

Abstract
Background
The utility of whole-exome sequencing (WES) for the diagnosis and management of adult-onset constitutional disorders has not been adequately studied. Genetic diagnostics may be advantageous in adults with chronic kidney disease (CKD), in whom the cause of kidney failure often remains unknown.
Objective
To study the diagnostic utility of WES in a selected referral population of adults with CKD.
Design
Observational cohort.
Setting
A major academic medical center.
Patients
92 adults with CKD of unknown cause or familial nephropathy or hypertension.
Measurements
The diagnostic yield of WES and its potential effect on clinical management.
Results
Whole-exome sequencing provided a diagnosis in 22 of 92 patients (24%), including 9 probands with CKD of unknown cause and encompassing 13 distinct genetic disorders. Among these, loss-of-function mutations were identified in PARN in 2 probands diagnosed respectively with tubulointerstitial fibrosis and CKD of unknown cause. PARN mutations have been implicated in a short telomere syndrome characterized by lung, bone marrow, and liver fibrosis; these findings extend the phenotype of PARN mutations to renal fibrosis. In addition, review of the American College of Medical Genetics actionable genes identified a pathogenic BRCA2 mutation in a proband who was diagnosed with breast cancer on follow-up. The results affected clinical management in most identified cases, including initiation of targeted surveillance, familial screening to guide donor selection for transplantation, and changes in therapy.
Limitation
The small sample size and recruitment at a tertiary care academic center limit generalizability of findings among the broader CKD population.
Conclusion
Whole-exome sequencing identified diagnostic mutations in a substantial number of adults with CKD of many causes. Further study of the utility of WES in the evaluation and care of patients with CKD in additional settings is warranted.
Primary Funding Source
New York State Empire Clinical Research Investigator Program, Renal Research Institute, and National Human Genome Research Institute of the National Institutes of Health.
Chronic kidney disease (CKD) affects an estimated 14% of Americans (1, 2). These persons have 10- to 15-fold higher morbidity and mortality rates than the general population (1, 3). In most patients with CKD, the diagnosis is based on standard office work-up and sometimes kidney biopsy findings. However, earlystage CKD is often clinically silent, and subtypes can be difficult to distinguish on the basis of clinical data alone. Thus, in many persons, the precise cause of kidney failure remains unknown. Approximately 10% to 25% of patients with CKD note a family history of nephropathy (4 – 6), suggesting that in many cases the disease has a hereditary component. Recent advances in genomic technologies, such as chromosomal microarray and massively parallel (“nextgeneration”) sequencing, enable genome-wide analysis at a modest cost and precise definition of the molecular cause of many complex diseases (7–13). Application of these methods has suggested opportunities for individualized diagnosis and risk stratification, including targeted work-up and surveillance for associated disease complications (11–13) and sometimes precision therapy (12–15). However, studies to date have focused mainly on a limited range of disorders in pediatric cohorts or on cancer in adults (7–17); thus, the clinical utility of these approaches for a broader spectrum of diseases, particularly among adults, remains unclear.
Applying chromosomal microarray analysis, we recently showed that 7.4% of 419 children with various forms of CKD had a major known pathogenic genomic imbalance that was not suspected after clinical assessment (18). These disorders were evenly distributed among patients clinically diagnosed with congenital and noncongenital forms of CKD, indicating that genetic analysis has utility across broad clinical categories. In most of these cases, the genetic findings either reclassified the disease or provided information that could guide subsequent clinical care, such as evaluation for metabolic or neuropsychiatric disease. Similarly, next-generation sequencing has been shown to have great utility for diagnosing genetic forms of nephrotic syndrome or congenital kidney defects in pediatric populations, albeit mainly in the context of targeted panels (19–21).
Whole-exome sequencing (WES) is a genome-wide testing approach that allows selective sequencing of the protein-coding regions of the genome, which are enriched for disease-associated variants (12–15). Because of its genome-wide coverage, WES enables screening of most genes associated with kidney disease and can therefore be applied across diverse categories of renal disorders. Moreover, it can potentially identify novel etiologic genes for nephropathy or detect actionable incidental mutations unrelated to the primary indications for testing. For these reasons, WES is emerging as a preferred diagnostic tool for hereditary disorders (12–15, 22, 23). In pediatric cohorts, WES recently identified diagnostic mutations in up to 11.5% of patients with congenital kidney anomalies and 26% of patients with steroid-resistant nephrotic syndrome, supporting its diagnostic utility for early-onset CKD (24, 25). However, the value of this sequencing method for the diagnosis and management of CKD in adults has not been adequately studied. We did a pilot study to test the utility of WES in adults referred for evaluation of CKD or hypertension.
Methods
Study Design
The results of WES in a convenience sample of patients referred for evaluation of CKD were reviewed for their potential to inform clinical practice. To facilitate diagnostic interpretation of WES data, we compiled a list of genes encompassing most common Mendelian forms of kidney and hypertensive disorders. We next annotated the exomes for diagnostic variants in nephropathy genes and then analyzed other genes, including =those recommended by the American College of Medical Genetics (ACMG), for return of medically actionable incidental findings (26).
Patient Population and Setting
The study sample was selected from a group of 344 patients seen at outpatient nephrology clinics between October 2013 and May 2014 at Columbia University Medical Center, a tertiary care medical center with a nephrology division offering highly specialized care for glomerular disorders. These 344 patients were referred for evaluation and management of kidney disease and consented to a general genetic research and biobanking protocol. Supplement Table 1 (available at Annals.org) presents the characteristics of these 344 patients. From this group, we selected 81 adult patients (aged >18 years) (Supplement Table 2, available at Annals.org) for WES who fulfilled 1 of the following inclusion criteria: a family history of kidney disease (defined as any family member with urinary abnormalities or impaired kidney function, as reported by the patient), undiagnosed kidney disease, or clinical suspicion of a genetic kidney disease (for example, in a proband with young age of onset and no family history of nephropathy). The PKD1 gene is not well-captured by WES because of gene duplication (27), so patients fulfilling clinical diagnostic criteria for autosomal dominant polycystic kidney disease were not included in the WES study.
In addition to these 81 patients from Columbia University Medical Center, we also included 11 patients referred for suspected inherited kidney disease or hypertension from outside institutions. Three patients with familial tubulointerstitial nephropathy and 1 with early-onset hypertension were referred from 3 local practices in the United States (New York University and nephrology practices in suburban New York and Delaware). Four were referred for evaluation of Mendelian hypertension from the Polish Kidney Genetics Network (POLYGENES, www.polygenes.org), centered in the Department of Genetics at Pozna University of Medical Sciences and The Center of Medical Genetics GENESIS (Poznan, Poland). Three other patients were referred from Gaslini Institute (Genova, Italy) for evaluation of glomerulonephritis with nondiagnostic kidney biopsies. All participants gave informed consent, and the study was approved by the Columbia University Institutional Review Board and local ethics committees.
WES and Sequence Interpretation
Staff extracted DNA from whole blood. Telomere length was measured using genomic DNA from whole blood, as previously described (28, 29). For WES, fragment libraries using 200 ng of genomic DNA were constructed from each sample, following the Agilent standard library preparation protocol for TruSeq (Illumina). Exome capture was done with the SureSelectXT Human All Exon V4 (51 Mb) kit (Agilent), and sequencing was done using the HiSeq 2000 or 2500 (Illumina) at the Columbia Genome Center. On average, 92.83 million independent paired-end reads (18.56-Gb bases) were generated per sample to provide an average coverage of 110-fold, with 99.17% of target regions being covered at least 10-fold. The paired-end reads (read size, 101 bp) were mapped to the human reference genome National Center for Biotechnology Information build 37 using Burrows–Wheeler Aligner, version 0.5.9. The Genome Analysis Toolkit, version 1.6–13, was used to call germline single nucleotide variants and insertions or deletions (indels).
Variants were annotated for predicted effect on protein function (using ANNOVAR and SnpEff); allele frequency in public databases (ExAC, dbSNP, and the 1000 Genomes Project); and predicted pathogenicity with in silico algorithms, including PolyPhen and Combined Annotation Dependent Depletion scores (30– 30). Evidence for disease causality was assessed using ClinVar and the Human Genome Mutation Database (Qiagen), followed by manual review of the cited primary literature (33, 36). In addition, we developed a curated “priority” list of 287 Online Mendelian Inheritance in Man (OMIM; http://omim.org) genes implicated in Mendelian forms of kidney disorders and hypertension to facilitate clinical annotation (hereon, we refer to this gene list as nephropathy genes; see Supplement Table 3, available at Annals.org). A known limitation of exome sequencing is that some segments of the genome are not amenable to capture (23). Among the 287 nephropathy genes, 29 were identified with at least 1 exon that is not captured by the Agilent kit, representing potential blind spots in the analysis (Supplement Table 3).
Variant interpretation was done by a panel of nephrologists or molecular geneticists with domain expertise in inherited kidney diseases (K.K., S.S.C., C.A., L.R., E.G., and A.G.G.), bioinformaticians (S.L. and D.A.F.), and a clinical molecular geneticist (V.J.), using the ACMG guidelines for clinical sequence interpretation (37). Detailed classification criteria for pathogenic and likely pathogenic variants are in Supplement Table 4 (available at Annals.org). We also reviewed potentially pathogenic mutations in OMIM genes associated with other heritable disorders and in the ACMG's 59 actionable genes (26). All diagnostic variants were confirmed by Sanger sequencing. Finally, we verified the distribution of potentially functional variants in nephropathy genes in each exome. These potentially functional variants were defined as missense, nonsense, splice site, or indel variants with a minor allele frequency less than 1% in ExAC (a database of genetic variation in >60 000 persons) and a Combined Annotation Dependent Depletion score greater than 10 (indicating a variant score in the top 10% of deleteriousness in a large reference data set of variants). We also verified allele frequencies using an anonymized in-house control data set derived from 9012 persons who had undergone WES for indications other than nephropathy; these control data included healthy parents of children with a developmental disorder and participants from genetic studies of amyotrophic lateral sclerosis or seizure disorders.
Role of the Funding Source
he study was funded by the New York State Empire Clinical Research Investigator Program, the Renal Research Institute, and the National Human Genome Research Institute of the National Institutes of Health. The funding sources had no role in the design, conduct, and analysis of the study or in the decision to submit the manuscript for publication.
Results
We performed WES in 92 adults with CKD with a clinical diagnosis compatible with a Mendelian genetic disease, a familial nephropathy of unclear cause, or unexplained kidney failure. The characteristics of the 92 participants are described in Table 1 and Supplement Table 2. Nineteen participants (20%) were of self-declared non-Caucasian ancestry, 53 (58%) had a family history of nephropathy, and 50 (54%) had a clinical diagnosis of glomerulopathy (Table 1).
Table 1.
Characteristics of Patients Having Exome Sequencing
| Clinical Category | Patients, n | Men, n | Women, n | Positive Family History, n (%) |
Mean Age at Presentation (SD), y |
|---|---|---|---|---|---|
| Glomerular disease | 50 | 21 | 29 | 26 (52) | 46 (16) |
| Tubulointerstitial disease | 10 | 6 | 4 | 8 (80) | 41 (15) |
| Developmental disorders | 11 | 10 | 1 | 7 (64) | 43 (16) |
| Hypertension | 5 | 4 | 1 | 3 (60) | 22 (7) |
| Undiagnosed disease/other | 16 | 9 | 7 | 9 (56) | 37 (11) |
| Total | 92 | 50 | 42 | 53 (58) | 42 (17) |
Diagnostic Variants in Known Nephropathy Genes
Using ACMG criteria, we identified 19 patients with a diagnostic mutation in 1 of the known nephropathy genes (Table 2): 9 had a pathogenic variant and 10 had a likely pathogenic variant. In 6 of these 19 cases, the genetic data confirmed the clinical diagnosis but were nonetheless valuable because they allowed discrimination of the mode of disease inheritance and enabled appropriate screening and counseling for family members. For example, in patients K009 and K024, WES helped to distinguish X-linked versus autosomal forms of Alport syndrome, thereby informing family counseling and selection of living related kidney donors. In the other 13 cases, including 7 patients presenting with CKD of unknown cause, WES clarified the clinical diagnosis or reclassified the patient's disease entirely, which significantly affected clinical decision making. For example, we found COL4A3, COL4A4, or COL4A5 mutations in probands with a clinical diagnosis of familial focal segmental glomerulosclerosis (K078 and K058) or familial nephropathy of unknown cause (K014 and K028). These genetic diagnoses had direct consequences for medical management, including avoidance of immunosuppressive agents, usually considered first-line therapy for familial focal segmental glomerulosclerosis; auditory and ophthalmologic screening; screening for mutation carriers among family members; optimal selection of living related organ donors; and in some cases, referral to an ongoing clinical trial targeting patients with type IV collagen mutations (ClinicalTrials.gov: NCT02855268). In another case, WES identified a diagnostic mutation for Dent disease in a patient (KGY1) with undiagnosed familial CKD who had had 2 nondiagnostic kidney biopsies as a child; implications included treatment with citrate and thiazide diuretics, prioritization for transplantation, and molecular diagnosis in a brother with recently diagnosed CKD. We also genetically diagnosed several rare diseases, including CHARGE (coloboma, heart defects, atresia choanae, growth retardation, genital abnormalities, and ear abnormalities) syndrome (K030) and renal cysts and diabetes syndrome (K064), prompting targeted work-up for associated extrarenal comorbid conditions and providing a unifying explanation for some organ defects.
Table 2.
Diagnostic Variants Identified in Exome Sequencing
| Patient Identification |
Sex | Age, y |
Race/E thnicit y |
Clinical Presentation |
Genetic Diagnosis | Gene Symbol |
Sequence Variant, RSID, and ExAC Frequency |
Clinical Implications of Genetic Information |
|---|---|---|---|---|---|---|---|---|
| Genetic diagnoses that confirmed the clinical diagnosis and their potential implications on clinical care | ||||||||
|
| ||||||||
| K008 | Female | 28 | White | ESRD of unknown cause, has undergone kidney transplantation, son has Alport syndrome | X-linked Alport syndrome (OMIM 301050) | COL4A5 | NM_000495:c.G3310T:p.G1104C† | Clarification of mode of inheritance; screening family members |
| RSID: NA | ||||||||
| ExAC frequency: 0 | ||||||||
|
| ||||||||
| K009 | Male | 26 | White | Biopsy-proven Alport syndrome, negative FHx | X-linked Alport syndrome (OMIM 301050) | COL4A5 | NM_000495:c.G2731A:p.G911R | Clarification of mode of inheritance; screening family members |
| RSID: rs281874704 | ||||||||
| ExAC frequency: 0 | ||||||||
|
| ||||||||
| K024 | Female | 31 | White | Biopsy-proven Alport syndrome, negative FHx | X-linked Alport syndrome (OMIM 301050) | COL4A5 | NM_000495:c.G5042A:p.C1681Y† | Clarification of mode of inheritance; screening family members |
| RSID: NA | ||||||||
| ExAC frequency: 0 | ||||||||
|
| ||||||||
| K079 | Male | 22 | White | Biopsy-proven Alport syndrome, positive FHx | X-linked Alport syndrome (OMIM 301050) | COL4A5 | NM_000495:c.G2474A:p.G825E† | Clarification of mode of inheritance; screening family members |
| RSID: NA | ||||||||
| ExAC frequency: 0 | ||||||||
|
| ||||||||
| K054 | Female | 31 | White | Nail–patella syndrome with biopsy-proven FSGS | Nail–patella syndrome (OMIM 161200) | LMX1B | NM_001174146: 775_776del: p.S259Cfs*38 | Do not consider immunosuppression; refer for transplant |
| RSID: NA | ||||||||
| ExAC frequency: 0 | ||||||||
|
| ||||||||
| K015 | Male | 37 | Asian | Fabry disease by biopsy and enzyme activity, ESRD | Fabry disease(OMIM 301500) | GLA | NM_000169:c.A886G:p.M296V | Screening family members; continue enzyme therapy |
| RSID: rs104894830 | ||||||||
| ExAC frequency: 4 × 10−6 | ||||||||
| Genetic diagnoses that provided new information and their potential implications on clinical care | ||||||||
|
| ||||||||
| K030 | Male | 19 | African American | CKD of unknown cause with bilateral renal hypoplasia, hypogonadism; history of repaired atrial septal defect; offspring of healthy parents | CHARGE syndrome (OMIM 214800) | CHD7 | NM_017780:c.G8554A:p.D2852N† | Screening for hearing and visual loss, cranial nerve dysfunction, and learning disability; caution during anesthesia because of upper airway maldevelopment |
| RSID: NA | ||||||||
| ExAC frequency: 0 | ||||||||
|
| ||||||||
| KGY1 | Male | 35 | White | Familial proteinuric CKD of unknown cause, inconclusive results on biopsies at age 6 and 12 y, brother with proteinuria (never had biopsy) | Dent disease 1 (OMIM 300009) | CLCN5 | NM_000084:c.2057delG:p.S686Tfs*5 | Therapy with thiazide diuretics and citrates; no posttransplant recurrence expected; brother screened and diagnosed with Dent disease 1 |
| RSID: NA | ||||||||
| ExAC frequency: 0 | ||||||||
|
| ||||||||
| K014 | Female | 22 | White | ESRD of unknown cause, has undergone kidney transplantation, positive FHx of microhematuria on father’s side | Autosomal recessive Alport syndrome (OMIM 203780) | COL4A3 |
NM_000091:c.G1558C:p.G520R
|
Posttransplant recurrence unlikely; compound heterozygosity confirmed by testing allele transmission in offspring; immunosuppressive therapy not indicated; family counseling |
NM_000091:c.T4421C:p.L1474P
| ||||||||
|
| ||||||||
| K078 | Female | 28 | White | FSGS by biopsy, has had kidney transplantation, sibling died at age 17 y with anasarca, FHx of hematuria | Autosomal recessive Alport syndrome (OMIM 203780) | COL4A4 |
NM_000092:c.G4288A:p.G1430R
|
Compound heterozygosity confirmed by testing allele transmission in offspring; immunosuppressive therapy not indicated; family counseling; patient opted to pursue pregnancy after genetic diagnosis clarified recessive inheritance |
NM_000092:c.213_239del:p.P72_G80delPQGPIGPLG
| ||||||||
|
| ||||||||
| K058 | Male | 28 | White | FSGS by biopsy, undergoing kidney transplant evaluation positive FHx for nephropathy | Autosomal dominant Alport syndrome (OMIM 104200) | COL4A4 | NM_000092:c.G1145C:p.G382A† | Immunosuppressive therapy not indicated; family screening for donor evaluation; referred for gene therapy trial |
| RSID: rs751952236 | ||||||||
| ExAC frequency: 4 × 10−5 | ||||||||
|
| ||||||||
| K028 | Female | 57 | White | mild CKD of unknown cause with hematuria. TBMD diagnosed as a child (self-reported), | X-linked Alport syndrome (OMIM 301050) | COL4A5 | NM_000495:c.G5030A:p.R1677Q | Auditory and ophthalmology screening; referred for clinical trial (NCT02855268) |
| RSID: rs104886308 | ||||||||
| ExAC frequency: 2.3 × 10−5 | ||||||||
|
| ||||||||
| K064 | Male | 48 | White | CKD of unknown cause with congenital small left kidney, diabetes, no prior kidney biopsy. | HNF1B-associated disease‡ (OMIM 137920) | HNF1B | NM_000458:c.C742T:p.Q248X | Insulin therapy; screening for hyperuricemia, hypomagnesemia, and hypoparathyroidism; family screening and counseling |
| RSID: NA | ||||||||
| ExAC frequency: 0 | ||||||||
|
| ||||||||
| K065 | Male | 20 | White | Presented with hypertension and hypokalemia at age 13 y | Liddle syndrome (OMIM 177200) | SCNN1G | NM_001039:c.C1874G:p.P625R† | Treat with amiloride |
| RSID: NA | ||||||||
| ExAC frequency: 0 | ||||||||
|
| ||||||||
| K027 | Female | 44 | White | Gitelman syndrome, type 2 diabetes | Gitelman syndrome (OMIM 263800) | SLC12A3 |
NM_000339: A2899G:p.R967G
|
– |
NM_000339:c.2986_2987insGCTC:p.Y999Afs*50
| ||||||||
|
| ||||||||
| K006 | Female | 42 | White | Biopsy-proven FSGS, mother with ESRD has undergone kidney transplantation | FSGS type 2(OMIM 603965) | TRPC6 | TRPC6:NM_004621: A434G:p.H145R† | Affected mother tested positive for mutation; immunosuppression is not indicated |
| RSID: NA | ||||||||
| ExAC frequency: 0 | ||||||||
|
| ||||||||
| K007 | Male | 55 | White | CKD with chronic tubulointerstitial nephropathy on biopsy, undergoing kidney transplant evaluation, Strong FHx for dominant transmission of CKD/ESRD | Autosomal dominant medullary cystic kidney disease type 2(OMIM 603860) | UMOD | NM_001008389:c.G317A:p.C106F† | Family screening and counseling; monitor uric acid levels |
| RSID: rs398123697 | ||||||||
| ExAC frequency: 3.7 × 10−5 | ||||||||
|
| ||||||||
| K016 | Male | 21 | White/Hispanic | Gout and CKD of unknown cause, no biopsy; positive FHx for dominant transmission of ESRD/gout | Autosomal dominant medullary cystic kidney disease type 2 (OMIM 603860) | UMOD | NM_001008389:c.G774C:p.W258C† | Family screening and counseling; monitor uric acid levels |
| RSID: NA | ||||||||
| ExAC frequency: 0 | ||||||||
|
| ||||||||
| K043 | Female | 45 | Asian | CKD with chronic tubulointerstitial nephropathy by biopsy, positive FHx for dominant transmission of CKD and gout | Autosomal dominant medullary cystic kidney disease type 2 (OMIM 603860) | UMOD | NM_001008389:c.T638A:p.M213K† | Family screening and counseling; monitor uric acid levels |
| RSID: NA | ||||||||
| ExAC frequency: 0 | ||||||||
CHARGE = coloboma, heart defects, atresia choanae, growth retardation, genital abnormalities, and ear abnormalities; CKD = chronic kidney disease; ESRD = end-stage renal disease; ExAC = Exome Aggregation Consortium; FHx = family history; FSGS = focal segmental glomerulosclerosis; NA = not applicable ; OMIM = Online Mendelian Inheritance in Man; RSID = Reference Single Nucleotide Polymorphism identification number ; TBMD = thin basement membrane disease.
Likely a pathogenic mutation.
Also known as renal cysts and diabetes syndrome or maturity-onset diabetes of the young type 5.
The genetic diagnostic rate was similar between glomerular and nonglomerular disorders (19% and 23%, respectively) and between persons of European and non-European ancestry (20% for both). The diagnostic rate in Columbia University Medical Center patients recruited during routine outpatient visits was 22%, compared with 9% among those referred for evaluation of genetic kidney disease from outside institutions. During the annotation process, we detected an average of 5 potentially functional variants in nephropathy genes per person, which is consistent with the known distribution of putatively functional variants in the general population (34). In many cases, these variants were in nephropathy genes that were not consistent with the clinical presentation and could therefore be eliminated from consideration; however, others were partially compatible with the clinical diagnosis and were classified as variants of unknown significance, pending additional corroborating clinical data. As an example, we list 5 cases where we detected rare, predicted, deleterious variants that potentially explained some aspects of the clinical presentation but were classified as variants of unknown significance because phenotypic evidence was insufficient to establish causality (Supplement Table 5).
Mutations in PARN in Patients With Tubulointerstitial Nephropathy
We next reviewed all genetically unresolved cases for mutations in OMIM genes. We identified the following 2 independent loss-of-function (LoF) mutations in PARN: a nonsense variant, p.Q215*, in a proband with early-onset nonproteinuric nephropathy of unclear cause, and a splice site variant, c.554+1G>A, in a parent–child pair with tubulointerstitial nephropathy (Table 3). PARN encodes a poly(A)-specific ribonuclease that participates in telomere maintenance (38). Heterozygous LoF mutations in PARN have recently been implicated in late-onset idiopathic pulmonary fibrosis; myelodysplastic syndrome; and, rarely, liver fibrosis. However, to our knowledge they have yet to be associated with kidney disease (28, 39, 40). The PARN mutations detected in the patients with CKD were absent in all public databases. In comparison, we detected 2 LoF mutations in PARN in WES data from 9012 control participants (0.02%) from our institution; these 2 represented a substantial portion of the 43 nonglomerular CKD cases (4.7%). Kidney biopsy specimens were available in the parent–child pair and were histologically concordant, revealing chronic tubulointerstitial nephropathy with glomeruli exhibiting minimal and nonspecific changes. The predominant pathologic abnormalities were seen in the medulla, including interstitial fibrosis with a disorganized architecture, resembling changes seen in renal dysplasia (Figure).
Table 3.
Characteristics of Patients with PARN (NM_002582) Mutations
| Characteristic | K017 | K018 | K060 |
|---|---|---|---|
| Clinical diagnosis | CKD of unknown cause, Tubulointerstitial nephropathy with dysplastic features on biopsy | CKD of unknown cause, Tubulointerstitial nephropathy with dysplastic features on biopsy | CKD of unknown cause, no kidney biopsy available |
| Sex | Male (father) | Female (daughter) | Female |
| Age at presentation | 44 y | 23 y | 48 y |
| Kidney outcome | ESRD, has undergone kidney transplantation | ESRD, has undergone kidney transplantation | ESRD, has undergone kidney transplantation |
| Follow-up clinical findings | Work-up for interstitial lung disease 3 y after study enrollment | None | Secondary hyperparathyroidism, normal findings on chest radiography at age 50 y, macrocytic anemia |
| Sequence variant† | c.554+1G>A | c.554+1G>A | c.C643T: p.Q215* |
CKD = chronic kidney disease; ESRD = end-stage renal disease.
Absent in the Short Genetic Variations (Single Nucleotide Polymorphism) and Exome Aggregation Consortium databases.
Figure. Kidney biopsy findings in a PARN mutation carrier (K018).
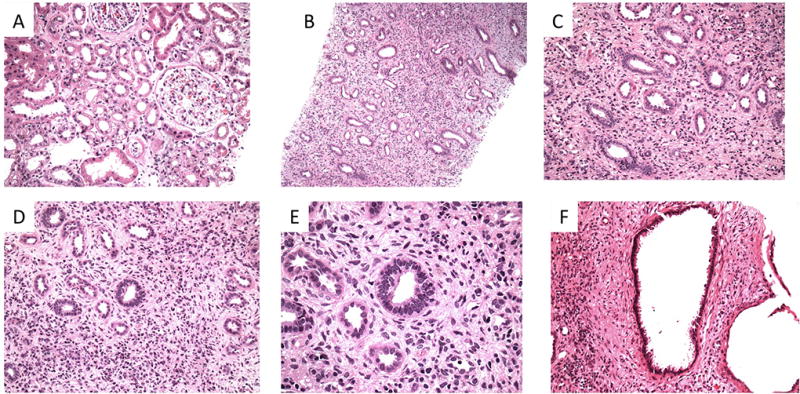
A. Low-power view of the renal cortex shows mild acute tubular injury and mild tubulointerstitial scarring. Glomeruli appear unremarkable (hematoxylin–eosin stain; original magnification, ×200). B. Low-magnification view of the renal medulla reveals more pronounced abnormalities. The tubular architecture is broadly disorganized, with a haphazard arrangement of tubules. The interstitium is notable for a diffuse increase in cellularity (hematoxylin–eosin stain; original magnification, ×100). C. The majority of tubules are lined by cuboidal to columnar epithelium. Flattened epithelium consistent with the thin limbs of the loop of Henle are difficult to discern (hematoxylin–eosin stain; original magnification, ×200). D. The interstitium is diffusely hypercellular, and the majority of the cellularity is composed of mesenchymal cells. Intermixed lymphocytes and plasma cells are also noted (hematoxylin–eosin stain; original magnification, ×200). E. High-magnification image shows columnar epithelium and apparent mesenchymal cuffing (hematoxylin–eosin stain; original magnification, ×600). F. A cyst within the medulla is lined by cuboidal to columnar epithelium (hematoxylin–eosin stain; original magnification, ×200).
We reviewed health records of mutation carriers for evidence of other organ dysfunction. At enrollment, none of the patients had evidence of extrarenal disease, but follow-up uncovered a new diagnosis of interstitial lung disease in 1 of the probands 3 years after enrollment (Table 3). The other proband had new evidence of macrocytic anemia, which in the presence of a PARN mutation prompted further investigation for possible incipient myelofibrosis. We also examined telomere length in PARN mutation carriers and, consistent with recent studies (29), did not detect differences compared with age-matched controls (Supplement Table 6).
hACMG Actionable Mutations
We searched for mutations in ACMG actionable genes and identified a BRCA2 nonsense mutation (c.T2151A:p.C717*) in a 68-year-old woman with fibrillary glomerulonephritis but no diagnostic mutation in kidney disease genes. Review of the medical records revealed that the patient had been diagnosed with breast cancer shortly after enrollment into our study. The BRCA2 variant was confirmed by Sanger sequencing in a clinical laboratory, and the patient and family were referred for additional cancer screening, genetic counseling, and cascade testing. Despite the lack of genetic explanation for the patient's nephropathy, the WES detection of a germline BRCA2 mutation informed the selection and intensity of immunosuppressive therapy for her renal disease. Moreover, the subsequent cascade testing of family members resulted in identification of 2 mutation carriers, who opted for prophylactic mastectomy.
Discussion
In this pilot study, we tested the utility of WES for diagnosis in adults with CKD. We identified many known and undiagnosed genetic disorders in 23% of a cohort of patients with familial or undiagnosed CKD. Diagnostic variants were detected across major clinical categories and among patients of both European and non-European ancestry. For most diagnosed cases, WES provided genetic information that subsequently affected clinical management and enabled family counseling (Table 2). Consistent with recent studies, we identified autosomal and X-linked forms of Alport syndrome among patients with a clinical diagnosis of focal segmental glomerulosclerosis, supporting the variable phenotypic expression of mutations in type IV collagen genes (41–43). These findings had immediate implications for surveillance for extrarenal complications, avoidance of immunosuppressive therapy in cases misdiagnosed as familial focal segmental glomerulosclerosis, and referral to a new clinical trial for Alport syndrome. Furthermore, we were able to obtain a genetic diagnosis in 9 probands with CKD of unknown cause, showing that WES has significant utility for diagnostic work-up in nephrology. In addition to detecting diagnostic variants in known nephropathy genes, WES identified PARN haploinsufficiency as a new genetic cause of CKD. PARN is required for maturation of the telomerase RNA component (38). Recessive mutations in PARN cause dyskeratosis congenita (44), a rare multisystem disorder presenting as severe bone marrow failure and abnormal cancer, skin, and mucosal pathology; many other organs, including the kidney, can also be affected (45). PARN haploinsufficiency produces variably penetrant pulmonary, bone marrow, and liver fibrosis in older adults, but the reason for interindividual differences in affected organs is unknown (28, 40, 46). The finding of 2 independent LoF mutations in 3 patients with nephropathy extends the phenotypes associated with PARN mutations and identifies renal tubulointerstitial fibrosis as another potential consequence of PARN haploinsufficiency, suggesting new avenues for investigating mechanisms of kidney injury. Of interest, the kidney biopsy revealed dysplastic features in the medulla, reminiscent of renal phenotypes associated with mutations in DNA repair genes in Fanconi anemia (47) and dyskeratosis congenita (44, 45). These findings further indicate that disorders of DNA maintenance may present predominantly as kidney dysfunction and fibrosis and have important implications for clinical care among PARN mutation carriers, including potential monitoring of kidney function and heightened awareness when dosing potentially nephrotoxic drugs.
Epidemiologic data suggest that hereditary or congenital disorders account for 10% of adult CKD (4–6), but the precise cause is frequently unknown. Recent investigations indicate that many late-onset constitutional disorders, such as amyotrophic lateral sclerosis (48) and pulmonary fibrosis (28, 40), also have a strong genetic basis that can be identified using these technologies. Our study similarly suggests that WES can provide a specific, molecular-level diagnosis, supporting its utility as part of the clinical diagnostic work-up. In comparison with other WES studies indexed in PubMed in the past 5 years, the diagnostic yield in our population was similar to those reported for pediatric developmental disorders (7–13). These findings motivate further examination of the utility of genomic technologies for diagnosis and stratification in the adult CKD population.
Our study has some limitations. The high diagnostic yield likely results from studying a cohort enriched for familial or suspected genetic forms of kidney disease, and the modest sample size limits generalizability to the broader CKD population. Hence, systematic WES analysis of larger, unselected CKD cohorts will provide a better assessment of its overall diagnostic yield in nephrology practice. Whole-exome sequencing also does not provide uniform coverage of all coding segments of the genome and may fail to capture some diagnostically relevant genomic regions. For example, because of inadequate capture of duplicated or repetitive segments in PKD1 and MUC1, WES has limited sensitivity for assessment of polycystic kidney disease and medullary cystic disease. Thus, the participants with autosomal dominant transmission of tubulointerstitial nephropathy and negative WES results in this study may have medullary cystic disease due to a MUC1 mutation. In addition, WES currently has limited ability to detect genomic imbalances and does not assess mutations in noncoding regions of the genome, leaving additional blind spots. Physician knowledge of these technical limitations will be important as WES is increasingly incorporated into clinical practice.
Constitutional genetic testing in adults is generally more complicated because family members are frequently not available to test inheritance of candidate variants (for example, to ascertain de novo status) and there is no pairwise comparison of diseased versus normal tissue to prioritize variants, unlike in cancer genomics. Other challenges include correct and consistent interpretation of genomic findings, integration of data into care in a clinically relevant time frame, development of test reports that are readily comprehensible to patients and providers to facilitate informed decisions, and demonstration that clinical sequencing is cost-effective and improves outcomes. Moreover, genome-wide tests like WES can discover actionable mutations unrelated to primary indications for testing, such as the BRCA2 mutation discovered in our study. The possibility of incidental findings leading to additional testing and therapy is a well-established challenge in clinical diagnostics, and the same principles of determining overall costs and benefits will need to be applied to genetic testing.
Large control databases, such as ExAC (34), are of great value for interpretation of WES data, helping prioritize predicted deleterious variants on the basis of their frequency in the population. New variant annotation algorithms that consider genomic context to assess mutation intolerance may also facilitate variant classification in adult singletons, independent of prior clinical reports (49–51), and help standardize clinical interpretation of genomes. Hence, many initiatives are currently examining the clinical relevance of genes and variants for use in genomic medicine (such as ClinGen, www.clinicalgenome.org) and will provide more evidence about the utility of genetic testing in diverse clinical settings (52, 53). Emerging data suggest that the introduction of genetic testing in the primary care setting does not improve or adversely affect standards of care and that some of the resulting increased health care use is clinically appropriate (54).
Altogether, WES offers the advantage of screening most relevant nephropathy genes at once, providing genetic diagnoses across diverse clinical categories, and enabling the identification of novel phenotypic extensions, as shown by the findings of PARN mutations in tubulointerstitial nephropathy. Because of its genome-wide approach, WES also enables periodic reevaluation of the sequence data for new genetic diagnoses as new disease genes are identified. In this study, WES had substantial diagnostic yield and affected clinical management in a referral population of adults with CKD, inviting more extensive investigation of the broader clinical utility of genetic testing for the CKD population.
Supplementary Material
Acknowledgments
The authors thank all study participants for contributing to this effort, David Goldstein and the Columbia Institute for Genomic Medicine for providing annotation tools and data from in-house controls, and Adam Platt for helpful comments.
Grant Support: By a New York State Empire Clinical Research Investigator Program grant, a research grant from the Renal Research Institute, and grant U01HG008680 from the National Human Genome Research Institute of the National Institutes of Health. Dr. Sanna-Cherchi is supported by grant 1R01DK103184 from the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health.
Dr. Newton reports grants from National Institutes of Health during the conduct of the study. Dr. Bomback reports grants from the National Institute on Minority Health and Health Disparities of the National Institutes of Health during the conduct of the study.
Footnotes
Disclosures: Authors not named here have disclosed no conflicts of interest. Disclosures can also be viewed at www.acponline.org/authors/icmje/ConflictOfInterestForms.do?msNum=M17-1309.
Reproducible Research Statement: Study protocol and data set: Available from Dr. Gharavi (ag2239@columbia.edu). Statistical code: Not applicable.
Contributor Information
Sneh Lata, Columbia University, New York, New York.
Maddalena Marasa, Columbia University, New York, New York.
Yifu Li, Columbia University, New York, New York.
David A. Fasel, Columbia University, New York, New York.
Emily Groopman, Columbia University, New York, New York.
Vaidehi Jobanputra, Columbia University, New York, New York.
Hila Rasouly, Columbia University, New York, New York.
Adele Mitrotti, Columbia University, New York, New York.
Rik Westland, Columbia University, New York, New York, and VU University Medical Center, Amsterdam, the Netherlands.
Miguel Verbitsky, Columbia University, New York, New York.
Jordan Nestor, Columbia University, New York, New York.
Lindsey M. Slater, Nephrology Associates, Newark, Delaware.
Vivette D’Agati, Columbia University, New York, New York.
Marcin Zaniew, Krysiewicza Children’s Hospital, Poznań, Poland.
Anna Materna-Kiryluk, Poznań University of Medical Sciences and Center for Medical Genetics GENESIS, Poznań, Poland.
Francesca Lugani, IRCCS Giannina Gaslini Children’s Hospital, Genova, Italy.
Gianluca Caridi, IRCCS Giannina Gaslini Children’s Hospital, Genova, Italy.
Luca Rampoldi, IRCCS San Raffaele Scientific Institute, Milan, Italy.
Aditya Mattoo, New York University School of Medicine, New York, New York.
Chad A. Newton, University of Texas Southwestern Medical Center, Dallas, Texas.
Maya K. Rao, Columbia University, New York, New York.
Jai Radhakrishan, Columbia University, New York, New York.
Wooin Ahn, Columbia University, New York, New York.
Pietro A. Canetta, Columbia University, New York, New York.
Andrew S. Bomback, Columbia University, New York, New York.
Gerald B. Appel, Columbia University, New York, New York.
Corinne Antignac, French Institute of Health and Medical Research (INSERM) U1163, Paris Descartes-Sorbonne Paris Cité University, Imagine Institute, and Necker Hospital, Paris, France.
Glen S. Markowitz, Columbia University, New York, New York.
Christine K. Garcia, University of Texas Southwestern Medical Center, Dallas, Texas.
Krzysztof Kiryluk, Columbia University, New York, New York.
Simone Sanna-Cherchi, Columbia University, New York, New York.
Ali G. Gharavi, Columbia University, New York, New York.
References
- 1.U.S. Renal Data System. USRDS 2016 Annual Data Report: Atlas of End-Stage Renal Disease in the United States. Bethesda: National Institutes of Health; 2016. [Google Scholar]
- 2.Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038–47. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- 3.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–305. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 4.Freedman BI, Wilson CH, Spray BJ, Tuttle AB, Olorenshaw IM, Kammer GM. Familial clustering of end-stage renal disease in blacks with lupus nephritis. Am J Kidney Dis. 1997;29:729–32. doi: 10.1016/s0272-6386(97)90126-8. [DOI] [PubMed] [Google Scholar]
- 5.Freedman BI, Soucie JM, Stone SM, Pegram S. Familial clustering of end-stage renal disease in blacks with HIV-associated nephropathy. Am J Kidney Dis. 1999;34:254–8. doi: 10.1016/s0272-6386(99)70352-5. [DOI] [PubMed] [Google Scholar]
- 6.Skrunes R, Svarstad E, Reisæter AV, Vikse BE. Familial clustering of ESRD in the Norwegian population. Clin J Am Soc Nephrol. 2014;9:1692–700. doi: 10.2215/CJN.01680214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiezun A, Garimella K, Do R, Stitziel NO, Neale BM, McLaren PJ, et al. Exome sequencing and the genetic basis of complex traits. Nat Genet. 2012;44:623–30. doi: 10.1038/ng.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–5. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–7. doi: 10.1038/nature13394. [DOI] [PubMed] [Google Scholar]
- 10.Sifrim A, Hitz MP, Wilsdon A, Breckpot J, Turki SH, Thienpont B, et al. INTERVAL Study. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet. 2016;48:1060–5. doi: 10.1038/ng.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Do R, Stitziel NO, Won HH, Jørgensen AB, Duga S, Angelica Merlini P, et al. NHLBI Exome Sequencing Project. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518:102–6. doi: 10.1038/nature13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312:1880–7. doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369:1502–11. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dixon-Salazar TJ, Silhavy JL, Udpa N, Schroth J, Bielas S, Schaffer AE, et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med. 2012;4:138ra78. doi: 10.1126/scitranslmed.3003544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–55. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 16.Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–53. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tarailo-Graovac M, Shyr C, Ross CJ, Horvath GA, Salvarinova R, Ye XC, et al. Exome sequencing and the management of neurometabolic disorders. N Engl J Med. 2016;374:2246–55. doi: 10.1056/NEJMoa1515792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verbitsky M, Sanna-Cherchi S, Fasel DA, Levy B, Kiryluk K, Wuttke M, et al. Genomic imbalances in pediatric patients with chronic kidney disease. J Clin Invest. 2015;125:2171–8. doi: 10.1172/JCI80877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, et al. SRNS Study Group. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015;26:1279–89. doi: 10.1681/ASN.2014050489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lovric S, Fang H, Vega-Warner V, Sadowski CE, Gee HY, Halbritter J, et al. Nephrotic Syndrome Study Group. Rapid detection of monogenic causes of childhood-onset steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2014;9:1109–16. doi: 10.2215/CJN.09010813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang DY, Dworschak GC, Kohl S, Saisawat P, Vivante A, Hilger AC, et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 2014;85:1429–33. doi: 10.1038/ki.2013.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldstein DB, Allen A, Keebler J, Margulies EH, Petrou S, Petrovski S, et al. Sequencing studies in human genetics: design and interpretation. Nat Rev Genet. 2013;14:460–70. doi: 10.1038/nrg3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katsanis SH, Katsanis N. Molecular genetic testing and the future of clinical genomics. Nat Rev Genet. 2013;14:415–26. doi: 10.1038/nrg3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bierzynska A, McCarthy HJ, Soderquest K, Sen ES, Colby E, Ding WY, et al. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. 2017;91:937–47. doi: 10.1016/j.kint.2016.10.013. [DOI] [PubMed] [Google Scholar]
- 25.Bekheirnia MR, Bekheirnia N, Bainbridge MN, Gu S, Coban Akdemir ZH, Gambin T, et al. Whole-exome sequencing in the molecular diagnosis of individuals with congenital anomalies of the kidney and urinary tract and identification of a new causative gene. Genet Med. 2017;19:412–20. doi: 10.1038/gim.2016.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–55. doi: 10.1038/gim.2016.190. [DOI] [PubMed] [Google Scholar]
- 27.Rossetti S, Hopp K, Sikkink RA, Sundsbak JL, Lee YK, Kubly V, et al. Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol. 2012;23:915–33. doi: 10.1681/ASN.2011101032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015;47:512–7. doi: 10.1038/ng.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newton CA, Batra K, Torrealba J, Kozlitina J, Glazer CS, Aravena C, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016;48:1710–20. doi: 10.1183/13993003.00308-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations [Letter] Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–8. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665–77. doi: 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moon DH, Segal M, Boyraz B, Guinan E, Hofmann I, Cahan P, et al. Poly(A)-specific ribonuclease (PARN) mediates 3'-end maturation of the telomerase RNA component. Nat Genet. 2015;47:1482–8. doi: 10.1038/ng.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kropski JA, Reiss S, Markin C, Brown KK, Schwartz DA, Schwarz MI, et al. Rare genetic variants in PARN are associated with pulmonary fibrosis in families. Am J Respir Crit Care Med. 2017 doi: 10.1164/rccm.201703-0635LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petrovski S, Todd JL, Durheim MT, Wang Q, Chien JW, Kelly FL, et al. An exome sequencing study to assess the role of rare genetic variation in pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196:82–93. doi: 10.1164/rccm.201610-2088OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malone AF, Phelan PJ, Hall G, Cetincelik U, Homstad A, Alonso AS, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014;86:1253–9. doi: 10.1038/ki.2014.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deltas C, Savva I, Voskarides K, Papazachariou L, Pierides A. Carriers of autosomal recessive Alport syndrome with thin basement membrane nephropathy presenting as focal segmental glomerulosclerosis in later life. Nephron. 2015;130:271–80. doi: 10.1159/000435789. [DOI] [PubMed] [Google Scholar]
- 43.Gast C, Pengelly RJ, Lyon M, Bunyan DJ, Seaby EG, Graham N, et al. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2016;31:961–70. doi: 10.1093/ndt/gfv325. [DOI] [PubMed] [Google Scholar]
- 44.Tummala H, Walne A, Collopy L, Cardoso S, de la Fuente J, Lawson S, et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest. 2015;125:2151–60. doi: 10.1172/JCI78963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Savage SA. Dyskeratosis congenita. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, et al., editors. GeneReviews. Seattle: Univ Washington; 1993. [PubMed] [Google Scholar]
- 46.Dhanraj S, Gunja SM, Deveau AP, Nissbeck M, Boonyawat B, Coombs AJ, et al. Bone marrow failure and developmental delay caused by mutations in poly(A)-specific ribonuclease (PARN) J Med Genet. 2015;52:738–48. doi: 10.1136/jmedgenet-2015-103292. [DOI] [PubMed] [Google Scholar]
- 47.Zhou W, Otto EA, Cluckey A, Airik R, Hurd TW, Chaki M, et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet. 2012;44:910–5. doi: 10.1038/ng.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. FALS Sequencing Consortium. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–41. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vassy JL, Christensen KD, Schonman EF, Blout CL, Robinson JO, Krier JB, et al. MedSeq Project. The impact of whole-genome sequencing on the primary care and outcomes of healthy adult patients: a pilot randomized trial. Ann Intern Med. 2017 doi: 10.7326/M17-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gussow AB, Petrovski S, Wang Q, Allen AS, Goldstein DB. The intolerance to functional genetic variation of protein domains predicts the localization of pathogenic mutations within genes. Genome Biol. 2016;17:9. doi: 10.1186/s13059-016-0869-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46:944–50. doi: 10.1038/ng.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bowdin S, Gilbert A, Bedoukian E, Carew C, Adam MP, Belmont J, et al. Recommendations for the integration of genomics into clinical practice. Genet Med. 2016;18:1075–84. doi: 10.1038/gim.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Green RC, Goddard KA, Jarvik GP, Amendola LM, Appelbaum PS, Berg JS, et al. CSER Consortium. Clinical sequencing Exploratory Research Consortium: accelerating evidence-based practice of genomic medicine. Am J Hum Genet. 2016;99:246. doi: 10.1016/j.ajhg.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.