

Abstract
Purpose of Review
The aim of this report is to review current literature regarding the work-up and management of congenital sensorineural hearing loss.
Recent Findings
Diagnostic evaluation of a newborn with sensorineural hearing loss begins with a complete audiologic evaluation and comprehensive history and physical exam. This review presents a diagnostic algorithm for the work-up of congenital hearing loss, focusing on the three following modalities: cytomegalovirus testing, genetic evaluation, and imaging.
Summary
Newborn hearing loss is a common problem and may be attributed to genetic and non-genetic factors. Complete diagnostic evaluation and treatment are essential for preventing delays in language development. Treatment consists of early intervention services and consideration of hearing aid amplification and cochlear implantation.
Keywords: Congenital hearing loss, newborn hearing screen, prelingual deafness, diagnosis, cytomegalovirus, cochlear implantation
1. Introduction
Hearing loss is the most common sensory disorder in newborns. It is a significant public health concern, as approximately 0.1–0.2% of infants are born deaf or hard-of-hearing (DHH)1. Neonatal hearing loss is attributed to a myriad of factors, including genetic causes, environmental exposures, and congenital infections, particularly cytomegalovirus. Identifying the underlying cause of hearing loss allows for targeted therapeutic decision making.
Unlike postlingual hearing loss, prelingual deafness delays development of auditory neural pathways, hindering normal speech and language acquisition. All DHH children benefit from early identification and subsequent timely intervention with hearing aids and cochlear implants, to prevent delays in speech and language production, and promote improved cognitive growth and developmental outcomes later in childhood.2,3
1.1. Hearing Screening and Audiologic Testing
In the United States, neonatal hearing screening is mandated by law. Guidelines outlined in the Joint Committee on Infant Hearing (JCIH) position statement published in 2007 emphasize early hearing detection and intervention (EHDI) through screening for all newborn infants within one month of age. A failed hearing screening warrants additional testing and audiologic diagnostic evaluation prior to 3 months of age with the goal of enrollment in early intervention services by 6 months of age.4 Additionally, the JCIH outlined a list of risk factors associated with delayed-onset or progressive hearing loss for which children are advised to undergo complete audiologic evaluation, regardless of the newborn hearing screening result.4
Newborn hearing screening consists primarily of two diagnostic examinations: (1) otoacoustic emissions (OAE) and (2) automated auditory brainstem response (aABR). Both OAE and aABR testing provide non-invasive recordings of physiologic activity underlying normal auditory function.
An OAE screening test is performed by transmitting a burst of sound to the inner ear, which causes the outer hair cells of the cochlea to emit soft sounds called OAEs in response to stimulus. OAE production decreases with increasing degrees of hearing loss; typically, OAE screening identifies hearing loss of 30–40 dB or more. Thus, OAEs can successfully identify middle ear pathology or moderate to severe sensorineural hearing loss. Since OAE testing requires no interactive feedback, it can be performed easily on newborns.
In aABR testing, the electrophysiological response of the auditory nerve and brainstem to repeated sound stimuli is recorded through surface electrodes. Unlike OAE screening, which identifies only cochlear and middle-ear dysfunction, aABR provides an assessment of auditory nerve transmission and thus is an effective screen for auditory neuropathy spectrum disorder (ANSD). Thus, for infants at risk for ANSD, including all babies in neonatal intensive care unit settings, aABR testing is strongly recommended.5
Infants who fail newborn hearing screening should undergo natural-sleep diagnostic ABR testing by 3 months of age. ABR testing is sufficient to recommend medical and audiologic treatment and early intervention services, although behavioral audiometric testing with visual reinforcement audiometry, which is typically possible beginning around 6 months of age, is the gold standard for definitive diagnosis. Though the exact mechanism by which early intervention services are administered is state-specific, all DHH children are entitled to government-provided services through IDEA Part C, with children 0–3 covered under an Individual Family Service Plan (IFSP) and children over 3 under an Individualized Educational Plan (IEP) or 504 Plan.6
While the implementation of newborn hearing screening has dramatically reduced the number of undiagnosed pediatric hearing loss, nearly 50% of newborns who fail hearing screens do not receive adequate surveillance or audiologic treatment.7 Delays in follow-up and intervention may be due in part to inadequate access to care, educational barriers, and poor understanding of the work-up of congenital hearing loss.
2. Medical Evaluation
2.1. Relevance of Diagnostic Testing
Once hearing loss is identified, a child is typically referred for medical evaluation. Medical diagnostic testing of hearing loss has multiple goals for children and their families. Identifying certain etiologies may affect management options and comprehensive medical evaluation may lead to the diagnosis of comorbid conditions that may warrant additional testing or work-up. Thorough diagnostic evaluation also provides information about the prognosis of hearing loss; common findings such as EVA and cCMV infection are associated with increased likelihood of progression of hearing loss. Identification of a heritable cause of hearing loss can have implications on family planning for the parents as well as, ultimately, the child. Finally, for many patients and their families, simply knowing the etiology of hearing loss provides psychological and emotional comfort. Despite its numerous benefits, diagnostic testing carries cost and some risk, and is not the choice of all patients. Ultimately, families and their providers share in decisions regarding diagnostic testing.
2.2. History and Physical Exam
Approximately 50% of all cases of congenital hearing loss are attributed to non-genetic causes, including a broad range of etiologies, such as prematurity, exposure to intrauterine infections (i.e. cytomegalovirus, rubella, toxoplasmosis, and syphilis), exposure to ototoxic medications (especially aminoglycoside antibiotics), trauma, and jaundice. Careful questioning about prenatal, perinatal, and postnatal history as well as a comprehensive review of systems may identify risk factors for hearing loss.
Of the genetic causes for neonatal hearing loss, roughly 70% are nonsyndromic, while the remaining 30% comprise syndromic causes of congenital hearing loss. In syndromic cases, patients typically present with other distinctive clinical features in addition to hearing loss. A thorough history and physical examination may reveal craniofacial and other systemic anomalies such as thyroid goiter (Pendred syndrome), pigmentation differences (Waardenburg syndrome), and branchial cleft anomalies (branchio-oto-renal syndrome) that lead to a diagnosis of an underlying syndromic etiology. Certain syndromes may portend worse prognoses or require additional treatment for potentially life-threatening abnormalities. For example, patients with Jervell and Lange-Nielsen syndrome have an increased incidence of syncope and sudden death due to a mutation in the potassium channel gene that leads to conduction abnormalities in the heart in addition to profound sensorineural hearing loss.8
Craniofacial anomalies and other phenotypic dysmorphisms are more often associated with autosomal dominant forms of syndromic hearing loss (i.e. Waardenburg, Stickler, and Treacher-Collins syndromes) than with autosomal recessive forms (i.e. Jervell and Lange-Nielsen, Pendred, and Usher syndromes). Genetic testing may be required in the latter group in order to establish a diagnosis. Over 400 syndromic forms of hearing loss have been identified; the most common syndromes are described in Table 1.
Table 1.
Syndromic causes of hearing loss
| Syndrome | Inheritance Pattern | Gene(s) | Clinical Features | Imaging Findings |
|---|---|---|---|---|
| Waardenburg | AD | PAX3, MITF, EDNRB, EDN3, SOX10 | Pigmentary abnormalities of skin, hair, and eyes (heterochromiairides), telecanthus | EVA |
| Branchiootorenal | AD | EYA1, S1X5 | Branchial fistulas, ear pits, renal defects | Cochlear dysplasia |
| CHARGE | AD | CDH7, SEMA3E | Heart defects, choanal atresia, colobomas, genital defects, growth delay | Cochlear and SCC dysplasia, auditory neuropathy |
| Neurofibromatosis | AD | NF2 | Variable; ophthalmic features and/ or skin lesions | Bilateral vestibular schwannoma |
| Stickler | AD/ AR | COL2A1, COL11A1, COL11A2, COL9A1, COL9A2 | Cleft palate, midfacial underdevelopment, spondyloepiphyseal dysplasia, ophthalmic features | None |
| Usher | AR | MYO7A, USH1B, CDH23, USH1C, GPR98, WHRN, USH2A, USH2C, CLRN1, HARS, CDH7, SEMA3E | Vestibular changes, retinitis pigmentosa | N/A |
| Pendred | AR | SLC26A4, FOX11 | Euthyroid goiter | EVA, cochlear dysplasia |
| Jervell and Lange- Nielsen | AR | KCNQ1, KCNE1 | Prolonged QT | None |
| Alport | AD/ AR/ X-linked | COL4A3, COL4A4, COL4A5 | Glomerulonephritis, cataracts | None |
2.3. Cytomegalovirus Testing
Congenital cytomegalovirus (CMV) is the leading non-genetic cause of sensorineural hearing loss. The incidence of hearing loss in infants born with congenital CMV is estimated to be 12.6%; conversely, 10–25% of pediatric hearing loss is estimated to be caused by cCMV.9,9 Congenital CMV (cCMV) infection demonstrates a wide range of phenotypes, with the majority of infected infants (85–90%) asymptomatic at birth. Progression and fluctuation of cCMV-associated hearing loss is quite common. In symptomatic infants, the clinical spectrum is variable, ranging from a mild sensorineural hearing loss to severe neurodevelopmental delay and potentially life-threatening disseminated disease.10
The importance of identifying cCMV as the cause of congenital or childhood-onset sensorineural hearing loss lies in the significant prognostic and, potentially, treatment options available given this identification. Among a cohort of children with otherwise asymptomatic cCMV infection followed through 18 years of life, 65% of those with sensorineural hearing loss demonstrated progression, and new-onset sensorineural hearing loss was identified into the late teenage years.11 Particularly significantly, 89% of children with unilateral congenital SNHL progressed to profound on that side, and 75% developed contralateral hearing loss. This prognostic information is highly important, particularly when considering cochlear implantation.
Historically, diagnosis of CMV infections was confirmed by viral culture of urine or saliva specimens. However, culture-based methods were often cumbersome and expensive. More recently, real-time PCR analyses for CMV have shown promising results, with assays for CMV in liquid-saliva samples having a sensitivity of 100% and a specificity of 99.9%.12
One of the major disadvantages of culture-based and real-time PCR methods is that both require testing before the child is 3 weeks of age. In children diagnosed after the first 3 weeks of life, direct testing of the infant has low specificity for congenital CMV infection owing to the high incidence of postnatal acquisition of CMV, which is not associated with hearing loss.13 Therefore, in children over 3 weeks of age, testing of dried blood spots (DBSs), which are obtained in most infants born in the United States within the first week of life as part of the Newborn Screening process, can be used to perform a retrospective CMV DNA analysis. DBS PCR testing has been shown to have a sensitivity of 34.4% and a specificity of 99.9%, making DBS CMV PCR testing unsuitable for use in large screening cohorts of neonates.14 Despite its poor performance as a screening tool, the high specificity of DBS PCR for detecting cCMV infection indicates high reliability, and this testing method remains the only way to definitively identify congenital CMV as the cause of hearing loss in children over 3 weeks of age.
Diagnosis of cCMV-associated hearing loss has implications not only in the use of hearing aids and cochlear implantation, but also in treatment with antiviral drugs. Kimberlin et al. (2003) showed prevention of hearing loss progression over 6 months when IV ganciclovir was administered for 6 weeks to infants with symptomatic CMV disease involving the central nervous system.15 Of note, the primary toxicity of ganciclovir is neutropenia, with 63% of infants developing neutropenic symptoms. A follow-up study evaluated treatment of symptomatic CMV with an orally administered prodrug valganciclovir, which has a significantly more benign risk profile, with only a 27% rate low-grade neutropenia, similar to that seen for CMV-infected infants receiving placebo. Compared to a 6-week course, when administered over 6 months, valganciclovir produced an improvement in audiologic outcomes in the long term at 12 and 24 months.16 As of this review, antiviral treatment with ganciclovir or valganciclovir is recommended only for symptomatic newborns with severe systemic disease or central nervous system involvement, though clinical trials are underway to examine the role of valganciclovir in otherwise asymptomatic children up to 4 years of age with congenital CMV-associated hearing loss.17
2.4. Genetic Testing
Nonsyndromic causes of hearing loss are subdivided into autosomal recessive (77%), autosomal dominant (22%), and X-linked or mitochondrial inheritance patterns (1%).18 Autosomal recessive nonsyndromic hearing loss tends to result in bilateral, prelingual hearing loss, while autosomal dominant nonsyndromic hearing loss is often progressive in nature and develops postlingually.19,20
Comprehensive genetic testing is an important part of the diagnostic algorithm of congenital hearing loss. New advances in high-throughput sequencing, such as next-generation sequencing (NGS) and massively parallel sequencing (MPS), allow sequencing of multiple genes simultaneously. At the time of this publication, over 150 genes known to cause nonsyndromic hearing loss and certain types of syndromic hearing loss that may masquerade as nonsyndromic hearing loss in early childhood, such as Pendred and Usher syndromes, are typically included in hearing-loss gene panels, and comprehensive testing is rapidly evolving into the standard of care for genetic testing in hearing loss.21,22
Although hundreds of genes have been identified to be associated with nonsyndromic forms of hearing loss, the more common genetic mutations are described below. Overall, genes associated with autosomal recessive nonsyndromic hearing loss account for the majority of cases.23
GJB2
The gene most frequently associated with congenital hearing loss is GJB2, which encodes Connexin 26, the major component of gap junctions in the cochlea. Mutations in the GJB2 gene account for close to 50% of patients with nonsyndromic deafness. A systematic review demonstrated that the worldwide prevalence of biallelic GJB2-associated hearing loss was 17.3%.24 Specific mutant alleles have increased prevalence in different regions of the world; for example, 35delG is prevalent in Europe and the Middle East, 235delC in Asia, V37I in Southeast Asia and W24X in India.23 A deletion in the GJB6 gene encoding connexin 30 can also cause sensorineural hearing loss by reducing the expression of Connexin 26; this phenotypic expression may occur in isolation or in combination with GJB2 mutations.
SLC26A4
Mutations in SLC26A4 are the second most frequent cause of nonsyndromic congenital hearing loss. SLC26A4 encodes a chloride and iodide transporter and may present in either a syndromic or nonsyndromic form of hearing loss. Pendred syndrome is characterized by presence of a thyroid goiter, congenital progressive hearing loss and either EVA or Mondini dysplasia. Irrespective of Pendred syndrome, mutations in SLC26A4 are also associated with findings of EVA. The presence of EVA is not always readily apparent in the neonate; often, diagnosis is made when an older child presents with sudden and profound hearing loss following minor head trauma.25
OTOF
Mutations in OTOF encodes otoferlin, a protein involved in vesicle membrane fusion, are associated with auditory neuropathy. Given the high association of OTOF mutations with auditory neuropathy, OTOF genetic testing should be performed when OAE responses are present in the absence of ABR responses.
CDH23
The gene CDH23 encodes for cadherin-23. Mutations in this gene are associated with Usher syndrome. Usher syndrome is the most common cause of deaf-blindness and is characterized by sensorineural hearing loss and retinitis pigmentosa with variable effects on the vestibular system. Usher syndrome has three distinct subtypes, distinguishable on the basis of severity or progression of hearing loss and extent of vestibular involvement.
STRC
STRC encodes stereocilin, a major protein component of the mechanotransducing stereocilia that sit atop hair cells. Copy-number variation of the STRC gene has been found to account for a significant fraction of pediatric hearing loss, and may escape detection in some next-generation sequencing protocols. Testing on a platform that explicitly assesses CNV is recommended to ensure capture of variants in genes as STRC.26,27
2.5 Ancillary Testing
Routine laboratory testing has low diagnostic yield in the workup of childhood SNHL, and is typically not ordered unless there are concerning clinical findings or the results of genetic testing raise concerns about another organ system.28 Ophthalmology evaluation is often recommended for neonates with newly diagnosed hearing loss both to assess for syndromes that may result in concurrent visual and hearing impairment, such as Usher syndrome, but also because all children with hearing loss will be more dependent on their vision for speech and language development. Routine cardiac and renal evaluation is not recommended, with the following exceptions: 1) An electrocardiogram should be performed in infants with bilateral severe or profound sensorineural hearing loss and family history of childhood sudden death or personal history of cardiac arrhythmia to evaluate for Jervell-Lange and Nielsen syndrome; 2) A renal ultrasound should be performed in infants with sensorineural hearing loss and concurrent ear pits or tags or branchial cleft anomalies to evaluate for presence branchio-oto-renal syndrome. Though historically perchlorate discharge testing was recommended for workup of thyroid dysfunction in Pendred syndrome, genetic testing has largely supplanted it.
3. Imaging
One of the common questions that arises during work-up of congenital sensorineural hearing loss is whether and when imaging studies should be performed. Other than for obtaining knowledge of the etiology for its own sake, imaging is performed in the evaluation of congenital sensorineural hearing loss for two primary management-related reasons: 1) to establish cochlear implant candidacy; and 2) to identify an anatomic cause for hearing loss for prognostic purposes. For example, a finding of an EVA has prognostic implications about the expected progression of hearing loss. Historically, head trauma has been thought to be associated with progression of hearing loss in patients with EVA, which has often led to specific recommendations regarding head trauma avoidance in these patients. A recent meta-analysis, however, suggests that this association is not strongly supported.29 In general, imaging is recommended for unilateral sensorineural hearing loss and as an adjunct assessment in bilateral sensorineural hearing loss when genetic testing has been negative.30
Previously, high-resolution computed tomography (CT) of temporal bones was the initial imaging study of choice. However, the increased awareness of risk of radiation exposure associated with CT scans has contributed to the rise in popularity of magnetic resonance imaging (MRI) scans.31 MRI has long been recommended as the initial diagnostic study for patients with suspected ANSD and is thought to provide improved resolution of intracranial abnormalities, cochlear nerve, and retrocochlear pathology. Now, there is increasing evidence to suggest that MRI provides sufficient anatomical information to guide treatment during work-up of congenital hearing loss. In particular, the 3-D FIESTA (fast imaging enabling steady-state acquisition) sequence thin-cut MRI performs as well as CT scan in the evaluation of congenital temporal bone anomalies, particularly in investigation of abnormalities of the membranous and osseous labyrinth (cochlear aplasia, common cavity deformity, incomplete partition), but also in identification of enlarged vestibular aqueduct.32
A frequently cited limitation of the use of MRI in the pediatric population is the need for sedation in infants given the increased duration of the MRI scan compared to CT. Several studies have found that a single anesthetic exposure before the age of 3 may increase the risk of long-term neurocognitive delay and that multiple exposures increase the risk for development of certain behavioral disorders, such as attention-deficit hyperactivity disorder.33,34,35 A recently developed rapid MRI protocol may allow infants to undergo an MRI scan of the temporal bone without sedation. In this protocol, only the high-resolution T2-weighted fast spin-echo sequence was used; the study could be performed in under 10 minutes, thus potentially avoiding the need for sedation in infants.36
4. Basic Algorithm for Evaluation of Congenital Sensorineural Hearing Loss
Early diagnostic evaluation of sensorineural hearing loss promotes early intervention, minimizing the potentially detrimental effects of hearing impairment on the cognitive, social, and emotional development during childhood. Here we present a simple algorithm for the diagnostic workup of non-syndromic SNHL in children (Figure 1).
Fig. 1.
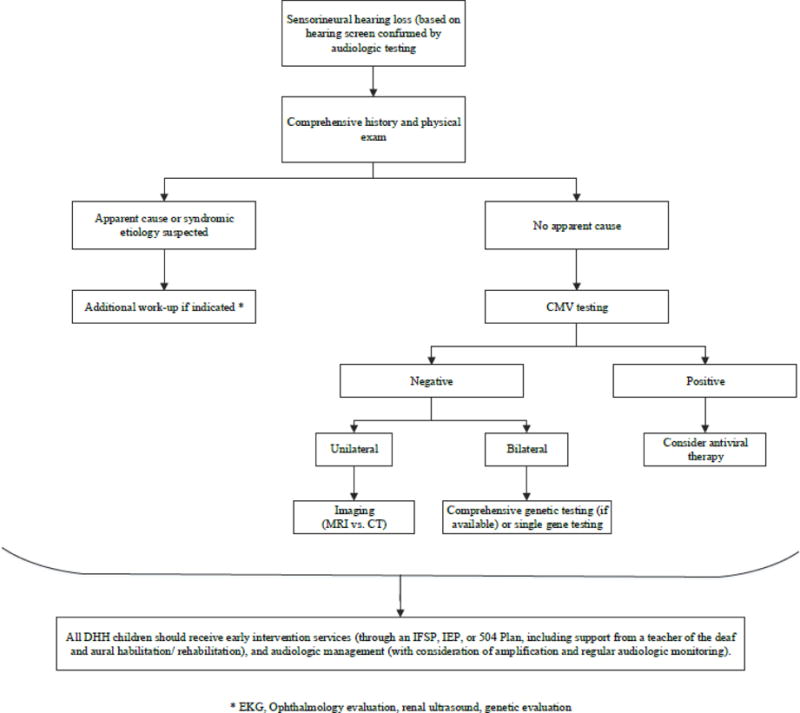
Basic diagnostic algorithm for congenital hearing loss
Cytomegalovirus (CMV)
All children without an obvious syndromic cause for hearing loss may begin work-up for congenital sensorineural hearing loss with CMV testing, if possible. Although not definitive for diagnosis of CMV, certain imaging characteristics are suggestive of CMV, including intracranial calcifications, periventricular cysts, and microcephaly.37
If the infant is under 3 weeks of age, he or she may undergo urine or saliva real-time PCR assays. Given the high specificity and sensitivity of this test, a positive finding confirms the presence of CMV.14 Children who are older than 3 weeks of age may undergo DBS testing. However, each state stores the residual samples of DBS for variable lengths of time; depending on the age of the child, DBS testing may not be a viable option.
If a diagnosis of CMV is confirmed through testing, treatment with antiviral medication may be considered. Although there are no current recommendations for initiation of antiviral therapy in asymptomatic children, clinical trials are in progress.18 Additionally, patients with unilateral hearing loss caused by CMV may undergo earlier consideration of cochlear implantation due to the high risk of progression to contralateral hearing loss. An informed discussion with the patient’s family should guide these management decisions.
Genetic Testing
Patients with bilateral sensorineural hearing loss in whom CMV testing has proven negative or inclusive may undergo genetic testing. Genetic testing for mutations in GBJ2, the gene most commonly affected in congenital hearing loss, has a diagnostic yield of 22%, whereas comprehensive gene testing has a diagnostic yield of 58%.38,39 Decision regarding which, if any, initial genetic test to undergo will depend on individual factors, including cost, family history, ethnic background, and family-planning priorities.
Imaging
Patients with unilateral sensorineural hearing loss with negative CMV testing may derive the greatest benefit from imaging studies. Systematic reviews concluded that the diagnostic yield of imaging studies is approximately 30% for CT and 26% for MRI, respectively.40,41 Imaging studies provide important information about anatomic variation in patients and can guide surgical planning for cochlear implantation; however, decision regarding timing of imaging must take into account risks attributable to either radiation exposure or sedation. If no significant management alterations would derive from imaging findings, one may consider deferring imaging until the child is old enough to undergo MRI without sedation, unless a non-sedated MRI protocol is available.
5. Conclusion
Early diagnosis and work-up of congenital hearing loss encourages prompt intervention with aural habilitation/rehabilitation services, hearing amplification, and, occasionally, medical and surgical treatment. It is well-established that early exposure to sound results in improved linguistic and social skills. A multi-disciplinary team approach that includes otolaryngologists, audiologists, geneticists, speech and language therapists, early educators, and family support is most beneficial for the ongoing evaluation and management of deaf and hard-of-hearing children.
Footnotes
Relevant financial disclosures: None.
Conflict of Interest: None.
Conflict of Interest
Dr. Chan and Dr. Chari have nothing to declare.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Mehra S, Eavey RD, Keamy DG. The epidemiology of hearing impairment in the United States: newborns, children, and adolescents. Otolaryngol-Head Neck Surg. 2009;140(4):461–72. doi: 10.1016/j.otohns.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy CR, McCann DC, Campbell MJ. Language ability after early detection of permanent childhood hearing impairment. N Engl J Med. 2006;354(20):2131–2141. doi: 10.1056/NEJMoa054915. [DOI] [PubMed] [Google Scholar]
- 3.Moeller MP. Early intervention and language development in children who are deaf and hard of hearing. Pediatrics. 2000;106(3):1–9. doi: 10.1542/peds.106.3.e43. [DOI] [PubMed] [Google Scholar]
- 4.Joint Committee on Infant Hearing. Year 2007 position statement: principles and guidelines for early hearing detection and intervention programs. Pediatrics. 2007;120(4):898–921. doi: 10.1542/peds.2007-2333. [DOI] [PubMed] [Google Scholar]
- 5.Balkany TJ, Berman SA, Simmons MA, Jafek BW. Middle ear effusions in neonates. Laryngoscope. 1978;88(3):398–405. doi: 10.1288/00005537-197803000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Listening and Spoken Language Knowledge Center. IDEA Part C. AG Bell Academy; Jul, 2017. Web. [Google Scholar]
- 7.Centers for Disease Control and Prevention. Early hearing detection and intervention among infants – hearing screening and follow-up survey, United States, 2005–2006 and 2009–2010. MMWR Surveill Summ. 2014;63(3):20–26. [PubMed] [Google Scholar]
- 8.Koffler T, Ushakov K, Avraham KB. Genetics of hearing loss: syndromic. Otolaryngol Clin North Am. 2015;48:1041–1061. doi: 10.1016/j.otc.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamaguchi A, Oh-ishi T, Arai T, et al. Screening for seemingly healthy newborns with congenital cytomegalovirus infection by quantitative real-time polymerase chain reaction using newborn urine: an observational study. BMJ Open. 2017;7:e013810. doi: 10.1136/bmjopen-2016-013810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dollard SC, Grosse SD, Ross DS. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol. 2007;17(5):355–363. doi: 10.1002/rmv.544. [DOI] [PubMed] [Google Scholar]
- 11.Lanzieri TM, Chung W, Flores M, et al. Hearing loss in children with asymptomatic congenital cytomegalovirus infection. Pediatrics. 2017;139(3):16. doi: 10.1542/peds.2016-2610. Epub 2017 Feb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boppana SB, Ross SA, Shimamura M, et al. Saliva polymerase-chain-reaction assays for cytomegalovirus screening in newborns. NEJM. 2011;364(22):2111–18. doi: 10.1056/NEJMoa1006561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peckham CS, Johnson C, Ades A, et al. Early acquisition of cytomegalovirus infection. Arch Dis Child. 1987;62(8):780–5. doi: 10.1136/adc.62.8.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boppana SB, Ross SA, Novak Z, et al. Dried blood spot real-time polymerase chain reaction assays to screen newborns for congenital cytomegalovirus infection. 2010;202(14):1375–82. doi: 10.1001/jama.2010.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimberlin DW, Lin CY, Sanchez PJ, et al. Effect of ganciclovir therapy on hearing in symptomatic congenital cytomegalovirus disease involving the central nervous system: A randomized controlled trial. J Pediatrics. 2003;134(1):16–25. doi: 10.1016/s0022-3476(03)00192-6. [DOI] [PubMed] [Google Scholar]
- 16••.Kimberlin DW, Jester PM, Sanchez PJ, et al. Valganciclovir for symptomatic congenital cytomegalovirus disease. NEJM. 2015;372(1):933–943. doi: 10.1056/NEJMoa1404599. The first randomized, placebo-controlled trial of valganciclovir treatment in infants with symptomatic congenital CMV disease assessing hearing outcomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimberlin D. Congenital CMV and hearing loss in children up to 4 years of age: Treating with valganciclovir therapy. ClinicalTrials.gov. 2017 May; Web July 2017. https://clinicaltrials.gov/ct2/show/NCT01649869.
- 18.ACMG Consensus Statement. Genetics evaluation guidelines for the etiologic diagnosis of congenital hearing loss. Genet Med. 2002;4(3):162–171. doi: 10.1097/00125817-200205000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu XZ, Xu LR, Zhang SL, et al. Epidemiological and genetic studies of congenital profound deafness. Am J Med Genet. 1994;53:192–5. doi: 10.1002/ajmg.1320530214. [DOI] [PubMed] [Google Scholar]
- 20.Chang KW. Genetics of hearing loss – nonsyndromic. Otolaryngol Clin North Am. 2015;48(6):1063–72. doi: 10.1016/j.otc.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 21.The University of Iowa- Molecular Otolaryngology and Renal Research Laboratories. OtoSCOPE Genetic Testing. Web July 2017 https://morl.lab.uiowa.edu/otoscope-genetic-testing.
- 22•.Shearer AE, Smith RJ. Massively parallel sequencing for genetic diagnosis of hearing loss: The new standard of care. Otolaryngol Head Neck Surg. 2015;153(2):175–82. doi: 10.1177/0194599815591156. Review article evaluating the use of new genetic sequencing techniques. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hilgert N, Smith RJ, Camp GV. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutation Research. 2009;681:189–196. doi: 10.1016/j.mrrev.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan DK, Chang KW. GJB2-associated hearing loss: systematic review of worldwide prevalence, genotype and auditory phenotype. Laryngoscope. 2014;124(2):E34–53. doi: 10.1002/lary.24332. [DOI] [PubMed] [Google Scholar]
- 25.Colvin IB, Beale T, Harrop-Griffiths Long-term follow-up of hearing loss in children and young adults with enlarged vestibular aqueducts: relationship to radiologic findings and Pendred syndrome diagnosis. Laryngoscope. 2006;116:2027–2036. doi: 10.1097/01.mlg.0000240908.88759.fe. [DOI] [PubMed] [Google Scholar]
- 26.Shearer AE, Kolbe DL, Azaiez H, et al. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 2014;6(5):37. doi: 10.1186/gm554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Francey LJ, Conlin LK, Kadesch HE, et al. Genome-wide SNP genotyping identifies the stereocilin (STRC) gene as a major contributor to pediatric bilateral sensorineural hearing impairment. Am J Med Genet A. 2012;158A(2):298–308. doi: 10.1002/ajmg.a.34391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mafong DD, Shin EJ, Lalwani AK. Use of laboratory evaluation and radiologic imaging in diagnostic evaluation of children with sensorineural hearing loss. Laryngoscope. 2002;112(1):1–7. doi: 10.1097/00005537-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 29.Alemi AS, Chan DK. Progressive hearing loss and head trauma in enlarged vestibular aqueduct: A systematic review and meta-analysis. Otolaryngol Head Neck Surg. 2015;153(4):512–7. doi: 10.1177/0194599815596343. [DOI] [PubMed] [Google Scholar]
- 30.Preciado DA, Lim LHY, Cohen AP, et al. A diagnostic paradigm for childhood idiopathic sensorineural hearing loss. Otolaryngol-Head Neck Surg. 2004;131(6):804–9. doi: 10.1016/j.otohns.2004.06.707. [DOI] [PubMed] [Google Scholar]
- 31.Miglioretti DL, Johnson E, Williams A, et al. The use of computed tomography in pediatrics and the associated radiation exposure and estimated cancer risk. JAMA Pediatr. 2013;167(8):700–7. doi: 10.1001/jamapediatrics.2013.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta SS, Maheshwari SR, Kirtane MV, Shrivastav N. Pictorial review of MRI/ CT scan in congenital temporal bone anomalies, in patients for cochlear implant. Indian J Radiol Imaging. 2009;19(2):99–106. doi: 10.4103/0971-3026.50825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sprung J, Flick RP, Katusic SK, et al. Attention-deficit/ hyperactivity disorder after early exposure to procedures requiring general anesthesia. Mayo Clin Proc. 2012;87:120–129. doi: 10.1016/j.mayocp.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flick RP, Katusic SK, Colligan RC, et al. Cognitive and behavioral outcomes after early exposure to anesthesia and surgery. Pediatrics. 2011:1281053–1061. doi: 10.1542/peds.2011-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ing C, DiMaggio CJ, Whitehouse A, et al. Long-term differences in language and cognitive function after childhood exposure to anesthesia. Pediatrics. 2012;130:476–485. doi: 10.1542/peds.2011-3822. [DOI] [PubMed] [Google Scholar]
- 36.Licameli G, Kenna MA. Is computed tomography (CT) or magnetic resonance imaging (MRI) more useful in the evaluation of pediatric sensorineural hearing loss. Laryngoscope. 2010;120:2358–2359. doi: 10.1002/lary.21363. [DOI] [PubMed] [Google Scholar]
- 37.Bale JF., Jr Cytomegalovirus infections. Semin Pediatr Neurol. 2012;19:101–106. doi: 10.1016/j.spen.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 38.Zazo Seco C, Wesdorp M, Feenstra I, et al. The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in The Netherlands. Eur J Hum Genet. 2017;25(3):308–314. doi: 10.1038/ejhg.2016.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sloan-Heggen CM, Bierer AO, Shearer AE, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135:441–450. doi: 10.1007/s00439-016-1648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen JX, Kachniarz B, Chin JJ. Diagnostic yield of computed tomography scan for pediatric hearing loss: a systematic review. Otolaryngol-Head Neck Surg. 2014;151(5):718–39. doi: 10.1177/0194599814545727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41••.Kachniarz B, Chen JX, Gilani S, Shin JJ. Diagnostic yield of MRI for pediatric hearing loss: a systematic review. Otolaryngol-Head Neck Surg. 2015;152(1):5–22. doi: 10.1177/0194599814555837. High quality systematic review and meta-analysis evaluating the diagnostic yield of MRI relative to CT for pediatric hearing loss with a subgroup analysis according to hearing loss severity and diagnostic imaging findings. [DOI] [PMC free article] [PubMed] [Google Scholar]