of ELQ-300 in PEG-400.
Table 2.
Inhibition of isolated P. falciparum cytochrome bc1.
In vivo efficacy of alkoxycarbonate ester prodrugs of ELQ-300 against murine malaria
In vivo experiments used a 4-day suppression test protocol in mice against P. yoelii, i.e., inoculation (day 0) followed by oral dosing on four sequential days, followed by microscopic inspection of Giemsa-stained blood smears on day 5 (Table 1). Like ELQ-300, four sequential low doses of ELQ-331 clear parasitemia; i.e., ED50= 0.02 mg/kg/day, ED90= 0.05 mg/kg/day, and both ELQ-300 and ELQ-331 prevent parasite recrudescence at an oral dose of 1 mg/kg/day.
While ELQ-331 performs similarly to its parent drug ELQ-300 in a low multi-dose regimen, it far outperforms ELQ-300 as a single-dose cure. All three alkoxycarbonate ester prodrugs of ELQ-300 deliver single-dose cures at or below 5 mg/kg, and ELQ-331 cured all four animals at a low dose of 3 mg/kg. ELQ-300 itself has not provided a single-dose cure without parasite recrudescence, even with a 20 mg/kg dose, which is the solubility limit of ELQ-300 in the vehicle PEG-400.
In vivo 24 hour prophylaxis of ELQ-300 and ELQ-331 against murine malaria
Single doses of ELQ-300 and ELQ-331 in PEG400 were administered orally to mice one day prior to infection with 10,000 sporozoites via intravenous inoculation, at doses of 3, 1, or 0.1 mg/kg. At 24, 48, and 72 hours after sporozoite inoculation, in vivo imaging of all infected mice treated with ELQ derivatives or control preparations was conducted with an IVIS Spectrum (Perkin Elmer, Hanover, MD) to assess parasite burden in the liver (Fig. 4). To assess blood stage parasitemia, parasite counts were measured by flow cytometry for 30 days. Atovaquone (2.5 mg/kg) and 4-methyl primaquine (5 mg/kg), which has 3-fold greater prophylactic efficacy compared to primaquine, were used as positive controls and a vehicle (PEG400) control group was also included as a negative control. All dosing solutions were prepared based on the body weight of mice. The minimum curative dose (ED100) is defined as the lowest dose that prevents the appearance of bloodstream parasites over the course of the 31-day study.
Figure 4.

Whole animal bioluminescence imaging of C57BL/6 albino mice infected with 10,000 luciferase-transfected P. berghei sporozoites. Mice (n = 5 per group) were treated with different doses of ELQ-300, prodrug ELQ-331, or atovaquone according to different dosing regimens: Panel A) ELQ-300 and ELQ-331 were administered respectively at 0.1, 1, and 3 mg/kg one day before sporozoite inoculation, and Panel B) Atovaquone was administered at 2.5 mg/kg on days -1, 0, and 1 post infections and at 2.5 and 5 mg/kg on the day before sporozoite inoculation. The bioluminescent signal represents the parasite burden over the body surface area where the red/bright yellow regions represent the most intense signal, followed by green and then blue which represents the weakest signal. A value reflecting the percentage suppression of parasite burden relative to controls at the same time point and based on overall bioluminescence signal appears below in vivo images of each group of animals. √ = Days in which drugs were administered.
Previously published results demonstrated that ELQ-300 is fully protective against liver stage infection at doses as low as 0.03 mg/kg when dosed 1 hour prior to sporozoite inoculation.14 Dosing 24 hours prior to sporozoite inoculation, animals in the ELQ-300 group had no detectable parasite burden in the liver at 3 and 1 mg/kg and no parasitemia in 5/5 mice at 3 mg/kg and 4/5 mice at 1 mg/kg. One mouse in the 1mg/kg group had positive parasitemia at day 10. At 0.1 mg/kg 2/5 mice had a positive IVIS signal at 24 hours while 5/5 had a positive IVIS signal at 48 hours. ELQ-331 demonstrated increased efficacy with an ED100 at 1 mg/kg, and at 0.1 mg/kg 0/5 mice had positive IVIS signal at 24 hours and 5/5 had a positive IVIS signal at 48 hours. Taken together, these data demonstrate the superior efficacy of ELQ-331 relative to the parent molecule, ELQ-300, and highlight the potential of prodrug ELQ-331 for single-dose prophylaxis and cure of malaria.
In vitro inhibition of Plasmodium falciparum cytochrome bc1 complex
ELQ-300 is a potent inhibitor of the P. falciparum cytochrome bc1 complex, more potent and more target-selective than atovaquone.14 Prodrugs of ELQ-300 are themselves poor inhibitors of the P. falciparum cytochrome bc1 complex; ELQ-331 has an EC50 that is 10,000 times higher than that of ELQ-300 against isolated P. falciparum mitochondria (Table 2). Because ELQ-331 shows nearly identical antiplasmodial activity to ELQ-300 in whole-parasite and mouse models (Table 1), these data together indicate that ELQ-331 is enzymatically converted to ELQ-300, which is the active inhibitor.
In vitro metabolism of ELQ-331 to ELQ-300
ELQ-331 was incubated briefly with pooled human microsomes at 37°C in the presence or absence of NADPH to test for P450-independent metabolism. At various times over the course of one hour, samples were analyzed by LC-MS-MS to quantitate the amount of prodrug present as ELQ-300 was liberated. Midazolam was used as a control with low metabolic stability. In both NADPH +/− tests, ELQ-331 converted to ELQ-300 at the same rate, consistent with the notion that host esterases are responsible for the cleavage of the alkoxycarbonate ester promoiety to liberate ELQ-300 (Table 3). That the conversion of ELQ-331 to ELQ-300 is comparably slow in plasma (Table 4) suggests that hepatic esterases are primarily responsible for cleavage of the promoiety in vivo.
Table 3.
In vitro microsomal intrinsic clearance assays for ELQ-331
| NADPH dependent | NADPH independent | ||||||
|---|---|---|---|---|---|---|---|
| Compound | Test Conc (μM) |
Test Species (1.0 mg/ml) | Clint (μl min−1 mg−1) |
T1/2 (min) |
Clint (μl min−1 mg−1) |
T1/2 (min) |
Comment |
| ELQ-331 | 1.0 | Human | 18.4 | 38 | 18.4 | 38 | Conversion to ELQ-300 followed by LC-MS/MS; plus/minus NADPH |
| Midazolam (control) | 1.0 | Human | 267 | 2.6 | <1 | >60 | Highly metabolized P450-dependent |
Table 4.
In vitro plasma metabolism assays for ELQ-331
| Compound | Test Conc (μM) |
Test Species | T1/2 (min) |
Comment |
|---|---|---|---|---|
| ELQ-331 | 1.0 | Human | >120 | slow conversion to ELQ-300 followed by LC-MS/MS |
| Propantheline bromide | 1.0 | Human | 37.5 | Esterase substrate |
Mitochondrial toxicity of ELQ-300 and ELQ-331
HepG2 cells were grown in galactose containing medium (as opposed to glucose), circumventing the Crabtree Effect and forcing cell mitochondria to operate at full capacity, thus imposing a higher sensitivity to mitochondrial inhibitors. Neither ELQ-300 or ELQ-331 inhibit or alter ATP production in HepG2 cells over a 72 hour incubation period at concentrations as high as 25 μM, more than 4000 times their IC50.
Conclusion
ELQ-300 is a promising preclinical drug candidate that is active against all life cycle stages of Plasmodium falciparum, but limitations in the physiochemical properties of ELQ-300 (limited aqueous solubility and high crystallinity) prevent the achievement of bloodstream concentrations of ELQ-300 necessary to provide a single-dose cure. Here we synthesized three new bioreversible alkoxycarbonate ester prodrugs of ELQ-300 to facilitate greater oral bioavailability of ELQ-300. Of the three alkoxycarbonate ester prodrugs of ELQ-300, we proceeded with ELQ-331 based on its excellent anti-plasmodial activity in vitro and its favorably low melting point. In a murine model of patent malaria infection, ELQ-331 delivered single-dose cures at a low dose of 3 mg/kg without any parasite recrudescence. Administered one day prior to inoculation with infectious sporozoites a single dose of 1 mg/kg ELQ-331 fully protected animals from liver and bloodstream infection in a murine model of causal prophylaxis against malaria. In vitro metabolism data confirm that the promoiety of ELQ-331 is cleaved to return ELQ-300 as the active drug compound.
Looking ahead, we are evaluating the feasibility of formulating ELQ-331 in tablet form, perhaps coformulated with atovaquone.20 Because ELQ-331 exhibits a lower degree of crystallinity relative to ELQ-337 we feel that it can be readily formulated for clinical use with the aid of established formulation technology including spray dried dispersion (SDD) and self-emulsifying drug delivery systems (SEDDS).29, 30 Thus ELQ-331 is a prodrug that appears to be readily absorbed following oral administration and is converted to ELQ-300 by host esterases to achieve superior bloodstream exposure and prophylactic as well as curative efficacy. Together, these findings indicate that ELQ-331 represents a promising new development toward a single-dose cure and protection against falciparum malaria.
Methods
Chemical Synthesis of ELQ-330
ELQ-300 (1.3 g, 2.7 mmol), tetrabutylammonium iodide (2.0 g, 5.5 mmol), and potassium carbonate (0.75 g, 5.5 mmol) were dissolved in anhydrous dimethylformamide (11 mL) in a flame-dried round bottom flask at 60°C under inert atmosphere. Chloromethyl isopropyl carbonate (0.7 mL, 5.5 mmol) was added dropwise and the reaction stirred under inert atmosphere at 60°C for two hours, until complete by thin layer chromatography. The reaction was cooled to room temperature and the reaction solvent evaporated under reduced pressure. The mixture was taken up in water (15 mL) and extracted with dichloromethane (3 × 25 mL). Combined organic layers were washed with brine (15 mL) and concentrated. Purification by silica column chromatography (EtOAc/DCM) followed by recrystallization in boiling hexanes yielded the title compound, ELQ-330, as white fluffy crystals (1.61 g, 72%). 1H NMR (400 MHz, CDCl3): δ 8.06 (s, 1H), 7.43 (s, 1H), 7.36 (m, 2H), 7.24 (s, 2H), 7.11 (m, 4H), 5.27 (s, 2H), 4.04 (s, 3H), 2.52 (s, 3H), 1.22 (s, 3H), 1.20 (s, 3H); M.P. (°C): 99.7-99.9; HRMS (ESI-TOF) m/z [M + H+]: Calculated for C29H26O7NClF3 592.13444; Found 529.13426 (0.30 ppm difference).
Chemical Synthesis of ELQ-331
ELQ-300 (0.85 g, 1.8 mmol), tetrabutylammonium iodide (1.33 g, 3.6 mmol) and potassium carbonate (0.50 g, 3.6 mmol) were dissolved anhydrous dimethylformamide (8 mL) in a flame-dried round bottom flask at 60 °C under inert atmosphere. Chloromethyl ethyl carbonate (0.5 g, 3.6 mmol) was added dropwise and the reaction stirred under inert atmosphere at 60 °C for two hours, at which point reaction completion was confirmed by thin layer chromatography. After cooling to room temperature, the reaction solvent was removed under reduced pressure and the mixture taken up in water (10 mL) and extracted with dichloromethane (3 × 20 mL). Combined organic layers were washed with brine (10 mL), dried over MgSO4, and the dichloromethane evaporated under reduced pressure. The resulting crude product was purified by flash chromatography (EtOAc/Hex) to yield the title compound, ELQ-331, as a white crystalline solid (560 mg, 54 %). 1H NMR (400 MHz, DMSO-d6): δ = 7.98 (s, 1H), 7.57 (s, 1H), 7.44 (m, 4H), 7.21 (m, 4H), 5.76 (s, 2H), 5.35 (s, 2H), 4.03 (s, 3H), 2.44 (s, 3H), 1.11 (t, 3H, J = 7.1 Hz); M.P. (°C): 103.5-103.7; HRMS (ESI-TOF) m/z [M + H+]: Calculated for C28H24O7NClF3 578.11879; Found 578.11872 (0.12 ppm difference).
Chemical Synthesis of ELQ-387
Tetrabutylammonium iodide (0.15 g, 0.42 mmol), potassium carbonate (0.06 g, 0.42 mmol), and 1-chloroethyl ethyl carbonate (0.06 mL, 0.42 mmol) were dissolved in anhydrous dimethylformamide (5 mL) in a flame-dried round bottom flask at 70°C under inert atmosphere. ELQ-300 (0.10 g, 0.21 mmol) was added and the reaction stirred under inert atmosphere at 70°C for four hours, until complete by thin layer chromatography. The reaction was cooled to room temperature and the reaction solvent evaporated under reduced pressure. The mixture was taken up in water (10 mL) and extracted with dichloromethane (3 × 15 mL). Combined organic layers were washed with brine (15 mL) and concentrated. Purification by silica column chromatography (EtOAc/DCM) yielded the title compound, ELQ-387, as a white crystalline solid (39 mg, 32%). 1H NMR (400 MHz, DMSO-d6): δ = 8.03 (s, 1H), 7.54 (s, 1H), 7.44 (m, 3H), 7.25 (m, 2H), 7.20 (m, 2H), 5.83 (q, 1H, J = 5.4 Hz), 4.02 (s, 3H), 3.79 (m, 2H), 2.44 (s, 3H), 1.19 (d, 3H, J = 5.3 Hz), 0.88 (t, 3H, J = 7.1 Hz); M.P. (°C): 135.0-135.7; HRMS (ESI-TOF) m/z [M + H+]: Calculated for C29H26O7NClF3 592.13444; Found 529.13458 (0.24 ppm difference).
In vitro metabolic stability assay
Controls and ELQ-331 were incubated at 37°C in a reaction mixture containing microsomal protein in 100 mM potassium phosphate, 3 mM MgCl2 and with or without 2mM NADPH at pH 7.4. At various times after beginning incubations, aliquots were removed and mixed with ice-cold stop solution of methanol and an internal standard to prevent further metabolism. To quantify the amount of unmetabolized parent drug at each time point, samples were centrifuged and the supernatants were analyzed using liquid chromatography/mass spectrometry (LC-MS-MS). Half-life (T1/2) was determined by fitting the data to a first-order decay model, and intrinsic clearance (CLint) is dependent on half-life and concentration of protein, calculated as: CLint= ln2/(T1/2[microsomal protein]).
Plasmodium falciparum isolation and culture
Laboratory strains of P. falciparum were cultured in human erythrocytes by standard methods.31 The parasites were grown in culture medium with fresh human erythrocytes maintained at 2% hematocrit at 37°C in low-oxygen conditions (5% O2, 5% CO2, 90% N2).32 The culture medium used was RPMI-1640 with 25 mg/L gentamicin sulfate, 45 mg/L Albumax II, 10 mM glucose, and 25 mM HEPES buffer. Cultures were maintained at less than 10% parasitemia by transfer of infected cells to fresh erythrocytes and culture medium every 3 or 4 days.
In vitro drug sensitivity
In vitro antimalarial activity was measured using the SYBR Green I fluorescence-based assay.32,33 ELQ-330, ELQ-331, and ELQ-387 were evaluated for antiplasmodial activity with chloroquine and ELQ-300 as controls. Experiments were set up in triplicate in 96-well plates with a total 100 μL total volume per well with 2% hematocrit, 0.2% parasitemia, and drug dilutions between 0.25 and 250 nM in complete culture medium described above. The plates were incubated at 37°C in low-oxygen conditions described previously for 72 hours, at which point 100 μL of fluorescent dye-detergent mixture (0.2 μL SYBR Green I : 1 mL lysing buffer) was added and the plates incubated in the dark at room temperature for one hour. After incubation with the fluorescent dye, a 96-well plate reader with excitation wavelength set at 497 nm, and emission at 520 nm was used to measure fluorescence of each well. Fluorescence readings were plotted as a function of drug concentration, and curve fitting by nonlinear regression analysis (Graphpad Prism software package) gave calculated drug concentration that gave 50% reduction in fluorescence compared to drug-free controls (50% inhibitory concentration, IC50).
In vivo efficacy against murine malaria
The in vivo efficacy of ELQ-331 was determined against blood-stage Plasmodium yoelii using a modified 4-day test.34 Female mice (Charles River Laboratories) were infected intravenously with 50,000 P. yoelii parasitized erythrocytes from a donor animal (Kenya MR4 MRA-428). One day after inoculation, drugs (dissolved in PEG-400, chloroquine as positive control) were administered by oral gavage once daily for 4 successive days. Five days after inoculation, percent parasitemia was calculated by examination of Giemsa-stained blood smears.
In vivo single dose efficacy against murine malaria
Female mice (Charles River Laboratories) were infected intravenously with 50,000 P. yoelii parasitized erythrocytes from a donor animal (Kenya MR4 MRA-428). One day after inoculation, drugs (dissolved in PEG-400, chloroquine as positive control) were administered by oral gavage. Five days after inoculation, blood smears were prepared for study animals (4 per group) and stained with Giemsa to assess percent parasitemia. Animals that remained parasite-free for 30 days following inoculation were considered cured of infection.
In vivo prophylaxis against murine malaria
Female 6-week-old C57BL/6 albino mice were obtained from Charles River Laboratories (Charles River Lab, MA). On arrival, the animals were acclimated in quarantine for 7 days. Mice were thus at 7 weeks of age at the initiation of these experiments. The animals were housed singly in a cage maintained in a room with a temperature range of 18-26 °C, 34-68% relative humidity and a 12-h light/dark cycles. Standard rodent maintenance food was provided ad libitum during quarantine and throughout the study. All animal research was conducted in compliance with federal statutes, Army regulations, and the Animal Welfare Act principles stated in the Guide for the Care and Use of Laboratory Animals, NRC Publication, 2011 edition.
In vivo imaging system (IVIS) studies
In vivo imaging studies of rodents infected with luciferase expressing P. berghei mice were conducted, as described previously, using a Perkin Elmer IVIS Spectrum. Bioluminescence assessments were conducted on all animals at 24, 48, and 72 hours after 10,000 of sporozoites injection. To conduct in vivo imaging studies, mice were injected intraperitoneally with 150 μL of luciferin at a concentration of 150 mg/kg body weight (Gold Biotechnology, St. Louis, MO). Three minutes after luciferin IP injection, the mice were anesthetized using inhaled isoflurane and positioned ventral side upon a heated platform inside the IVIS instrument. The mice were administered isoflurane through nose cones throughout the IVIS imaging study. The IVIS camera exposure times utilized were 1 and 5 minutes for the 24, 48, and 72 hour time points with a large binning setting and an f-stop = 1. The IVIS Living Image software (version 4.0) was used to quantitate the bioluminescence in photons per second observed from the whole animal or the liver region. 3-D bioluminescent imaging tomography was performed using sequential images taken with filters ranging from 580 to 660 nm.35
Drug dosing and formulations
All drug solutions were prepared fresh before drug administration by dissolving the needed quantity of drug in cold (4°C) 0.5% (w/v) hydroxyethyl cellulose and 0.2% (v/v) Tween-80 (0.5% HECT) or PEG400. If needed, drugs were ground using a ProScientific 300D homogenizer and the particle size was measured using a Horiba LA-950V2 particle size analyzer. Formulations of ELQ-300 and ELQ-331 were prepared in PEG400 and administered orally (PO) at 3, 1, and 0.1 mg/kg one day prior to animal infections. Atovaquone was dissolved in 0.5% HECT and was administered PO at 2.5 and 5 mg/kg on day-1 before infections or at 2.5 mg/kg on days -1, 0, and 1 post infections. A vehicle control (PEG400) or HECT group was included as a negative control in both studies. Positive control consisted on three doses of 4-methyl primaquine which was dissolved in 0.5% HECT and was given at 5 mg/kg on days -1, 0, and 1 post infections. All dosing solutions were given based on mice body weight measured on the day before infections took place.
Flow cytometry (FCM)
Starting at 6 days after infection, all mice were assessed for blood stage parasitemia which was quantitated using flow cytometry conducted with an FC500 MPL flow cytometer (Beckman Coulter, Fullerton, CA). The green photomultiplier tube and filter setting were used for these studies (520-555 nm filter settings for the green PMT and greater than 580 nm settings for the red PMT). Infected erythrocytes, uninfected erythrocytes, and leukocytes were gated on logarithmic forward/side dot plots. The method of FCM sample preparation has been previously described previously.36, 37 In brief, a small 3 μl sample of blood was obtained from the tails of all mice. The blood was transferred into 0.3 ml of 1% heparinized isotonic buffer (PBS saline). 1 ml of 0.04% of glutaraldehyde was added to fix each sample, and samples were incubated for 60 minutes at 4°C followed by centrifugation at 450 g for 5 minutes. The supernatant was removed by aspiration, and the cells were re-suspended in 0.5 ml PBS buffer supplemented with 0.25% (v/v) Triton X-100 for 10-minute incubation at room temperature. After centrifugation, the permeabilized cells were re-suspended in 0.5 ml of RNAase at 1 mg/ml concentrations and incubated for at least 2 hours at 37°C to ensure complete digestion of reticulocytes which are at high concentrations due to anemia associated with P. berghei infection. 20 μL of YOYO-1 dye at a concentration of 2500 ng/ml was added to the 0.5 ml sample volume to create a final dye concentration of 100 ng/mL of YOYO-1. Parasitemia was monitored for up to 31-days post infections. Blood samples for parasitemia determination were taken every week or prior to euthanizing the animals sick with malaria that were being removed from the study. Mice that tested negative for parasitemia on day 31 post infection were considered cured. The minimum curative dose (ED100) in 100% of animals dosed was defined as the lowest dose which cured all animals for the entire 31-days follow-up period.
Isolation of P. falciparum mitochondria and ubiquinol cytochrome c oxidoreductase assay
Plasmodium falciparum mitochondria were isolated using the protocol developed by Mather, et. al.38 To measure ubiquinol-cytochrome c oxidoreductase activity, mitochondria were diluted and dispersed in 2 mg/mL n-dodecyl β-D-maltoside. After 45 minute incubation at 0°C, the mixture was centrifuged for 5 minutes at 10000 g. Enzymatic activity was measured using a UV/visible (Agilent 8453 diode array) spectrophotometer measuring absorbance at 550 nm with single reference wavelength 542 nm over 80 seconds at 30°C. The reaction buffer (pH = 8.0) used was 50 mM Tricine, 100 mM KCl, 4 mM NaN3, 50 μM cytochrome c, 0.1 mg/mL n-dodecyl β-D-maltoside. Background cytochrome c reductase measurements were recorded after addition of decylubiquinol (prepared by reduction of decylubiquinone with sodium borohydride followed by neutralization with HCl) to the reaction buffer for 5-10 seconds. After this short time, 4 μL mitochondria were added to the cuvette and the reaction was left to proceed for the remaining 80 seconds of data collection. Activity was calculated as the fractional activity of enzyme under inhibitory conditions relative to uninhibited enzyme.
Mitochondrial toxicity of ELQ-300 and ELQ-331
The mitochondrial toxicity of ELQ-331 and relevant controls were tested in triplicate against human HepG2 cells using the Promega CellTiter-Glo Kit as described by Swiss and Will in 2011.39 HepG2 cells (ATCC) were plated at 10,000 cells per well in 96-well plates. In a standard 96-well plate, compound and standard dilutions were made in DMSO. 4 μL of compound or standard dilutions were added to 396 μL of glucose or galactose medium in standard 96-well plates. After removing media from the cell plates, 100 μL of the medium/compound mixture was added per well and the plates were incubated for 72 hours at 37°C at 5% CO2. After incubation, Promega CellTiter-Glo Kit reagents were prepared according to manufacturer instructions, and 50 μL was added to each well. After incubating for 30 minutes at room temperature, the luminescent signal was measured using a plate reader.
Acknowledgments
Research reported in this publication was supported by the United States National Institutes of Health under award numbers AI100569 (MKR), and AI028398 (ABV). This project was also supported by funds from the Veterans Affairs Merit Review Program Award number i01 BX003312 (MKR). Research funds were also provided by the US Department of Defense Peer Reviewed Medical Research Program (Log #PR130649; Contract #W81XWH2142120447) (MKR). Additional support was provided by the Military Infectious Disease Research Program. We thank Zeng Qiang, Norma Roncal, Jing Zhang, Ping Zhang, Hsiuling Lin and Chad Black for excellent technical and managerial contributions. Analytical support was provided by the Bioanalytical Shared Resource/Pharmacokinetics Core Facility which is part of the University Shared Resource Program at Oregon Health and Sciences University. We thank Lisa Bleyle for her assistance with the LC-MS/MS analysis.
References
- 1.World Health Organization. World Malaria Report 2015. WHO; 2015. [Google Scholar]
- 2.World Health Organization. World Malaria Report 2016. WHO; 2016. [Google Scholar]
- 3.Bruxvoort K, Katia B, Catherine G, Patrick Kachur S, David S. How Patients Take Malaria Treatment: A Systematic Review of the Literature on Adherence to Antimalarial Drugs. PLoS One. 2014;9:e84555. doi: 10.1371/journal.pone.0084555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yeung S, Shunmay Y, White NJ. How do Patients use Antimalarial Drugs? A Review of the Evidence. Trop Med Int Health. 2005;10:121–138. doi: 10.1111/j.1365-3156.2004.01364.x. [DOI] [PubMed] [Google Scholar]
- 5.Bloland PB, World Health Organization Drug Resistance in Malaria. 2001 [Google Scholar]
- 6.Vliegenthart-Jongbloed K, de Mendonça Melo M, van Wolfswinkel ME, Koelewijn R, van Hellemond JJ, van Genderen PJJ. Severity of Imported Malaria: Protective Effect of Taking Malaria Chemoprophylaxis. Malar J. 2013;12:265. doi: 10.1186/1475-2875-12-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Talisuna AO, Bloland P, D’Alessandro U. History, Dynamics, and Public Health Importance of Malaria Parasite Resistance. Clin Microbiol Rev. 2004;17:235–254. doi: 10.1128/CMR.17.1.235-254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harinasuta T, Suntharasamai P, Viravan C. Chloroquine-resistant Falciparum Malaria in Thailand. Lancet. 1965;286:657–660. doi: 10.1016/S0140-6736(65)90395-8. [DOI] [PubMed] [Google Scholar]
- 9.Kyaw MP, Nyunt MH, Chit K, Aye MM, Aye KH, Aye MM, Lindegardh N, Tarning J, Imwong M, Jacob CG, et al. Reduced Susceptibility of Plasmodium falciparum to Artesunate in Southern Myanmar. PLoS One. 2013;8:e57689. doi: 10.1371/journal.pone.0057689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashley EA, et al. Spread of Artemisinin Resistance in Plasmodium falciparum Malaria. N Engl J Med. 2014;371:786–786. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burrows JN, van Huijsduijnen RH, Möhrle JJ, Oeuvray C, Wells TNC. Designing the Next Generation of Medicines for Malaria Control and Eradication. Malar J. 2013;12:187. doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.malERA Consultative Group on Drugs. A Research Agenda for Malaria Eradication: Drugs. PLoS Med. 2011;8:e1000402. doi: 10.1371/journal.pmed.1000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salzer W, Timmler H, Andersag H. Über einen neuen, gegen Vogelmalaria wirksamen Verbindungstypus. Chem Ber. 1948;81:12–19. [Google Scholar]
- 14.Nilsen A, LaCrue AN, White KL, Forquer IP, Cross RM, Marfurt J, Mather MW, Delves MJ, Shackleford DM, Saenz FE, et al. Quinolone-3-diarylethers: a New Class of Antimalarial Drug. Sci Transl Med. 2013;5:177ra37. doi: 10.1126/scitranslmed.3005029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nilsen A, Miley GP, Forquer IP, Mather MW, Katneni K, Li Y, Pou S, Pershing AM, Stickles AM, Ryan E, et al. Discovery, Synthesis, and Optimization of Antimalarial 4(1H)-quinolone-3-diarylethers. J Med Chem. 2014;57:3818–3834. doi: 10.1021/jm500147k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birth D, Kao W-C, Hunte C. Structural Analysis of Atovaquone-inhibited Cytochrome bc1 Complex Reveals the Molecular Basis of Antimalarial Drug Action. Nat Commun. 2014;5:4029. doi: 10.1038/ncomms5029. [DOI] [PubMed] [Google Scholar]
- 17.Happi CT, Gbotosho GO, Folarin OA, Milner D, Sarr O, Sowunmi A, Kyle DE, Milhous WK, Wirth DF, Oduola AMJ. Confirmation of Emergence of Mutations Associated with Atovaquone-proguanil Resistance in Unexposed Plasmodium falciparum Isolates from Africa. Malar J. 2006;5:82. doi: 10.1186/1475-2875-5-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korsinczky M, Chen N, Kotecka B, Saul A, Rieckmann K, Cheng Q. Mutations in Plasmodium falciparum Cytochrome b That Are Associated with Atovaquone Resistance Are Located at a Putative Drug-Binding Site. Antimicrob Agents Chemother. 2000;44:2100–2108. doi: 10.1128/AAC.44.8.2100-2108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stickles AM, de Almeida MJ, Morrisey JM, Sheridan KA, Forquer IP, Nilsen A, Winter RW, Burrows JN, Fidock DA, Vaidya AB;, et al. Subtle Changes in Endochin-like Quinolone Structure Alter the Site of Inhibition Within the Cytochrome bc1 Complex of Plasmodium falciparum. Antimicrob Agents Chemother. 2015;59:1977–1982. doi: 10.1128/AAC.04149-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stickles AM, Smilkstein MJ, Morrisey JM, Li Y, Forquer IP, Kelly JX, Pou S, Winter RW, Nilsen A, Vaidya AB, et al. Atovaquone and ELQ-300 Combination Therapy as a Novel Dual-Site Cytochrome bc1 Inhibition Strategy for Malaria. Antimicrob Agents Chemother. 2016;60:4853–4859. doi: 10.1128/AAC.00791-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stickles AM, Ting L-M, Morrisey JM, Li Y, Mather MW, Meermeier E, Pershing AM, Forquer IP, Miley GP, Pou S, et al. Inhibition of Cytochrome bc1 as a Strategy for Single-dose, Multi-stage Antimalarial Therapy. Am J Trop Med Hyg. 2015;92:1195–1201. doi: 10.4269/ajtmh.14-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piplani M, Rana AC, Sharma PC. Prodrugs of Antiinfective Agents: A Review. J Pharm Pharm Sci. 2016;19:82–113. doi: 10.18433/J3X61S. [DOI] [PubMed] [Google Scholar]
- 23.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Järvinen T, Savolainen J. Prodrugs: design and clinical applications. Nat Rev Drug Discov. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 24.Chung MC, Ferreira EI, Santos JL, Giarolla J, Rando DG, Almeida AE, Bosquesi PL, Menegon RF, Blau L. Prodrugs for the Treatment of Neglected Diseases. Molecules. 2007;13:616–677. doi: 10.3390/molecules13030616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jornada DH, dos Santos Fernandes GF, Chiba DE, de Melo TRF, dos Santos JL, Chung MC. The Prodrug Approach: A Successful Tool for Improving Drug Solubility. Molecules. 2015;21:42. doi: 10.3390/molecules21010042.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miley GP, Pou S, Winter R, Nilsen A, Li Y, Kelly JX, Stickles AM, Mather MW, Forquer IP, Pershing AM, et al. ELQ-300 Prodrugs for Enhanced Delivery and Single-dose Cure of Malaria. Antimicrob Agents Chemother. 2015;59:5555–5560. doi: 10.1128/AAC.01183-15.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bodin NO, Ekström B, Forsgren U, Jalar LP, Magni L, Ramsay CH, Sjöberg B. Bacampicillin: a new orally well-absorbed derivative of ampicillin. Antimicrob Agents Chemother. 1975;8:518–525. doi: 10.1128/AAC.8.5.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Becker MW. Prodrugs of Phosphonate Nucleotide Analogues and Methods for Selecting and Making Same. US Patent Application WO2002008241 A2. 2002 Jan 31;
- 29.Porter CJ, Tervaskis NL, Charman WN. Lipids and Lipid-Based Formulations: Optimizing the Oral Delivery of Lipophilic Drugs. Nat Rev Drug Discov. 2007;6:231–248. doi: 10.1038/nrd2197. [DOI] [PubMed] [Google Scholar]
- 30.Feeney OM, Crum MF, McEvoy CL, Trevaskis NL, Williams HD, Pouton CW, Charman WN, Bergström CAS, Porter CJH. 50 Years of Oral Lipid-Based Formulations: Provenance, Progress, and Future Perspectives. Adv Drug Deliv Rev. 2016;101:167–194. doi: 10.1016/j.addr.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 31.Schuster FL. Cultivation of Plasmodium spp. Clin Microbiol Rev. 2002;15(3):355–364. doi: 10.1128/CMR.15.3.355-364.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trager W, Jensen JB. Human Malaria Parasites in Continuous Culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 33.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and Inexpensive Fluorescence-based Technique for High-throughput Antimalarial Drug Screening. Antimicrob Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ager AL. Rodent Malaria Models. Handbook of Experimental Pharmacology. 1984:225–264. [Google Scholar]
- 35.Li Q, O’Neil M, Xie L, Caridha D, Zeng Q, Zhang J, Pybus B, Hickman M, Melendez V. Assessment of the prophylactic activity and pharmacokinetic profile of oral tafenoquine compared to primaquine for inhibition of liver stage malaria infections. Malar J. 2014;13:141. doi: 10.1186/1475-2875-13-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie L, Li Q, Johnson J, Zhang J, Milhous W, Kyle D. Development and Validation of Flow Cytometric Measurement for Parasitemia Using Autofluorescence and YOYO-1 in Rodent Malaria. Parasitology. 2007;134:1151–1162. doi: 10.1002/cyto.a.20380. [DOI] [PubMed] [Google Scholar]
- 37.Li Q, Gerena L, Xie L, Zhang J, Kyle D, Milhous W. Development and Validation of Flow Cytometric Measurement for Parasitemia in Cultures of P. falciparum Vitally Stained with YOYO-1. Cytometry A. 2007;71A:296–307. doi: 10.1002/cyto.a.20380. [DOI] [PubMed] [Google Scholar]
- 38.Mather MW, Morrisey JM, Vaidya AB. Hemozoin-free Plasmodium falciparum Mitochondria for Physiological and Drug Susceptibility Studies. Mol Biochem Parasitol. 2010;174:150–153. doi: 10.1016/j.molbiopara.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swiss R, Will Y. Assessment of Mitochondrial Toxicity in HepG2 Cells Cultured in High-Glucose- or Galactose-Containing Media. Current Protocols in Toxicology. 2011 doi: 10.1002/0471140856.tx0220s49. [DOI] [PubMed] [Google Scholar]